

Abstract
Nitroxyl (HNO) is a molecule of significant interest due to its unique pharmacological properties, particularly within the cardiovascular system. A large portion of HNO biological effects can be attributed to its reactivity with protein thiols, where it can generate disulfide bonds. Evidence from studies in erythrocytes suggests that the activity of GLUT1 is enhanced by the formation of an internal disulfide bond. However, there are no reports that document the effects of HNO on glucose uptake. Therefore, we examined the acute effects of Angeli’s salt (AS), a HNO donor, on glucose uptake activity of GLUT1 in L929 fibroblast cells. We report that AS stimulates glucose uptake with a maximum effective concentration of 5.0 mM. An initial 7.2-fold increase occurs within 2 min, which decreases and plateaus to a 4.0-fold activation after 10 min. About 60% of the 4.0-fold activation recovers within 10 min, and 40% remains after an hour. The activation is blocked by the pretreatment of cells with thiol-reactive compounds, iodoacetamide (0.75 mM), cinnamaldehyde (2.0 mM), and phenylarsine oxide (10 μM). The effects of AS are not additive to the stimulatory effects of other acute activators of glucose uptake in L929 cells, such as azide (5 mM), berberine (50 μM), or glucose deprivation. These data suggest that GLUT1 is acutely activated in L929 cells by the formation of a disulfide bond, likely within GLUT1 itself.
Keywords: Angeli’s salt, Nitroxyl (HNO), GLUT1, Glucose uptake, L929 fibroblast cells
1. Introduction
The unique biological effects of nitroxyl (HNO) and its potential to be a cell-signaling molecule distinct from nitric oxide (NO) are receiving increasing attention [1–6]. For example, HNO has documented efficacy for the treatment of heart failure [7–11] and alcoholism [12]. The physiological effects of HNO appear to be mediated by its reactions with hemeproteins and with thiols [1,2,13,14]. In particular, the propensity of HNO to react with biological thiols, such as cysteine residues, is well established. It can react with a single cysteine residue to produce a sulfinamide as the endproduct, or it can react with two cysteine residues to produce a disulfide. This chemistry accounts for the observations that HNO inhibits aldehyde dehydrogenase [12,15], glyceraldehyde-3-phosphate dehydrogenase [16,17], cysteine proteases such as papain [18] and cathepsin B [19], and tubulin polymerization [20]. There has been significant interest in HNO’s unique and positive effects on cardiac muscle contractility [1,4]. HNO has been shown to react with several sarcoplasmic proteins involved in Ca2+ release and uptake, including the ryanodine receptor [21], Ca2+-ATPase (SERCA) [11], and phospholamban [9], a protein that regulates SERCA. It is not clear, at this time, if HNO is produced by cells as a physiological signal or if it is purely a pharmacological agent [2,6].
The ubiquitously expressed glucose transporter GLUT1 long thought to be responsible only for basal glucose uptake. However, there is mounting evidence that GLUT1 can be acutely activated. Cell stressors such as azide [22,23], osmotic stress [24,25], methylene blue [26], C-peptide [27] and glucose deprivation [28,29] all increase the transport activity of GLUT1. In contrast to GLUT4 activation, the activation of GLUT1 occurs without a change in the membrane concentration of the transporter [22]. Currently it is not clear what alteration in GLUT1 structure accounts for its enhanced activity. Based on work in erythrocytes, the Carruthers’ lab [30–34] suggests that GLUT1 is activated by the formation of an internal disulfide bond. This triggers a conformational change in GLUT1 and leads to the oligomerization (tetramer) and activation of the transporter. This mechanism of activation has not been confirmed in other cell lines. However, in support of this mechanism, we have found that two thiol-active agents, phenylarsine oxide [35] and cinnamaldehyde [36], activate glucose uptake in L929 fibroblast cells, a cell line that express only the GLUT1 isoform [37]. If GLUT1 is activated in L929 cells in a manner similar to erythrocytes, we would predict that HNO would trigger a disulfide bond formation in GLUT1, which appears to be the preferred HNO-induced product in a hydrophobic environment [38], and thereby activate glucose uptake. Therefore, the specific purpose of this study was to systematically investigate the acute effects of Angeli’s salt (AS), an in situ producer of HNO, on glucose uptake in L929 fibroblast cells.
2. Materials and Methods
2.1. Chemicals
AS was a generous gift of Dr. John P. Toscano (Johns Hopkins University). Phenylarsine oxide (PAO), cinnamaldehyde, cysteine, iodoacetamide, berberine, 2-deoxy-D-glucose-[1,2-3H] (2DG) and D-mannitol-1-14C were purchased from the Sigma–Aldrich Chemical Company (St. Louis, MO, USA).
2.2. Cell culture
L929 mouse fibroblast cells were obtained from the American Type Culture Collection. To initiate each experiment, approximately 1.0 × 105 L929 fibroblast cells were plated into each well of a 24-well culture-treated plate in 1.0 mL of low-glucose (5.5 mM) DMEM (Dulbecco’s Modified Eagle Medium) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. The cells were grown overnight at 37 °C in an incubator supplied with humidified room air with 5% CO2.
2.3. General experimental design
In general, experiments consisted of either one or two phases, either a treatment phase followed by a glucose uptake phase, or only the glucose uptake phase. Times for each phase are indicated in the figure legends. To initiate an experiment with a treatment phase, the media from cells in 24-well plates were replaced with 0.4 mL of fresh treatment media consisting of either low-glucose DMEM alone (0% FBS) (basal), low-glucose DMEM plus the chemical of interest (see figure and table legends), or glucose-free DMEM (0% FBS) (activation by glucose deprivation). Cells were maintained at 37 °C for the times indicated. If there was no treatment phase, cells were immediately incubated in glucose uptake media (see below). AS was stored at −4 °C under nitrogen. In all experiments using AS, the solid compound was quickly dissolved in media at room temperature and distributed to the cells in the 24-well plate (process took about 30 s). The cells were then returned to the incubator and the media allowed to warm to 37 °C. All other reagents were added to the media from 100 to 200× stock aqueous (sodium azide, iodoacetamide, cysteine), ethanol (cinnamaldehyde) or DMSO (berberine, phenylarsine oxide) solutions. Ethanol and DMSO have no effect on glucose uptake [35,36].
In experiments designed to measure recovery from the effects of AS, the treatment phase was followed by a recovery period in which cells were washed and returned to low-glucose media (0.8 mL) without AS for varying times as indicated in figure legend. Glucose uptake was measured as described below.
2.4. Glucose uptake assay
Glucose uptake was measured using the radiolabeled glucose analog 2-deoxyglucose (2DG) as previously described [39]. Briefly, the media was replaced with 0.2 mL of glucose-free HEPES buffer (140 mM NaCl, 5 mM KCl, 20 mM HEPES/Na pH = 7.4, 2.5 mM MgSO4, 1 mM CaCl2, 2 mM NaPyruvate, 1 mM mannitol) supplemented with 1.0 mM (0.3 μCi/mL) 2-DG (1,2-3H) and 1.0 mM (0.02 μCi/mL) mannitol (1-14C). Uptake media was supplemented with additional compounds, such as AS, as indicated in the figure and table legends. After a 10-min incubation, cells were washed twice with cold glucose-free HEPES. The cells were lysed in 0.5 mL lysis buffer (10 mM Tris pH = 7.4, 150 mM NaCl, 5 mM EDTA, 1.0% triton X-100, 0.4% SDS) and the 3H-2 DG uptake with 14C-mannitol as the extracellular marker was measured using scintillation spectrometry. Uptake of 14C-mannitol only occurs if the cell membrane is compromised. Therefore, the use of a double-labeled uptake solution allows us to both measure surface binding and monitor potential toxic effects of the experimental treatments that would compromise the cell membrane.
2.5. Statistical analysis
Experimental conditions were repeated in triplicate or quadruplicate and glucose uptake was measured and reported as nmol/10 min/well ± standard error. Statistical significance was determined by either ANOVA followed by a post-hoc Dunnett test or a two-tailed t-test. Statistical significance is reported at P < 0.01 or P < 0.05. Experiments were repeated several times and results from representative experiments are reported.
3. Results
3.1. Angeli’s salt activates glucose uptake in a dose-dependent manner
Experiments with erythrocytes suggest that GLUT1 can be activated by internal disulfide bond formation, which leads to an oligomerization of the transporter [32,33]. Because AS stimulates disulfide bond formation via an in situ release of HNO [1,2], we were curious to determine if AS could activate glucose uptake in L929 fibroblast cells, which express only GLUT1 [37]. In our initial experiment, L929 fibroblast cells were exposed to 10 mM Angeli’s salt either (1) only during a 30-min treatment phase, (2) during the treatment phase and again during the 10-min uptake phase, or (3) only during the uptake phase. We chose this AS concentration based on previous work, which had shown that AS in the range of 0.25–10 mM stimulated the dimerization of the cardiac sacroplasmic reticulum protein phospholamban [9]. The results, shown in Fig. 1, reveal that the greatest activation of glucose uptake by L929 fibroblast cells is observed when AS is included only during the uptake phase (uptake increased 4.2×). In a subsequent experiment, the dose dependency of glucose uptake was determined by exposing cells to concentrations of AS ranging from 0 to 20 mM only during the 10-min uptake. The results are shown in Fig. 2. There was a significant 2.5× activation of uptake (P < 0.01) at 1.0 mM, which reached a 3.8× maximum at 5.0 mM. There was a small, but significant decrease in the activation at 20 mM AS. AS did not produce any visible toxic effects, as assessed by cell morphology and cell attachment, at any concentration tested.
Fig. 1.
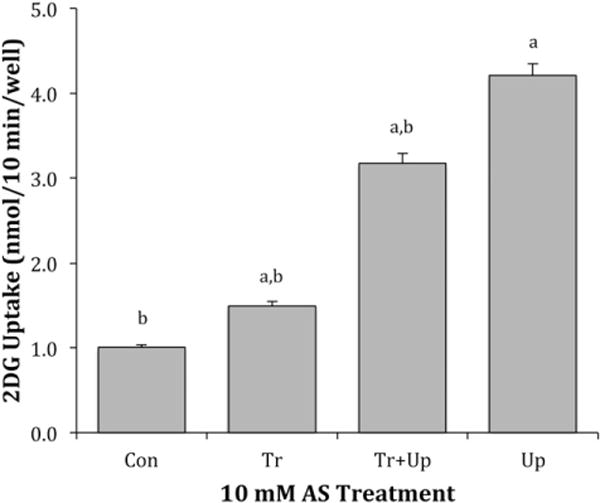
Effects of AS on 2DG uptake. L929 fibroblast cells were incubated at 37 °C for 30 min in DMEM media (5.5 mM glucose) with or without 10.0 mM AS. Ten-minute 2DG uptakes were then measured as described in materials and methods in the presence and absence of 10 mM AS. Con (control) represents cells not exposed to AS, (Tr) represent cells exposed with AS during the 30-min treatment phase, (Tr + Up) represents cells treated to AS during both the treatment and uptake phase, and (Up) represents cells exposed to AS only during the 10-min uptake phase. Data are means ± S.E from four wells. aSignificantly greater than control and bsignificantly different than maximum effect (Up) at P < 0.01.
Fig. 2.
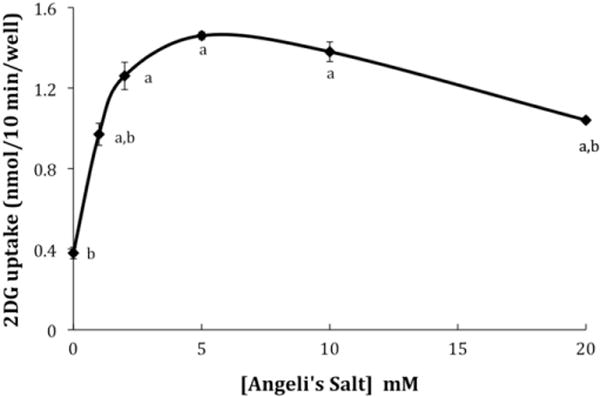
Dose-dependent effects of AS on 2DG uptake. Ten-minute 2DG uptakes were measured in the presence of AS concentrations ranging from 0 to 20 mM as described in materials and methods. Data are means ± S.E. of four wells from a representative experiment. aSignificantly different than 0 mM exposure to AS and bsignificantly different than maximum effect at 5.0 mM AS at P < 0.01.
3.2. Control experiments
AS decomposes with a half-life of about 2.5 min to release HNO and nitrite [3]. HNO itself is not stable, but forms a dimer that decomposes to nitrous oxide (N2O) and water [2]. To insure that the activity that we were observing was a result of HNO rather than nitrite, we compared the stimulatory effects of 5.0 mM AS when it was added to uptake media just prior to measurement of glucose uptake to its effects when it was incubated in the uptake media overnight (room temperature), allowing AS to decompose. The results, normalized to control, are shown in Fig. 3. The overnight incubation did reduce uptake from 3.04 ± 0.01 times control to 2.19 ± 0.06, but surprisingly, uptake remained significantly elevated over control. This residual activity could not be attributed to nitrite, because in a separate experiment, 5 mM nitrite had no effect on glucose uptake (see Fig. 3). However, incubating the AS with excess cysteine overnight to react with HNO did completely block the stimulatory effects of AS (Fig. 3). Interestingly, cysteine’s ability to HNO at this concentration (2× concentration of AS) appears to be time dependent. Adding cysteine with fresh AS to cells only modestly reduced the stimulatory effect of AS (data not shown). Higher concentrations of cysteine may be required to scavenge HNO more quickly [3].
Fig. 3.
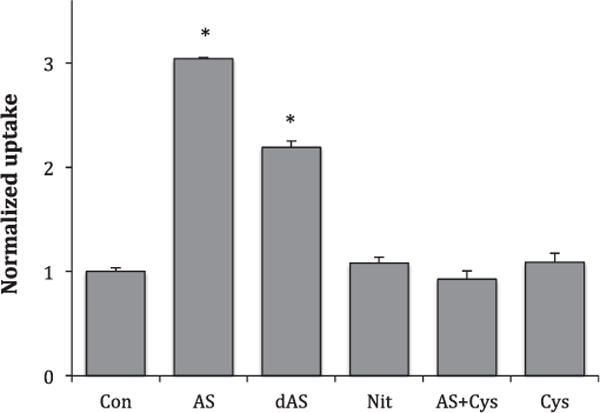
Control experiments. 2DG uptakes were measured as described in materials and methods in the presence of 5.0 mM AS added just before uptake (AS), 5.0 mM AS allowed to decompose overnight at room temperature (dAS), 5.0 mM sodium nitrite (Nit), 5.0 mM AS plus 10.0 mM cysteine incubated overnight (AS + Cys), or 10.0 mM cysteine (Cys). Data are means ± S.E. normalized to control for 10–14 samples. *Significantly elevated from control at P < 0.01.
3.3. Time course of activation and recovery of AS-activated glucose uptake
It seems possible that the much lower activation observed when AS was placed only in the 30-min treatment phase (see Fig. 1) could actually represent activation followed by recovery as HNO is depleted. Therefore, to explore time course of activation, L929 cells were exposed to 5 mM AS in the radioactive uptake buffer for times ranging from 2 to 20 min. The results, expressed as nmols glucose uptake per minute, are shown in Fig. 4. Uptake at time represents the uptake per minute for control cells that had been exposed to uptake media without AS for 20 min. The data shows a burst of activation in the first 3 min followed by a steady rate of uptake that remains elevated (4.0×) over control for the remainder of the 20 min. This burst of activity correlates well with the half-life for the release of HNO from AS (2.5 min).
Fig. 4.
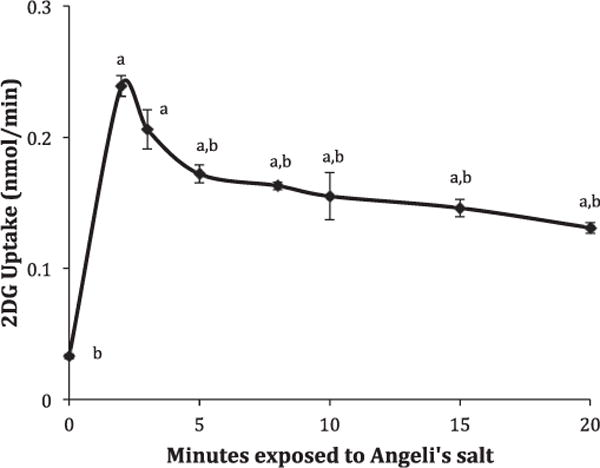
Time course for AS activation of 2DG uptake. 2DG uptakes in the presence of 5.0 mM AS were measured for 2, 3, 5, 8, 10, 15, and 20 min as described in materials and methods and expressed as the average nmol 2DG/minute ± S.E from four wells of a representative experiment. The uptake at 0 min exposed to AS represents the per minute-uptake for a 20-min uptake for cells not exposed to AS. aSignificantly different than zero time exposure to AS and bsignificantly different than maximum effect at 2 min exposure to AS at P < 0.01.
To measure the recovery from the effects of AS, L929 cells were exposed to AS for 10 min, then incubated in fresh DMEM media (5.5 mM glucose) without AS. Uptakes were measured either immediately or after 5, 10, 25 or 50 min. As shown in Fig. 5, about 60% of the activity recovered within 10 min, but the remaining 40% was maintained even after 50 min. This experiment was repeated multiple times and all results revealed a fast recovery phase lasting between 5 and 10 min with a residual activation of 20–50% that lasted for up to an hour.
Fig. 5.
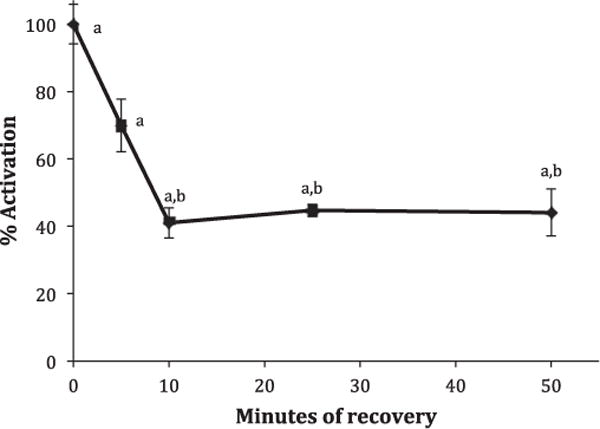
Effect of recovery time on AS-activated 2DG uptake. L929 fibroblast cells were incubated at 37 °C for 10 min in DMEM media (5.5 mM glucose) supplemented with 5.0 mM AS. Ten-minute 2DG uptakes were then measured immediately or media were replaced with AS-free media and uptakes were measured after 5, 10, 25, or 50 min. Data are expressed as a percentage of full activation (0 min recovery) ± S.E. from three wells of a representative experiment. aSignificantly different than basal uptake (0% activation) and bsignificantly different than 100% activation at P < 0.01.
3.4. Effects of pretreatment with thiol-reactive compounds on AS activation
If the activation of glucose uptake by AS is triggered by a reaction of HNO with thiols, we ought to be able to inhibit the activation by pretreating cells with a thiol-reactive compound. To test this, we pretreated L929 cells with 0.75 mM iodoacetamide for 20 min before stimulating with AS. The results are shown in Fig. 6. Pretreatment with iodoacetamide by itself did not affect glucose uptake, but essentially completely inhibited activation by AS. When iodoacetamide was incubated overnight (room temperature) with an equal concentration of cysteine, the iodoacetamide no longer inhibited AS activation of glucose uptake. Cysteine itself did not alter glucose uptake (see Fig. 3).
Fig. 6.
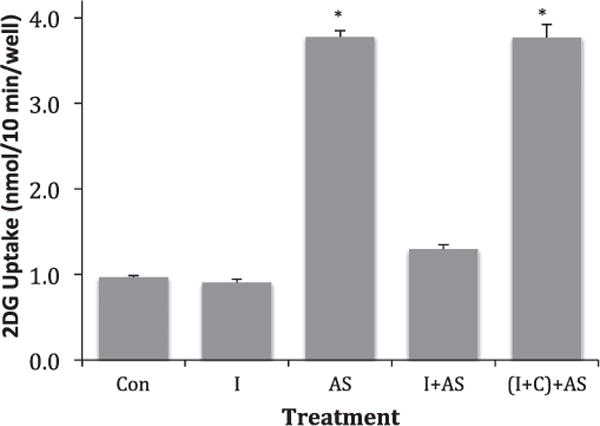
Effect of iodoacetamide on AS-activated 2DG uptake. L929 fibroblast cells were treated for 20 min at 37 °C in DMEM media (5.5 mM glucose) alone or supplemented with 0.75 mM iodoacetamide or supplemented with a mixture of 0.75 mM iodoacetamide and 0.75 mM cysteine that had been incubated overnight. Ten-minute 2DG uptakes were then measured in the presence and absence of 5 mM AS. (Con) represents control cells not treated with iodoacetamide or AS, (I) represents cells exposed to iodoacetamide during the 20-min treatment, (AS) represents cells exposed to AS during the 10-min uptake, (I + AS) represents cells pre-treated with iodoacetamide and exposed to AS during the uptake phase, and ((I + C)+AS) represents cells pretreated with an iodoacetamide–cysteine mixture and then exposed to AS. Data are means ± S.E. from four wells of a representative experiment. *Significantly elevated from control at P < 0.01.
We also investigated the effects of two other thiol-reactive compounds, cinnamaldehyde (CA) and phenylarsine oxide (PAO). Previous work had shown that these two agents can activate glucose uptake in L929 cells, but that they also inhibit subsequent activation by more robust activators of glucose uptake [35,36]. We treated cells with either 2 mM CA or 10 μM PAO for 30 min and then measured glucose uptake in the presence and absence of 5 mM AS. The results are shown in Table 1. As previously reported, treatment with either CA or PAO activated glucose uptake [35,36], but not to the same degree as AS. However, treatment with CA or PAO prior to treatment with AS blocked the more robustly activating effects of AS.
Table 1.
Effects of CA and PAO treatment on AS activation of glucose uptake.
| Treatment | 2DG uptake (w/o AS) (nmol/10 min/well) | 2DG uptake (+5.0 mM AS) (nmol/10 min/well) |
|---|---|---|
| Control | 1.41 ± 0.03 | 2.84 ± 0.08 |
| 2.0 mM CA | 1.99 ± 0.07* | 1.51 ± 0.15* |
| 10.0 μM PAO | 2.17 ± 0.06* | 2.09 ± 0.09# |
L929 fibroblast cells were incubated for 30 minutes at 37° in DMEM media (5.5 mM glucose) plus either no additions (control), 2.0 mM CA or 10.0 μM PAO. Glucose uptake was measured as described in materials and methods in the presence and absence of 5.0 mM AS. Data are the means from quadruplicate samples of 2DG uptake in nmol/10 min/well ± standard error. Significance from respective control at P < 0.01 (*), or at P < 0.05 (#).
3.5. Combined effects of activators on glucose uptake
We had previously shown in L929 fibroblast cells that glucose uptake is acutely activated by multiple agents [26,29,40]. In order to gain some insight into to the activation pathway of AS, we conducted a series of additivity experiments with three of these activators: azide, berberine and glucose deprivation. L929 cells were incubated for 30 min with maximally effective concentrations of sodium azide (5 mM), berberine (50 μM), or media lacking glucose. Uptakes were then measured in the presence and absence of maximally effective concentration of AS (5.0 mM). The results are shown in Table 2. As expected, each stimulant significantly activated glucose uptake. However, AS did not significantly increase the effects of the other stimulants. These results suggest that the stimulalants, azide, berberine, glucose deprivation, and AS, share a common, rate-determining step in their mechanisms of action.
Table 2.
Combined effects of AS with other activators.
| [AS] | Control | +Azide | +Berb | +No glucose |
|---|---|---|---|---|
| 0 mM | 0.59 ± 0.03 | 1.88 ± 0.06* | 1.74 ± 0.08* | 2.82 ± 0.07* |
| 5.0 mM | 1.83 ± 0.09# | 2.21 ± 0.09 | 1.88 ± 0.04 | 3.00 ± 0.10 |
L929 fibroblast cells were incubated for 30 minutes at 37° in DMEM media containing either 0 mM glucose (no glucose), or 5.5 mM glucose containing no additions (control), 5.0 mM sodium azide, or 50 μM berberine. Glucose uptake was measured as described in materials and methods in the presence and absence of 5.0 mM AS. Data are the means from quadruplicate samples of 2DG uptake in nmol/10 min/well ± standard error. Significance at P ± 0.01 from 0 mM AS control (*), or from respective conditions without AS (#).
4. Discussion
The role of nitric oxide (NO) as a biological second messenger has been well established leading to increased interest in the physiological action of other redox forms of NO [3]. HNO, the product of a one-electron reduction of NO, in particular has garnered much attention because it has interesting pharmacological properties that differ distinctly from NO [1–5,41–43]. Of particular interest is the reaction of HNO with thiols. HNO can react with a thiol to produce an N-hydroxysulfenamide which can either rearrange to form a sulfinamide or react with a second thiol to produce a disulfide plus hydroxylamine [2]. This chemistry appears to account for much of the biological activity of HNO [3,44,45].
In this study we report for the first time that AS very quickly activates glucose transport activity in L929 fibroblast cells, which express only the GLUT1 isoform of the GLUT family [37]. A 7.2-fold increase in activity occurs within 2 min of exposure to AS (see Fig. 4), which decreases to an average 4.0-fold activation after 10 min (see Figs. 1, 2 and 4). The maximum effective concentration is about 5.0 mM and higher concentrations (20 mM) reduce this activation, likely due to additional reactions of HNO (see dose-dependent data in Fig. 2 and the twice-exposed data in Fig. 1). GLUT1 activation by AS can be blocked by three thiol-active reagents: iodoacetamide, cinnamaldehye, and phenylarsine oxide (Fig. 6, Table 1), suggesting the activation is caused by a reaction of HNO with cysteine residues. Possible targets for HNO are cysteine residues within GLUT1 itself. Previous work in erythrocytes has shown that GLUT1 can be activated by disulfide bond formation within GLUT1 [31], and computational studies have indicated that within a hydrophobic environment such as the cell membrane, the disulfide product of HNO reaction with cysteine residues is the preferred product over the sulfinamide [38]. It is not known if HNO is the actual agent responsible for physiological activation of GLUT1 or if this is purely a pharmacological phenomenon. The millimolar concentrations required to activate GLUT1 are higher than the concentrations needed to inhibit GAP [16,17], but are similar to the range of concentrations shown to enhance cardiac function [11]. Regardless, if HNO enhances glucose uptake via activation of GLUT1 in cardiac tissue as well as L929 fibroblast cells, that would be an added benefit in the use of HNO donors in the treatment of heart failure. While HNO appears to enhance cardiac contractility by affecting calcium concentrations, and calcium is also known to be involved in the regulation of glucose uptake in some cell types, this does not appear to be the mechanism of AS activation of glucose uptake in L929 cells. Previous work in this cell line has shown that altering calcium concentrations in the media and treatment with dantrolene does not affect glucose uptake [39]. Future investigations should be done to determine if HNO does indeed activate glucose uptake in cardiac cells.
Angeli’s salt can produce nitric oxide as well as HNO [5]. This study does not directly test for the potential involvement of the nitric oxide signaling pathway in AS action. However, it seems unlikely that the causative agent for the activation of glucose uptake in these cells is nitric oxide. A previous study showed that the nitric oxide donor, sodium nitroprusside, did activate glucose uptake in L929 cells, but it was about half as effective as AS (2× activation rather than 4×) and required a much longer activation time (30–45 min) [39].
In addition to the interesting potential pharmacological effects of AS on glucose uptake, this study also sheds light on a potential mechanism for the acute activation of GLUT1 in L929 fibroblast cells. Once thought to be responsible only for basal glucose uptake, it has been shown by a number of studies that this ubiquitously expressed transporter can be acutely activated by a variety of reagents [22–29]. It is known that the activation of GLUT1 occurs without a change in the concentration of the transporter in the plasma membrane [22]. However, the change in GLUT1 that accounts for its increased activity has not been established. One intriguing possible mechanism, based on Carruthers’ results with erythrocytes [30–34] and consistent with our data in L929 fibroblast cells, is outlined in Fig. 7. This model proposes that GLUT1 can exist in multiple states ranging from monomers containing reduced thiols to oligomers stabilized by internal disulfide bond formation. The reduced monomer displays the lowest activity and the oligomer the highest. Carruthers has demonstrated in erthryocytes that an internal disulfide bond forms between Cys 347 and Cys 421 that stabilizes the more active GLUT1 oligomer, likely a tetramer [31,32]. Furthermore, they have shown that Cys 421 can be modified by iodoacetamide. While our study in L929 cells does not provide direct evidence that GLUT1 is the target for HNO, the ability to block HNO activation with iodoacetamide suggest that activation requires a disulfide bond formation within some protein. The speed at which activation occurs makes it attractive to suggest that GLUT1 itself is the target. Also, cinnamaldehye, a known Michael acceptor for thiols, partially activates glucose uptake, but prevents the full, subsequent activation by AS. This model explains these observations by suggesting that cinnamaldehye adds to a thiol in GLUT1, partially activating the transporter, but effectively blocking the disulfide bond formation required for full activation of GLUT1 [36]. In addition, phenylarsine oxide, a substance that reacts with vicinal thiols also activates glucose uptake, but prevents full activation by AS. Interestingly, PAO is typically a more robust activator than cinnamaldehye, and like AS, activates glucose uptake within minutes [35]. This suggests that the reaction of PAO with vicinal thiols mimics a disulfide bond. It may be that the bulky phenyl ring prevents stabilization of the tetramer required for full activation of GLUT1.
Fig. 7.
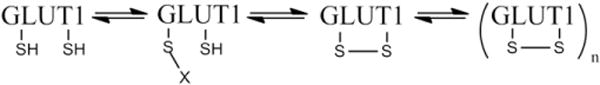
Model mechanism for activation of GLUT1. In this model for activation of GLUT1 in L929 fibroblast cells, GLUT1 exists in a number of states in the membrane ranging from a fully reduced monomer form, which would have the lowest transport activity, to an oligomer (tetramers), which would have the highest activity.
We also show that AS does not further enhance the activation of other, slower acting stimulators such as sodium azide, berberine, or glucose deprivation (see Table 2). A logical interpretation of this data is that these other methods of activation also stimulate disulfide bond formation. Future studies should look for more direct evidence of disulfide bond formation in GLUT1 and subsequent oligomerization.
5. Conclusions
This study demonstrates that AS, likely through the release of HNO and subsequent disulfide bond formation, very quickly activates the transport activity of GLUT1. These data are consistent with a proposed mechanism for GLUT1 activation that involves the formation of a disulfide bond within GLUT1.
Acknowledgments
This research was supported by a NIH R15 grant (DK08193-1A1). Special thanks to Darla McCarthy for her critique of this manuscript.
References
- 1.Fukuto JM, Bianco CL, Chavez TA. Nitroxyl (HNO) signaling. Free Radic Biol Med. 2009;47:1318–1324. doi: 10.1016/j.freeradbiomed.2009.06.014. [DOI] [PubMed] [Google Scholar]
- 2.Fukuto JM, Carrington SJ. HNO signaling mechanisms. Antioxid Redox Signal. 2011;14:1649–1657. doi: 10.1089/ars.2010.3855. [DOI] [PubMed] [Google Scholar]
- 3.Irvine JC, Ritchie RH, Favaloro JL, Andrews KL, Widdop RE, Kemp-Harper BK. Nitroxyl (HNO): the Cinderella of the nitric oxide story. Trends Pharmacol Sci. 2008;29:601–608. doi: 10.1016/j.tips.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 4.Switzer CH, Flores-Santana W, Mancardi D, Donzelli S, Basudhar D, Ridnour LA, Miranda KM, Fukuto JM, Paolocci N, Wink DA. The emergence of nitroxyl (HNO) as a pharmacological agent. Biochim Biophys Acta. 2009;1787:835–840. doi: 10.1016/j.bbabio.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paolocci N, Jackson MI, Lopez BE, Miranda K, Tocchetti CG, Wink DA, Hobbs AJ, Fukuto JM. The pharmacology of nitroxyl (HNO) and its therapeutic potential: not just the Janus face of NO. Pharmacol Ther. 2007;113:442–458. doi: 10.1016/j.pharmthera.2006.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reisz JA, Bechtold E, King SB. Oxidative heme protein-mediated nitroxyl (HNO) generation. Dalton Trans. 2010;39:5203–5212. doi: 10.1039/c000980f. [DOI] [PubMed] [Google Scholar]
- 7.Dai T, Tian Y, Tocchetti CG, Katori T, Murphy AM, Kass DA, Paolocci N, Gao WD. Nitroxyl increases force development in rat cardiac muscle. J Physiol. 2007;580:951–960. doi: 10.1113/jphysiol.2007.129254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feelisch M. Nitroxyl gets to the heart of the matter. Proc Natl Acad Sci U S A. 2003;100:4978–4980. doi: 10.1073/pnas.1031571100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Froehlich JP, Mahaney JE, Keceli G, Pavlos CM, Goldstein R, Redwood AJ, Sumbilla C, Lee DI, Tocchetti CG, Kass DA, Paolocci N, Toscano JP. Phospholamban thiols play a central role in activation of the cardiac muscle sarcoplasmic reticulum calcium pump by nitroxyl. Biochemistry. 2008;47:13150–13152. doi: 10.1021/bi801925p. [DOI] [PubMed] [Google Scholar]
- 10.Paolocci N, Saavedra WF, Miranda KM, Martignani C, Isoda T, Hare JM, Espey MG, Fukuto JM, Feelisch M, Wink DA, Kass DA. Nitroxyl anion exerts redox-sensitive positive cardiac inotropy in vivo by calcitonin gene-related peptide signaling. Proc Natl Acad Sci U S A. 2001;98:10463–10468. doi: 10.1073/pnas.181191198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tocchetti CG, Wang W, Froehlich JP, Huke S, Aon MA, Wilson GM, Di Benedetto G, O’Rourke B, Gao WD, Wink DA, Toscano JP, Zaccolo M, Bers DM, Valdivia HH, Cheng H, Kass DA, Paolocci N. Nitroxyl improves cellular heart function by directly enhancing cardiac sarcoplasmic reticulum Ca2+ cycling. Circ Res. 2007;100:96–104. doi: 10.1161/01.RES.0000253904.53601.c9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeMaster EG, Redfern B, Nagasawa HT. Mechanisms of inhibition of aldehyde dehydrogenase by nitroxyl, the active metabolite of the alcohol deterrent agent cyanamide. Biochem Pharmacol. 1998;55:2007–2015. doi: 10.1016/s0006-2952(98)00080-x. [DOI] [PubMed] [Google Scholar]
- 13.Kumar MR, Fukuto JM, Miranda KM, Farmer PJ. Reactions of HNO with heme proteins: new routes to HNO–heme complexes and insight into physiological effects. Inorg Chem. 2010;49:6283–6292. doi: 10.1021/ic902319d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller TW, Cherney MM, Lee AJ, Francoleon NE, Farmer PJ, King SB, Hobbs AJ, Miranda KM, Burstyn JN, Fukuto JM. The effects of nitroxyl (HNO) on soluble guanylate cyclase activity: interactions at ferrous heme and cysteine thiols. J Biol Chem. 2009;284:21788–21796. doi: 10.1074/jbc.M109.014282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shoeman DW, Shirota FN, DeMaster EG, Nagasawa HT. Reaction of nitroxyl, an aldehyde dehydrogenase inhibitor, with N-acetyl-L-cysteine. Alcohol. 2000;20:55–59. doi: 10.1016/s0741-8329(99)00056-7. [DOI] [PubMed] [Google Scholar]
- 16.Lopez BE, Rodriguez CE, Pribadi M, Cook NM, Shinyashiki M, Fukuto JM. Inhibition of yeast glycolysis by nitroxyl (HNO): mechanism of HNO toxicity and implications to HNO biology. Arch Biochem Biophys. 2005;442:140–148. doi: 10.1016/j.abb.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 17.Lopez BE, Wink DA, Fukuto JM. The inhibition of glyceraldehyde-3-phosphate dehydrogenase by nitroxyl (HNO) Arch Biochem Biophys. 2007;465:430–436. doi: 10.1016/j.abb.2007.06.017. [DOI] [PubMed] [Google Scholar]
- 18.Vaananen AJ, Kankuri E, Rauhala P. Nitric oxide-related species-induced protein oxidation: reversible, irreversible, and protective effects on enzyme function of papain. Free Radic Biol Med. 2005;38:1102–1111. doi: 10.1016/j.freeradbiomed.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 19.Vaananen AJ, Salmenpera P, Hukkanen M, Miranda KM, Harjula A, Rauhala P, Kankuri E. Persistent susceptibility of cathepsin B to irreversible inhibition by nitroxyl (HNO) in the presence of endogenous nitric oxide. Free Radic Biol Med. 2008;45:749–755. doi: 10.1016/j.freeradbiomed.2008.05.025. [DOI] [PubMed] [Google Scholar]
- 20.Landino LM, Koumas MT, Mason CE, Alston JA. Modification of tubulin cysteines by nitric oxide and nitroxyl donors alters tubulin polymerization activity. Chem Res Toxicol. 2007;20:1693–1700. doi: 10.1021/tx7001492. [DOI] [PubMed] [Google Scholar]
- 21.Cheong E, Tumbev V, Abramson J, Salama G, Stoyanovsky DA. Nitroxyl triggers Ca2+ release from skeletal and cardiac sarcoplasmic reticulum by oxidizing ryanodine receptors. Cell Calcium. 2005;37:87–96. doi: 10.1016/j.ceca.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 22.Shetty M, Loeb JN, Vikstrom K, Ismail-Beigi F. Rapid activation of GLUT-1 glucose transporter following inhibition of oxidative phosphorylation in clone 9 cells. J Biol Chem. 1993;268:17225–17232. [PubMed] [Google Scholar]
- 23.Rubin D, Ismail-Beigi F. Distribution of Glut1 in detergent-resistant membranes (DRMs) and non-DRM domains: effect of treatment with azide. Am J Physiol Cell Physiol. 2003;285:C377–C383. doi: 10.1152/ajpcell.00060.2003. [DOI] [PubMed] [Google Scholar]
- 24.Barnes K, Ingram JC, Porras OH, Barros LF, Hudson ER, Fryer LG, Foufelle F, Carling D, Hardie DG, Baldwin SA. Activation of GLUT1 by metabolic and osmotic stress: potential involvement of AMP-activated protein kinase (AMPK) J Cell Sci. 2002;115:2433–2442. doi: 10.1242/jcs.115.11.2433. [DOI] [PubMed] [Google Scholar]
- 25.Barros LF, Barnes K, Ingram JC, Castro J, Porras OH, Baldwin SA. Hyper-osmotic shock induces both activation and translocation of glucose transporters in mammalian cells. Pflugers Arch. 2001;442:614–621. doi: 10.1007/s004240100577. [DOI] [PubMed] [Google Scholar]
- 26.Louters LL, Dyste SG, Frieswyk D, Tenharmsel A, Vander Kooy TO, Walters L, Whalen T. Methylene blue stimulates 2-deoxyglucose uptake in L929 fibroblast cells. Life Sci. 2006;78:586–591. doi: 10.1016/j.lfs.2005.05.082. [DOI] [PubMed] [Google Scholar]
- 27.Meyer JA, Froelich JM, Reid GE, Karunarathne WK, Spence DM. Metal-activated C-peptide facilitates glucose clearance and the release of a nitric oxide stimulus via the GLUT1 transporter. Diabetologia. 2008;51:175–182. doi: 10.1007/s00125-007-0853-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kumar A, Xiao YP, Laipis PJ, Fletcher BS, Frost SC. Glucose deprivation enhances targeting of GLUT1 to lipid rafts in 3T3-L1 adipocytes. Am J Physiol Endocrinol Metab. 2004;286:E568–E576. doi: 10.1152/ajpendo.00372.2003. [DOI] [PubMed] [Google Scholar]
- 29.Roelofs B, Tidball A, Lindborg AE, TenHarmsel A, Vander Kooy TO, Louters LL. Acute activation of glucose uptake by glucose deprivation in L929 fibroblast cells. Biochimie. 2006;88:1941–1946. doi: 10.1016/j.biochi.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 30.Graybill C, van Hoek AN, Desai D, Carruthers AM, Carruthers A. Ultra-structure of human erythrocyte GLUT1. Biochemistry. 2006;45:8096–8107. doi: 10.1021/bi060398x. [DOI] [PubMed] [Google Scholar]
- 31.Carruthers A, DeZutter J, Ganguly A, Devaskar SU. Will the original glucose transporter isoform please stand up! Am J Physiol Endocrinol Metab. 2009;297:E836–E848. doi: 10.1152/ajpendo.00496.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zottola RJ, Cloherty EK, Coderre PE, Hansen A, Hebert DN, Carruthers A. Glucose transporter function is controlled by transporter oligomeric structure. A single, intramolecular disulfide promotes GLUT1 tetramerization. Biochemistry. 1995;34:9734–9747. doi: 10.1021/bi00030a011. [DOI] [PubMed] [Google Scholar]
- 33.Hebert DN, Carruthers A. Glucose transporter oligomeric structure determines transporter function. Reversible redox-dependent interconversions of tetrameric and dimeric GLUT1. J Biol Chem. 1992;267:23829–23838. [PubMed] [Google Scholar]
- 34.Pessino A, Hebert DN, Woon CW, Harrison SA, Clancy BM, Buxton JM, Carruthers A, Czech MP. Evidence that functional erythrocyte-type glucose transporters are oligomers. J Biol Chem. 1991;266:20213–20217. [PubMed] [Google Scholar]
- 35.Scott J, Opejin A, Tidball A, Stehouwer N, Rekman J, Louters LL. Dual action of phenylarsine oxide on the glucose transport activity of GLUT1. Chem Biol Interact. 2009;182:199–203. doi: 10.1016/j.cbi.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 36.Plaisier C, Cok A, Scott J, Opejin A, Bushhouse KT, Salie MJ, Louters LL. Effects of cinnamaldehyde on the glucose transport activity of GLUT1. Biochimie. 2011;93:339–344. doi: 10.1016/j.biochi.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liong E, Kong SK, Au KK, Li JY, Xu GY, Lee YL, Kwok TT, Choy YM, Lee CY, Fung KP. Inhibition of glucose uptake and suppression of glucose transporter 1 mRNA expression in L929 cells by tumour necrosis factor-alpha. Life Sci. 1999;65:PL215–220. doi: 10.1016/s0024-3205(99)00408-7. [DOI] [PubMed] [Google Scholar]
- 38.Sherman MP, Grither WR, McCulla RD. Computational investigation of the reaction mechanisms of nitroxyl and thiols. J Org Chem. 2010;75:4014–4024. doi: 10.1021/jo100172t. [DOI] [PubMed] [Google Scholar]
- 39.Van Dyke DA, Walters L, Frieswyk D, Kokmeyer D, Louters LL. Acute effects of troglitazone and nitric oxide on glucose uptake in L929 fibroblast cells. Life Sci. 2003;72:2321–2327. doi: 10.1016/s0024-3205(03)00119-x. [DOI] [PubMed] [Google Scholar]
- 40.Cok A, Plaisier C, Salie MJ, Oram DS, Chenge J, Louters LL. Berberine acutely activates the glucose transport activity of GLUT1. Biochimie. 2011;93:1187–1192. doi: 10.1016/j.biochi.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fukuto JM, Dutton AS, Houk KN. The chemistry and biology of nitroxyl (HNO): a chemically unique species with novel and important biological activity. Chembiochem. 2005;6:612–619. doi: 10.1002/cbic.200400271. [DOI] [PubMed] [Google Scholar]
- 42.Miranda KM, Paolocci N, Katori T, Thomas DD, Ford E, Bartberger MD, Espey MG, Kass DA, Feelisch M, Fukuto JM, Wink DA. A biochemical rationale for the discrete behavior of nitroxyl and nitric oxide in the cardiovascular system. Proc Natl Acad Sci U S A. 2003;100:9196–9201. doi: 10.1073/pnas.1430507100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wink DA, Miranda KM, Katori T, Mancardi D, Thomas DD, Ridnour L, Espey MG, Feelisch M, Colton CA, Fukuto JM, Pagliaro P, Kass DA, Paolocci N. Orthogonal properties of the redox siblings nitroxyl and nitric oxide in the cardiovascular system: a novel redox paradigm. Am J Physiol Heart Circ Physiol. 2003;285:H2264–H2276. doi: 10.1152/ajpheart.00531.2003. [DOI] [PubMed] [Google Scholar]
- 44.Donzelli S, Espey MG, Thomas DD, Mancardi D, Tocchetti CG, Ridnour LA, Paolocci N, King SB, Miranda KM, Lazzarino G, Fukuto JM, Wink DA. Discriminating formation of HNO from other reactive nitrogen oxide species. Free Radic Biol Med. 2006;40:1056–1066. doi: 10.1016/j.freeradbiomed.2005.10.058. [DOI] [PubMed] [Google Scholar]
- 45.Shen B, English AM. Mass spectrometric analysis of nitroxyl-mediated protein modification: comparison of products formed with free and protein-based cysteines. Biochemistry. 2005;44:14030–14044. doi: 10.1021/bi0507478. [DOI] [PubMed] [Google Scholar]