

ABSTRACT
β-site APP-cleaving enzyme 1 (BACE1) has become infamous for its pivotal role in the pathogenesis of Alzheimer's disease (AD). Consequently, BACE1 represents a prime target in drug development. Despite its detrimental involvement in AD, it should be quite obvious that BACE1 is not primarily present in the brain to drive mental decline. In fact, additional functions have been identified. In this review, we focus on the regulation of ion channels, specifically voltage-gated sodium and KCNQ potassium channels, by BACE1. These studies provide evidence for a highly unexpected feature in the functional repertoire of BACE1. Although capable of cleaving accessory channel subunits, BACE1 exerts many of its physiologically significant effects through direct, non-enzymatic interactions with main channel subunits. We discuss how the underlying mechanisms can be conceived and develop scenarios how the regulation of ion conductances by BACE1 might shape electric activity in the intact and diseased brain and heart.
KEYWORDS: ataxia, BACE1, β-subunit, direct interaction, epilepsy, hippocampus, IKs, KCNQ channels, NaV channels, resurgent current
BACE1 – a major culprit in Alzheimer's disease
Alzheimer's disease (AD)
Alzheimer's disease (AD) is a devastating neurodegenerative illness with increasing socioeconomic relevance.1 Despite some favorable trends,2 prevalence might triple by 2050 in developed countries like the US, which will demand vast effort of the health care systems and caregivers. As first reported in the beginning of the 20th century by Alois Alzheimer, symptoms of AD include cognitive decline, in particular memory deficits, spatial disorientation, aphasia, apraxia, and affective disturbances.3 Histopathological hallmarks in the brains of AD patients are intracellular neurofibrillary tangles, which are composed of hyperphosphorylated tau protein, and extracellular amyloid deposits.3,4 The latter mainly consist of aggregated Aβ peptides, which are generated from the amyloid precursor protein (APP) in a proteolytic cascade.5 Once considered the evil moiety, amyloid plaques are now rather seen as inert deposits. Instead, oligomeric assemblies of Aβ have been identified as the neurotoxic substrate.6
Role of BACE1 in AD
In 1999, BACE1 (also: β-secretase 1 or memapsin 2) was identified as a β-site APP-cleaving enzyme,7-11 and, 2 years later, confirmed as the major β-secretase in vivo.12 BACE1 initiates the amyloidogenic pathway of APP processing. Sequential cleavage of APP by BACE1 and the γ-secretase complex creates Aβ moieties, which are prone to aggregation.5 Competitively, α-secretase cleaves APP instead of BACE1 and directs proteolysis toward the non-amyloidogenic pathway with shorter metabolites. Interdependence with further APP-cleaving secretases, i.e. δ-secretase and η-secretase, is just beginning to emerge.13,14 In healthy brain, non-amyloidogenic and amyloidogenic processing are balanced, with Aβ peptides being important effectors in synaptic transmission and plasticity.15,16 In 2003, Yang et al. reported enhanced BACE1-mediated amyloidogenic cleavage in AD.17 Recent evidence suggests that BACE1 activity can be elevated much earlier and may foster the conversion from mild cognitive impairment to AD.18,19 Recent support for the amyloid hypothesis of AD came from a study which recapitulated the disease in a 3D human neuronal cell culture model, demonstrating that the appearance of prominent neurofibrillary tangles requires Aβ generation,20 a causality that is still controversial.21
While APP triplication in trisomy 21 and mutations in APP and PSEN1 or PSEN2 of the γ-secretase complex are known to engender familial AD, contributions of BACE1 polymorphisms remain to be established.22 Nevertheless, the importance of BACE1 in AD pathogenesis has been demonstrated by mutations in APP, which either promote β-site cleavage causing a familial form of early-onset AD, or, contrarily, hinder APP β-cleavage and thus protect against the disease and even mild cognitive impairment.23,24 As a note of caution against a too straightforward interpretation of these findings, it is worth mentioning that the above APP mutations might also affect the aggregation kinetics of Aβ.25,26
In view of its pathogenic impact, inhibition of BACE1 is considered a highly promising strategy against AD. Almost a decade before BACE1 has been identified as β-secretase, protease inhibition by competing peptides had been proposed.27 Today, enormous effort is devoted to the design of small molecule BACE1 inhibitors, some of which are currently in Phase II/III clinical trials.28 Alternative strategies aim to inhibit BACE1 allosterically by BACE1-binding antibodies or peptides, to target Aβ by antibodies, or to develop vaccines against Aβ.28,29
Early alterations of network activity in AD
Long unrecognized in the clinical care of AD patients, in particular at early stages of the disease, epileptiform activity is now receiving increasing attention, as it appears to play an important role in AD pathogenesis and progression.30,31 AD is associated with an enhanced risk of seizures, and patients displaying (subclinical) epileptiform activity or overt seizures show earlier onset of cognitive decline and faster transition into severe dementia.32 Pharmacological suppression of aberrant network activity in AD should therefore offer more than symptomatic treatment as it is thought to interfere with a mechanism that contributes to and propels cognitive decline. Support for this hypothesis comes from a study with APP-over-expressing mice, in which the second-generation anti-epileptic drug levetiracetam blocked abnormal EEG activity and reversed synaptic and cognitive deficits.33 In contrast, other established anticonvulsant drugs failed to produce similar beneficial effects. Notably, the Na+ channel blocker phenytoin even exacerbated the aberrant EEG pattern, which would be consistent with the concept that neuronal hyperexcitability in AD results from decreased levels of NaV1.1 channels in inhibitory interneurons.34 These findings emphasize the need to identify and specifically target the ionic mechanisms directly involved in the altered network activity in AD.
BACE1-deficient mice
The first BACE1-deficient mouse lines, which were generated, seemed to lack appreciable phenotypic abnormalities.35,36 This was a highly welcomed finding as it appeared to indicate that inhibition of BACE1 would not engender unwarranted side effects, thereby lending strong momentum to anti-BACE strategies. Subsequent studies on BACE1-deficient mice, however, revealed significant postnatal lethality, reduced body weight, decreased anxiety-related behavior, and hyperactivity.37 Further investigations found decreased thermal pain threshold, impaired motor coordination, myelination deficits, impaired spatial learning and memory, and altered synaptic function and neurotransmitter metabolism.38-40 As a putative correlate of their learning and memory deficits, Wang et al. found that activity-dependent potentiation at the mossy-fiber - CA3 synapse in the hippocampus of BACE1-deficient mice is substantially impaired.41,42 Given that AD patients, in which BACE1 activity should be elevated, often display lowered seizure threshold (see above), it appeared counterintuitive at first that BACE1 knockout mice develop epileptic seizures, too, as reported by 2 groups.43,44
BACE1 - electrophysiological effects beyond Alzheimer's disease
Evidence accumulates that BACE1 does not exclusively affect the brain by cleavage of APP. Rather, a multitude of additional substrates that undergo β-site proteolysis has emerged, in part with remarkably higher affinity for BACE1 compared to APP.45 Among them are ion channel proteins, suggesting that BACE1 might directly influence the electrical behavior of excitable cells. Upon closer examination of how BACE1 interferes with ion channels, an entirely unexpected finding was made, going far beyond the starting point of these studies: Whereas the initial work had been based on the biochemical finding that some accessory β-subunits of Na+ channels can be cleaved by BACE1, it later became obvious that BACE1 can also act as an accessory subunit itself, directly interacting with the pore-forming α-subunit in a non-enzymatic fashion. In the following sections, we will review the different forms of how BACE1 interacts with ion channels, discuss the underlying mechanisms and elaborate on the functional consequences.
BACE1 and neuronal Nav channels
Nav function, structure, and regulation by β-subunits
A decade ago, voltage-gated sodium channels (Nav) were the first ion channels to be identified as targets of BACE1.46,47 Nav channels are best known for the fast, transient inward current underlying the upstroke of the action potential. In addition, they generate persistent and resurgent Na+ currents, which have been implicated in the regulation of firing threshold and frequency, membrane oscillations, and synaptic integration (see below). The main, pore-forming α-subunit comes in 9 subtypes (Nav1.1–1.9, encoded by the genes SCN1A-SCN5A, SCN8A-SCN11A). Accessory subunits (β1–4, SCN1B-SCN4B) can associate with the α-subunit and influence channel expression, trafficking, and gating.48-50 All β-subunits and Nav1.1, Nav1.2, Nav1.3, and Nav1.6 are expressed in the brain.51 Mutations in both α- or β-subunits have been associated, among other disorders, with epilepsy.52-54
β2 cleavage and Nav expression
In 2005, Wong et al. and Kim et al. demonstrated that β-subunits are substrates of BACE1 and γ-secretase in model systems and in vivo.46,55 Five years later, BACE1 cleavage sites were mapped in the β2 subunit, which plays an important role in hippocampus and cortex.56 Our group then showed that proteolysis of β2 by BACE1 caused a leftward shift in the voltage dependence of reconstituted Nav1.2 current, demonstrating that the cleavage bears significance for channel gating.57 In BACE1-transgenic mice and in temporal cortex of AD patients, increased BACE1 levels were found to be associated with enhanced β2 cleavage.58 Moreover, that study revealed that the generated β2 intracellular domain acted as transcriptional activator for its Nav1.1 α-subunit and increased Nav1.1 protein, which was, however, retained intracellularly. Consistent with the findings in BACE1-over-expressing mice, Corbett et al. reported similar changes in APP-over-expressing mice.59 They observed increased BACE1 levels and β2 cleavage, accompanied by increased Nav1.1 total, but decreased surface expression in cortical pyramidal cells and interneurons, accompanied by aberrant EEG activity. Verret et al. found reduced total Nav1.1 levels in another strain of APP-over-expressing mice and the parietal cortex of AD patients, which was in line with reduced functional Nav1.1 in the previous studies.60 The authors related abnormal EEG activity to reduced gamma oscillations and impaired interneuron function. Restoring Nav1.1 levels in these APP-over-expressing mice augmented the function of inhibitory interneurons and rescued cognitive deficits. These findings place the interplay between BACE1, β2, and Nav1.1 center stage because the effects on channel expression and trafficking can be causally linked to cognitive deterioration, as had been hypothesized earlier.61
Compared to their wild type counterparts, neurons from BACE1-deficient mice have lower levels of Nav1.1 and less channel protein is found in the outer cell membrane, so that, with respect to functional Nav1.1, BACE1-deficiency and -over-expression appear to produce strikingly similar effects.62 As an additional feature, BACE1−/− mice have increased Nav1.2 surface levels, possibly compensating for reduced Nav1.1. However, the issue of whether BACE1-deficiency leads to enhanced Nav1.2 surface levels and, as a consequence, to neuronal hyperexcitability remains controversial, with groups reporting evidence in favor or against effects of BACE1 on Nav1.2 levels.43,44,62
β4 cleavage and resurgent Na+ currents
Resurgent currents (INaR) have been first reported from cerebellar Purkinje cells, where they are mainly carried by Nav1.6.63 Mechanistically, they result from transient channel re-openings upon repolarization, when an unorthodox open-channel block, most likely exerted by parts of the β4 subunit, is relieved.64,65 Since BACE1, which is known to cleave β4, is highly expressed in cerebellum, an obvious question was whether Purkinje cells from BACE1-deficient mice would exhibit altered firing pattern.66 When compared to wild type neurons, the spontaneous firing of Purkinje cells from mutant mice was indeed reduced (Fig. 1A1). The reduced discharge activity was ascribed to a faster decay of the resurgent current (Fig. 1A2). Thus, in the absence of BACE1, reduced cleavage of β4 coincides with impaired open channel block and aberrant firing (Fig. 1A3). This finding suggests that proteolysis by BACE1 specifically tailors the β4 subunit to become the high-affinity blocking particle, which is required for resurgent current generation.
Figure 1.

BACE1 modifies gating of Na+ and K+ channels. (A1) Spontaneous firing of acutely isolated cerebellar Purkinje cells (aligned to first action potential) showed reduced frequency in BACE1−/− cells (red trace) compared to wild type (black trace). (A2) Acutely isolated Purkinje cells were held at −90 mV, then depolarized to +30 mV, which opened Na+ channels (peak truncated) that quickly inactivated. Repolarization to −22.5 mV evoked prominent resurgent current (INaR, normalized to peak INaR) that decayed faster in BACE1-deficient cells (red) than in cells from wild type mice (black). Solid lines in blue show single-exponential fits to rise and decay of INaR. (A3) Western blot (left) of cerebellar protein lysates from wild type or BACE1−/− mice at postnatal day 15 shows levels of BACE1, full-length β4 (FL), β4 C-terminal fragment (CTF), β4 intracellular domain (ICD), and β-actin. Densitometric analysis (right) demonstrated reduced β4-CTF in BACE1-deficient mice. (B1) Membrane potential of CA1 pyramidal neurons in hippocampal slices was set to −70 mV, then depolarized by 50 pA current injection (blue inset). Cells from BACE1-deficient mice (red trace) fired more frequently and showed less frequency adaptation, compared to wild type (black trace). (B2) In voltage-clamp mode, acutely isolated CA1 pyramidal cells were hyperpolarized from 30 mV to 45 mV, 55 mV, or 65 mV (inset). Compared to wild type neurons (black current traces), M-current was reduced in BACE1-deficient cells (red traces). (B3) Proximity ligation assay was performed on transiently transfected HEK-T cells expressing BACE1 and KCNQ2-V5 (schematic drawing on the left). Right-hand side: Sites of close proximity are indicated by fluorescent spots (orange, yellow outlines), which were assigned to the respective nucleus (gray, white outline) and cell borders (green outline). A1-3 adapted, with permission, from ref. 66; B1-3 adapted, with permission, from ref. 96.
Does the impaired firing of Purkinje cells engender motor abnormalities in BACE1-deficient mice? When subjected to elevated beam walking, BACE1-deficient mice showed indeed signs of ataxia (Fig. 2B). Furthermore, video analysis of their footprint pattern revealed that, with their hind paws, they significantly stepped outside their body line, which was not observed in wild type mice (Fig. 2C). The motor deficits in BACE1−/− mice strongly resemble those reported from mice with a Purkinje cell-specific ablation of BK potassium channels,67 suggesting that the symptoms after Bace1 ablation are of cerebellar origin. It is worth noting, however, that, as a consequence of the unprocessed BACE1 substrate neuregulin 1, BACE1-deficient mice also show muscle spindle defects and reduced myelination of peripheral nerves, which also give rise to impaired motor coordination.68 Thus, the contributions of cerebellar dysfunction and peripheral deficits to the motor symptoms of BACE1−/− mice remain to be determined in future studies.
Figure 2.
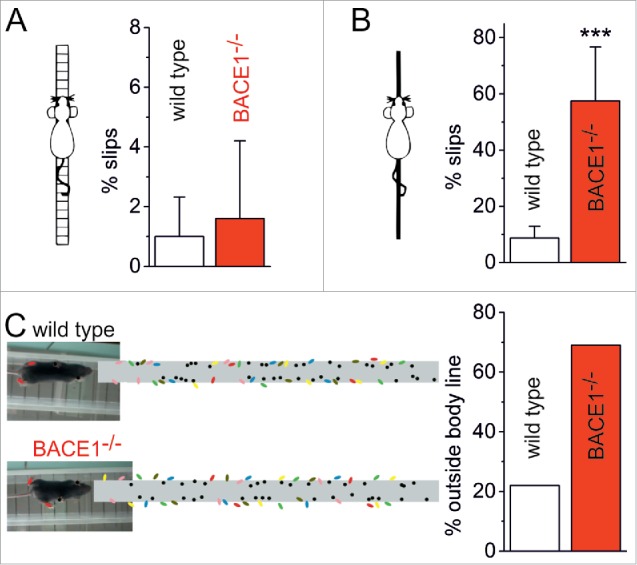
BACE1-deficient mice show ataxia-like motor phenotype. Distinct alterations in motor performance were analyzed in 3 different tasks. (A) In the ladder walking test, the mice had to cross a ladder composed of 38 staves with a diameter of 2 mm placed at intervals of 2 cm. The runs were stored on video tapes and slips of the forepaws and hindpaws were counted off-line in a frame-by-frame analysis. (B) In the elevated beam test, mice had to balance on a circular beam with a diameter of 1 cm and a length of 50 cm. Runs were recorded and analyzed as described above. Whereas both wild type (n = 9) and BACE1-deficient mice (n = 5) performed the ladder walking test with almost the same precision (A), the more challenging elevated beam task revealed a highly significant difference in performance (B, p < 0.0001, One-Way ANOVA). The number of slips was 8.7 ± 4.2 % (SD, n = 10) for wild type mice and 57.8 ± 19.2 % (SD, n = 8) for BACE1-deficient mice. (C) Foot print analysis was performed by detecting the positions of forepaws (black) and hindpaws (colored) of the mice while running along a transparent bridge. In wild type mice, the foot prints of both forepaws and hindpaws were almost exclusively confined to a path of a width corresponding to the transverse body diameter (8/36 steps of the hindpaws out of n = 6 mice outside the body line). By contrast, in BACE1-deficient mice 25/36 steps of the hindpaws (out of n = 6 mice) were placed outside the body diameter. The positions of the forepaws were not affected. Tests were approved by the local authorities.
Once considered a peculiarity of Purkinje cells, resurgent currents have since been observed in other neurons of the cerebellum as well as in neurons of other parts of the nervous system, most of them expressing β4 and showing high frequency firing.64 It therefore seems plausible to assume that, by altering INaR, BACE1 is capable of influencing the discharge properties of neurons other than Purkinje cells.
Direct interaction and Nav gating
In addition to Nav expression and trafficking, BACE1 has also direct impact on channel gating.57 As expected, cleavage of β2 or β4 by BACE1 affected the gating of Nav1.2 currents. Most surprisingly, however, current modulation was not entirely dependent on BACE1 proteolytic activity. Analyses with proteolytically inactive BACE1 suggested, for the first time, that BACE1 may act as an accessory subunit at Nav1.2, mimicking β2 effects at the Nav1.2/β4 complex.57
Although non-enzymatic actions of a protease that has been evolutionally conserved for β-cleavage may seem enigmatic, BACE1 is by far not the only enzyme endowed with such activity. For example, the G protein-coupled receptor (GPCR) kinase 2 (GRK2) rapidly desensitizes GPCR-coupled potassium currents independent of its phosphorylation capacity.69 A whole collection of structural effects has been put together for transglutaminase 2.70 Further examples are non-enzymatic functions of matrix metalloproteinase 12 and the γ-secretase component presenilin 1.71,72 BACE1 itself has been discussed as a non-enzymatic binding partner for a set of proteins, including BRI3, which is, like BACE1, processed by furin.73 While in Nav channels, non-enzymatic interactions between BACE1 and channel proteins were clearly discernible but small, such effects became much more prominent when explored in members of the KCNQ family.
BACE1 and neuronal KCNQ currents
KCNQ structure and regulation by β-subunits
The family of KCNQ (KV7) channels has 5 members, KCNQ1-5 (KV7.1–7.5, KCNQ1-5). The pore-forming α-subunits form homo- or hetero-tetramers.74,75 KCNQ1 (also: KVLQT, long QT) is expressed in heart, gastrointestinal tract, and inner ear.76 KCNQ2 and KCNQ3 are widely found in the central and peripheral nervous system.77,78 KCNQ4 seems to fulfil specific functions in the inner ear and in peripheral sensors, while KCNQ4/Q5 heteromers are important for vessel smooth muscle diameter control and in the vestibular system.79-81 KCNQ5 has further been identified as a neuronal KCNQ member.82-84
A complex molecular network is regulating the expression, localization, and gating of KCNQ channels. The family of the widely expressed β-subunits KCNE1-5 (KCNE1-5) modulates a variety of ion channels and is part of the KCNQ regulation system.85 The best studied interaction between a KCNQ α-subunit and an accessory KCNE subunit is that of the KCNQ1/KCNE1 complex, which forms the IKs and contributes to action potential repolarization in cardiomyocytes.86,87 Neuronal KCNQ channels are currently thought to operate without accessory β-subunits.74 This view is challenged by our recent work and will be discussed below.
Neuronal KCNQ function
KCNQ2 and KCNQ3 are the main constituents of neuronal M-current, a non-inactivating potassium current with slow activation and deactivation kinetics, which gives rise to a standing outward current below firing threshold.77 M-current is suppressed by muscarinic receptor activation (hence its name) owing to depletion of PIP2.88,77 Depending on its subcellular localization, M-current may regulate various aspects of intrinsic excitability. At the axon initial segment, M-current counteracts the propagation of subthreshold inputs to the axon, while it promotes action potential amplitude at nodes of the distal axon by increasing the availability of Nav channels.89 Somatodendritic M-current mediates spike frequency adaptation during repetitive firing.90 M-current further contributes to subthreshold theta frequency oscillations and regulates coherent spiking of neuron populations.91,92
The important role of M-current in keeping the balance between neuronal excitation and inhibition is emphasized by KCNQ2 and KCNQ3 mutations, which lead to an infantile epilepsy syndrome (benign familial neonatal convulsions, BFNC).93 Studies with conditional KCNQ2 or KCNQ3 knockout mice indicated that KCNQ2, rather than KCNQ3, is the important player in the regulation of neuronal excitability.94 A prominent role of KCNQ2 has been demonstrated in mice with conditional neuronal KCNQ2 knockout, which show neuronal hyperactivity and premature death.94 Transgenic mice with a dominant-negative variant of KCNQ2 in the nervous system are epileptic and have impaired spatial memory.95 Males are hyperactive and show morphological changes in the CA1 and the mossy fiber region. Intriguingly, these phenotypes closely resemble those of BACE1-deficient mice, raising the possibility that neuronal KCNQ channels might be a target of BACE1, too.
Direct interaction and KCNQ gating
A recent study indeed demonstrated a functionally significant interaction between BACE1 and neuronal KCNQ channels, which may well explain the striking phenotypic similarities between KCNQ2-deficient and BACE1-deficient mice.96 In CA1 pyramidal neurons from wild type mice, depolarizing current injection elicited a train of action potentials (APs), which showed frequency adaption due to M-current activation.90,97 By contrast, BACE1-deficient neurons fired APs at higher frequency (Fig. 1B1), similar to neurons in which M-current was suppressed by drugs or genetic manipulations.94,98,99 Furthermore, the M-current blocker XE991, which increased input resistance and firing frequency, and reduced latency to first AP in control neurons, failed to do so in mutant neurons. In voltage-clamp recordings, M-currents of acutely isolated CA1 pyramidal neurons from BACE1-deficient mice were reduced by 2/3 and displayed faster decay kinetics when compared to their wild type counterparts (Fig. 1B2). The prominent current decrease in knockout animals markedly exceeds the reduction in human BFNC, where a 25% decrease in M-current seems enough to cause hyperexcitability.100,101 Therefore, it is conceivable that the strongly impaired M-current in BACE1-deficient mice is responsible for their epileptic phenotype.
To elucidate the BACE1/KCNQ interplay, we examined KCNQ2 and KCNQ3 in a heterologous expression system.96 Co-expression of BACE1 increased KCNQ2/3 current amplitude, shifted activation to slightly more negative potentials, accelerated activation, and decelerated deactivation. Most importantly, the effects of BACE1 were non-proteolytic in nature, since they were reproduced by a proteolytically inactive BACE1 mutant.102 Thus, the non-enzymatic interaction between BACE1 and the α-subunit of voltage-dependent ion channels first reported for Nav1.2 (see above) seems to emerge as a more widely applicable mechanism through which BACE1 can influence neuronal excitability. Co-immunoprecipitation and proximity ligation assay served as independent means to confirm direct BACE1/KCNQ interaction (Fig. 1B3). Consistent with findings from KCNQ knockout mice (see above), the effects of BACE1 on reconstituted M-current were mainly due to its interaction with KCNQ2 rather than with KCNQ3. Notably, BACE1 was also capable of increasing currents mediated by heterologously expressed KCNQ4 or KCNQ5.96
Since the functionality of KCNQ channels strongly depends on adequate PIP2 levels, an obvious question is whether BACE1, as it becomes part of KCNQ channel complexes, enhances their affinity for PIP2, thereby promoting channel openings. However, BACE1 did not alter the decrease of KCNQ2/3 currents upon PIP2 depletion, nor did PIP2 displace BACE1.96,103 Thus, BACE1 appears to act differently than KCNE1, which sensitizes KCNQ1 to PIP2.104 Furthermore, the current-promoting effect of BACE1 was different from that of the established M-current activator retigabine.96 Taken together, these data suggest that the long-held view of M-current arising from neuronal KCNQ channel assemblies lacking auxiliary subunits requires modification. In the absence of KCNEs as binding partners for neuronal KCNQ channels, BACE1 may step in and become an essential constituent of M-current.
BACE1 and KCNQ1/KCNE1 (IKs) currents
BACE1 is present in murine heart and in human iPSC-derived cardiomyocytes.105 Underscoring the significance of BACE1 for cardiac electrophysiology, atrial cardiomyocytes from BACE1−/− mice show only about half the IKs of wild type cells.105 Since IKs is thought to be mediated by KCNQ1/KCNE1 channel complexes, these findings strongly point to physiologically relevant BACE1/KCNQ interactions also in the heart. Compared to neurons, however, matters in the heart are complicated by the fact that KCNE1 (and KCNE2) were identified as substrates to sequential cleavage by BACE1 or α-secretase, and γ-secretase.106 This situation leaves us with a number of possibilities of how BACE1 may influence IKs. For example, cleavage of KCNE1 by BACE1 may be a prerequisite for proper IKs. In addition, or alternatively, BACE1 may physically interact with KCNQ1 to augment IKs, analogous to the boosting of M-current in neurons. If a direct, non-enzymatic interaction holds true, we will have to ask next how BACE1 competes with KCNE1 for binding to KCNQ1 channels. First answers to these questions come from a study, in which KCNQ1, KCNE1, and BACE1 (including its proteolytically inactive variant) were heterologously expressed in various combinations.105 Major findings from this work are: First, BACE1 modulates currents mediated by KCNQ1 alone and by KCNQ1/KCNE1 complexes independent of its proteolytic function. Second, BACE1 slows activation and inactivation kinetics of KCNQ1/KCNE1 currents, and third, the effects of BACE1 critically depend on cellular ATP levels: BACE1 reduced reconstituted IKs in well-supplied cells, but counteracts current decline when ATP levels fall.
With BACE1 emerging as a β-subunit-like binding partner of KCNQ1, the issue of competition between BACE1 and KCNE subunits comes to the fore. Lundquist et al. suggested complex channel assemblies when different KCNE subunits are present in a tissue.85 BACE1 now further complicates the assembly options and may temper with KCNQ1/KCNE1 stoichiometry, which has been vividly debated in the past years.107-111
The interaction between BACE1 and KCNQ1/KCNE1 also raises a number of clinically interesting points. For example, can the increased lethality of BACE1-deficient mice be linked to abnormal cardiac electrophysiology? In the CNS, cellular stress was reported to elevate BACE1 expression and activity. Does this also hold for cardiac stress, and if so, would it protect against arrhythmias, as suggested by the strengthened effect of BACE1 when ATP is depleted?
Outlook
The finding that β-subunits of several voltage-dependent channel families can be split by BACE1 was the starting point to focus on the enzyme in the context of cellular excitability. The more recent finding that BACE1 can act as a β-subunit itself, thereby regulating channel function in a non-enzymatic fashion, offers an entirely new look at the role of this protein in the healthy and diseased brain and heart (summarized in Fig. 3). It is well conceivable that the list of channels, which can be influenced by BACE1 through proteolytic and, perhaps more so, through non-proteolytic effects, will be growing. Given that BACE1 is a rather ubiquitous protein and channels such as KCNQ1 are also expressed in various non-excitable cells, multiple organ systems and tissues might rely on BACE1/channel interactions. So far, hypotheses related to the physiological and pathogenic implications of the interplay between BACE1 and ion channels have been mainly derived from studies of BACE1-deficient preparations. From a translational point of view, it would be particularly important to gain a deeper understanding of how increases in BACE1 expression and activity, which are associated with Alzheimer´s disease, might influence the spectrum of electrophysiological effects related to BACE1. So far, BACE1 is mainly seen as a major culprit in AD owing to its pivotal role in the amyloidogenic pathway. But if BACE1 at the same time augments M-current, wouldn´t this non-proteolytic effect help to dampen the heightened propensity of AD patients to develop seizure activity? Another endogenous, putatively anti-epileptic mechanism has already been described for hippocampus and superior cervical ganglia, where KCNQ2 and KCNQ3 mRNA is upregulated as a response to induced seizures.112 If a synergistic and beneficial effect of BACE1 holds true, can we design drugs that suppress the noxious enzymatic actions of BACE1 while at the same time preserving its beneficial actions on certain ion channels? In this context, strategies that aim to render BACE1 harmless by other means, such as blocking peptides that bind to APP and inhibit the fatal BACE1/APP interaction,113 may gain importance. With issues like these, it becomes evident that studies on the electrophysiological impact of BACE1 hold great promise not only for elucidating the amazingly rich repertoire of targets and actions of this truly protean protein under physiological and pathological conditions, but also to exploit that knowledge to develop specifically tailored therapies.
Figure 3.

Schematic model of proteolytic and non-proteolytic effects of BACE1 on Na+ and K+ channels. Acting like an accessory β-subunit, BACE1 may attach itself to α-subunits of Nav and KCNQ channels in a non-enzymatic fashion (configurations on left- and right-hand side). In addition, BACE1 may cleave β-subunits (Navβs and KCNEs) of the respective channels (configurations at center). The different forms of interaction with BACE1 all influence channel function and are thus expected to have an impact on cellular excitability and network activity.
Funding Statement
This work was supported by Deutsche Forschungsgemeinschaft INST 90/675-1 FUGG to C.A., the Johannes und Frieda Marohn-Stiftung to T.H. and C.A., the Staedtler-Stiftung to C.A., the ELAN program of the Universitätsklinikum Erlangen to T.H., and the Studienstiftung des deutschen Volkes to S.L. and S.Ha.
Abbreviations
- Aβ
amyloid β
- AD
Alzheimer's disease
- AP
action potential
- APP
amyloid precursor protein
- ATP
adenosine triphosphate
- BACE1
β-site APP-cleaving enzyme 1
- BFNC
benign familial neonatal convulsions
- BK
large-conductance voltage- and calcium-activated potassium channel
- BRI3
brain protein I3
- CA1/3
cornu ammonis areas 1/3
- CNS
central nervous system
- EEG
electroencephalogram
- GPCR
G protein-coupled receptor
- GRK2
G protein-coupled receptor kinase 2
- IKs
slow delayed rectifier potassium current
- INaR
resurgent sodium current
- iPSC
induced pluripotent stem cell
- KCNE
voltage-gated potassium channel subfamily E
- KCNQ
voltage-gated potassium channel subfamily Q (Kv7.1-5)
- KV
voltage-gated potassium channel
- NaV
voltage-gated sodium channel
- PIP2
phosphatidylinositol-4,5-bisphosphate
- PSEN1/2
presenilin 1/2
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Alzheimer's Association . 2015 Alzheimer's disease facts and figures. Alzheimers Dement 2015; 11:332-84; PMID:25984581; http://dx.doi.org/ 10.1016/j.jalz.2015.02.003 [DOI] [PubMed] [Google Scholar]
- [2].Doblhammer G, Fink A, Fritze T. Short-term trends in dementia prevalence in Germany between the years 2007 and 2009. Alzheimers Dement 2015; 11:291-9; PMID:25301681; http://dx.doi.org/ 10.1016/j.jalz.2014.02.006 [DOI] [PubMed] [Google Scholar]
- [3].Alzheimer A. Ueber eine eigenartige Erkrankung der Hirnrinde. Allg Zeitschrift fuer Psychiatr und Psych Medizin 1907; 64:146-8; http:/dx.doi.org/ 10.1002/ca.980080612 [DOI] [Google Scholar]
- [4].Selkoe DJ. The molecular pathology of Alzheimer's disease. Neuron 1991; 6:487-98; PMID:1673054; http://dx.doi.org/ 10.1016/0896-6273(91)90052-2 [DOI] [PubMed] [Google Scholar]
- [5].Vassar R, Kuhn PH, Haass C, Kennedy ME, Rajendran L, Wong PC, Lichtenthaler SF. Function, therapeutic potential and cell biology of BACE proteases: current status and future prospects. J Neurochem 2014; 130:4-28; PMID:24646365; http://dx.doi.org/ 10.1111/jnc.12715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ferreira ST, Lourenco MV, Oliveira MM, De Felice FG. Soluble amyloid-β oligomers as synaptotoxins leading to cognitive impairment in Alzheimer's disease. Front Cell Neurosci 2015; 9:191; PMID:26074767; http://dx.doi.org/ 10.3389/fncel.2015.00191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sinha S, Anderson JP, Barbour R, Basi GS, Caccavello R, Davis D, Doan M, Dovey HF, Frigon N, Hong J, et al.. Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature 1999; 402:537-40; PMID:10591214; http://dx.doi.org/ 10.1038/990114 [DOI] [PubMed] [Google Scholar]
- [8].Yan R, Bienkowski MJ, Shuck ME, Miao H, Tory MC, Pauley AM, Brashier JR, Stratman NC, Mathews WR, Buhl AE, et al.. Membrane-anchored aspartyl protease with Alzheimer's disease beta-secretase activity. Nature 1999; 402:533-7; PMID:10591213; http://dx.doi.org/ 10.1038/990107 [DOI] [PubMed] [Google Scholar]
- [9].Hussain I, Powell D, Howlett DR, Tew DG, Meek TD, Chapman C, Gloger IS, Murphy KE, Southan CD, Ryan DM, et al.. Identification of a novel aspartic protease (Asp 2) as beta-secretase. Mol Cell Neurosci 1999; 14:419-27; PMID:10656250; http://dx.doi.org/ 10.1006/mcne.1999.0811 [DOI] [PubMed] [Google Scholar]
- [10].Lin X, Koelsch G, Wu S, Downs D, Dashti A, Tang J. Human aspartic protease memapsin 2 cleaves the beta-secretase site of beta-amyloid precursor protein. Proc Natl Acad Sci U S A 2000; 97:1456-60; PMID:10677483; http://dx.doi.org/ 10.1073/pnas.97.4.1456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, et al.. Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999; 286:735-41; PMID:10531052; http://dx.doi.org/ 10.1126/science.286.5440.735 [DOI] [PubMed] [Google Scholar]
- [12].Cai H, Wang Y, McCarthy D, Wen H, Borchelt DR, Price DL, Wong PC. BACE1 is the major beta-secretase for generation of Abeta peptides by neurons. Nat Neurosci 2001; 4:233-4; PMID:11224536; http://dx.doi.org/ 10.1038/85064 [DOI] [PubMed] [Google Scholar]
- [13].Willem M, Tahirovic S, Busche MA, Ovsepian SV, Chafai M, Kootar S, Hornburg D, Evans LDB, Moore S, Daria A, et al.. η-Secretase processing of APP inhibits neuronal activity in the hippocampus. Nature 2015; 526:443-7; PMID:26322584; http://dx.doi.org/ 10.1038/nature14864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zhang Z, Song M, Liu X, Su Kang S, Duong DM, Seyfried NT, Cao X, Cheng L, Sun YE, Ping Yu S, et al.. Delta-secretase cleaves amyloid precursor protein and regulates the pathogenesis in Alzheimer's disease. Nat Commun 2015; 6:8762; PMID:26549211; http://dx.doi.org/ 10.1038/ncomms9762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Parihar MS, Brewer GJ. Amyloid-β as a modulator of synaptic plasticity. J Alzheimers Dis 2010; 22:741-63; PMID:20847424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron 2003; 37:925-37; PMID:12670422; http://dx.doi.org/ 10.1016/S0896-6273(03)00124-7 [DOI] [PubMed] [Google Scholar]
- [17].Yang LB, Lindholm K, Yan R, Citron M, Xia W, Yang XL, Beach T, Sue L, Wong P, Price D, et al.. Elevated beta-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat Med 2003; 9:3-4; PMID:12514700; http://dx.doi.org/ 10.1038/nm0103-3 [DOI] [PubMed] [Google Scholar]
- [18].Cheng X, He P, Lee T, Yao H, Li R, Shen Y. High activities of BACE1 in Brains with mild cognitive impairment. Am J Pathol 2014; 184:141-7; PMID:24332014; http://dx.doi.org/ 10.1016/j.ajpath.2013.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Jansen WJ, Ossenkoppele R, Knol DL, Tijms BM, Scheltens P, Verhey FRJ, Visser PJ, Aalten P, Aarsland D, Alcolea D, et al.. Prevalence of Cerebral Amyloid Pathology in Persons Without Dementia: A Meta-analysis. JAMA 2015; 313:1924-38; PMID:25988462; http://dx.doi.org/ 10.1001/jama.2015.4668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Choi SH, Kim YH, Hebisch M, Sliwinski C, Lee S, D'Avanzo C, Chen H, Hooli B, Asselin C, Muffat J, et al.. A three-dimensional human neural cell culture model of Alzheimer's disease. Nature 2014; 515:274-8; PMID:25307057; http://dx.doi.org/ 10.1038/nature13800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Stefanova NA, Muraleva NA, Korbolina EE, Kiseleva E, Maksimova KY, Kolosova NG. Amyloid accumulation is a late event in sporadic Alzheimer's disease-like pathology in nontransgenic rats. Oncotarget 2015; 6:1396-413; PMID:25595891; http://dx.doi.org/ 10.18632/oncotarget.2751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Yu M, Liu Y, Shen J, Lv D, Zhang J. Meta-Analysis of BACE1 Gene rs638405 Polymorphism and the risk of Alzheimer's Disease in Caucasion and Asian population. Neurosci Lett 2016; 616:189-96; PMID:26828303; http://dx.doi.org/ 10.1016/j.neulet.2016.01.059 [DOI] [PubMed] [Google Scholar]
- [23].Tanzi RE, Bertram L. Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective. Cell 2005; 120:545-55; PMID:15734686; http://dx.doi.org/ 10.1016/j.cell.2005.02.008 [DOI] [PubMed] [Google Scholar]
- [24].Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, Bjornsson S, Stefansson H, Sulem P, Gudbjartsson D, Maloney J, et al.. A mutation in APP protects against Alzheimer's disease and age-related cognitive decline. Nature 2012; 488:96-9; PMID:22801501; http://dx.doi.org/ 10.1038/nature11283 [DOI] [PubMed] [Google Scholar]
- [25].Benilova I, Gallardo R, Ungureanu A-A, Castillo Cano V, Snellinx A, Ramakers M, Bartic C, Rousseau F, Schymkowitz J, De Strooper B. The Alzheimer disease protective mutation A2T modulates kinetic and thermodynamic properties of amyloid-β (Aβ) aggregation. J Biol Chem 2014; 289:30977-89; PMID:25253695; http://dx.doi.org/ 10.1074/jbc.M114.599027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Maloney JA, Bainbridge T, Gustafson A, Zhang S, Kyauk R, Steiner P, van der Brug M, Liu Y, Ernst JA, Watts RJ, et al.. Molecular mechanisms of Alzheimer disease protection by the A673T allele of amyloid precursor protein. J Biol Chem 2014; 289:30990-1000; PMID:25253696; http://dx.doi.org/ 10.1074/jbc.M114.589069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Carrell RW. Alzheimer's disease. Enter a protease inhibitor. Nature 1988; 331:478-9; PMID:2448646; http://dx.doi.org/ 10.1038/331478a0 [DOI] [PubMed] [Google Scholar]
- [28].Qian X, Hamad B, Dias-Lalcaca G. The Alzheimer disease market. Nat Rev Drug Discov 2015; 14:675-6; PMID:26388231; http://dx.doi.org/ 10.1038/nrd4749 [DOI] [PubMed] [Google Scholar]
- [29].Wang W, Liu Y, Lazarus RA. Allosteric inhibition of BACE1 by an exosite-binding antibody. Curr Opin Struct Biol 2013; 23:797-805; PMID:23998983; http://dx.doi.org/ 10.1016/j.sbi.2013.08.001 [DOI] [PubMed] [Google Scholar]
- [30].Vossel KA, Beagle AJ, Rabinovici GD, Shu H, Lee SE, Naasan G, Hegde M, Cornes SB, Henry ML, Nelson AB, et al.. Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol 2013; 70:1158-66; PMID:23835471; http://dx.doi.org/ 10.1001/jamaneurol.2013.136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Born HA. Seizures in Alzheimer's disease. Neuroscience 2015; 286:251-63; PMID:25484360; http://dx.doi.org/ 10.1016/j.neuroscience.2014.11.051 [DOI] [PubMed] [Google Scholar]
- [32].Spencer D. Seizures and epileptiform activity in early Alzheimer disease: how hard should we be looking? Epilepsy Curr 2014; 14:73-5; PMID:24872782; http://dx.doi.org/ 10.5698/1535-7597-14.2.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Sanchez PE, Zhu L, Verret L, Vossel KA, Orr AG, Cirrito JR, Devidze N, Ho K, Yu G-Q, Palop JJ, et al.. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer's disease model. Proc Natl Acad Sci 2012; 109:E2895-903; PMID:22869752; http://dx.doi.org/ 10.1073/pnas.1121081109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Huang Y, Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell 2012; 148:1204-22; PMID:22424230; http://dx.doi.org/ 10.1016/j.cell.2012.02.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Luo Y, Bolon B, Kahn S, Bennett BD, Babu-Khan S, Denis P, Fan W, Kha H, Zhang J, Gong Y, et al.. Mice deficient in BACE1, the Alzheimer's β-secretase, have normal phenotype and abolished β-amyloid generation. Nat Neurosci 2001; 4:231-2; PMID:11224535; http://dx.doi.org/ 10.1038/85059 [DOI] [PubMed] [Google Scholar]
- [36].Luo Y, Bolon B, Damore MA, Fitzpatrick D, Liu H, Zhang J, Yan Q, Vassar R, Citron M. BACE1 (beta-secretase) knockout mice do not acquire compensatory gene expression changes or develop neural lesions over time. Neurobiol Dis 2003; 14:81-8; PMID:13678669; http://dx.doi.org/ 10.1016/S0969-9961(03)00104-9 [DOI] [PubMed] [Google Scholar]
- [37].Dominguez D, Tournoy J, Hartmann D, Huth T, Cryns K, Deforce S, Serneels L, Camacho IE, Marjaux E, Craessaerts K, et al.. Phenotypic and biochemical analyses of BACE1- and BACE2-deficient mice. J Biol Chem 2005; 280:30797-806; PMID:15987683; http://dx.doi.org/ 10.1074/jbc.M505249200 [DOI] [PubMed] [Google Scholar]
- [38].Hu X, Hicks CW, He W, Wong P, Macklin WB, Trapp BD, Yan R. Bace1 modulates myelination in the central and peripheral nervous system. Nat Neurosci 2006; 9:1520-5; PMID:17099708; http://dx.doi.org/ 10.1038/nn1797 [DOI] [PubMed] [Google Scholar]
- [39].Laird FM, Cai H, Savonenko AV, Farah MH, He K, Melnikova T, Wen H, Chiang H-C, Xu G, Koliatsos VE, et al.. BACE1, a major determinant of selective vulnerability of the brain to amyloid-beta amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J Neurosci 2005; 25:11693-709; PMID:16354928; http://dx.doi.org/ 10.1523/JNEUROSCI.2766-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Harrison SM, Harper AJ, Hawkins J, Duddy G, Grau E, Pugh PL, Winter PH, Shilliam CS, Hughes ZA, Dawson LA, et al.. BACE1 (beta-secretase) transgenic and knockout mice: identification of neurochemical deficits and behavioral changes. Mol Cell Neurosci 2003; 24:646-55; PMID:14664815; http://dx.doi.org/ 10.1016/S1044-7431(03)00227-6 [DOI] [PubMed] [Google Scholar]
- [41].Wang H, Song L, Laird F, Wong PC, Lee H-K. BACE1 knock-outs display deficits in activity-dependent potentiation of synaptic transmission at mossy fiber to CA3 synapses in the hippocampus. J Neurosci 2008; 28:8677-81; PMID:18753368; http://dx.doi.org/ 10.1523/JNEUROSCI.2440-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wang H, Megill A, Wong PC, Kirkwood A, Lee HK. Postsynaptic Target Specific Synaptic Dysfunctions in the CA3 Area of BACE1 Knockout Mice. PLoS One 2014; 9:e92279; PMID:24637500; http://dx.doi.org/ 10.1371/journal.pone.0092279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Hu X, Zhou X, He W, Yang J, Xiong W, Wong P, Wilson CG, Yan R. BACE1 deficiency causes altered neuronal activity and neurodegeneration. J Neurosci 2010; 30:8819-29; PMID:20592204; http://dx.doi.org/ 10.1523/JNEUROSCI.1334-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hitt BD, Jaramillo TC, Chetkovich DM, Vassar R. BACE1−/− mice exhibit seizure activity that does not correlate with sodium channel level or axonal localization. Mol Neurodegener 2010; 5:31; PMID:20731874; http://dx.doi.org/ 10.1186/1750-1326-5-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kuhn PH, Koroniak K, Hogl S, Colombo A, Zeitschel U, Willem M, Volbracht C, Schepers U, Imhof A, Hoffmeister A, et al.. Secretome protein enrichment identifies physiological BACE1 protease substrates in neurons. EMBO J 2012; 31:3157-68; PMID:22728825; http://dx.doi.org/ 10.1038/emboj.2012.173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wong HK, Sakurai T, Oyama F, Kaneko K, Wada K, Miyazaki H, Kurosawa M, De Strooper B, Saftig P, Nukina N. beta Subunits of voltage-gated sodium channels are novel substrates of beta-site amyloid precursor protein-cleaving enzyme (BACE1) and gamma-secretase. J Biol Chem 2005; 280:23009-17; PMID:15824102; http://dx.doi.org/ 10.1074/jbc.M414648200 [DOI] [PubMed] [Google Scholar]
- [47].Huth T, Alzheimer C. Voltage-dependent Na+ channels as targets of BACE1 - implications for neuronal firing and beyond. Curr Alzheimer Res 2012; 9:184-8; PMID:22455479; http://dx.doi.org/ 10.2174/156720512799361619 [DOI] [PubMed] [Google Scholar]
- [48].Isom LL, Ragsdale DS, De Jongh KS, Westenbroek RE, Reber BF, Scheuer T, Catterall WA. Structure and function of the beta 2 subunit of brain sodium channels, a transmembrane glycoprotein with a CAM motif. Cell 1995; 83:433-42; PMID:8521473; http://dx.doi.org/ 10.1016/0092-8674(95)90121-3 [DOI] [PubMed] [Google Scholar]
- [49].Namadurai S, Yereddi NR, Cusdin FS, Huang CLH, Chirgadze DY, Jackson AP. A new look at sodium channel β subunits. Open Biol 2015; 5:140192; PMID:25567098; http://dx.doi.org/ 10.1098/rsob.140192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Yu FH, Westenbroek RE, Silos-Santiago I, McCormick KA, Lawson D, Ge P, Ferriera H, Lilly J, DiStefano PS, Catterall WA, et al.. Sodium channel beta4, a new disulfide-linked auxiliary subunit with similarity to beta2. J Neurosci 2003; 23:7577-85; PMID:12930796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Vacher H, Mohapatra DP, Trimmer JS. Localization and targeting of voltage-dependent ion channels in mammalian central neurons. Physiol Rev 2008; 88:1407-47; PMID:18923186; http://dx.doi.org/ 10.1152/physrev.00002.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Wimmer VC, Reid CA, Mitchell S, Richards KL, Scaf BB, Leaw BT, Hill EL, Royeck M, Horstmann M-T, Cromer BA, et al.. Axon initial segment dysfunction in a mouse model of genetic epilepsy with febrile seizures plus. J Clin Invest 2010; 120:2661-71; PMID:20628201; http://dx.doi.org/ 10.1172/JCI42219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Volkers L, Kahlig KM, Verbeek NE, Das JHG, van Kempen MJA, Stroink H, Augustijn P, van Nieuwenhuizen O, Lindhout D, George AL, et al.. Nav 1.1 dysfunction in genetic epilepsy with febrile seizures-plus or Dravet syndrome. Eur J Neurosci 2011; 34:1268-75; PMID:21864321; http://dx.doi.org/ 10.1111/j.1460-9568.2011.07826.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].O'Malley HA, Isom LL. Sodium channel β subunits: emerging targets in channelopathies. Annu Rev Physiol 2015; 77:481-504; PMID:Can't; http://dx.doi.org/ 10.1146/annurev-physiol-021014-071846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Kim DY, Ingano LAM, Carey BW, Pettingell WH, Kovacs DM. Presenilin/gamma-secretase-mediated cleavage of the voltage-gated sodium channel beta2-subunit regulates cell adhesion and migration. J Biol Chem 2005; 280:23251-61; PMID:15833746; http://dx.doi.org/ 10.1074/jbc.M412938200 [DOI] [PubMed] [Google Scholar]
- [56].Gersbacher MT, Kim DY, Bhattacharyya R, Kovacs DM. Identification of BACE1 cleavage sites in human voltage-gated sodium channel beta 2 subunit. Mol Neurodegener 2010; 5:61; PMID:21182789; http://dx.doi.org/ 10.1186/1750-1326-5-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Huth T, Schmidt-Neuenfeldt K, Rittger A, Saftig P, Reiss K, Alzheimer C. Non-proteolytic effect of beta-site APP-cleaving enzyme 1 (BACE1) on sodium channel function. Neurobiol Dis 2009; 33:282-9; PMID:19056495; http://dx.doi.org/ 10.1016/j.nbd.2008.10.015 [DOI] [PubMed] [Google Scholar]
- [58].Kim DY, Carey BW, Wang H, Ingano LA, Binshtok AM, Wertz MH, Pettingell WH, He P, Lee VM, Woolf CJ, et al.. BACE1 regulates voltage-gated sodium channels and neuronal activity. Nat Cell Biol 2007; 9:755-64; PMID:17576410; http://dx.doi.org/ 10.1038/ncb1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Corbett BF, Leiser SC, Ling HP, Nagy R, Breysse N, Zhang X, Hazra A, Wood A, Pangalos MN, Reinhart PH, et al.. Sodium channel cleavage is associated with aberrant neuronal activity and cognitive deficits in a mouse model of Alzheimer's disease. J Neurosci 2013; 33:7020-6; PMID:23595759; http://dx.doi.org/ 10.1523/JNEUROSCI.2325-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Verret L, Mann EO, Hang GB, Barth AM, Cobos I, Ho K, Devidze N, Masliah E, Kreitzer AC, Mody I, et al.. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 2012; 149:708-21; PMID:22541439; http://dx.doi.org/ 10.1016/j.cell.2012.02.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Kovacs DM, Gersbacher MT, Kim DY. Alzheimer's secretases regulate voltage-gated sodium channels. Neurosci Lett 2010; 486:68-72; PMID:20817076; http://dx.doi.org/ 10.1016/j.neulet.2010.08.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Kim DY, Gersbacher MT, Inquimbert P, Kovacs DM. Reduced sodium channel Na(v)1.1 levels in BACE1-null mice. J Biol Chem 2011; 286:8106-16; PMID:21190943; http://dx.doi.org/ 10.1074/jbc.M110.134692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Raman IM, Bean BP. Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J Neurosci 1997; 17:4517-26; PMID:9169512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Lewis AH, Raman IM. Resurgent current of voltage-gated Na(+) channels. J Physiol 2014; 592:4825-38; PMID:25172941; http://dx.doi.org/ 10.1113/jphysiol.2014.277582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Barbosa C, Tan Z-Y, Wang R, Xie W, Strong JA, Patel RR, Vasko MR, Zhang J-M, Cummins TR. Navβ4 regulates fast resurgent sodium currents and excitability in sensory neurons. Mol Pain 2015; 11:60; PMID:26408173; http://dx.doi.org/ 10.1186/s12990-015-0063-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Huth T, Rittger A, Saftig P, Alzheimer C. beta-Site APP-cleaving enzyme 1 (BACE1) cleaves cerebellar Na+ channel beta4-subunit and promotes Purkinje cell firing by slowing the decay of resurgent Na+ current. Pflugers Arch 2011; 461:355-71; PMID:21246381; http://dx.doi.org/ 10.1007/s00424-010-0913-2 [DOI] [PubMed] [Google Scholar]
- [67].Chen X, Kovalchuk Y, Adelsberger H, Henning HA, Sausbier M, Wietzorrek G, Ruth P, Yarom Y, Konnerth A. Disruption of the olivo-cerebellar circuit by Purkinje neuron-specific ablation of BK channels. Proc Natl Acad Sci U S A 2010; 107:12323-8; PMID:20566869; http://dx.doi.org/ 10.1073/pnas.1001745107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Cheret C, Willem M, Fricker FR, Wende H, Wulf-Goldenberg A, Tahirovic S, Nave KA, Saftig P, Haass C, Garratt AN, et al.. Bace1 and Neuregulin-1 cooperate to control formation and maintenance of muscle spindles. EMBO J 2013; 32:2015-28; PMID:23792428; http://dx.doi.org/ 10.1038/emboj.2013.146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Raveh A, Cooper A, Guy-David L, Reuveny E. Nonenzymatic rapid control of GIRK channel function by a G protein-coupled receptor kinase. Cell 2010; 143:750-60; PMID:21111235; http://dx.doi.org/ 10.1016/j.cell.2010.10.018 [DOI] [PubMed] [Google Scholar]
- [70].Kanchan K, Fuxreiter M, Fésüs L. Physiological, pathological, and structural implications of non-enzymatic protein-protein interactions of the multifunctional human transglutaminase 2. Cell Mol Life Sci 2015; 72:3009-35; PMID:25943306; http://dx.doi.org/ 10.1007/s00018-015-1909-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Marchant DJ, Bellac CL, Moraes TJ, Wadsworth SJ, Dufour A, Butler GS, Bilawchuk LM, Hendry RG, Robertson AG, Cheung CT, et al.. A new transcriptional role for matrix metalloproteinase-12 in antiviral immunity. Nat Med 2014; 20:493-502; PMID:24784232; http://dx.doi.org/ 10.1038/nm.3508 [DOI] [PubMed] [Google Scholar]
- [72].McBrayer M, Nixon RA. Lysosome and calcium dysregulation in Alzheimer's disease: partners in crime. Biochem Soc Trans 2013; 41:1495-502; PMID:24256243; http://dx.doi.org/ 10.1042/BST20130201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Wickham L, Benjannet S, Marcinkiewicz E, Chretien M, Seidah NG. Beta-amyloid protein converting enzyme 1 and brain-specific type II membrane protein BRI3: binding partners processed by furin. J Neurochem 2005; 92:93-102; PMID:15606899; http://dx.doi.org/ 10.1111/j.1471-4159.2004.02840.x [DOI] [PubMed] [Google Scholar]
- [74].Brown DA, Passmore GM. Neural KCNQ (Kv7) channels. Br J Pharmacol 2009; 156:1185-95; PMID:19298256; http://dx.doi.org/ 10.1111/j.1476-5381.2009.00111.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Bal M, Zhang J, Zaika O, Hernandez CC, Shapiro MS. Homomeric and heteromeric assembly of KCNQ (Kv7) K+ channels assayed by total internal reflection fluorescence/fluorescence resonance energy transfer and patch clamp analysis. J Biol Chem 2008; 283:30668-76; PMID:18786918; http://dx.doi.org/ 10.1074/jbc.M805216200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Abbott GW. KCNE1 and KCNE3: The yin and yang of voltage-gated K(+) channel regulation. Gene 2016; 576:1-13; PMID:26410412; http://dx.doi.org/ 10.1016/j.gene.2015.09.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE, McKinnon D. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science 1998; 282:1890-3; PMID:9836639; http://dx.doi.org/ 10.1126/science.282.5395.1890 [DOI] [PubMed] [Google Scholar]
- [78].King CH, Lancaster E, Salomon D, Peles E, Scherer SS. Kv7.2 regulates the function of peripheral sensory neurons. J Comp Neurol 2014; 522:3262-80; PMID:24687876; http://dx.doi.org/ 10.1002/cne.23595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Kubisch C, Schroeder BC, Friedrich T, Lutjohann B, El-Amraoui A, Marlin S, Petit C, Jentsch TJ. KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell 1999; 96:437-46; PMID:10025409; http://dx.doi.org/ 10.1016/S0092-8674(00)80556-5 [DOI] [PubMed] [Google Scholar]
- [80].Spitzmaul G, Tolosa L, Winkelman BH, Heidenreich M, Frens MA, Chabbert C, de Zeeuw CI, Jentsch TJ. Vestibular role of KCNQ4 and KCNQ5 K+ channels revealed by mouse models. J Biol Chem 2013; 288:9334-44; PMID:23408425; http://dx.doi.org/ 10.1074/jbc.M112.433383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Brueggemann LI, Mackie AR, Cribbs LL, Freda J, Tripathi A, Majetschak M, Byron KL. Differential protein kinase C-dependent modulation of Kv7.4 and Kv7.5 subunits of vascular Kv7 channels. J Biol Chem 2014; 289:2099-111; PMID:24297175; http://dx.doi.org/ 10.1074/jbc.M113.527820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].King CH, Scherer SS. Kv7.5 is the primary Kv7 subunit expressed in C-fibers. J Comp Neurol 2012; 520:1940-50; PMID:22134895; http://dx.doi.org/ 10.1002/cne.23019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Lerche C, Scherer CR, Seebohm G, Derst C, Wei AD, Busch AE, Steinmeyer K. Molecular cloning and functional expression of KCNQ5, a potassium channel subunit that may contribute to neuronal M-current diversity. J Biol Chem 2000; 275:22395-400; PMID:10787416; http://dx.doi.org/ 10.1074/jbc.M002378200 [DOI] [PubMed] [Google Scholar]
- [84].Fidzinski P, Korotkova T, Heidenreich M, Maier N, Schuetze S, Kobler O, Zuschratter W, Schmitz D, Ponomarenko A, Jentsch TJ. KCNQ5 K(+) channels control hippocampal synaptic inhibition and fast network oscillations. Nat Commun 2015; 6:6254; PMID:25649132; http://dx.doi.org/ 10.1038/ncomms7254 [DOI] [PubMed] [Google Scholar]
- [85].Lundquist AL, Turner CL, Ballester LY, George AL. Expression and transcriptional control of human KCNE genes. Genomics 2006; 87:119-28; PMID:16303284; http://dx.doi.org/ 10.1016/j.ygeno.2005.09.004 [DOI] [PubMed] [Google Scholar]
- [86].Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, Keating MT. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature 1996; 384:80-3; PMID:8900283; http://dx.doi.org/ 10.1038/384080a0 [DOI] [PubMed] [Google Scholar]
- [87].Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature 1996; 384:78-80; PMID:8900282; http://dx.doi.org/ 10.1038/384078a0 [DOI] [PubMed] [Google Scholar]
- [88].Brown DA, Adams PR. Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neurone. Nature 1980; 283:673-6; PMID:6965523; http://dx.doi.org/ 10.1038/283673a0 [DOI] [PubMed] [Google Scholar]
- [89].Battefeld A, Tran BT, Gavrilis J, Cooper EC, Kole MH. Heteromeric Kv7.2/7.3 channels differentially regulate action potential initiation and conduction in neocortical myelinated axons. J Neurosci 2014; 34:3719-32; PMID:24599470; http://dx.doi.org/ 10.1523/JNEUROSCI.4206-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Gu N, Vervaeke K, Hu H, Storm JF. Kv7/KCNQ/M and HCN/h, but not KCa2/SK channels, contribute to the somatic medium after-hyperpolarization and excitability control in CA1 hippocampal pyramidal cells. J Physiol 2005; 566:689-715; PMID:15890705; http://dx.doi.org/ 10.1113/jphysiol.2005.086835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Guan D, Higgs MH, Horton LR, Spain WJ, Foehring RC. Contributions of Kv7-mediated potassium current to sub- and suprathreshold responses of rat layer II/III neocortical pyramidal neurons. J Neurophysiol 2011; 106:1722-33; PMID:21697446; http://dx.doi.org/ 10.1152/jn.00211.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Ocker GK, Doiron B. Kv7 Channels Regulate Pairwise Spiking Covariability in Health and Disease. J Neurophysiol 2014; 112:340-52; PMID:24790164; http://dx.doi.org/ 10.1152/jn.00084.2014 [DOI] [PubMed] [Google Scholar]
- [93].Maljevic S, Wuttke TV, Seebohm G, Lerche H. KV7 channelopathies. Pflugers Arch 2010; 460:277-88; PMID:20401729; http://dx.doi.org/ 10.1007/s00424-010-0831-3 [DOI] [PubMed] [Google Scholar]
- [94].Soh H, Pant R, LoTurco JJ, Tzingounis AV. Conditional deletions of epilepsy-associated KCNQ2 and KCNQ3 channels from cerebral cortex cause differential effects on neuronal excitability. J Neurosci 2014; 34:5311-21; PMID:24719109; http://dx.doi.org/ 10.1523/JNEUROSCI.3919-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Peters HC, Hu H, Pongs O, Storm JF, Isbrandt D. Conditional transgenic suppression of M channels in mouse brain reveals functions in neuronal excitability, resonance and behavior. Nat Neurosci 2005; 8:51-60; PMID:15608631; http://dx.doi.org/ 10.1038/nn1375 [DOI] [PubMed] [Google Scholar]
- [96].Hessler S, Zheng F, Hartmann S, Rittger A, Lehnert S, Volkel M, Nissen M, Edelmann E, Saftig P, Schwake M, et al.. Beta-Secretase BACE1 Regulates Hippocampal and Reconstituted M-Currents in a Beta-Subunit-Like Fashion. J Neurosci 2015; 35:3298-311; PMID:25716831; http://dx.doi.org/ 10.1523/JNEUROSCI.3127-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Madison DV, Lancaster B, Nicoll RA. Voltage clamp analysis of cholinergic action in the hippocampus. J Neurosci 1987; 7:733-41; PMID:3559710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Otto JF, Yang Y, Frankel WN, White HS, Wilcox KS. A spontaneous mutation involving Kcnq2 (Kv7.2) reduces M-current density and spike frequency adaptation in mouse CA1 neurons. J Neurosci 2006; 26:2053-9; PMID:16481438; http://dx.doi.org/ 10.1523/JNEUROSCI.1575-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Zhou P, Yu H, Gu M, Nan F, Gao Z, Li M. Phosphatidylinositol 4,5-bisphosphate alters pharmacological selectivity for epilepsy-causing KCNQ potassium channels. Proc Natl Acad Sci U S A 2013; 110:8726-31; PMID:23650395; http://dx.doi.org/ 10.1073/pnas.1302167110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Gribkoff VK. The therapeutic potential of neuronal K V 7 (KCNQ) channel modulators: an update. Expert Opin Ther Targets 2008; 12:565-81; PMID:18410240; http://dx.doi.org/ 10.1517/14728222.12.5.565 [DOI] [PubMed] [Google Scholar]
- [101].Schroeder BC, Kubisch C, Stein V, Jentsch TJ. Moderate loss of function of cyclic-AMP-modulated KCNQ2/KCNQ3 K+ channels causes epilepsy. Nature 1998; 396:687-90; PMID:9872318; http://dx.doi.org/ 10.1038/25367 [DOI] [PubMed] [Google Scholar]
- [102].Jin S, Agerman K, Kolmodin K, Gustafsson E, Dahlqvist C, Jureus A, Liu G, Falting J, Berg S, Lundkvist J, Lendahl U. Evidence for dimeric BACE-mediated APP processing. Biochem Biophys Res Commun 2010; 393:21-7; PMID:20097169; http://dx.doi.org/ 10.1016/j.bbrc.2010.01.064 [DOI] [PubMed] [Google Scholar]
- [103].Kim KS, Duignan KM, Hawryluk JM, Soh H, Tzingounis AV. The Voltage Activation of Cortical KCNQ Channels Depends on Global PIP2 Levels. Biophys J 2016; 110:1089-98; PMID:26958886; http://dx.doi.org/ 10.1016/j.bpj.2016.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Li Y, Zaydman MA, Wu D, Shi J, Guan M, Virgin-Downey B, Cui J. KCNE1 enhances phosphatidylinositol 4,5-bisphosphate (PIP2) sensitivity of IKs to modulate channel activity. Proc Natl Acad Sci U S A 2011; 108:9095-100; PMID:21576493; http://dx.doi.org/ 10.1073/pnas.1100872108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Agsten M, Hessler S, Lehnert S, Volk T, Rittger A, Hartmann S, Raab C, Kim DY, Groemer TW, Schwake M, et al.. BACE1 modulates gating of KCNQ1 (Kv7.1) and cardiac delayed rectifier KCNQ1/KCNE1 (IKs). J Mol Cell Cardiol 2015; 89:335-48; PMID:26454161; http://dx.doi.org/ 10.1016/j.yjmcc.2015.10.006 [DOI] [PubMed] [Google Scholar]
- [106].Sachse CC, Kim YH, Agsten M, Huth T, Alzheimer C, Kovacs DM, Kim DY. BACE1 and presenilin/gamma-secretase regulate proteolytic processing of KCNE1 and 2, auxiliary subunits of voltage-gated potassium channels. FASEB J 2013; 27:2458-67; PMID:23504710; http://dx.doi.org/ 10.1096/fj.12-214056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Murray CI, Westhoff M, Eldstrom J, Thompson E, Emes R, Fedida D. Unnatural amino acid photo-crosslinking of the IKs channel complex demonstrates a KCNE1:KCNQ1 stoichiometry of up to 4:4. Elife 2016; 5:e11815; PMID:26802629; http:/dx.doi.org/ 10.7554/eLife.11815.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Nakajo K, Ulbrich MH, Kubo Y, Isacoff EY. Stoichiometry of the KCNQ1 - KCNE1 ion channel complex. Proc Natl Acad Sci U S A 2010; 107:18862-7; PMID:20962273; http://dx.doi.org/ 10.1073/pnas.1010354107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Morin TJ, Kobertz WR. Counting membrane-embedded KCNE beta-subunits in functioning K+ channel complexes. Proc Natl Acad Sci U S A 2008; 105:1478-82; PMID:18223154; http://dx.doi.org/ 10.1073/pnas.0710366105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Plant LD, Xiong D, Dai H, Goldstein SA. Individual IKs channels at the surface of mammalian cells contain two KCNE1 accessory subunits. Proc Natl Acad Sci U S A 2014; 111:E1438-46; PMID:24591645; http://dx.doi.org/ 10.1073/pnas.1323548111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Yu H, Lin Z, Mattmann ME, Zou B, Terrenoire C, Zhang H, Wu M, McManus OB, Kass RS, Lindsley CW, et al.. Dynamic subunit stoichiometry confers a progressive continuum of pharmacological sensitivity by KCNQ potassium channels. Proc Natl Acad Sci U S A 2013; 110:8732-7; PMID:23650380; http://dx.doi.org/ 10.1073/pnas.1300684110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Zhang J, Shapiro MS. Activity-dependent transcriptional regulation of M-Type (Kv7) K(+) channels by AKAP79/150-mediated NFAT actions. Neuron 2012; 76:1133-46; PMID:23259949; http://dx.doi.org/ 10.1016/j.neuron.2012.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Yang SG, Wang SW, Zhao M, Zhang R, Zhou WW, Li YN, Su YJ, Zhang H, Yu XL, Liu RT. A Peptide Binding to the beta-Site of APP Improves Spatial Memory and Attenuates Abeta Burden in Alzheimer's Disease Transgenic Mice. PLoS One 2012; 7:e48540; PMID:23133641; http://dx.doi.org/ 10.1371/journal.pone.0048540 [DOI] [PMC free article] [PubMed] [Google Scholar]