

Abstract
The recent and precipitous increase in opioid analgesic abuse and overdose has inspired investigation of the dopamine D3 receptor (D3R) as a target for therapeutic intervention. Metabolic instability or predicted toxicity has precluded successful translation of previously reported D3R-selective antagonists to clinical use for cocaine abuse. Herein, we report a series of novel and D3R crystal structure-guided 4-phenylpiperazines with exceptionally high D3R affinities and/or selectivities with varying efficacies. Lead compound 19 was selected based on its in vitro profile: D3R Ki = 6.84 nM, 1700 fold D3R versus D2R binding selectivity, and its metabolic stability in mouse microsomes. Compound 19 inhibited oxycodone-induced hyperlocomotion in mice and reduced oxycodone-induced locomotor sensitization. In addition, pretreatment with 19 also dose-dependently inhibited the acquisition of oxycodone-induced conditioned place preference (CPP) in rats. These findings support the D3R as a target for opioid dependence treatment and compound 19 as a new lead molecule for development.
Keywords: Dopamine D3 receptor, antagonist, partial agonist, eticlopride, phenylpiperazine, oxycodone, opiate, opioid addiction, sensitization, locomotor activity, dependence, conditioned place preference
INTRODUCTION
Dopamine is a major neurotransmitter in the central nervous system responsible for many neurological processes, including emotion, cognition, reward, motivation and fine motor control. Dopamine signaling is mediated by D1-like (D1 and D5) and D2-like (D2, D3 and D4) receptor subtypes. Dopamine D3 receptors (D3R) have been implicated as potential pharmacotherapeutic targets for substance use disorders because of their restricted localization to limbic brain regions, effectiveness in animal models of drug abuse and upregulation in the brains of cocaine addicts.1 Inspired by early reports that the D3R appeared to be involved in the reinforcing effects of cocaine and therefore a potential target for drug development,2,3 an enormous effort has been made to develop D3R selective antagonists or partial agonists targeting psychostimulant abuse.4–7 However, the recent discovery that the investigational D3R-selective antagonist, GSK598,809 (1, Fig. 1) caused significant hypertension in dogs in the presence of cocaine, may preclude further development of these agents toward cocaine addiction.8 Nevertheless, efficacy in both animal models of other substance use disorders, including heroin, alcohol and nicotine support continued efforts toward developing D3R-selective antagonists and/or partial agonists that are metabolically stable and have appropriate drug-like properties for potential translation to specific drug abusing populations.1,9,10
Figure 1.

Representative D3R antagonists
The recent availability of the high-resolution structures of G-protein coupled receptors (GPCRs) has aided in structure-based drug design.11,12 Specifically, the high-resolution crystal structure of D3R can be used to guide the design of novel D3R-selective compounds with predicted efficacy.11,13,14 In the crystal structure, eticlopride occupies the orthosteric binding site (OBS) defined by side chains from transmembrane helices II, III, IV, V, VI and VII. Out of 18 eticlopride contact residues in the D3R structure, 17 are identical in the D2R and one is similar (Valine 350 corresponds to Isoleucine in D2R).13 Thus, the dopamine D2-like receptors are largely homologous, with key differences allowing for exploitation of D3R selectivity over D2R.
Several classes of chemical structures have been explored in the search for highly D3R selective ligands.15,16 However, with the exception of 117 these molecules have only served as preclinical tools, and none are available clinically. One challenge is that many of these preclinical tools lacked the appropriate ADME (absorption, distribution, metabolism, and excretion) properties to be deemed clinically useful.18 Indeed, one of our lead compounds, R-PG648 (2, Fig. 1) failed to clear the bar to clinical investigation due to metabolic instability and failure to show efficacy in nonhuman primates, despite promise in rodent models of methamphetamine self-administration and relapse.14,18,19 In addition, the preponderance of published studies have focused on developing these target molecules toward cocaine addiction, and yet, a recent report suggests that toxic hypertension may result from treatment with a D3R antagonist in the presence of cocaine (i.e., if the patient relapsed to cocaine taking), further dampening enthusiasm for developing these molecules for human psychostimulant abusers.8
Nevertheless, there remains a need to identify novel templates and better molecules to target the D3R. First, there are several drugs of abuse (e.g., heroin) that do not produce hypertension and preclinical data support the efficacy of D3R antagonists in attenuating their self-administration and relapse behaviors.14,20 Secondly, structural determinants of affinity, selectivity and efficacy at D3R can be used for the highly homologous but functionally distinct D2R, for which there are no highly selective agonists or antagonists.21 Recent reports of D2R bitopic ligands can trace their SAR to the D3R crystal structure and molecular determinant studies done with D3R-selective ligands and their synthons.14 These reports highlight the importance of characterizing primary and secondary pharmacophores and the function of the respective OBS and secondary binding pocket (SBP), as well as potential allosteric sites that can have a significant impact on the pharmacology of these agents. Indeed, many new and exciting avenues of drug design pursuit have been aided by GPCR crystal structures and homology modeling.22,23 Hence, the combination of small molecule SAR and clues from the D3R crystal structure can produce structurally and pharmacologically unique molecules with D3R or potentially D2R selectivities and differing functional efficacies.12
Eticlopride (3, Fig. 2) binds with sub-nanomolar affinity to both D2R (Ki = 0.086 nM) and D3R (Ki = 0.134 nM) but without selectivity (D2R>D3R; ratio = 0.64).18 The high-resolution crystal structure of D3R was solved with eticlopride and showed its primary pharmacophore (PP) to include the highly decorated 2-hydroxy, 3-ethyl, 5-chloro, 6-methoxy substituted benzamide function. Another commonly used D2-like antagonist, raclopride24 (4, Fig. 2), is structurally similar to eticlopride but has a 3-chloro group instead of the 3-ethyl substituent. This simple substitution dramatically decreases its affinity at these receptor subtypes (D3R Ki = 13.4 nM, D2R Ki = 12.7 nM). Thus the ethyl substitution, present in eticlopride, results in an increase of affinity for D3R of ~100 times and nearly 150-times at D2R, compared to raclopride.
Figure 2.

Design of the primary pharmacophore (PP)
When the D3R-selective antagonist 2 was docked into the D3R crystal structure, the 2,3-dichlorophenylpiperazine overlayed onto eticlopride and confirmed that this was the PP that binds to the OBS in the D3R.13,25 Subsequent synthon studies and SAR corroborated the molecular models and explained the roles of the OBS and SBP in binding affinities, selectivities and efficacies.9,14 Herein, we reasoned that if the 2,3-dichloro-substituted phenylpiperazine was replaced with substituents borrowed from the eticlopride structure that the new templates might serve as PPs with potentially improved D3R affinities, selectivities and/or metabolic stability. Hence, we incorporated another privileged D3R PP, the 2-methoxyphenylpiperazine, into our design to make the 3-chloro-5-ethyl-2-methoxyphenylpiperazine PP (Fig. 2), which includes all but the 2-OH substituent from eticlopride and the 5-chloro, 6-methoxy substituent found in both eticlopride and raclopride. In addition, we designed a simpler hybrid PP, using the 2-Cl substituent from the 2,3-dichlorophenylpiperazine and the 3-ethyl group from eticlopride that gives rise to high affinity binding at D2R and D3R to give 2-chloro-3-ethylphenylpiperazine PP (Fig. 2). Moreover, we explored the secondary pharmacophore with different heteroaryl amides, and further investigated the 2-OH and 3-OH substituted butyl-linking chain. We had previously shown that although the 2-OH substituent retained high affinity binding at D3R, the 3-OH gave higher D3R selectivity.26 Nevertheless, we wanted to determine if this remained the case when the PP was modified. After assessing in vitro binding and functional profiles, the most D3R-selective compound in the series (19) was evaluated for metabolic stability in mouse microsomes. In addition, compound 19 was evaluated for its effects on locomotion in mice and its effects on increased locomotor activity induced by the prescription opioid analgesic, oxycodone, as well as oxycodone-induced conditioned place preference (CPP) in rats.
CHEMISTRY
The synthetic strategy used for the arylpiperazinebutyl carboxamide derivatives is shown in Scheme 1. Nitration of 3-chloro-4-methoxybenzoic acid (5) to 6 was followed by reduction to the alcohol 7, using borane dimethyl sulfide complex, and oxidation to the aldehyde 8a, in 84% yield. 27,28 Conversion of the aldehyde 8a, to the alkene 9a, was conducted under Wittig reaction conditions and the nitro group was reduced to the aniline 10a via catalytic hydrogenation.29 Compound 10a was cyclized to the N-aryl piperazine 11a and N-alkylation was carried out either through reductive amination to give 12a or reacting with the appropriate alkyl halide under basic conditions to give 13a.30 Compound 14a was generated by reacting the aryl piperazine 11a with 4-bromobutyl phthalimide. The alcohol intermediate 15a was synthesized via epoxide opening of 2-(2-oxiran-2-yl)ethyl)isoindoline-1,3-dione with 11a, in 56% yield, using a previously reported procedure.31,32 Deprotection of 14a and 15a, using hydrazine, gave the free amines 16a and 17a respectively, which were reacted with the respective aryl carboxylic acid or ester to afford the 3-chloro-5-ethyl-2-methoxyphenylpiperazinebutylarylamide derivatives (18-21).33,34 A similar strategy was used to synthesize the 2-chloro-3-ethylphenylpiperazinebutylarylamide derivatives (22-31) starting from 8b.35,36
Scheme 1. Synthesis of analogues synthons 11–13 and full-length analogues 18–31a.

aReagents and conditions: (a) fuming HNO3, 0 °C to room temperature, 2h; (b) BH3.CS2, 24h; (c) PCC, CH2Cl2, overnight; (d) Ph3P+CH3Br-, n-BuLi, THF; (e) 10% Pd/C, H2, 50 psi, EtOH/EtOAc, 3h; (f) bis(2-chloroethyl)amine. HCl, diethyleneglycol monoethylether, 150 °C, 7h; (g) HCHO, NaBH(OAc)3, AcOH; (h) 1-bromobutane, K2CO3, acetone, reflux, 7h; (i) n-bromobutyl phthalimide, K2CO3, acetone, reflux, 7h; (j) 2-(2-oxiran-2-yl)ethyl)isoindoline-1,3-dione, IPA, reflux, overnight; (k) hydrazine, EtOH, reflux, overnight; (l) ArCOOH, CDI, THF/DMF, 0 °C to room temperature; (m) ethyl 4-ethyl-1H-imidazole-2-carboxylate, (CH3)3Al, CH2Cl2, room temperature.
In Scheme 2, 11b was N-alkylated with 4-bromobutenephthalimide (32),37 to afford 33, in 98% yield. Deprotection using hydrazine gave the free amine 34, which was reacted with 4-methyl-1H-imidazole-2-carboxylic acid in presence of CDI (1,1′ carbonyldiimidazole) to give 35.
Scheme 2. Synthesis of 35a.

aReagents and conditions: (a) 11b, K2CO3, acetone, reflux, overnight; (b) hydrazine, EtOH, reflux, overnight; (c) 4-methyl-1H-imidazole-2-carboxylic acid, CDI, THF, 0 °C to room temperature, overnight.
In Scheme 3, 11b was N-alkylated with 2-(2-bromoethyl)oxirane (36)38 to give epoxide 37, in 41% yield. The epoxide was regioselectively opened with sodium azide to 38 in 53% yield and reduced via catalytic hydrogenation to the amino-alcohol 39, which was coupled with indole-2-carboxylic acid to give the 2-OH substituted analogue, 40.39,40 Note, all final compounds with hydroxylated linking chains are the racemic mixtures.
Scheme 3. Synthesis of 40a.

aReagents and conditions: (a) 11b, K2CO3, acetone, reflux, overnight; (b) NaN3, NH4Cl, DMF, 100 °C, 6 h; (c) 10% Pd/C, H2, 50 psi, 2 h; (d) Indole-2-COOH, CDI, THF, 0 °C to room temperature.
PHARMACOLOGICAL RESULTS AND DISCUSSION
To further dissect the contributions of individual pharmacophore components to D3R selectivity and efficacy, we extended our previous SAR studies by modifying all three segments of the classical D3R antagonist template (Fig. 2). We first evaluated the binding affinities of this series of compounds, by performing competition binding studies with [3H]N-methylspiperone using membranes prepared from HEK293 cells stably expressing either the human D2R, D3R or D4R (Tables 1 and 2.) In addition, in Table 2, cLogP values and polar surface area (PSA) were calculated to provide measures of lipophilicity and predicted brain penetration respectively, for all compounds.41
Table 1.
Human D2-like receptor binding data for synthonsa
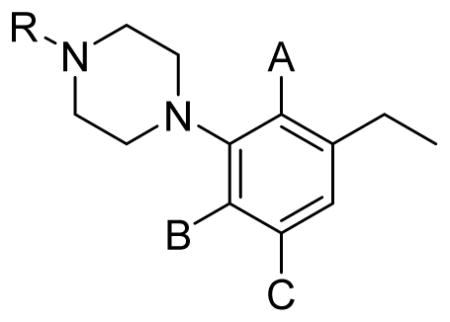
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compd. | R | A | B | C | D2R | D3R | D4R | D2/D3 | D4/D3 | D4/D2 |
|
| ||||||||||
| Ki±S.E.M. (nM) | ||||||||||
| 11a | H | H | OMe | Cl | 619±97.6 | 446±25.2 | 2300±820 | 1.4 | 5.2 | 3.7 |
| 11b | H | Cl | H | H | 139±11.9 | 31.1±0.462 | 316±47.1 | 4.5 | 10 | 2.3 |
| 12a | Me | H | OMe | Cl | 233±22.4 | 179±18.1 | 400±83.4 | 1.3 | 2.2 | 1.7 |
| 12b | Me | Cl | H | H | 58.1±3.09 | 13.6±0.488 | 89.9±6.81 | 4.3 | 6.6 | 1.5 |
| 13a | n-But | H | OMe | Cl | 29.8±4.58 | 22.5±4.86 | 322±76.0 | 1.3 | 14 | 11 |
| 13b | n-But | Cl | H | H | 2.93±0.316 | 0.363±0.052 | 65.1±10.0 | 8.1 | 179 | 22 |
Binding inhibition values determined using HEK 293 cells transfected with hD2LR, hD3R or hD4.4 and [3H]N-methylspiperone radioligand as described.59
Table 2.
Human D2-like receptor binding data on full-length analoguesa
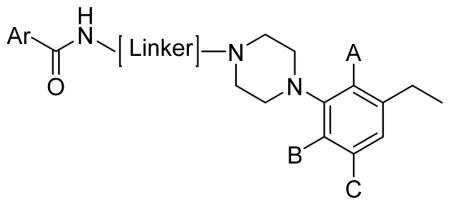
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compd | Series | Ar | A | B | C | Linker | cLogPb | PSAc | D2LR | D3R | D4.4R | D2R/D3R |
| Ki ± S.E.M. (nM) | ||||||||||||
| 18 | a |
|
H | OMe | Cl |
|
5.53 | 56.84 | 27.8±6.71 | 0.341±0.031 | 1050±279 | 82 |
| 19 | a |
|
H | OMe | Cl |
|
4.93 | 77.07 | 11400±3270 | 6.84±1.18 | 13900±1350 | 1700 |
| 20 | a |
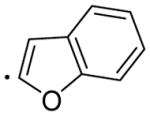
|
H | OMe | Cl |
|
5.50 | 54.04 | 137±9.00 | 3.19±0.268 | 714±293 | 43 |
| 21 | a |
|
H | OMe | Cl |
|
4.91 | 74.27 | 1620±339 | 36.1±5.00 | 5290±319 | 45 |
| 22 | b |
|
Cl | H | H |
|
5.76 | 47.61 | 5.50±0.805 | 0.142±0.025 | 334±113 | 39 |
| 23 | b |
|
Cl | H | H |
|
5.17 | 67.84 | 151±25.4 | 0.362±0.047 | 5520±1660 | 417 |
| 24 | b |
|
Cl | H | H |
|
5.74 | 44.81 | 6.34±0.959 | 0.153±0.005 | 356±64.4 | 41 |
| 25 | b |
|
Cl | H | H |
|
5.14 | 65.04 | 164±32.4 | 0.985±0.105 | 2380±110 | 166 |
| 26 | b |
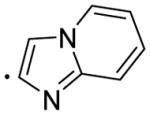
|
Cl | H | H |
|
5.52 | 51.18 | 5.64±0.98 | 0.331±0.085 | 819±175 | 17 |
| 27 | b |
|
Cl | H | H |
|
4.93 | 71.41 | 193±40.5 | 4.23±0.84 | >10000 | 46 |
| 28 | b |
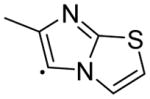
|
Cl | H | H |
|
5.43 | 51.18 | 4.65±1.24 | 0.941±0.13 | 143±3.18 | 5 |
| 29 | b |
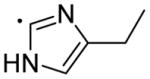
|
Cl | H | H |
|
5.38 | 59.97 | 12.5±1.07 | 1.14±0.17 | 500±30.9 | 11 |
| 30 | b |
|
Cl | H | H |
|
4.39 | 59.97 | 12.2±2.58 | 0.621±0.096 | 529±42.3 | 20 |
| 31 | b |
|
Cl | H | H |
|
3.79 | 80.2 | 150±13.1 | 11.2±2.66 | 1960±344 | 13 |
| 35 | b |
|
Cl | H | H |
|
4.31 | 59.97 | 16.1±1.2 | 1.28±0.22 | 503±157 | 13 |
| 40 | b |
|
Cl | H | H |
|
5.05 | 67.84 | 6.83±0.53 | 0.200±0.018 | 305±70.5 | 34 |
The privileged PP, 2,3-dichlorophenylpiperazine, was replaced with eticlopride-inspired synthons (11a and 11b). These new PPs showed moderate binding affinities for both D3R and D2R receptors with relatively low binding affinities to D4R and little or no selectivity, as shown in Table 1. As previously observed, when the alkyl linking chain was extended from the N-phenylpiperazine to N-methyl (12a and 12b) and N-n-butyl analogues (13a and 13b), binding affinities were improved for both D3R and D2R.14,22,42 Of this set of synthons, compound 13b demonstrated the highest D3R affinity (Ki=0.363 nM) and was the most D3R selective (D2R/D3R=8.1; D4R/D3R=179).
Binding data for the full-length substituted ligands are shown in Table 2. The majority of analogues demonstrated binding affinities in the low to sub-nanomolar range for D3R. In the 1-(2-chloro-3-ethylphenyl)piperazine PP based series (22-31), both 22 and 23 showed sub-nanomolar affinities for D3R (Ki = 0.142 and 0.362 nM, respectively). Moreover, 23 was highly D2R/D3R selective (>400 fold.) When the indole moiety of 22 and 23 was bioisoterically replaced with benzofuran, subnanomolar D3R binding affinities were maintained for both 24 and 25. The indole ring was also replaced with other heteroaryl ring systems, such as 2-(imidazo[1,2-a]pyridine) (26 and 27), 5-(6-methylimidazo[2,1-b]thiazole) (28), 2-(4-ethyl-1H-imidazole) (29), 2-(4-methyl-1H-imidazole) (30 and 31). However, in most cases, although high D3R binding affinities were retained, relative improvements in D2R binding affinities reduced D3R selectivity. No major change in the affinity and selectivity of 30 was observed when a trans-butenyl linker was introduced between the aryl amide and piperazine moiety to afford 35.
In the more highly decorated 1-(3-chloro-5-ethyl-2-methoxyphenyl)piperazine-based series (18-21), the indole 18 showed a similar binding profile to its analogue 22, but with slightly higher D3R selectivity (D2R/D3R=82 versus 39). Compound 19 exhibited lower affinity to D3R (Ki = 6.84 nM) but this analogue had the greatest D3R versus D2R binding selectivity, (1700 fold) of all the 4-phenylpiperazines that we have synthesized and evaluated to date. The benzofuran derivative 20 showed reduced D3R binding affinity (Ki = 3.19 nM) compared to 24 (Ki = 0.153 nM) but was similarly D3R selective. Whereas 21 (Ki = 36.1 nM) showed significantly reduced D3R affinity compared to 25 (Ki = 0.985 nM), and was less D3R selective. As expected, the introduction of a 3-OH group in the butyl linking chain of both series resulted in reduced binding affinities at D2R and D3R as well as somewhat lower cLogP values, predicting decreased lipophilicity, compared to corresponding parent aliphatic compounds. What was unexpected was the dramatic decrease in affinities in the ‘a’ series as compared to the ‘b’ series, when the 3-OH was introduced. In contrast, compared to all the 3-OH-butyl analogues, the 2-OH-butyl derivative 40 showed high D3R affinity (Ki = 0.20 nM) but as previously observed for other 2-OH-substituted 4-phenylpiperazine analogues,26 it exhibited lower D3R selectivity, similar to compounds that had no OH substitution in the linking chain. None of the compounds demonstrated high binding affinity for D4R. PSA values ranged from 45–80 with cLogP values in the range of 3.8–5.8, suggesting relatively high lipophilicity predictive of good blood-brain barrier penetration.
Functional Data and Off-Target Actions
D3R efficacy was measured in a cell-based mitogenesis assay wherein a subset of compounds was tested for stimulation or inhibition of quinpirole-stimulated mitogenesis in CHO cells (Table 3.) Interestingly, all of the synthons (11-13) demonstrated partial agonist profiles with the 2-chloro-3-ethyl-phenylpiperazine ‘b’ series being more efficacious than the 3-chloro-5-ethyl-2-methoxy-phenylpiperazine ‘a’ series. This profile was repeated in the full-length molecules wherein the more highly decorated ‘a’ series (more eticlopride-like) analogues (18-21) were all antagonists in the mitogenesis assay. As described previously, the 2-OCH3-substituted 4-phenylpiperazine precludes the formation of H-bonds with Transmembrane (TM) 5 Serines (5.42 and 5.43) in contrast to the 3-Cl substituent in the 2,3-dichlorophenylpiperazine that positions these molecules in the OBS driving its transition to an active state. These residues are the same that form H-bonds with the catechol hydroxyl groups of dopamine and give rise to an agonist profile.9,14 In contrast, the ‘b’ series analogues 22-25 were all partial agonists at D3R wherein there is no 3-Cl substituent, but rather the PP is a 2-chloro-3-ethylphenylpiperazine. Molecular simulations studies are underway to address the molecular mechanism underlying the efficacy profiles of this series of compounds.
Table 3.
Functional data for selected compounds using stimulation or inhibition of quinpirole-stimulated mitogenesis in CHO cells with human dopamine D3Ra
| Compd | Agonist EC50 ± S.E.M., nM | % Stimulation | Antagonist IC50 ± S.E.M., nM |
|---|---|---|---|
| 11b | 17.9 ± 0.48 | 64.7 | >10000 |
| 12a | >9000 | 15.2 | 1840 ± 700 |
| 12b | 710 ± 230 | 69.2 | NDb |
| 13a | >6600 | 17.2 | 930 ± 170 |
| 13b | 590 ± 210 | 29.1 | 63 ± 11 |
| 18 | >8300 | 5.8 | 330 ± 100 |
| 19 | >8300 | 6.7 | 360 ± 100 |
| 20 | >6600 | 4.9 | 420 ± 130 |
| 21 | >7100 | 3.6 | 950 ± 350 |
| 22 | 4.70 ± 0.57 | 42.3 | 19.1 ± 3.5 |
| 23 | 2.58 ± 0.87 | 17.9 | 50.5 ± 7.2 |
| 24 | 410 ± 130 | 31.2 | 18.5 ± 6.3 |
| 25 | 196 ± 64 | 22.3 | 230 ± 27 |
Data were obtained through the NIDA Addiction Treatment Discovery Program contract (ADA151001) with Oregon Health & Science University, using published methods.21
ND = Not determined.
The same set of analogues that were evaluated in the D3R mitogenesis assay was also tested for binding affinities at 5HT1A, 5HT2A and 5HT2C receptors. In general, the ‘a’ series of synthons demonstrated lower affinities at these 5HT receptor subtypes than the ‘b’ series and this pattern was repeated in the full-length molecules (18-21) versus (22-25). Also, the ‘b’ series synthons were functional agonists at 5HT1A with moderately high potency, as were corresponding full-length molecules 22, 24 and 25. All of the full-length analogues were selective for D3R over the 5HT receptor subtypes, with compound 19 demonstrating the most selective profile in the series: (5HT1A/D3R=>1000; 5HT2A/D3R=27; 5HT2C/D3R=320). Hence this compound was selected for further evaluation. It is important to note that compound 19 does not bind to mu, delta or kappa opioid receptors at a concentration of 10 μM (Data obtained through the NIDA Addiction Treatment Discovery Program contract (ADA151001) with Oregon Health & Science University) and thus, there is no direct effect of this compound on the opioid receptor system.
Mouse Microsomal Metabolism Results
Compound 19 was tested for phase I metabolism following procedures previously described to predict the susceptibility to metabolism following in vivo administration.43 Briefly, compound 19 was incubated in mouse liver microsomal incubations in the presence of NADPH, and compound disappearance was measured over time using HPLC with tandem mass spectrometry (LC/MS/MS). As depicted in Fig. 3 (see also S.I. Table 2), compound 19 showed excellent metabolic stability with >80% remaining over 1 h. These data supported testing compound 19 in a preclinical behavioral model of opioid dependence.
Figure 3. Mouse Microsomal Phase I metabolism data for Compound 19.

Compound 19 was incubated in mouse liver microsomes with NADPH regenerating system and compound disappearance was measured over time via LC/MS/MS. Compound 19 showed excellent metabolic stability substantiating its use in in vivo efficacy studies.
Locomotor Activity Studies in Mice with Compound 19 in the Absence and Presence of Oxycodone
Dopamine D3R antagonists are effective in attenuating the self-administration of heroin, alcohol or nicotine, but not cocaine or methamphetamine under low fixed-ratio reinforcement schedules (e.g. FR1).1,9 However, little is known as to whether D3R antagonists are similarly effective in attenuating the addiction-related behaviors of prescription opioids. Opioid-induced locomotor sensitization and conditioned place preference (CPP) are commonly used animal models to evaluate pharmacological agents for their utility in the treatment of addiction.44,45 Given the rapid increase in non-medical use of prescription opioids such as oxycodone and hydrocodone, the development of new and effective medications for the treatment of opioid abuse and addiction is urgent.46,47 Indeed, the recent rise in opioid overdose in the United States has been referred to as “epidemic”. 48,49
Oxycodone is the most commonly used prescription opioid analgesic. Like other opioid agonists such as morphine or heroin, acute (or single dose) administration of oxycodone produces a significant increase in locomotor activity, in mice. Moreover, repeated administration of these opioid agonists produces a progressive increase in locomotion over time, i.e., locomotor sensitization.50 In this study, we sought to 1) determine if compound 19 alone had any effect on basal locomotor activity, 2) if it would attenuate oxycodone-stimulated locomotor activity and sensitization, and 3) if this effect was long-lasting. The results of this evaluation are shown in Fig. 4 (A–D). Figure 4A shows the overall locomotor effects of repeated, daily administration of oxycodone (4 mg/kg, i.p.) in the presence of vehicle (25% beta-cyclodextrin) or one dose of compound 19 (5, 15 mg/kg, i.p. administered 15 min before oxycodone injection). Compound 19 alone had no effect on basal locomotor activity in mice when tested on day 1 (Fig. 4A and B). In contrast, oxycodone caused significant increases in locomotor activity (Fig. 4 A and C). Repeated oxycodone administration produced a progressive increase in locomotion (sensitization) over time (from day 2 to day 6 in the vehicle pretreatment group). Strikingly, pretreatment with compound 19 not only attenuated acute oxycodone-induced hyperactivity (Fig. 4A, day 2), but also blocked the acquisition of repeated oxycodone-induced locomotor sensitization (Figure 4A, day 2-day 6) and the expression of oxycodone prime-induced locomotor sensitization after 2 days of withdrawal (Fig. 4A, day 9). Two-way ANOVA for repeated measures over time (Fig. 4A) revealed a statistically significant drug treatment main effect (F2,21=12.24, p<0.001), time main effect (F6, 49 =9.14, p<0.001), and treatment X time interaction (F12, 98 =31.51, p<0.001). Figure 4B–D shows the time courses of oxycodone-induced change in locomotor activity when tested on day 1 (Fig. 4B: F2,21=0.12, p=0.88), day 2 (Fig. 4C: F2,21=5.22, p<0.05 and day 9 (Fig. 4D, F2,21=5.95, p<0.01), indicating that compound 19 pretreatment significantly attenuated oxycodone-induced increases in locomotion on each test day.
Figure 4.

Effects of compound 19 (0, 5, 15 mg/kg, i.p.) on basal and oxycodone-induced increases in locomotor activity in mice. A: The effects of oxycodone on locomotion over time (from day 2 to day 9) in the presence or absence of compound 19 treatment. On day 1, compound 19 alone (5, 15 mg/kg, 15 min before saline) was administered and had no effect on locomotion. From day 2 to day 6, each animal received one daily dose of 19 (vehicle, 5, 15 mg/kg, 15 min prior to oxycodone) and oxycodone (4 mg/kg) administration. In the vehicle treatment group, repeated daily administration of oxycodone produced a progressive increase in locomotion (i.e., locomotor sensitization) (#p<0.05, ##p<0.01, compared to the level of locomotor activity on day 2). This locomotor sensitization was dose-dependently blocked by 19 (*p<0.05, **p<0.01, compared to the vehicle control group at each time point labeled). After 5 days of the (vehicle/19 + oxycodone) co-administration, animals underwent 2 days of withdrawal, followed by a 4 mg/kg oxycodone challenge injection (without 19 pretreatment) on day 9, indicating that oxycodone-induced locomotor sensitization was still present in the vehicle control group, which was blocked in the 19 pretreatment groups. B: The time course of 19-induced changes in locomotion (day 1), indicating that it has no effect on locomotor activity by itself. C: Time course of oxycodone-induced changes in locomotion after the first injection of oxycodone (acute effect on day 2), indicating that 19 pretreatment dose-dependently inhibited oxycodone-induced hyperactivity (acute effect). D: The time course of oxycodone priming-induced changes in locomotion in mice after 2 days of withdrawal from the last (vehicle/19 + oxycodone) co-administration (day 9), indicating that repeated 19 pretreatment from day 2 to day 6 produced a long-lasting inhibition in oxycodone-induced increases in locomotion. N=8 mice in each group.
Effects of compound 19 on Oxycodone-induced CPP in Rats
Figure 5 shows the effects of compound 19 on oxycodone-induced CPP. Four days of oxycodone (e.g., Vehicle + oxycodone group) (3 mg/kg, i.p.) versus saline conditioning produced significant CPP compared to the (Vehicle + saline) control group of rats. Pretreatment with 19 (5, 15 mg/kg, i.p., 15 min before each oxycodone injection) dose-dependently attenuated oxycodone-induced CPP (Fig. 5). One-way ANOVA revealed a statistically significant treatment main effect (F4,45 =4.29, p<0.01). Post-hoc individual group comparisons indicated a significant reduction in oxycodone-induced CPP after 15 mg/kg 19 (p<0.05, compared to (Vehicle + oxycodone) group. Compound 19 alone, at 15 mg/kg, did not produce CPP, suggesting a lack of reward by itself.
Figure 5.

Effects of compound 19 on oxycodone-induced conditioned place preference. Oxycodone (3 mg/kg, i.p.) conditioning in the (Vehicle + oxycodone) produced a robust place preference to the oxycodone-paired compartment. Pretreatment with 19 (5, 15 mg/kg, i.p., 15 min before oxycodone injection) dose-dependently blocked the acquisition of oxycodone-induced CPP. Compound 19 alone (15 mg/kg, i.p.) in the (19 + saline) group did not produce significant place preference. ** p<0.01, compared to the (Vehicle + vehicle) group; ## p<0.01, compared to the (Vehicle + oxycodone) group. The number on each bar shows the animal number in each experimental group.
CONCLUSION
In the present study we combined small molecule SAR with clues from the D3R crystal structure to design two novel series of D3R-selective antagonists and partial agonists. In the synthon study, PPs were synthesized including their alkyl homologated analogues extending our previous findings to show that these compounds generally bind to both D2R and D3R, but with varying affinities and modest, if any, D3R selectivity, depending on the phenyl ring substitutions and the N-alkyl group. The 3-ethyl-2-chloro-substituted ‘b’ series typically showed higher affinity binding at D3R than the more highly substituted ‘a’ series with compound 13b showing the highest D3R affinity and selectivity. In addition, the ‘b’ series synthons were far more efficacious in the D3R mitogenesis functional assay, suggesting a novel interaction in the OBS that clearly dictates the efficacy of the full-length molecules, in this series. In general, the full- length molecules based on both the new PPs, 3-chloro-5-ethyl-2-methoxyphenylpiperazine and 2-chloro-3-ethylphenylpiperazine, showed high D3R binding affinities and selectivities over D2R and D4R. However, the 2-chloro-3-ethylphenylpiperazines had higher affinities for both D2R and D3R and as such were not as D3R selective compared to their 3-chloro-5-ethyl-2-methoxyphenylpiperazine counterparts. Nevertheless, several of these analogues were quite D3R selective (e.g. 23 and 25) and given their partial agonist profile will be evaluated in vivo, in due course.
The replacement of the indole secondary pharmacophore with other heteroaryl ring systems showed no improvement in D3R selectivity in either series of molecules; based on these and other SAR studies the indole amide should be considered a privileged secondary pharmacophore for D3R.22 In addition, the indole-2-carboxamide scaffold occupies a secondary binding pocket between TM2 and TM7 within D2R giving rise to a negative allosteric modulator profile.22 In the present study, no attempt to elucidate allosterism at D3R was made, but it is intriguing to consider that depending on the PP, these molecules may show a bitopic profile. Identifying the structural determinants of these potentially bitopic agents will be of future interest.
The lead compound 19 was chosen in the present study based on its highly D3R-selective profile in vitro. Indeed, this compound appears to be the most selective D3R antagonist reported in the literature to date. This compound demonstrated exceptional metabolic stability in mouse microsomes that was significantly improved over our previous lead compound 2.18
Locomotor sensitization is a commonly-used animal model to determine the potential use of novel compounds for the treatment of substance use disorders.51–54 In this model, compound 19 alone had no effect on basal locomotor activity. However, pretreatment with compound 19 significantly inhibited oxycodone-induced hyperlocomotion and reduced repeated oxycodone-induced locomotor sensitization in a dose-dependent manner, which provides in vivo evidence that the D3R is a viable target for opioid use disorder medication development. To further determine the potential utility of compound 19 in treatment of prescription opioid abuse and dependence, we observed the effects of repeated 19 pretreatment for 4 consecutive days on the development of oxycodone-induced CPP. We found that compound 19 also dose-dependently blocked the acquisition or ability of oxycodone to establish CPP, suggesting that 19 pretreatment may prevent the development of oxycodone dependence. This is consistent with our recent finding that blockade of D3Rs by another set of D3R-selective partial agonists also inhibited intravenous heroin self-administration in wild-type mice, but not in D3R-knockout mice.9 Given compound 19’s exceptional D3R-selectivity profile, the behavioral effects observed in the present study are likely mediated via blockade of central D3Rs. This hypothesis is supported by recent findings that genetic deletion of D3R in D3-knockout mice blocked morphine-induced hyperactivity and repeated morphine-induced locomotor sensitization.55
Oxycodone is one of the most commonly used prescription opioids in clinical pain management. Nevertheless, a severe adverse effect that can arise with oxycodone’s chronic use is its potential to produce dependence in patients. Epidemic-like increases in prescription opioid addiction and overdose in the U.S. and abroad strongly support consideration of novel mechanisms for treatment.56–58 Of course, somatic symptoms associated with opioid overdose are unlikely to be modified by this class of drugs. These sometimes fatal consequences will require other medication strategies, such as the opioid antagonist naloxone. Nevertheless, discovery of medications that might prevent relapse to drug seeking or ideally, the development of dependence for patients that require long term opioid therapy is of vital importance. The studies herein point to the D3R as a potential therapeutic target and compound 19 as a new lead molecule for development.
EXPERIMENTAL METHODS
Synthesis
Reaction conditions and yields were not optimized. Anhydrous solvents were purchased from Aldrich and were used without further purification except for tetrahydrofuran, which was freshly distilled from sodium-benzophenone ketyl. All other chemicals and reagents were purchased from Sigma-Aldrich Co. LLC, Combi-Blocks, TCI America, Acros Organics, Maybridge, and Alfa Aesar. All amine final products were converted into the oxalate salt. Spectroscopic data and yields refer to the free base form of all compounds. Teledyne ISCO CombiFlash Rf or glass flash column chromatography were performed using silica gel (EMD Chemicals, Inc.; 230–400 mesh, 60 Å). 1H and 13C NMR spectra were acquired using a Varian Mercury Plus 400 spectrometer at 400 MHz and 100 MHz, respectively. Chemical shifts are reported in parts-per-million (ppm) and referenced according to deuterated solvent for 1H spectra (CDCl3, 7.26, CD3OD, 3.31 or DMSO-d6, 2.50) and 13C spectra (CDCl3, 77.2, CD3OD, 49.0 or DMSO-d6, 39.5). Gas chromatography-mass spectrometry (GC/MS) data were acquired (where obtainable) using an Agilent Technologies (Santa Clara, CA) 6890N GC equipped with an HP-5MS column (cross-linked 5% PH ME siloxane, 30 m × 0.25 mm i.d. × 0.25 μm film thickness) and a 5973 mass-selective ion detector in electron-impact mode. Ultrapure grade helium was used as the carrier gas at a flow rate of 1.2 mL/min. The injection port and transfer line temperatures were 250 and 280 °C, respectively, and the oven temperature gradient used was as follows: the initial temperature (100 °C) was held for 3 min and then increased to 295 °C at 15 °C/min over 13 min, and finally maintained at 295 °C for 10 min. Combustion analysis was performed by Atlantic Microlab, Inc. (Norcross, GA) and the results agree within ±0.4% of calculated values. cLogP and polar surface area (PSA)values were calculated using ChemDraw Professional Ultra 15.0. Melting point determination was conducted using a Thomas-Hoover melting point apparatus and are uncorrected. On the basis of NMR and combustion data, all final compounds are≥95% pure.
3-Chloro-4-methoxy-5-nitrobenzoic acid (6)
3-Chloro-4-methoxybenzoic acid (5, 5.0 g, 26.8 mmol) was added in small portions to cold fuming HNO3 (90%, 25 mL) at 0–5°C. The reaction mixture was allowed to warm to 20 °C and stirred for additional 2 h. Cold water (50 mL) was added, and the precipitated product was filtered under the vacuum. The solid product was dissolved in CHCl3 and washed with brine solution and concentrated. The product was purified by flash chromatography using 8% MeOH/CHCl3 as eluent to provide 4.87 g (79%) of product. 1H NMR (400 MHz, CD3OD) 8.27 (s, 1H), 8.22 (s, 1H), 4.02 (s, 3H); 13C NMR (100 MHz, CD3OD) δ 165.9, 153.9, 146.4, 136.3, 131.56, 125.9, 63.2. GC-MS (EI) m/z 231 (M+).
(3-Chloro-4-methoxy-5-nitrophenyl)methanol (7)
Borane dimethyl sulfide complex (10 M, 3.07 g, 40.4 mmol) was added dropwise to a solution of 6 (4.46 g, 19.3 mmol) in THF (60 mL) at 0–5°C. The mixture was allowed to come to room temperature and stirred overnight. The reaction mixture was cooled to 0–5°C and quenched carefully by dropwise addition of MeOH. The reaction mixture was evaporated (4.7 g) and used directly in the next step without further purification. 1H NMR (400 MHz, CDCl3) 8.49-8.38 (m, 2H), 5.50 (s, 2H), 4.82 (m, 3H), 3.70 (bs, 1H); 13C NMR (100 MHz, CDCl3) δ 148.9, 145.2, 137.9, 132.5, 130.6, 121.3, 63.1, 62.5. GC-MS (EI) m/z 217 (M+).
3-Chloro-4-methoxy-5-nitrobenzaldehyde (8a)28
Pyridinium chlorochromate (PCC, 9.31 g, 43.2 mmol) was added to a solution of 7 (4.70 g, 21.6 mmol) in CH2Cl2 (100 mL). The reaction mixture was stirred overnight and filtered through the celite bed. The product was purified by flash chromatography using 20% EtOAc/hexanes as eluent to provide 3.92 g (84%) of the product as a yellow solid.1H NMR (400 MHz, CDCl3) 9.94 (s, 1H), 8.19 (d, J = 2.0 Hz, 1H), 8.13 (d, J = 2.0 Hz, 1H), 4.10 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 187.8, 154.4, 145.7, 134.5, 132.2, 131.9, 124.8, 63.0.GC-MS (EI) m/z 215 (M+).
1-Chloro-2-methoxy-3-nitro-5-vinylbenzene (9a)
N-BuLi (12.0 mL, 19.2 mmol, 1.6 M in hexane) was added dropwise to a suspension of methyltriphenylphosponium bromide (7.20 g, 20.2 mmol) in dry THF (100 mL) at −78 °C under argon. The reaction mixture was allowed to warm to 0°C and stirred for 1h. The reaction mixture was again cooled to −78°C and the solution of 8a (3.95 g, 18.3 mmol) in THF (50 mL) was added dropwise over 30 min. The reaction mixture was allowed to warm to 0 °C and stirred for 1 h. The reaction mixture was quenched with sat. NH4Cl solution (50 mL) and the crude product was extracted with EtOAc (3 × 25 mL). The organic layer was combined, dried, concentrated and purified using flash chromatography with 5% EtOAc/hexanes as eluent to provide 1.27 g (33%) of the product as a yellow solid. 1H NMR (400 MHz, CDCl3) 7.59 (s, 1H), 7.51 (s, 1H), 6.55-6.47 (m, 1H), 5.69 (d, J = 16 Hz, 1H), 5.32 (d, J = 10.8 Hz, 1H), 3.92 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 148.7, 145.2, 134.5, 133.1, 131.6, 130.5, 120.8, 117.2, 62.3. GC-MS (EI) m/z 213 (M+).
2-Chloro-1-nitro-3-vinylbenzene (9b)
The same procedure was used as described for 9a, starting from 8b to give the desired product in 70% yield. 1H NMR (400 MHz, CDCl3) 7.74 (dd, J = 8.0, 1.6 Hz, 1H), 7.64 (dd, J = 8.0, 1.6 Hz, 1H), 7.37 (t, J = 8.0 Hz, 1H), 7.12 (dd, J = 17.2, 11.0 Hz, 1H), 5.81 (dd, J = 17.2, 0.8 Hz, 1H), 5.54 (dd, J = 10.8, 0.8 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 149.6, 138.9, 132.1, 129.9, 127.2, 124.8, 123.9, 119.6.
3-Chloro-5-ethyl-2-methoxyaniline (10a)
A mixture of 9a (1.27 g, 5.94 mmol) and 10% Pd/C (0.20 g) in EtOH (30 mL) was stirred under an atmosphere of hydrogen (50 psi) at room temperature for 3h. The reaction mixture was filtered through a Celite pad and evaporated under vacuum. The reaction mixture was sufficiently pure to be used for the next step without further purification. 1H NMR (400 MHz, CDCl3) 6.60 (d, J = 2.0 Hz, 1H), 6.44 (d, J = 2.0 Hz, 1H), 3.96 (bs, 2H), 3.83 (s, 3H), 2.48 (q, J = 7.6 Hz, 2H), 1.19 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 141.3, 141.0, 140.9, 127.0, 118.3, 113.7, 59.4, 28.1, 15.2. GC-MS (EI) m/z 185 (M+).
2-Chloro-3-ethylaniline (10b)
The same procedure was used as described for 10a using EtOAc as the solvent. The product was sufficiently pure to be used for the next step without further purification. 1H NMR (400 MHz, CDCl3) 6.99 (t, J = 7.6 Hz, 1H), 6.66-6.62 (m, 2H), 4.04 (bs, 2H), 2.72 (q, J = 7.6 Hz, 2H), 1.22 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 143.1, 142.4, 126.9, 119.2, 118.9, 113.4, 27.1, 13.9.
1-(3-Chloro-5-ethyl-2-methoxyphenyl)piperazine (11a)
The reaction mixture of 10a (0.63 g, 3.39 mmol) and bis(2-chloroethyl)amine hydrochloride (0.67 g, 3.73 mmol) in diethylene glycol monomethyl ether (2.0 mL) was heated at 150 °C for 7 h. The reaction mixture was allowed to come to room temperature and dissolved in MeOH (5 mL) followed by dilution with ether (250 mL). The salt was filtered, suspended in CHCl3 (20 mL) and neutralized with 2N NaOH to pH 8–9. The organic layer was collected, concentrated and purified by column chromatography using 2% MeOH/CHCl3 as eluent to provide 0.36 g (56%) of solid product. 1H NMR (400 MHz, CDCl3) δ 6.84 (d, J = 2 Hz, 1H), 6.61 (d, J = 2.4 Hz, 1H), 3.85 (s, 3H), 3.08-3.01 (m, 8H), 2.55 (q, J = 8 Hz, 2H), 1.20 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 146.8, 146.6, 140.9, 128.4, 122.4, 116.9, 59.2, 51.9, 46.8, 28.6, 15.6. GC-MS (EI) m/z 254.2 (M+). The oxalate salt was precipitated from acetone; Mp 190–191°C. Anal. C13H19ClN2O•C2H2O4•0.5H2O) C, H, N.
1-(2-Chloro-3-ethylphenyl)piperazine (11b)
The same procedure was used as described for 11a. The product was purified by flash chromatography using 2% MeOH/CHCl3/0.1% NH4OH as eluent to give the desired product in 46% yield. 1H NMR (400 MHz, CDCl3) δ 7.16-7.12 (m, 1H), 6.94-6.89 (m, 2H), 3.05-2.95 (m, 8H), 2.76 (q, J = 7.6 Hz, 2H), 1.21(t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 150.0, 143.2, 128.7, 126.9, 123.9, 118.0, 53.0, 46.3, 27.5, 14.1. The oxalate salt was precipitated from acetone; Mp 166–167 °C. Anal. (C12H17ClN2•1.5C2H2O4•0.1NH4OH) C, H, N.
1-(3-Chloro-5-ethyl-2-methoxyphenyl)-4-methylpiperazine (12a)
Sodium triacetoxyborohydride (0.30 g, 1.2 mmol) and AcOH (0.1 mL) were added to a solution of 11a (0.118 g, 0.46 mmol) and formaldehyde (0.042 g, 1.4 mmol) in 1,2-dichloroethane (10 mL) under an argon atmosphere. The reaction mixture was stirred overnight at room temperature and quenched with a 2N NaOH (10 mL) solution. The reaction mixture was extracted with EtOAc (3 × 15 mL). The organic layer was combined, dried and concentrated. The crude product was purified by flash column chromatography using 15% acetone/CHCl3 as eluent to afford 0.053 g (yield = 43%) of pure product as an oil. 1H NMR (400 MHz, CDCl3) δ 6.83 (d, J = 2 Hz, 1H), 6.61 (d, J = 2.4 Hz, 1H), 3.83 (s, 3H), 3.13 (s, 4H), 2.56 (s, 4H), 2.53 (q, J = 7.6 Hz, 2H), 2.33 (s, 3H), 1.19 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 146.5, 146.2, 140.9, 128.3, 122.3, 116.8, 59.1, 55.8, 50.3, 46.3, 28.6, 15.5. The oxalate salt was precipitated from acetone; Mp 185–186 °C. Anal. (C14H21ClN2O•1.5C2H2O4•H2O) C, H, N.
1-(2-Chloro-3-ethylphenyl)-4-methylpiperazine (12b)
The same procedure was used as described for 12a, employing 11b. The product was purified by flash chromatography using 25% acetone/CHCl3 as eluent to afford 12b in 94% yield. 1H NMR (400 MHz, CDCl3) δ 7.14 (d, J = 7.6 Hz, 1H), 6.94-6.91(m, 2H), 3.06 (s, 4H), 2.76 (q, J = 7.6 Hz, 2H), 2.61 (s, 4H), 2.36 (s, 3H), 1.21(t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 149.7, 143.3, 128.8, 126.9, 124.1, 118.1, 55.5, 51.6, 46.2, 27.6, 14.2. The oxalate salt was precipitated from acetone; Mp 158–159 °C.; Anal. (C13H19ClN2•1.5C2H2O4•0.5H2O) C, H, N.
1-Butyl- 4-(3-chloro-5-ethyl-2-methoxyphenyl)piperazine (13a)
1-Bromobutane (0.11 g, 0.78 mmol) was added to the reaction mixture of 11a (0.10 g, 0.39 mmol) and K2CO3 (0.16 g, 1.2 mmol) in acetone (5 ml) and stirred at reflux for 17 h. The reaction mixture was filtered, concentrated and purified using flash chromatography with 15% acetone/CHCl3 as eluent to provide 0.10 g (81%) of product as an oil. 1H NMR (400 MHz, CDCl3) δ 6.82 (d, J = 2 Hz, 1H), 6.07 (d, J = 2 Hz, 1H), 3.82 (s, 3H), 3.13 (s, 4H), 2.61 (s, 4H), 2.58-2.50 (m, 2H), 2.40-2.36 (m, 2H), 1.52-1.47 (m, 2H), 1.37-1.31 (m, 2H), 1.19 (t, J = 7.2 Hz, 3H), 0.92 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 146.5, 146.3, 140.8, 128.3, 122.2, 116.7, 59.0, 58.7, 53.9, 50.4, 29.1, 28.6, 20.9, 15.5, 14.1. The oxalate salt was precipitated from acetone; Mp 210–211 °C. Anal. (C17H27ClN2O•C2H2O4) C, H, N.
1-Butyl-4-(2-chloro-3-ethylphenyl)piperazine (13b)
The same procedure was used as described for 13a, employing 11b. The product was purified by flash chromatography using 20% EtOAc/hexanes as eluent to afford 13b in 63% yield. 1H NMR (400 MHz, CDCl3) 7.14 (d, J = 8 Hz, 1H), 6.93(m, 2H), 3.06 (s, 4H), 2.72 (q, J = 7.6 Hz, 2H), 2.64 (s, 4H), 2.43-2.39 (m, 2H), 1.56-1.49 (m, 2H), 1.48-1.32 (m, 2H), 1.22 (t, J = 7.6 Hz, 3H), 0.94 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 149.8, 143.3, 128.8, 126.9, 123.9, 118.1, 58.7, 53.6, 51.7, 29.2, 27.6, 20.9, 14.2, 14.2. The p-toluenesulfonic acid salt was precipitated from ether; Mp 145–146 °C. Anal (C16H25ClN2•C7H8O3S•0.25H2O) C, H, N.
2-(4-(4-(3-Chloro-5-ethyl-2-methoxyphenyl)piperazin-1-yl)butyl)isoindoline-1,3-dione (14a)
N-(4-Bromobutyl)phthalimide (0.38 g, 1.34 mmol) was added to a reaction mixture of 11a (0.31 g, 1.22 mmol) and K2CO3 (0.51 g, 3.65 mmol) in acetone (15 mL) and stirred at reflux overnight. The crude product was filtered, concentrated and purified by flash chromatography using 12% EtOAc/hexanes as eluent to provide 0.47 g (84%) of the product as an oil. 1H NMR (400 MHz, CDCl3) δ 7.68 (dd, J = 5.6, 2.8 Hz, 2H), 7.57 (dd, J = 5.6, 3.2 Hz, 2H), 6.68 (d, J = 2.4 Hz, 1H), 6.49(d, J = 2.0 Hz, 1H), 3.69 (s, 3H), 3.60 (t, J = 7.0 Hz, 2H), 3.01 (bs, 4H), 2.48 (bs, 4H), 2.40 (q, J = 7.6 Hz, 2H), 2.32 (t, J = 7.0 Hz, 2H), 1.62 (quintet, J = 7.6 Hz, 2H), 1.48-1.45 (m, 2H), 1.06 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 168.2, 146.3, 146.0, 140.6, 133.8, 132.0, 128.0, 123.0, 122.0, 116.6, 58.8, 57.9, 53.6, 50.0, 37.7, 28.3, 26.5, 23.9, 15.3.
2-(4-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)butyl)isoindoline-1,3-dione (14b)
The same procedure was used as described for 14a, employing 11b. The product was purified by flash chromatography using 20% EtOAc/hexanes as eluent to afford 14b in 64% yield. 1H NMR (400 MHz, CDCl3) 7.85-7.82 (m, 2H), 7.71-7.69 (m, 2H), 7.15 (dt, J = 7.6, 2.4 Hz, 1H), 6.95-6.91 (m, 2H), 3.75 (dt, J = 6.4, 0.8 Hz, 2H), 3.06 (bs, 4H), 2.77 (dq, J = 7.6, 2.0 Hz, 2H), 2.64 (bs, 4H), 2.47 (t, J = 6.8 Hz, 2H), 1.80-1.75 (m, 2H), 1.63-1.59 (m, 2H), 1.27-1.21 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 167.9, 149.4, 142.8, 133.6, 131.9, 128.4, 126.6, 123.6, 122.8, 117.7, 57.8, 53.2, 51.3, 37.6, 27.2, 26.4, 24.0, 13.9.
2-(4-(4-(3-Chloro-5-ethyl-2-methoxyphenyl)piperazin-1-yl)-3-hydroxybutyl)isoindoline-1,3-dione (15a)
A mixture of compounds 11a (0.31 g, 1.21 mmol) and 2-(2-(oxiran-2-yl)ethyl)isoindoline-1,3-dione (0.26 g, 1.21 mmol) was stirred at reflux in 2-PrOH (20 mL) overnight. The reaction mixture was concentrated and purified by flash chromatography using 20% EtOAc/hexanes as eluent to provide 0.52 g (91%) of product. 1H NMR (400 MHz, CDCl3) δ 7.85 (dd, J = 5.2, 3.2 Hz, 2H), 7.71 (dd, J = 5.6, 2.8 Hz, 2H), 6.84 (d, J = 2.0 Hz, 1H), 6.60(d, J = 2.0 Hz, 1H), 3.93-3.79 (m, 6H), 3.13 (bs, 4H), 2.81-2.79 (m, 2H), 2.60-2.58 (m, 2H), 2.55 (q, J = 8.0 Hz, 2H), 2.45-2.41 (m, 2H), 1.79 (q, J = 7.2 Hz, 2H), 1.19 (t, J = 8.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 168.5, 146.4, 145.9, 140.8, 133.9, 132.2, 128.2, 123.2, 122.4, 116.7, 64.5, 63.9, 59.1, 53.8, 50.3, 35.1, 33.6, 28.4, 15.4.
2-(4-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)-3-hydroxybutyl)isoindoline-1,3-dione (15b)
The same procedure was used as described for 15a, employing 11b. The product was purified by flash chromatography using 30% EtOAc/hexanes as eluent to afford 15b in 57.2% yield. 1H NMR (400 MHz, CDCl3) 7.85 (dd, J = 7.2, 5.6 Hz, 2H), 7.71 (dd, J = 5.6, 3.2 Hz, 2H), 7.15 (t, J = 8.0 Hz, 1H), 6.92 (dq, J = 7.6, 1.6 Hz, 2H), 3.94-3.77 (m, 4H), 3.03 (bs, 4H), 2.84-2.81 (m, 2H), 2.76 (q, J = 7.6 Hz, 2H), 2.59 (m, 2H), 2.46-2.38 (m 2H), 1.79 (q, J = 7.0, 2H), 1.22 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 168.7, 151.7, 149.6, 143.4, 134.0, 132.4, 128.8, 127.0, 124.2, 123.4, 118.1, 64.6, 64.0, 51.8, 35.3, 33.8, 27.6, 14.2.
4-(4-(3-Chloro-5-ethyl-2-methoxyphenyl)piperazin-1-yl)butan-1-amine (16a)
Hydrazine (0.097 g, 3.02 mmol) was added to a solution of compound 14a (0.460 g, 1.00 mmol) in EtOH (15 mL) and stirred at reflux for 7 h. The solvent was evaporated and the reaction mixture was diluted with 20% aq K2CO3 solution (15 mL) and extracted in CHCl3 (2 × 15 mL). The organic layer was combined, dried and concentrated to afford the product as a yellow oil, which was sufficiently pure to be used in the next step without further purification. 1H NMR (400 MHz, CDCl3) δ 6.70 (d, J = 2.0 Hz, 1H), 6.49 (d, J = 2.0 Hz, 1H), 3.70 (s, 3H), 3.01 (bs, 4H), 2.59 (s, 2H), 2.47-2.38 (m, 6H), 2.27 (t, J = 7.6 Hz, 2H), 1.46-1.33 (m, 4H), 1.07 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 146.3, 146.1, 140.6, 128.0, 122.0, 116.5, 58.8, 58.5, 53.7, 50.2, 28.3, 24.2, 15.3.
4-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)butan-1-amine (16b)
The same procedure was used as described for 16a, employing 14b to afford 16b in 85.1% yield. The product was sufficiently pure to be used for the next step without further purification. 1H NMR (400 MHz, CDCl3) 6.99-6.95 (m, 1H), 6.77-6.74 (m, 2H), 2.89 (bs, 4H), 2.63-2.54 (m, 4H), 2.47 (bs, 4H), 2.27-2.23 (m, 2H), 1.43-1.28 (m, 4H), 1.08-1.04 (m, 5H); 13C NMR (100 MHz, CDCl3) δ 149.3, 142.7, 128.3, 126.5, 123.5, 117.6, 58.2, 53.1, 51.2, 41.8, 31.5, 27.1, 24.0, 13.8.
4-Amino-1-(4-(3-chloro-5-ethyl-2-methoxyphenyl)piperazin-1-yl)butan-2-ol (17a)
The same procedure was used as described for 16a, employing 15a to afford 16a in 88.5% yield. The product was sufficiently pure to be used in the next step without further purification. 1H NMR (400 MHz, CDCl3) δ 6.77 (d, J = 2.0 Hz, 1H),6.55 (d, J = 1.6 Hz, 1H),3.85-3.81 (m, 1H), 3.76 (s, 3H), 3.06 (bs, 4H), 2.86 (m, 2H), 2.72-2.70 (m, 2H), 2.53-2.44 (m, 4H), 2.37-2.28 (m, 3H), 1.53-1.44 (m,2H), 1.13 (t, J = 8.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 146.3, 146.0, 140.7, 128.1, 122.2, 116.6, 66.2, 64.6, 58.9, 53.8, 50.3, 39.6, 37.7, 28.4, 15.4.
4-Amino-1-(4-(2-chloro-3-ethylphenyl)piperazin-1-yl)butan-2-ol (17b)
The same procedure was used as described for 16a, employing 15b to afford 17b in 89.6% yield. The product was sufficiently pure to be used in the next step without further purification. 1H NMR (400 MHz, CDCl3) 7.03-7.0 (m, 1H), 6.82-6.77 (m, 2H), 3.80-3.71 (m, 1H), 2.92-2.61 (m, 10H), 2.49-2.24 (m, 7H), 1.46-1.40 (m, 2H), 1.10 (t, J = 7.6, 0.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 149.2, 142.8, 128.3, 126.6, 123.6, 117.7, 65.8, 64.4, 51.3, 39.3, 37.5, 27.1, 13.8.
General Amidation Procedure
CDI (1 equiv) was added to a solution of the carboxylic acid (1 equiv) in dry THF (10 mL/mmol) and stirred for 3 h at room temperature. The reaction mixture was cooled to 0 °C and the amine (1 equiv) was added dropwise after diluting with dry THF (10 mL/mmol). The reaction mixture was allowed to come to room temperature and stirred overnight, concentrated, diluted with H2O (20 mL) and extracted in CHCl3 (3 × 10 mL). The organic layer was concentrated and the product was purified by flash column chromatography to provide the desired amide product.
N-(4-(4-(3-Chloro-5-ethyl-2-methoxyphenyl)piperazin-1-yl)butyl)-1H-indole-2-carboxamide (18)
Compound 18 was prepared from indole-2-carboxylic acid and 16a according to the general amidation procedure. The crude product was purified by flash chromatography using 25% acetone/CHCl3 as eluent to give the desired product in 57.6% yield. 1H NMR (400 MHz, CDCl3) δ 10.25 (s, 1H), 7.62 (d, J = 8.0 Hz, 1H), 7.45 (dd, J = 8.0, 0.8 Hz, 1H), 7.28-7.24 (m, 1H), 7.14-7.10 (m, 1H), 6.93 (m, 1H), 6.90 (s, 1H), 6.84 (d, J = 1.6 Hz, 1H), 6.58 (d, J = 1.6 Hz, 1H), 3.83 (s, 3H), 3.54 (q, J = 6.0 Hz, 2H), 3.14 (bs, 4H), 2.60 (bs, 4H), 2.53 (q, J = 7.6 Hz, 2H), 2.45 (d, J = 7.0 Hz, 2H), 1.73-1.64 (m, 4H), 1.19 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 162.0, 146.4, 146.0, 140.9, 136.6, 131.0, 128.2, 127.6, 124.3, 122.3, 121.8, 120.5, 116.7, 112.1, 102.3, 59.0, 58.0, 53.7, 50.1, 39.6, 28.4, 27.5, 24.2, 15.4. The oxalate salt was precipitated from acetone and crystallized in MeOH/ether; Mp 220–221 °C. Anal (C26H33ClN4O2•C2H2O4•0.5H2O) C, H, N.
N-(4-(4-(3-Chloro-5-ethyl-2-methoxyphenyl)piperazin-1-yl)-3-hydroxybutyl)-1H-indole-2-carboxamide (19)
Compound 19 was prepared from indole-2-carboxylic acid and 17a according to the general amidation procedure. The crude product was purified by flash chromatography using 15% acetone/CHCl3 as eluent to give the desired product in 22.2% yield. 1H NMR (400 MHz, CDCl3) δ 9.90 (s, 1H), 7.63 (d, J = 8.0 Hz, 1H), 7.51-7.48 (m, 1H), 7.46 (dd, J = 8.0, 0.8 Hz, 1H), 7.28-7.24 (m, 1H), 7.14-7.10 (m, 1H), 6.87 (dd, J = 6.0, 2.0 Hz, 2H), 6.61 (d, J = 2.0 Hz, 1H),3.96-3.84 (m, 2H), 3.84 (s, 3H), 3.57-3.51 (m, 1H), 3.15 (bs, 4H), 2.85-2.81 (m, 2H), 2.58-2.52 (m, 4H), 2.43 (d, J = 6.8 Hz, 2H), 1.86-1.82 (m, 1H), 1.67-1.62 (m, 1H), 1.21 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 161.8, 146.4, 146.0, 140.9, 136.4, 131.1, 128.3, 127.7, 124.2, 122.4, 121.8, 120.4, 116.7, 112.0, 102.0, 66.3, 63.8, 59.1, 53.7, 50.4, 38.0, 33.4, 28.5, 15.4. The oxalate salt was precipitated from acetone and crystallized in hot MeOH; Mp 188–189 °C. Anal (C26H33ClN4O3•C2H2O4•H2O) C, H, N.
N-(4-(4-(3-Chloro-5-ethyl-2-methoxyphenyl)piperazin-1-yl)butyl)benzofuran-2-carboxamide (20)
Compound 20 was prepared from benzofuran-2-carboxylic acid and 16a according to the general amidation procedure. The crude product was purified by flash chromatography using 20% acetone/CHCl3 as eluent to give the desired product in 76.5% yield. 1H NMR (400 MHz, CDCl3) δ 7.63 (dd, J = 7.2, 1.2 Hz, 1H), 7.44-7.42 (m, 2H), 7.36 (dt, J = 7.0, 1.2 Hz, 1H), 7.28-7.24 (m, 1H), 7.13 (m, 1H), 6.82 (d, J = 2.0 Hz, 1H), 6.59 (d, J = 2.0 Hz, 1H), 3.82 (s, 3H), 3.51 (q, J = 6.4 Hz, 2H), 3.15 (bs, 4H), 2.61 (bs, 4H), 2.51 (q, J = 7.6 Hz, 2H), 2.45 (t, J = 7.0 Hz, 2H), 1.73-1.63 (m, 4H), 1.17 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 158.9, 154.7, 149.0, 146.4, 146.1, 140.8, 128.2, 127.7, 126.7, 123.7, 122.7, 122.2, 116.7, 111.6, 110.2, 59.0, 58.0, 53.8, 50.2, 39.2, 28.4, 27.5, 24.3, 15.4. The oxalate salt was precipitated from acetone and crystallized in MeOH/ether; Mp 126–127 °C. Anal (C26H32ClN3O3• 2C2H2O4.H2O) C, H, N.
N-(4-(4-(3-Chloro-5-ethyl-2-methoxyphenyl)piperazin-1-yl)-3-hydroxybutyl)benzofuran-2-carboxamide (21)
Compound 21 was prepared from benzofuran-2-carboxylic acid and 17a according to the general amidation procedure. The crude product was purified by flash chromatography using 20% acetone/CHCl3 as eluent to give the desired product in 50.5% yield. 1H NMR (400 MHz, CDCl3) δ 7.64 (dd, J = 7.6, 0.8 Hz, 1H), 7.53 (m, 1H), 7.49 (dd, J = 8.4. 0.8 Hz, 1H), 7.44 (d, J = 0.8 Hz, 1H), 7.40-7.36 (m, 1H), 7.29-7.25 (m, 1H), 6.85 (d, J = 2.0 Hz, 1H), 6.61 (d, J = 2.0 Hz, 1H), 3.95-3.81 (m, 5H), 3.56-3.48 (m, 1H), 3.14 (bs, 4H), 2.84-2.81 (m, 2H), 2.62-2.51 (m, 4H), 2.46-2.42 (m, 2H), 1.87-1.80 (m, 1H), 1.68-1.61 (m, 1H), 1.20 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 159.0, 154.8, 149.0, 146.4, 146.0, 140.8, 128.3, 127.7, 126.7, 123.6, 122.6, 122.4, 116.7, 111.8, 110.1, 65.9, 63.8, 59.1, 53.7, 50.4, 37.3, 33.7, 28.5, 15.4. The oxalate salt was precipitated from acetone and crystallized in 2-PrOH/ether; Mp 154–155 °C. Anal (C26H32ClN3O4•1.5C2H2O4•H2O) C, H, N.
N-(4-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)butyl)-1H-indole-2-carboxamide (22)
Compound 22 was prepared from indole-2-carboxylic acid and 16b according to the general amidation procedure. The crude product was purified by flash chromatography using 30% acetone/CHCl3 as eluent to give the desired product in 51.0% yield. 1H NMR (400 MHz, CDCl3) 10.35 (s, 1H), 7.63 (d, J = 7.6 Hz, 1H), 7.47 (d, J = 8.8 Hz, 1H), 7.27 (dt, J = 8.0, 1.2 Hz, 1H), 7.13 (sextet, J = 4.0 Hz, 2H), 6.96-6.87 (m, 4H), 3.56 (dd, J = 12.8, 6.4 Hz, 2H), 3.06 (bs, 4H), 2.78 (q, J = 7.6 Hz, 2H), 2.65 (bs, 4H), 2.47 (t, J = 7.2 Hz, 2H), 1.72-1.64 (m, 4H), 1.23 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 162.1, 149.6, 143.3, 136.7, 131.1, 128.7, 127.6, 127.0, 124.3, 124.1, 121.9, 120.6, 118.1, 112.2, 102.2, 58.1 53.5, 51.5, 39.8, 27.7, 27.6, 24.5, 14.2. The oxalate salt was precipitated from acetone; Mp 228–229 °C. Anal (C25H31ClN4O•C2H2O4•0.75H2O) C, H, N.
N-(4-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)-3-hydroxybutyl)-1H-indole-2-carboxamide (23)
Compound 23 was prepared from indole-2-carboxylic acid and 17b according to the general amidation procedure. The crude product was purified by flash chromatography using 30% acetone/CHCl3 as eluent to give the desired product in 61.1% yield. 1H NMR (400 MHz, DMSO) 11.52 (s, 1H), 8.43 (t, J = 5.6 Hz, 1H), 7.58 (d, J = 8.4 Hz, 1H), 7.40 (d, J = 8.4 Hz, 1H), 7.16 (quintet, J = 8 Hz, 2H), 7.07 (d, J = 1.6 Hz, 1H), 7.99 (m, 3H), 4.48 (d, J = 4.8 Hz, 1H), 3.73 (m, 1H), 3.45-3.33 (m, 2H), 2.91 (bs, 4H), 2.64 (q, J = 7.6 Hz, 2H), 2.58 (bs, 4H), 2.40-2.30 (m, 2H), 1.78 (m, 1H), 1.53 (m, 1H), 1.13 (t, J = 8 Hz, 3H); 13C NMR (100 MHz, DMSO) δ 160.0, 149.4, 142.4, 136.3, 131.9, 127.6, 127.3, 127.1, 123.9, 123.1, 121.4, 119.6, 118.4, 112.2, 102.1, 65.4, 64.4, 53.6, 51.2, 36.1, 35.4, 26.8, 14.1. The oxalate salt was precipitated from acetone; Mp 208–209 °C. Anal (C25H31ClN4O2•C2H2O4•0.75H2O) C, H, N.
N-(4-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)butyl)benzofuran-2-carboxamide (24)
Compound 24 was prepared from benzofuran-2-carboxylic acid and 16b according to the general amidation procedure. The crude product was purified by flash chromatography using 25% EtOAc/hexanes as eluent to give the desired product in 76.5% yield. 1H NMR (400 MHz, CDCl3) 7.65-7.63 (m, 1H), 7.46 (bs, 1H), 7.44 (d, J = 0.8 Hz, 1H), 7.38 (dt, J = 7.2, 1.2 Hz, 1H), 7.29-7.25 (m, 1H), 7.15-7.11 (m, 2H), 6.91 (dq, J = 7.2, 1.6 Hz, 2H), 3.53 (dd, J = 12.4, 6.4 Hz, 2H), 3.08 (bs, 4H), 2.77 (q, J = 7.6 Hz, 2H), 2.67 (bs, 4H), 2.48 (t, J = 7.0 Hz, 2H), 1.74-1.65 (m, 4H), 1.22 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 158.9, 154.7, 149.6, 149.0, 143.2, 128.7, 127.7, 126.9, 126.8, 124.0, 123.7, 122.7, 118.0, 111.7, 110.2, 58.0, 53.5, 51.5, 39.2, 27.6, 27.5, 24.4, 14.1. The oxalate salt was precipitated from acetone; Mp 119–120 °C. Anal (C25H30ClN3O2•C2H2O4•0.5H2O) C, H, N.
N-(4-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)-3-hydroxybutyl)benzofuran-2-carboxamide (25)
Compound 25 was prepared from benzofuran-2-carboxylic acid and 17b according to the general amidation procedure. The crude product was purified by flash chromatography using 25% acetone/CHCl3 as eluent to give the desired product in 52.1% yield. 1H NMR (400 MHz, CDCl3) 7.64 (d, J = 7.2 Hz, 1H), 7.58 (t, J = 5.0 Hz, 1H), 7.50-7.45 (m, 2H), 7.38 (dt, J = 7.2, 1.2 Hz, 1H), 7.28-7.24 (m, 1H), 7.15 (t, J = 7.6 Hz, 1H), 6.92 (dq, J = 8.0, 1.2 Hz, 2H), 3.94-3.84 (m, 3H), 3.55-3.50 (m, 1H), 3.06 (bs, 4H), 2.87-2.85 (m, 2H), 2.76 (q, J = 7.6 Hz, 2H), 2.62-2.61 (m, 2H), 2.48-02.43 (m, 2H), 1.85-1.80 (m, 1H), 1.66-1.63 (m, 1H), 1.22 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 159.0, 154.8, 149.4, 149.1, 143.3, 128.7, 127.7, 127.0, 126.7, 124.2, 123.6, 122.7, 118.0, 111.9, 110.1, 66.0, 63.9, 51.7, 37.5, 33.7, 27.5, 14.2. The oxalate salt was precipitated from ether; Mp 147–148 °C. Anal (C25H30ClN3O3. C2H2O4•H2O) C, H, N.
N-(4-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)butyl)imidazo[1,2-a]pyridine-2-carboxamide (26)
Compound 26 was prepared from imidazo[1,2-a]pyridine-2-carboxylic acid and 16b according to the general amidation procedure. The crude product was purified by flash chromatography using 3% MeOH/CHCl3 as eluent to give the desired product in 78.4% yield. 1H NMR (400 MHz, CDCl3) δ 8.11-8.09 (m, 2H), 7.53-7.49 (m, 2H), 7.21-7.17 (m, 1H), 7.10 (t, J = 8.0 Hz, 1H), 6.89 (t, J = 7.2 Hz, 2H), 6.81-6.77 (m, 1H), 3.48 (q, J = 6.0 Hz, 2H), 3.03 (bs, 4H), 7.72 (q, J = 7.6 Hz, 2H), 2.62 (m, 4H), 2.45-2.42 (m, 2H), 1.65 (m, 4H), 1.18 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 162.7, 149.6, 144.4, 143.1, 140.0, 128.6, 126.9, 126.5, 126.0, 123.9, 118.0, 114.1, 113.3, 58.1, 53.4, 51.4, 39.0, 27.7, 27.4, 24.3, 14.1. The oxalate salt was precipitated from acetone; Mp 144–145 °C. Anal (C24H30ClN5O•2C2H2O4•1.5H2O) C, H, N.
N-(4-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)-3-hydroxybutyl)imidazo[1,2-a]pyridine-2-carboxamide (27)
Compound 27 was prepared from imidazo[1,2-a]pyridine-2-carboxylic acid and 17b according to the general amidation procedure. The crude product was purified by flash chromatography using 2% MeOH/CHCl3 as eluent to give the desired product in 46.3% yield. 1H NMR (400 MHz, CDCl3) δ 8.13-8.11 (m, 2H), 7.90 (bt, J = 6.0 Hz, 1H), 7.56 (dd, J = 7.2, 0.8 Hz, 1H), 7.23-7.19 (m, 1H), 7.14 (t, J = 8.0 Hz, 1H), 6.93 (dq, J = 7.6, 1.6 Hz, 2H), 6.82 (dt, J = 6.4, 1.2 Hz, 1H), 3.91-3.78 (m, 2H), 3.56-3.48 (m, 1H), 3.04 (bs, 4H), 2.86-2.83 (m, 2H), 2.75 (q, J = 7.6 Hz, 2H), 2.62-2.60 (m, 2H), 2.45-2.40 (m, 2H), 1.84-1.76 (m, 1H), 1.70-1.61 (m, 1H), 1.21 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 162.9, 149.5, 144.6, 143.2, 140.2, 128.7, 126.9, 126.4, 125.8, 124.0, 118.2, 118.0, 114.1, 113.3, 65.3, 63.9, 53.5, 51.6, 36.6, 34.4, 27.4, 14.1. The oxalate salt was precipitated from acetone; Mp 137–138 °C. Anal (C24H30ClN5O2. 2C2H2O4•H2O•0.75CHCl3) C, H, N.
N-(4-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)butyl)-6-methylimidazo[2,1-b]thiazole-5-carboxamide (28)
Compound 28 was prepared from 6-methylimidazo[2,1-b]thiazole-5-carboxylic acid and 16b according to the general amidation procedure. The crude product was purified by flash chromatography using 3% MeOH/CHCl3 as eluent to give the desired product in 48.9% yield. 1H NMR (400 MHz, CDCl3) δ 8.19 (d, J = 4.0 Hz, 1H), 7.13-7.09 (m, 1H), 6.92-6.82 (m, 3H), 5.98 (m, 1H), 3.50-3.45 (m, 2H), 3.03 (bs, 4H), 2.73 (q, J = 7.6 Hz, 2H), 2.64 (bs, 4H), 2.58 (s, 3H), 2.48-2.45 (m, 2H), 1.66 (m, 4H), 1.19 (t, J = 8.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 160.7, 150.9, 149.4, 145.1, 143.2, 128.7, 126.9, 124.0, 121.4, 118.7, 117.9, 112.4, 58.1, 53.4, 51.3, 39.4, 27.9, 27.4, 24.2, 16.5, 14.1. The oxalate salt was precipitated from acetone; Mp 113–114 °C. Anal (C23H30ClN5OS•2C2H2O4•2.5H2O) C, H, N.
N-(4-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)butyl)-4-ethyl-1H-imidazole-2-carboxamide (29)
A solution of trimethylaluminium (2M in hexane) (0.18 mL, 3.54 mmol) was added dropwise to a solution of 16b (0.105 g, 3.54 mmol) in CH2Cl2 (10 mL) under argon at room temperature. The reaction mixture was stirred at room temperature for 15 min and ethyl 4-ethyl-1H-imidazole-2-carboxylate solution in CH2Cl2 (10 mL) was added dropwise and stirred for 6 h. The reaction mixture was quenched with 10% HCl (15 mL). The organic layer was extracted, dried over Na2SO4, concentrated and purified using flash chromatography with 4% MeOH/CHCl3 as eluent to provide 0.148 g (14.9%) of the product. 1H NMR (400 MHz, CDCl3) δ 7.56 (bs, 1H), 7.13 (t, J = 7.6 Hz, 1H), 6.92 (t, J = 8.0 Hz, 2H), 6.82 (s, 1H), 3.46-3.43 (m, 2H), 3.06 (s, 4H), 2.75 (q, J = 7.6 Hz, 2H), 2.66 (m, 5H), 2.48-2.46 (m, 2H), 2.18-2.15 (m, 1H), 1.65 (s, 5H), 1.29-1.18 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 159.1, 149.5, 143.2, 140.0, 128.7, 126.9, 124.0, 118.0, 58.0, 53.4, 51.2, 39.1, 27.5, 27.4, 24.1, 14.1, 13.5. The oxalate salt was precipitated from acetone; Mp 167–168 °C. Anal (C22H32ClN5O•2.5C2H2O4•H2O) C, H, N.
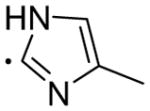
N-(4-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)butyl)-4-methyl-1H-imidazole-2-carboxamide (30)
Compound 30 was prepared from 4-methyl-1H-imidazole-2-carboxylic acid and 16b according to the general amidation procedure. The crude product was purified by flash chromatography using 3% MeOH/CHCl3 as eluent to give the desired product in 65.2% yield. 1H NMR (400 MHz, CDCl3) δ 7.76 (bs, 1H), 7.13 (t, J = 8.0 Hz, 1H), 6.94-6.89 (m, 2H), 6.82 (s, 1H), 3.46 (q, J = 6.4 Hz, 2H), 3.06 (bs, 4H), 2.75 (q, J = 8.0 Hz, 2H), 2.64 (bs, 4H), 2.45 (t, J = 7.2 Hz, 2H), 2.28 (s, 3H), 1.68-1.61 (m, 4H), 1.20 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 159.3, 149.5, 143.2, 140.1, 128.6, 126.9, 124.0, 118.0, 58.1, 53.4, 51.4, 39.2, 27.5, 24.2, 14.1. The oxalate salt was precipitated from acetone; Mp 175–176 °C. Anal (C21H30ClN5O•2C2H2O4•H2O) C, H, N.
N-(4-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)-3-hydroxybutyl)-4-methyl-1H-imidazole-2-carboxamide (31)
Compound 31 was prepared from 4-methyl-1H-imidazole-2-carboxylic acid and 17b according to the general amidation procedure. The crude product was purified by flash chromatography using 3% MeOH/CHCl3 as eluent to give the desired product in 39.8% yield. 1H NMR (400 MHz, CDCl3) δ 12.40 (bs, 1H), 8.11 (bs, 1H), 8.01 (s, 1H), 7.14 (t, J = 8.0 Hz, 1H), 6.95-6.88 (m, 2H), 6.83 (bs, 1H), 3.90-3.84 (m, 1H), 3.79-3.70 (m, 1H), 3.57-3.49 (m, 1H), 3.03 (bs, 4H), 2.83-2.81 (m, 2H), 2.75 (q, J = 8.0 Hz, 2H), 2.58 (bs, 2H), 2.42 (d, J = 6.6 Hz, 2H), 2.29 (bs, 3H), 1.81-1.73 (m, 1H), 1.68-1.59 (m, 1H), 1.20 (t, J = 8.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 162.6, 159.3, 149.4, 143.2, 140.0, 128.6, 126.9, 124.1, 118.0, 65.0, 63.9, 53.5, 51.6, 36.8, 34.1, 27.5, 14.1. The oxalate salt was precipitated from acetone; Mp 130–131 °C. Anal (C21H30ClN5O2•2C2H2O4•3H2O) C, H, N.
(E)-2-(4-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)but-2-en-1-yl)isoindoline-1,3-dione (33)
Compound 3237 (0.592 g, 2.11 mmol) was added to the reaction mixture of 11b (0.475 g, 2.11 mmol) and K2CO3 (1.458 g, 10.56 mmol) in acetone (25 ml) and stirred at reflux overnight. The reaction mixture was filtered, concentrated and purified using flash chromatography with 15% acetone/CHCl3 as eluent to provide 0.880 g (98.2%) of product as an oil. 1H NMR (400 MHz, CDCl3) δ 7.67 (dd, J = 5.2, 3.2 Hz, 2H), 7.53 (dd, J = 5.2, 3.2 Hz, 2H), 6.98 (t, J = 8.0 Hz, 1H), 6.76 (t, J = 6.4 Hz, 2H), 5.63 (d, J = 4.4 Hz, 1H), 5.61 (d, J = 4.4 Hz, 1H), 4.16 (d, J = 4.0 Hz, 2H), 2.90 (m, 6H), 2.59 (q, J = 8.0 Hz, 2H), 2.47 (bs, 4H), 1.06 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 167.6, 149.5, 142.9, 133.8, 132.0, 130.1, 128.5, 126.9, 126.8, 123.8, 123.1, 117.9, 60.1, 53.3, 51.4, 38.9, 27.4, 14.1.
(E)-4-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)but-2-en-1-amine (34)
The same procedure was used as described for 16a. The product was isolated in 91.2% yield and was sufficiently pure to be used for the next step without further purification. 1H NMR (400 MHz, CDCl3) δ 6.96 (t, J = 8.0 Hz, 1H), 6.75 (dd, J = 8.0. 1.6 Hz, 2H), 5.63-5.57 (m, 1H), 5.53-5.46 (m, 1H), 3.13 (d, J = 5.6 Hz, 2H), 2.88-2.87 (m, 6H), 2.59 (q, J = 7.6 Hz, 2H), 2.47 (bs, 4H), 1.16 (bs, 2H), 1.05 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 149.5, 142.9, 135.3, 128.5, 126.8, 126.0, 123.8, 117.9, 60.4, 53.2, 51.4, 43.7, 27.4, 14.1. GC-MS (EI) m/z 293.1 (M+).
(E)-N-(4-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)but-2-en-1-yl)-4-methyl-1H-imidazole-2-carboxamide (35)
Compound 35 was prepared from 4-methyl-1H-imidazole-2-carboxylic acid and 34 according to the general amidation procedure. The crude product was purified by flash chromatography using 3% MeOH/CHCl3 as eluent to give the desired product in 54.7% yield. 1H NMR (400 MHz, CDCl3) δ 12.70-12.63 (m, 1H), 7.90 (bs, 1H), 7.13 (t, J = 8.0 Hz, 1H), 6.94-6.89 (m, 2H), 6.80 (s, 1H), 5.76 (d, J = 5.0, 1H), 5.73 (d, J = 5.0, 1H), 4.06 (t, J = 5.0, 2H), 3.05-3.04 (m, 6H), 2.75 (q, J = 7.6, 2H), 2.62 (bs, 4H), 2.31 (s, 2H), 2.24 (s, 1H), 1.20 (t, J = 8.0, 3H); 13C NMR (100 MHz, CDCl3) δ 159.1, 149.5, 143.2, 139.9, 129.3, 129.2, 128.7, 126.9, 124.0, 118.0, 60.3, 53.4, 51.4, 40.8, 27.5, 14.1. The oxalate salt was precipitated from acetone and crystallized in MeOH/ether; Mp 166–167 °C. Anal (C21H28ClN5O•2C2H2O4•1.25H2O) C, H, N.
1-(2-Chloro-3-ethylphenyl)-4-(2-(oxiran-2-yl)ethyl)piperazine (37)
2-(2-Bromoethyl)oxirane (36, 0.269 g, 1.78 mmol) was added to a reaction mixture of 11b (0.266 g, 1.18 mmol) and K2CO3 (0.491 g, 3.56 mmol) in acetone (20 mL) and stirred at reflux overnight. The crude product was filtered, concentrated and purified by flash chromatography using 12% acetone/CHCl3 as eluent to provide 0.145 g (41.4%) of the product as an oil. 1H NMR (400 MHz, CDCl3) δ 7.15 (t, J = 7.6 Hz, 1H), 6.95-6.91 (m, 2H), 3.06 (bs, 4H), 3.02-2.95 (m, 1H), 2.80-2.73 (m, 3H), 2.66 (bs, 4H), 2.62-2.55 (m, 2H), 2.53-2.51 (m, 1H), 1.87-1.79 (m, 1H), 1.76-1.67 (m, 1H), 1.22 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 149.6, 143.2, 128.7, 126.9, 124.0, 118.0, 55.0, 53.5, 51.6, 50.9, 47.1, 30.2, 27.5, 14.2. GC-MS (EI) m/z 294.1 (M+).
1-Azido-4-(4-(2-chloro-3-ethylphenyl)piperazin-1-yl)butan-2-ol (38)
A reaction mixture of 37 (0.170 g, 0.57 mmol), NaN3 (0.056 g, 0.86 mmol) and NH4Cl (0.062 g, 1.15 mmol) in DMF (5 mL) was heated at 100 °C for 6 h. The solvent was evaporated and the reaction mixture was diluted with water (15 mL) and extracted in EtOAc (3 × 15 mL). The organic layer was combined, dried, concentrated and purified by flash chromatography using 7% acetone/CHCl3 as eluent to provide 0.103 g (52.9%) of the product as an oil.1H NMR (400 MHz, CDCl3) δ 7.17 (t, J = 7.6 Hz, 1H), 6.97 (dd, J = 7.6, 1.2 Hz, 1H), 6.91 (dd, J = 7.6, 1.2 Hz, 1H), 4.07-4.02 ((m, 1H), 3.28 (m, 2H), 3.07 (bs, 4H), 2.85-2.71 (m, 6H), 2.64-2.61 (m, 2H), 1.87-1.77 (m, 1H), 1.60-1.54 (m, 1H), 1.26-1.22 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 149.2, 143.3, 128.7, 127.0, 124.3, 118.1, 73.0, 57.3, 56.6, 53.4, 51.5, 28.5, 27.5, 14.1.
1-Amino-4-(4-(2-chloro-3-ethylphenyl)piperazin-1-yl)butan-2-ol (39)
A mixture of compound 38 (0.123 g, 0.36 mmol) and 10% Pd/C (0.050 g) in EtOAc (10 mL) was stirred under an atmosphere of hydrogen (50 psi) at room temperature for 2 h. The reaction mixture was filtered through a Celite pad and evaporated under vacuum. The reaction mixture was sufficiently pure to be used for the next step without further purification. 1H NMR (400 MHz, CD3OD+CDCl3) δ 7.07 (t, J = 7.6 Hz, 1H), 6.89 (dd, J = 13.6, 8.4 Hz, 2H), 4.89 (bs, 2H), 4.11 (bs, 1H), 3.33-3.17 (m, 10H), 3.04-2.99 (m, 1H), 2.64 (q, J = 7.2 Hz, 2H), 2.06-1.97 (m, 2H), 1.12 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 147.5, 143.3, 128.5, 127.1, 125.1, 118.3, 65.5, 54.2, 52.7, 48.7, 44.8, 28.8, 27.2, 13.8.
N-(4-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)-2-hydroxybutyl)-1H-indole-2-carboxamide (40)
Compound 40 was prepared from indole-2-carboxylic acid and 39 according to the general amidation procedure using DMF as solvent. The crude product was purified by flash chromatography using 2% MeOH/CHCl3 as eluent to give the desired product in 16.4% yield. 1H NMR (400 MHz, CDCl3) δ 9.73 (s, 1H), 7.63 (d, J = 7.6 Hz, 1H), 7.43 (dd, J = 8.4, 0.8 Hz, 1H), 7.28-7.23 (m, 1H), 7.16-7.09 (m, 2H), 6.95 (dd, J = 7.6, 1.2 Hz, 2H), 6.91 (d, J = 1.2 Hz, 1H), 6.86 (dd, J = 7.6, 1.6 Hz, 1H), 4.10-4.06 (m, 1H), 3.76-3.71 (m, 1H), 3.43-3.37 (m, 1H), 3.04 (bs, 4H), 2.93-2.81 (m, 2H), 2.79-2.69 (m, 4H), 2.62 (m, 2H), 1.82-1.78 (m, 1H), 1.62-1.57 (m, 1H), 1.21 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 161.9, 149.1, 143.3, 136.3, 130.8, 128.7, 127.7, 126.9, 124.3, 124.3, 121.9, 120.5, 118.0, 112.0 102.3, 72.6, 57.3, 53.4, 51.4, 45.3, 30.9, 28.6, 27.4, 14.0. The oxalate salt was precipitated from acetone; Mp 225–226 °C. Anal (C25H31ClN4O2•1.5C2H2O4•1.75H2O) C, H, N.
Radioligand binding assays
Binding at dopamine D2-like receptors was determined using previously described methods.59 Membranes were prepared from HEK293 cells expressing human D2LR, D3R or D4.4R, grown in a 50:50 mix of DMEM and Ham’s F12 culture media, supplemented with 20 mM HEPES, 2 mM L-glutamine, 0.1 mM non-essential amino acids, 1X antibiotic/antimycotic, 10% heat-inactivated fetal bovine serum, and 200 μg/mL hygromycin (Life Technologies, Grand Island, NY) and kept in an incubator at 37°C and 5% CO2. Upon reaching 80–90% confluence, cells were harvested using pre-mixed Earle’s Balanced Salt Solution (EBSS) with 5 μM EDTA (Life Technologies) and centrifuged at 3000 rpm for 10 min at 21 °C. The supernatant was removed and the pellet was resuspended in 10 mL hypotonic lysis buffer (5 mM MgCl2 · 6 H2O, 5 mM Tris, pH 7.4 at 4 °C) and centrifuged at 20,000 rpm for 30 min at 4 °C. The pellet was then resuspended in fresh EBSS buffer made from 8.7 g/L Earle’s Balanced Salts without phenol red (US Biological, Salem, MA), 2.2 g/L sodium bicarbonate, pH to 7.4. A Bradford protein assay (Bio-Rad, Hercules, CA) was used to determine the protein concentration and membranes were diluted to 500 μg/mL and stored in a −80 °C freezer for later use.
Radioligand competition binding experiments were conducted using thawed membranes. Test compounds were freshly dissolved in 30% DMSO and 70% H2O to a stock concentration of 1 mM or 100 μM. To assist the solubilization of free-base compounds, 10 μl of glacial acetic acid was added along with the DMSO. Each test compound was then diluted into 13 half-log serial dilutions using 30% DMSO vehicle; final test concentrations ranged from 100 μM to 100 pM or from 10 μM to 10 pM. Previously frozen membranes were diluted in fresh EBSS to a 100 μg/mL (for hD2LR or hD3R) or 200 μg/mL (hD4.4R) stock for binding. Radioligand competition experiments were conducted in glass tubes containing 300 μl fresh EBSS buffer with 0.2 mM sodium metabisulfite, 50 μl of diluted test compound, 100 μl of membranes (10 μg total protein for hD2LR or hD3R, 20 μg total protein for hD4.4R), and 50 μl of [3H]N-methylspiperone (0.4 nM final concentration; Perkin Elmer). Nonspecific binding was determined using 10 μM butaclamol (Sigma-Aldrich, St. Louis, MO) and total binding was determined with 30% DMSO vehicle. All compound dilutions were tested in triplicate and the reaction incubated for one hour at room temperature. The reaction was terminated by filtration through Whatman GF/B filters, presoaked for one hour in 0.5% polyethylenimine, using a Brandel R48 filtering manifold (Brandel Instruments, Gaithersburg, MD). The filters were washed 3 times with 3 mL of ice-cold EBSS buffer and transferred to scintillation vials. 3 mL CytoScint liquid scintillation cocktail (MP Biomedicals, Solon, OH) was added and vials were counted using a Perkin Elmer Tri-Carb 2910 TR liquid scintillation counter (Waltham, MA). IC50 values for each compound were determined from dose-response curves and Ki values were calculated using the Cheng-Prusoff equation; these analyses were performed using GraphPad Prism version 5.00 for Windows (GraphPad Software, San Diego, CA). Reported Ki values were determined from least three independent experiments.
Mouse microsomal stability assay
The phase I metabolic stability assay for compound 19 was conducted in mouse liver microsomes as previously described with minor modifications.43 In brief, the reaction was carried out with 100 mM potassium phosphate buffer, pH 7.4, in the presence of NADPH regenerating system, (compound final concentration was 10 μM; and 0.5 mg/mL microsomes). Positive controls for phase I metabolism (testosterone) were also evaluated. Compound disappearance was monitored over time using a liquid chromatography and tandem mass spectrometry (LC/MS/MS) method. All reactions were sampled in triplicate.
Chromatographic analysis was performed using an Accela™ ultra high-performance system consisting of an analytical pump and an autosampler coupled with a TSQ Vantage mass spectrometer (Thermo Fisher Scientific Inc., Waltham, MA). Separation of the analyte from potentially interfering material was achieved at ambient temperature using Agilent Eclipse Plus column (100 × 2.1mm i.d.) packed with a 1.8 μm C18 stationary phase. The mobile phase used was composed of 0.1% formic acid in acetonitrile and 0.1% formic acid in H2O with gradient elution, starting with 10% (organic) linearly increasing to 99% up to 2 min, maintaining at 99% (2–2.5 min) and reequlibrating to 10% by 2.7 min. The total run time for each analyte was 4.5 min. The mass transitions used for compounds for LC/MS/MS analysis are given in Table S2 (Supporting Information).
Locomotor Activity studies
Twenty-four male mice (22–25 g) with a C57BL/6J genetic background were purchased from the Charles River Laboratories (Raleigh, NC). After arrival, they were group housed in the animal facility under a reversed 12 h light-dark cycle (light on at 7:00 PM) with free access to food and water, and allowed to acclimate to the new environment for 7 days prior to initiating the experiment. All procedures were in accordance with the “Principles of Laboratory Animal Care” outlined by National Institute of Health (NIH publication 86-23, 1996).
Locomotor activity tests were conducted in open-field chambers (43 × 43 × 30 cm) (Accuscan Instruments, Inc., Columbus, OH, USA). Before testing, the mice were habituated to the locomotor chamber (2 hr/day for 3 consecutive days), and then were randomly divided into 3 dose groups (vehicle, 5, 15 mg/kg compound 19). On habituation days, the animals were moved to the test room, acclimated there for 10 min, and then placed in their assigned locomotor chambers. On test day 1, each group of mice was pretreated with either vehicle (25% of 2-hydroxypropyl-β-cyclodextrin) or one dose of compound 19 (5, 15 mg/kg, i.p.) without an oxycodone injection. Fifteen min later, the animals were placed into the test chambers. Their locomotor activities were recorded every 20 min for 2 h using VersaMax version 3.0 software (Accuscan Instruments, Inc.). On test days 2–6, the three groups of mice first received vehicle or one dose of compound 19, followed by an oxycodone injection 15 min after compound 19 pretreatment. After 72 h of withdrawal in the home cages in the animal facility, each animal was challenged with a 4 mg/kg oxycodone injection again (day 9) but without compound 19 pretreatment. The locomotor activities were recorded for 2 h/day after each oxycodone injection.
The locomotor behavioral data are expressed as means (±S.E.M.). The traveled distance (cm) within 2 h (Fig. 4A) or every 20 min (Fig. 4B–D) was used to evaluate the locomotor effects of 19 and/or oxycodone in mice. Two-way ANOVA with repeated measures over time was used to evaluate the statistical significance of the changes in locomotor activity after compound 19 and/or oxycodone administration. Post-hoc Fisher’s least significant difference was used for multiple comparisons. p<0.05 was considered statistically significant.
Oxycodone-induced conditioned place preference (CPP)
Fifty male Long-Evans rats (275–300 g) were purchased from the Charles River Laboratories (Raleigh, NC) and housed in the animal facility at NIDA IRP under a reversed 12 h light-dark cycle (light on at 7:00 PM) with free access to food and water. All procedures were in accordance with the “Principles of Laboratory Animal Care” outlined by National Institute of Health (NIH publication 86-23, 1996).
CPP was tested in the place-conditioning apparatus (Med Associates, St Albans, VT) consisting of two side compartments (21 × 28 cm2) and a central gray connecting area (21 × 12.5 cm2); a sliding door separated each compartment from the connecting area. The two side compartments differed in wall color (black vs white), floor type (net vs grid), and illumination. The animals were then divided randomly into 5 treatment groups as shown in Figure 5 legend. Each rat was first exposed in the CPP apparatus for 15 min (preconditioning) and then followed by oxycodone CPP conditioning. Animals displayed significant bias in one compartment (defined by the time difference between two compartments ≥ 200 seconds) during preconditioning were excluded from the study. Oxycodone CPP procedures consisted of 2 days of oxycodone (3 mg/kg, i.p.) conditioning in one compartment and 2 days of saline conditioning in another compartment, alternatively (e.g., oxycodone – saline – oxycodone – saline). The compartments paired with oxycodone or saline were counterbalanced. On the conditioning days, each animal received saline or compound 19 (5, 15 mg/kg, i.p.) pretreatment 15 min prior to oxycodone injection. After oxycodone injection, animals were immediately confined in the assigned compartments for 40 min. To potentiate oxycodone CPP formation, animals received four days of oxycodone (3 mg/kg, i.p., twice daily at 8:00 and 18:00) treatment at home cages.60,61 In the (19 + vehicle) group, saline was paired with one compartment and compound 19 (15 mg/kg) was paired with another compartment. In the (Vehicle + vehicle) group, saline was paired with each compartment. 24 hours after the last saline or oxycodone conditioning injection, animals were placed in the same 3-chamber CPP apparatus for 15 min and the time spent in each compartment was recoded. The CPP score was calculated by the time difference (seconds) that animal spent in drug-paired compartment versus saline-paired compartment.
Supplementary Material
Table 4.
Additional in vitro binding and functional data for selected compounds at 5HT1A, 5HT2A and 5HT2C receptorsa
| Compd | 5-HT1A [3H]-8-OH-DPAT Ki ± SEM, nM | 5-HT2A [125I]DOI Ki ± SEM, nM | 5-HT2C [125I]DOI Ki ± SEM, nM | 5HT1A [35S]GTPγS binding
|
|
|---|---|---|---|---|---|
| Agonist EC50 ± SEM, nM | % Stimulation | ||||
| 11a | 3560 ± 490 | 90.4 ± 4.7 | 22.4 ± 1.9 | NDb | - |
| 11b | 70.3 ± 4.2 | 7.00 ± 2.1 | 5.40 ± 1.6 | 77.0 ± 23 | 90.1 |
| 12a | 4400 ± 1000 | 140 ± 14 | 17.2 ± 5.3 | NDb | - |
| 12b | 40.3 ± 8.9 | 3.22 ± 0.93 | 4.00 ± 1.3 | 27.7 ± 8.3 | 89 |
| 13a | 2260 ± 490 | 470 ± 130 | 1000 ± 190 | NDb | - |
| 13b | 37.0 ± 8.9 | 53.0 ± 15 | 222 ± 59 | 48.0 ± 13 | 90.8 |
| 18 | 2330 ± 700 | 60.1 ± 9.5 | 144 ± 37 | NDb | - |
| 19 | >7600 | 188 ± 22 | 2190 ± 460 | NDb | - |
| 20 | 560 ± 170 | 25.0 ± 6.6 | 55.0 ± 17 | NDb | - |
| 21 | 2410 ± 830 | 42.9 ± 6.8 | 268 ± 34 | NDb | - |
| 22 | 59 ± 13 | 5.22 ± 0.98 | 18.3 ± 2.2 | 149 ± 47 | 94.3 |
| 23 | 880 ± 160 | 28.1 ± 9.7 | 113 ± 33 | NDb | - |
| 24 | 26.0 ± 8.1 | 2.41 ± 0.82 | 18.7 ± 4.5 | 31.0 ± 10 | 99.4 |
| 25 | 27.6 ± 5.3 | 3.20 ± 1.1 | 89.0 ± 22 | 118 ± 39 | 101.1 |
Data were obtained through the NIDA Addiction Treatment Discovery Program contract (ADA151001) with Oregon Health & Science University.
ND = Not determined; Functional assays for each receptor was not conducted if the Ki value for the binding assay was >250 nM for 5-HT receptors.
Acknowledgments
Support for this research was provided by the National Institute on Drug Abuse - Intramural Research Program, NIDA Addiction Treatment Discovery Program contract (ADA151001) with Oregon Health & Science University and Johns Hopkins Drug Discovery (P30MH075673 to BSS). We thank Dr. Anerbasha Shaik for help with the TOC Graphic.
ABBREVIATIONS USED
- DA
dopamine
- SAR
structure activity relationship
- TM
transmembrane
- D2R
dopamine D2 receptor
- D3R
dopamine D3 receptor
- D4R
dopamine D4 receptor
- OBS
orthosteric binding site
- SBP
secondary binding pocket
- 5-HT
5-hydroxytryptamine (serotonin)
- CMA
chloroform/methanol/ammonium hydroxide
Footnotes
Supporting information. Elemental analysis results and additional metabolism data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Heidbreder CA, Newman AH. Current perspectives on selective dopamine D3 receptor antagonists as pharmacotherapeutics for addictions and related disorders. Ann NY Acad Sci. 2010;1187:4–34. doi: 10.1111/j.1749-6632.2009.05149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Caine SB, Koob GF. Modulation of cocaine self-administration in the rat through D-3 dopamine receptors. Science. 1993;260:1814–1816. doi: 10.1126/science.8099761. [DOI] [PubMed] [Google Scholar]
- 3.Pilla M, Perachon S, Sautel F, Garrido F, Mann A, Wermuth CG, Schwartz JC, Everitt BJ, Sokoloff P. Selective inhibition of cocaine-seeking behaviour by a partial dopamine D3 receptor agonist. Nature. 1999;400:371–375. doi: 10.1038/22560. [DOI] [PubMed] [Google Scholar]
- 4.Pich EM, Collo G. Pharmacological targeting of dopamine D3 receptors: Possible clinical applications of selective drugs. Eur Neuropsychopharmacol. 2015;25:1437–1447. doi: 10.1016/j.euroneuro.2015.07.012. [DOI] [PubMed] [Google Scholar]
- 5.Le Foll B, Collo G, Rabiner EA, Boileau I, Merlo Pich E, Sokoloff P. Dopamine D3 receptor ligands for drug addiction treatment: update on recent findings. Prog Brain Res. 2014;211:255–275. doi: 10.1016/B978-0-444-63425-2.00011-8. [DOI] [PubMed] [Google Scholar]
- 6.Paterson NE, Vocci F, Sevak RJ, Wagreich E, London ED. Dopamine D3 receptors as a therapeutic target for methamphetamine dependence. Am J Drug Alcohol Abuse. 2014;40:1–9. doi: 10.3109/00952990.2013.858723. [DOI] [PubMed] [Google Scholar]
- 7.Newman AH, Blaylock BL, Nader MA, Bergman J, Sibley DR, Skolnick P. Medication discovery for addiction: translating the dopamine D3 receptor hypothesis. Biochem Pharmacol. 2012;84:882–890. doi: 10.1016/j.bcp.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Appel NM, Li SH, Holmes TH, Acri JB. Dopamine D3 receptor antagonist (GSK598809) potentiates the hypertensive effects of cocaine in conscious, freely-moving dogs. J Pharmacol Exp Ther. 2015;354:484–492. doi: 10.1124/jpet.115.224121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boateng CA, Bakare OM, Zhan J, Banala AK, Burzynski C, Pommier E, Keck TM, Donthamsetti P, Javitch JA, Rais R, Slusher BS, Xi ZX, Newman AH. High affinity dopamine D3 receptor (D3R)-selective antagonists attenuate heroin self-administration in wildtype but not D3R knockout mice. J Med Chem. 2015;58:6195–6213. doi: 10.1021/acs.jmedchem.5b00776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Foll BL, Collo G, Rabiner EA, Boileau I, Pich EM, Sokoloff P. Dopamine D3 receptor ligands for drug addiction treatment: update on recent findings. Prog Brain Res. 2014;211:255–275. doi: 10.1016/B978-0-444-63425-2.00011-8. [DOI] [PubMed] [Google Scholar]
- 11.Michino M, Beuming T, Donthamsetti P, Newman AH, Javitch JA, Shi L. What can crystal structures of aminergic receptors tell us about designing subtype-selective ligands? Pharmacol Rev. 2015;67:198–213. doi: 10.1124/pr.114.009944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keck TM, Burzynski C, Shi L, Newman AH. Beyond small-molecule SAR: using the dopamine D3 receptor crystal structure to guide drug design. Adv Pharmacol. 2014;69:267–300. doi: 10.1016/B978-0-12-420118-7.00007-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V, Stevens RC. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science. 2010;330:1091–1095. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Newman AH, Beuming T, Banala AK, Donthamsetti P, Pongetti K, LaBounty A, Levy B, Cao J, Michino M, Luedtke RR, Javitch JA, Shi L. Molecular determinants of selectivity and efficacy at the dopamine D3 receptor. J Med Chem. 2012;55:6689–6899. doi: 10.1021/jm300482h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Micheli F. Recent advances in the development of dopamine D3 receptor antagonists: a medicinal chemistry perspective. Chem Med Chem. 2011;6:1152–1162. doi: 10.1002/cmdc.201000538. [DOI] [PubMed] [Google Scholar]
- 16.Micheli F, Heidbreder C. Dopamine D3 receptor antagonists: a patent review (2007 – 2012) Expert Opin Ther Pat. 2013;23:363–381. doi: 10.1517/13543776.2013.757593. [DOI] [PubMed] [Google Scholar]
- 17.Micheli F, Bonanomi G, Blaney FE, Braggio S, Capelli AM, Checchia A, Curcuruto O, Damiani F, Fabio RD, Donati D, Gentile G, Gribble A, Hamprecht D, Tedesco G, Terreni S, Tarsi L, Lightfoot A, Stemp G, Macdonald G, Smith A, Pecoraro M, Petrone M, Perini O, Piner J, Rossi T, Worby A, Pilla M, Valerio E, Griffante C, Mugnaini M, Wood M, Scott C, Andreoli M, Lacroix L, Schwarz A, Gozzi A, Bifone A, Ashby JCR, Hagan JJ, Heidbreder C. 1,2,4-Triazol-3-yl-thiopropyl-tetrahydrobenzazepines: a series of potent and selective dopamine D(3) receptor antagonists. J Med Chem. 2007;50:5076–5089. doi: 10.1021/jm0705612. [DOI] [PubMed] [Google Scholar]
- 18.Keck TM, John WS, Czoty PW, Nader MA, Newman AH. Identifying medication targets for psychostimulant addiction: unraveling the dopamine D3 receptor hypothesis. J Med Chem. 2015;58:5361–5380. doi: 10.1021/jm501512b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Newman AH, Grundt P, Cyriac G, Deschamps JR, Taylor M, Kumar R, Ho D, Luedtke RR. N-(4-(4-(2,3-dichloro- or 2-methoxyphenyl)piperazin-1-yl)butyl)heterobiarylcarboxamides with functionalized linking chains as high affinity and enantioselective D3 receptor antagonists. J Med Chem. 2009;52:2559–2570. doi: 10.1021/jm900095y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Galaj E, Manuszak M, Babic S, Ananthan S, Ranaldi R. The selective dopamine D3 receptor antagonist, SR 21502, reduces cue-induced reinstatement of heroin seeking and heroin conditioned place preference in rats. Drug Alcohol Depend. 2015;156:228–233. doi: 10.1016/j.drugalcdep.2015.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zou MF, Keck TM, Kumar V, Donthamsetti P, Michino M, Burzynski C, Schweppe C, Bonifazi A, Free RB, Sibley DR, Janowsky A, Shi L, Javitch JA, Newman AH. Novel analogues of (R)-5-(methylamino)-5,6-dihydro-4H-imidazo[4,5,1-ij]quinolin-2(1H)-one (sumanirole) provide clues to dopamine D2/D3 receptor agonist selectivity. J Med Chem. 2016;59:2973–2988. doi: 10.1021/acs.jmedchem.5b01612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mistry SN, Shonberg J, Draper-Joyce CJ, Herenbrink CK, Michino M, Shi L, Christopoulos A, Capuano B, Scammells PJ, Lane JR. Discovery of a novel class of negative allosteric modulator of the dopamine D2 receptor through fragmentation of a bitopic ligand. J Med Chem. 2015;58:6819–6843. doi: 10.1021/acs.jmedchem.5b00585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carlsson J, Coleman RG, Setola V, Irwin JJ, Fan H, Schlessinger A, Sali A, Roth BL, Shoichet BK. Ligand discovery from a dopamine D3 receptor homology model and crystal structure. Nat Chem Biol. 2011;7:769–778. doi: 10.1038/nchembio.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hall H, Köhler C, Gawell L, Farde L, Sedvall G. Raclopride, a new selective ligand for the dopamine-D2 receptors. Prog Neuropsychopharmacol Biol Psychiatry. 1988;12:559–568. doi: 10.1016/0278-5846(88)90001-2. [DOI] [PubMed] [Google Scholar]
- 25.Ehrlich K, Gotz A, Bollinger S, Tschammer N, Bettinetti L, Harterich S, Hubner H, Lanig H, Gmeiner P. Dopamine D2, D3, and D4 selective phenylpiperazines as molecular probes to explore the origins of subtype specific receptor binding. J Med Chem. 2009;52:4923–4935. doi: 10.1021/jm900690y. [DOI] [PubMed] [Google Scholar]
- 26.Grundt P, Prevatt KM, Cao J, Taylor J, Floresca CZ, Choi JK, Jenkins BG, Luedtke RR, Newman AH. Heterocyclic analogues of N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)-butyl)-aryl-carboxamides with functionalized linking chains as novel dopamine D3 receptor ligands: potential substance abuse therapeutic agents. J Med Chem. 2007;50:4135–4146. doi: 10.1021/jm0704200. [DOI] [PubMed] [Google Scholar]
- 27.Menzella HG, Tran TT, Carney JR, Lau-Wee J, Galazzo J, Reeves CD, Carreras C, Mukadam S, Eng S, Zhong Z, Timmermans PB, Murli S, Ashley GW. Potent non-benzoquinone ansamycin heat shock protein 90 inhibitors from genetic engineering of Streptomyces hygroscopicus. J Med Chem. 2009;26:1518–1521. doi: 10.1021/jm900012a. [DOI] [PubMed] [Google Scholar]
- 28.Mizuhara T, Kato T, Hirai A, Kurihara H, Shimada Y, Taniguchi M, Maeta H, Togami H, Shimura K, Matsuoka M, Okazaki S, Takeuchi T, Ohno H, Oishi S, Fujii N. Structure-activity relationship study of phenylpyrazole derivatives as a novel class of anti-HIV agents. Bioorg Med Chem Lett. 2013;23:4557–4561. doi: 10.1016/j.bmcl.2013.06.026. [DOI] [PubMed] [Google Scholar]
- 29.White JD, Wang G, Quaranta L. Studies on the synthesis of gymnodimine. Construction of the spiroimine portion via Diels-Alder cycloaddition. Org Lett. 2003;5:4983–4986. doi: 10.1021/ol035939e. [DOI] [PubMed] [Google Scholar]
- 30.Liu KG, Robichaud AJ. A general and convenient synthesis of N-aryl piperazines. Tetrahedron Lett. 2005;46:7921–7922. [Google Scholar]
- 31.Newman AH, Grundt P, Cyriac G, Deschamps JR, Taylor M, Kumar R, Ho D, Luedtke RR. N-(4-(4-(2,3-Dichloro- or 2-methoxyphenyl)piperazin-1-yl)butyl)heterobiarylcarboxamides with functionalized linking chains as high affinity and enantioselective D3 receptor antagonists. J Med Chem. 2009;52:2559–2570. doi: 10.1021/jm900095y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kumar V, Banala AK, Garcia EG, Cao J, Keck TM, Bonifazi A, Deschamps JR, Newman AH. Chiral resolution and serendipitous fluorination reaction for the selective dopamine D3 receptor antagonist BAK2-66. ACS Med Chem Lett. 2014;5:647–651. doi: 10.1021/ml500006v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Banala AK, Levy BA, Khatri SS, Furman CA, Roof RA, Mishra Y, Griffin SA, Sibley DR, Luedtke RR, Newman AH. N-(3-fluoro-4-(4-(2-methoxy or 2,3-dichlorophenyl)piperazine-1-yl)butyl)arylcarboxamides as selective dopamine D3 receptor ligands: critical role of the carboxamide linker for D3 receptor selectivity. J Med Chem. 2011;54:3581–3594. doi: 10.1021/jm200288r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Basha A, Lipton M, Weinreb SM. A mild, general method for conversion of esters to amides. Tetrahedron Lett. 1977;48:4171–4174. [Google Scholar]
- 35.Rovnyak G, Andersen N, Gougoutas J, Hedberg A, Kimball SD, Malley M, Moreland S, Porubcan M, Pudzianowski A. Studies directed toward ascertaining the active conformation of 1,4-dihydropyridine calcium entry blockers. J Med Chem. 1988;31:936–944. doi: 10.1021/jm00400a008. [DOI] [PubMed] [Google Scholar]
- 36.Sasaki Y, Murphy WA, Heiman ML, Lance VA, Coy DH. Solid-phase synthesis and biological properties of psi [CH2NH] pseudopeptide analogues of a highly potent somatostatin octapeptide. J Med Chem. 1987;30:1162–1166. doi: 10.1021/jm00390a008. [DOI] [PubMed] [Google Scholar]
- 37.Gagnon H, Beauchemin S, Kwiatkowska A, Couture F, D’Anjou F, Levesque C, Dufour F, Desbiens AR, Vaillancourt R, Bernard S, Desjardins R, Malouin F, Dory YL, Day R. Optimization of furin inhibitors to protect against the activation of influenza hemaglutinin H5 and Shiga toxin. J Med Chem. 2014;57:29–41. doi: 10.1021/jm400633d. [DOI] [PubMed] [Google Scholar]
- 38.Cruickshank PA, Fishman M. Studies in alkylation. II. Reactions of epoxyalkyl bromides. J Org Chem. 1969;34:4060–4065. [Google Scholar]
- 39.Grundt P, Prevatt KM, Cao J, Taylor M, Floresca CZ, Choi JK, Jenkins BG, Luedtke RR, Newman AH. Heterocyclic analogues of N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butyl)arylcarboxamides with functionalized linking chains as novel dopamine D3 receptor ligands: potential substance abuse therapeutic agents. J Med Chem. 2007;50:4135–4146. doi: 10.1021/jm0704200. [DOI] [PubMed] [Google Scholar]
- 40.Duan H, Ning M, Zou Q, Ye Y, Feng Y, Zhang L, Leng Y, Shen J. Discovery of intestinal targeted TGR5 agonists for the treatment of type 2 diabetes. J Med Chem. 2015;58:3315–3328. doi: 10.1021/jm500829b. [DOI] [PubMed] [Google Scholar]
- 41.Ertl P, Rohde B, Selzer P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J Med Chem. 2000;43:3714–3717. doi: 10.1021/jm000942e. [DOI] [PubMed] [Google Scholar]
- 42.Furman CA, Roof RA, Moritz AE, Miller BN, Doyle TB, Free B, Banala AK, Paul NM, Kumar V, Sibley CD, Newman AH, Sibley DR. Investigation of the binding and functional properties of extended length D3 dopamine receptor-selective antagonists. Eur Neuropsychopharmacol. 2015;25:1448–1461. doi: 10.1016/j.euroneuro.2014.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rais R, Thomas AG, Wozniak K, Wu Y, Jaaro-Peled H, Sawa A, Strick CA, Engel SJ, Brandon NJ, Rojas C, Slusher BS, Tsukamoto T. Pharmacokinetics of oral D-serine in D-amino acid oxidase knockout mice. Drug Metab Dispos. 2012;40:2067–2073. doi: 10.1124/dmd.112.046482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robinson TE, Berridge KC. The incentive sensitization theory of addiction: some current issues. Philos Trans R Soc London. 2008;363:3137–3146. doi: 10.1098/rstb.2008.0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gardner EL. Use of animal models to develop antiaddiction medications. Curr Psychiatry Rep. 2008;10:377–384. doi: 10.1007/s11920-008-0061-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Compton WM, Volkow ND. Major increases in opioid analgesic abuse in the United States: concerns and strategies. Drug Alcohol Depend. 2006;81:103–107. doi: 10.1016/j.drugalcdep.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 47.Compton WM, Jones CM, Baldwin GT. Relationship between nonmedical prescription-opioid use and heroin use. N Engl J Med. 2016;374:154–163. doi: 10.1056/NEJMra1508490. [DOI] [PubMed] [Google Scholar]
- 48.Wilkerson RG, Kim HK, Windsor TA, Mareiniss DP. The opioid epidemic in the United States. Emerg Med Clin North Am. 2016;34:e1–e23. doi: 10.1016/j.emc.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 49.Von Korff MR, Franklin G. Responding to America’s iatrogenic epidemic of prescription opioid addiction and overdose. Med Care. 2016;54:426–429. doi: 10.1097/MLR.0000000000000537. [DOI] [PubMed] [Google Scholar]
- 50.Niikura K, Ho A, Kreek MJ, Zhang Y. Oxycodone-induced conditioned place preference and sensitization of locomotor activity in adolescent and adult mice. Pharmacol Biochem Behav. 2013;110:112–116. doi: 10.1016/j.pbb.2013.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Song R, Yang RF, Wu N, Su RB, Li J, Peng XQ, Li X, Gaál J, Xi ZX, Gardner EL. YQA14: a novel dopamine D3 receptor antagonist that inhibits cocaine self-administration in rats and mice, but not in D3 receptor-knockout mice. Addict Biol. 2012;17:259–273. doi: 10.1111/j.1369-1600.2011.00317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sun L, Song R, Chen Y, Yang RF, Wu N, Su RB, Li J. A selective D3 receptor antagonist YQA14 attenuates methamphetamine-induced behavioral sensitization and conditioned place preference in mice. Acta Pharmacol Sin. 2016;37:157–165. doi: 10.1038/aps.2015.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Le Foll B, Diaz J, Sokoloff P. Increased dopamine D3 receptor expression accompanying behavioral sensitization to nicotine in rats. Synapse. 2003;47:176–183. doi: 10.1002/syn.10170. [DOI] [PubMed] [Google Scholar]
- 54.Filip M, Papla I, Czepiel K. Role of dopamine D3 receptors in controlling the expression of cocaine sensitization in rats. Pol J Pharmacol. 2002;54:687–691. [PubMed] [Google Scholar]
- 55.Li T, Hou Y, Yan CX, Chen T, Zhao Y, Li SB. Dopamine D3 receptor knock-out mice display deficits in locomotor sensitization after chronic morphine administration. Neurosci Lett. 2010;485:256–260. doi: 10.1016/j.neulet.2010.09.025. [DOI] [PubMed] [Google Scholar]
- 56.Volkow ND, McLellan AT. Opioid abuse in chronic pain-misconceptions and mitigation strategies. N Engl J Med. 2016;374:1253–1263. doi: 10.1056/NEJMra1507771. [DOI] [PubMed] [Google Scholar]
- 57.Compton WM, Volkow ND. Major increases in opioid analgesic abuse in the United States: concerns and strategies. Drug Alcohol Depend. 2006;81:103–107. doi: 10.1016/j.drugalcdep.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 58.Compton WM, Jones CM, Baldwin GT. Relationship between nonmedical prescription-opioid use and heroin use. N Engl J Med. 2016;374:154–163. doi: 10.1056/NEJMra1508490. [DOI] [PubMed] [Google Scholar]
- 59.Chen J, Levant B, Jiang C, Keck TM, Newman AH, Wang S. Tranylcypromine substituted cis-hydroxycyclobutylnaphthamides as potent and selective dopamine D(3) receptor antagonists. J Med Chem. 2014;57:4962–4968. doi: 10.1021/jm401798r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gaiardi M, Bartoletti M, Bacchi A, Gubellini C, Costa M, Babbini M. Role of repeated exposure to morphine in determining its affective properties: place and taste conditioning studies in rats. Psychopharmacology. 1991;103:183–186. doi: 10.1007/BF02244201. [DOI] [PubMed] [Google Scholar]
- 61.Lett BT. Repeated exposures intensify rather than diminish the rewarding effects of amphetamine, morphine, and cocaine. Psychopharmacology. 1989;98:357–362. doi: 10.1007/BF00451687. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.