

Abstract
Background
In combination with gene expression profiles, the protein interaction network (PIN) constructs a dynamic network that includes multiple functional modules. Previous studies have demonstrated that rifampin can influence drug metabolism by regulating drug-metabolizing enzymes, transporters, and microRNAs (miRNAs). Rifampin induces gene expression, at least in part, by activating the pregnane X receptor (PXR), which induces gene expression; however, the impact of rifampin on global gene regulation has not been examined under the molecular network frameworks.
Methods
In this study, we extracted rifampin-induced significant differentially expressed genes (SDG) based on the gene expression profile. By integrating the SDG and human protein interaction network (HPIN), we constructed the rifampin-regulated protein interaction network (RrPIN). Based on gene expression measurements, we extracted a subnetwork that showed enriched changes in molecular activity. Using the Kyoto Encyclopedia of Genes and Genomes (KEGG), we identified the crucial rifampin-regulated biological pathways and associated genes. In addition, genes targeted by miRNAs that were significantly differentially expressed in the miRNA expression profile were extracted based on the miRNA-gene prediction tools. The miRNA-regulated PIN was further constructed using associated genes and miRNAs. For each miRNA, we further evaluated the potential impact by the gene interaction network using pathway analysis.
Results and Disccussion
We extracted the functional modules, which included 84 genes and 89 interactions, from the RrPIN, and identified 19 key rifampin-response genes that are associated with seven function pathways that include drug response and metabolism, and cancer pathways; many of the pathways were supported by previous studies. In addition, we identified that a set of 6 genes (CAV1, CREBBP, SMAD3, TRAF2, KBKG, and THBS1) functioning as gene hubs in the subnetworks that are regulated by rifampin. It is also suggested that 12 differentially expressed miRNAs were associated with 6 biological pathways.
Conclusions
Our results suggest that rifampin contributes to changes in the expression of genes by regulating key molecules in the protein interaction networks. This study offers valuable insights into rifampin-induced biological mechanisms at the level of miRNAs, genes and proteins.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-2909-6) contains supplementary material, which is available to authorized users.
Background
Protein-protein interactions are intrinsic to most biological processes [1]. Expanded knowledge of the protein interaction network (PIN) may shed light on basic cellular mechanisms. An expression profile is a dynamic collection of data used to deduce a gene’s function, state, environment, etc. With the increasing availability of genome and proteome data, the PIN can be integrated with gene expression profiles to create conditional network modules within a specific biological state. This method has been used to explore cellular mechanisms associated with multiple diseases [2], including cancer. For instance, Zhang et al. [3] analysed the genes and crucial modules associated with coronary artery diseases (CAD), and suggested that two proteins were critical for the development of CAD. Lin et al. [2] studied dynamic functional modules and co-expressed protein interaction networks in cases of dilated cardiomyopathy. Previous studies suggest that the integrated analysis of PIN and gene expression profile information may contribute to the identification of the functional modules and key genes that are relevant to important biological pathways.
Rifampin is a drug that is usually used to treat tuberculosis and inactive meningitis [4]. The molecular mechanisms and functions of rifampin-regulation have previously been identified. Our previous study has confirmed that rifampin altered expression level of miRNAs and many cytochrome P450 enzymes (CYPs), which are the major metabolic enzymes that control the metabolism of most clinically important drugs, and some of the changes exist in associated relationships that suggest that some of CYP mRNAs are targeted by some miRNAs [5–8]. Rifampin is also a typical ligand of the pregnane X receptor (PXR), which is a transcription factor and a key regulator of the CYPs and other genes involved in drug disposition [9, 10]. Furthermore, rifampin can rapidly downregulate hepatic angiogenesis- and mitogenesis-related genes. Therefore, it shows favorable antiproliferative effects on endothelial cell, which is make it potentially beneficial for targeting hepatobiliary cancer cells [11, 12].
Previous studies have demonstrated that the drug-metabolizing enzymes [6], transporters, and microRNAs (miRNAs) are regulated by rifampin [11, 12], and the mechanisms of the regulation of some of these genes are well-studied; however, little has been done to put the global gene expression effects of rifampin into biological pathways and interactive networks. Protein interaction network can depict and integrate information pertaining to domain architecture, post-translational modification, interaction networks and disease association for each protein in the human proteome [13]. Furthermore, by combining with mRNA expression profiles, they can be used to identify specific correlations of between the genes, and to identify the key genes and functional modules associated with critical biological pathways. In addition, the integration of the miRNA expression profiles can depict relationship between the altered expression of miRNAs and their targeted-mRNA. The implementation of an integrative method that incorporates protein interaction networks and gene expression profiles to reveal conditional network modules associated with the rifampin-regulated biological processes becomes increasingly important in clarifying the regulatory mechanisms responsible for the rifampin effects on gene expression.
In this study, we focused on identifying the key genes, miRNAs, and the regulatory relationships among them. We also explored the rifampin-induced biological pathways by integrating the protein interaction networks and the miRNA and mRNA expression profiles. In this study, we propose a method which can be used to identify the rifampin-regulated functional modules in the protein interaction network of human hepatocytes, and can also be used to further analyze the rifampin-induced miRNAs and their functions. A schematic of the overall method is illustrated in Fig. 1. In this model, the gene expression profile and PIN are integrated to construct the rifampin-regulated protein interaction network (RrPIN). Then, in order to analyse the crucial biological pathways, we identify the functional modules that participate in a common biological function within the protein-protein interaction network. Next, the functional modules are extracted using BioNet and jActiveModules, and the rifampin-induced significant differentially expressed key genes are identified based on an analysis of Gene Ontology (GO) and the Kyoto Encyclopedia of Genes and Genomes (KEGG). Finally, the miRNA-regulated PINs are established using these key genes and gene-targeted miRNAs based on the miRNA expression profile and miRNA-target prediction databases, and the functions of the miRNAs are revealed based on GO and KEGG. The proposed analysis enables us to uncover rifampin-induced biological mechanisms in human hepatocytes.
Fig. 1.
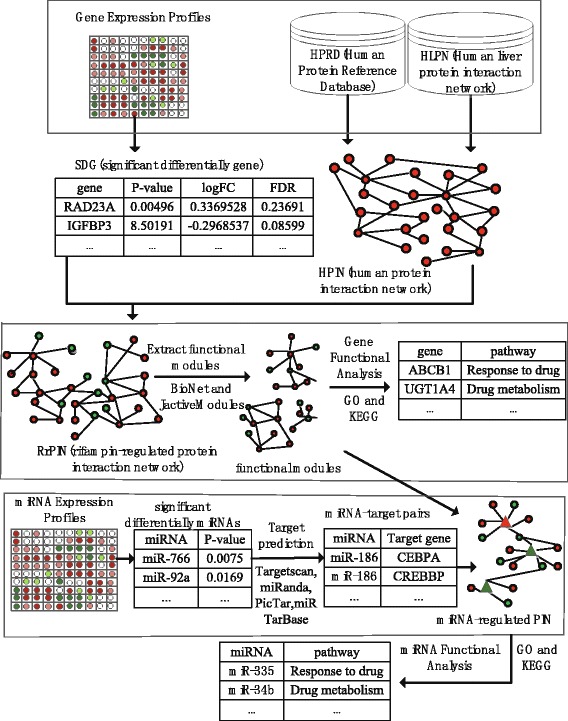
The hierarchical chart of identification of rifampin-regulated functional modules and related miRNAs
Methods
Data
The gene expression dataset and miRNA expression dataset were performed as our previous study in Ramamoorthy et al. [8]. In the current study, the miRNA and mRNA expression profiles were obtained from primary human hepatocyte cultures (obtained from CellzDirect) from 7 donors, each treated with rifampin or vehicle for a total of 14 datasets. Cultures from each subject were treated as biologic replicates (n = 7). The hepatocytes were treated with rifampin or vehicle for 24 h and the total RNAs were isolated using a miRNeasy kit. The mRNA expression profile included 12,780 genes. The miRNA expression profile, which included 334 miRNAs, was measured using the Taqman OpenArray Human miRNA Panel using a NT Cycler. The mRNAs expression was measured using a standard method including EZBead preparation, Next-Gene sequencing, read quality assessment, sequence alignment, and RNA-Seq differential expression analysis.
Construction of RrPIN
The PPI data was downloaded from the Human Protein Reference Database (HPRD) [13], which contains experimentally validated interactions within the human proteome. The human liver protein interaction network (HLPN) [14] contains proteome-scale protein interaction maps of the human liver. It is comprised of 3484 interactions among 2582 proteins and provides substantial new insights into systems biology, disease research, and drug discovery. To construct the human protein interaction network (HPIN), all the proteins and non-overlapped interactions in the HPLN and HPRD were merged as the nodes and interactions of HPIN.
To construct the rifampin-regulated gene network, we integrated the gene expression profile and HPIN as follows: the SDGs which were included in the HPIN were used as RrPIN’s nodes, and the interactions of RrPIN’s nodes in the HPIN were used as the RrPIN’s interactions. Cytoscape version 3.0.2 software (http://chianti.ucsd.edu/cytoscape-3.2.0/) [15] was used to generate the network.
Identification of the functional modules
Particular interest of BioNet and jActiveModules were the identification of functional modules in the network in which the nodes have significant P-values by means of detecting differentially expressed regions in networks. This indicates a group of nodes which are densely connected and have significant differences in expression level, suggesting a module whose activity is influenced by the experimental context of the expression data. The functional modules tend to correspond to shared common cellular function beyond the scope of classical pathways [16–18]. The maximally scoring optimal module was identified using BioNet [17, 18]. And the jActiveModules plug-in of cytoscape was used to further identify multiple significant modules in the PPI network [16].
Enrichment analysis of functional modules
The gene-annotation enrichment analysis was performed using the Database for Annotation, Visualization, and Integrated Discovery (DAVID), which provides a comprehensive set of functional annotation tools for biological interpretation of large gene lists. GO and KEGG are included in the set of functional annotation tools of DAVID. To study the rifampin-regulated biological process, we used DAVID’s GOTERM_BP_FAT (lower levels of biological process ontology), and KEGG pathway analysis to identify enriched biological themes, particularly GO terms [19].
Identification of miRNAs and analysis of their functions
MiRNAs with p < 0.05 were regarded as significant differentially expressed miRNAs. We identified these miRNAs’ target genes using the R library RmiR.Hs.miRNA [20] which collects information from different miRNA target databases. In this study, Targetscan [21, 22], miRanda [23], PicTar [24] and miRTarBase [25, 26] were choosen. The BiomaRt [27] library, which provides a wide range of online queries from gene annotation to database mining, was used to convert gene IDs to gene symbols based on the hsapiens_gene_ensembl database. For each miRNA, the miRNA-targeted genes belonging to functional modules were considered as the nodes of the miRNA-regulated PIN. The interactions of these genes in PPI and of each miRNA with its target genes were considered as the interactions of the miRNA-regulated PIN. As a result, we obtained the miRNA-regulated PIN. For each miRNA, we analysed its potential functions by analysing the miRNA’s target genes based on GO and KEGG.
Results
SDGs and RrPIN
The mRNA expression profile was obtained from RNA-seq data from primary hepatocytes from 7 subjects treated with rifampin or vehicle. In order to focus on cellular responses that are triggered by the rifampin treatment, we pre-selected the genes that are differentially expressed with a loose p-value threshold at p-value < 0.01 without multiple hypothesis correction. Our further analysis focuses on 1866 differentially expressed genes that pass the threshold. We mapped all the differentially expressed genes on the combined human protein interaction network, which consists of 10,210 proteins with 42,521 interactions. As shown in Fig. 2, the resultant rifampin interaction network includes 663 proteins with 1024 interactions.
Fig. 2.

The workflow of the RrPIN. Color is according to the fold change where red denotes upregulated and green denotes downregulated
Identification and analysis of the functional modules
The aforementioned network contains candidate differentially expressed genes with a flexible p-value cutoff. This is intentional since our network analysis will be further used to identify a cluster of interacting molecules that tend to be collectively differentially expressed, and therefore will reduce false positives. We used BioNet [17, 18], a bioconductor package for the functional analysis of biological networks, which uses the p-values obtained from differential expressed genes from RNA-seq data. The goal of this algorithm is to identify functional modules, or significantly differentially expressed subnetworks, within large networks [17]. This was achieved by computing a score for each node, reflected by its p-value, and used a network search algorithm to find the highest-scoring subgraph.
In this study, the maximally functional module was identified by computing optimal scores based on the p-values from the RNA-seq data to evaluate how molecular activity changes were correlated with rifampin regulation. False discovery rate (FDR) is an adjustment parameter for controlling the resultant subnetwork size. Since FDR can be used for fine-tuning of the signal noise decomposition, we scan a large range of FDRs and evaluate the obtained modules according to true-positive rate and precision (ratio of true positives to all positively classify). As a result, a threshold value of >0.0001 was used, because others thresholds lead to either too small or too large size of the module. The derived module captures the characteristically differently expressed interactions associated with rifampin treatment. There were 84 genes and 89 interactions in the maximally functional module. P-values, fold-changes, and false discovery rates (FDR) for the genes of the maximally functional module are shown in Additional file 1.
To avoid bias and to ensure generality of our results, besides the maximally functional module, we identified the multiple functional modules and key genes that also demonstrated the significant enrichment on differentially expressed genes; this analysis was done using the jActiveModules plug-in in cytoscape [15]. The maximum depth from the start node was set as 2 and the overlap threshold was set as 0. There were 31 nodes and 36 interactions within the five functional modules. P-values, Fold Changes, and false discovery rates (FDR) for the genes within the five functional modules are shown in Additional file 2. The maximally functional module and five functional modules are shown in Fig. 3.
Fig. 3.
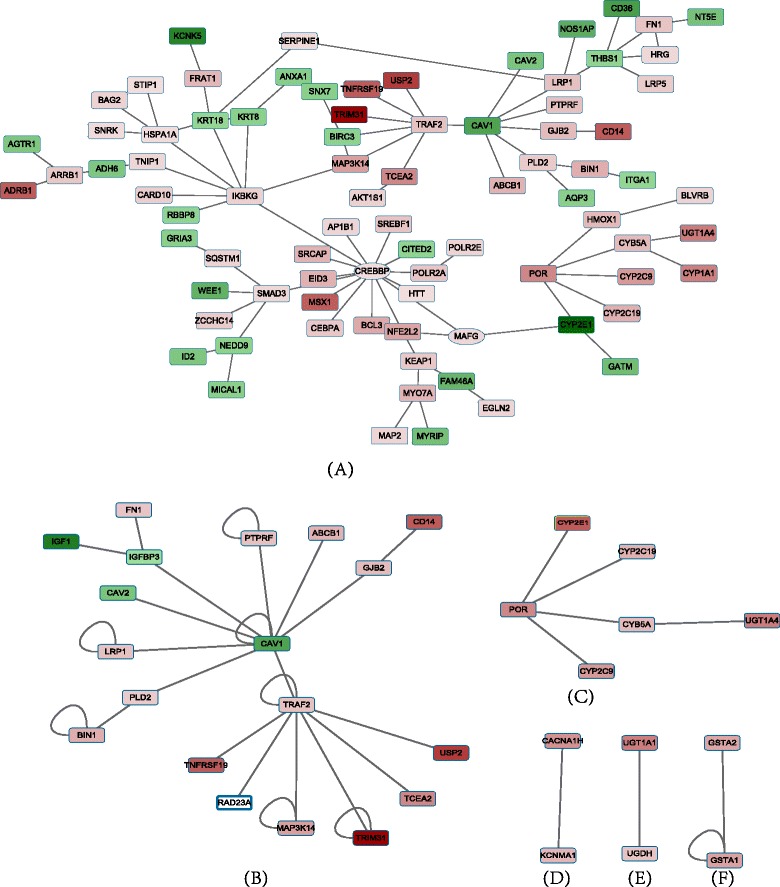
The functional modules of RrPIN. a The maximally functional module of PPI network. Color is according to the fold change where red denotes upregulated and green denotes downregulated. The shape of the nodes depicts the aggregate score: circles indicate a negative score, rectangles denotes a positive score. b, c, d, e and f are the five functional modules of RrPIN. The regulatory relationships are denoted by colours in which red indicates upregulated genes, and green indicates downregulated. As well, the depth of the colour explains the size of fold change
As expected, the results from BioNet are essentially in agreement with the results from the jActiveModules plug-in. The maximally functional module included all the nodes and interactions of the five functional modules.
Enrichment analysis of functional modules
To systematically determine the roles of genes in the functional modules, the online biological classification tool DAVID was used to carry out the functional classification based on GO and key signal pathways from KEGG. Since most of the genes within the five modules were included in the maximally functional module, we primarily focused on the analysis of the maximally functional module. Table of top 20 GO terms and top 10 KEGG terms for the genes of functional modules were shown in Table 1. Due to the redundant nature of the ontology analysis, functional annotation clustering was also derived from DAVID. A table of the top 20 functional annotations clustering for the genes of functional modules assessed by DAVID are provided in Additional file 3. Since the evidence suggests that rifampin have a broad spectrum of effect on enhancing drug metabolism, specifically, we focused on the GO terms “drug” and “metabolism,” and the top six listed KEGG pathways were extracted and the duplicates were eliminated. The p-value was used to evaluate the significance of the GO terms and KEGG pathways. Table 2 shows the results of the enrichment analysis of the maximally functional module in the RrPIN.
Table 1.
Table of top 20 GO terms and top 10 KEGG terms for the genes of functional modules
| Category | Term | Count | Percent | P-value | Benjiamini |
|---|---|---|---|---|---|
| GOTERM_BP_FAT | regulation of apoptosis | 19 | 22.6 | 5.10E-07 | 7.30E-04 |
| GOTERM_BP_FAT | regulation of programme cell death | 19 | 22.6 | 5.90E-07 | 4.20E-04 |
| GOTERM_BP_FAT | regulation of cell death | 19 | 22.6 | 6.20E-07 | 3.00E-04 |
| GOTERM_BP_FAT | negative regulation of apoptosis | 13 | 15.5 | 7.90E-07 | 2.80E-04 |
| GOTERM_BP_FAT | negative regulation of programmed cell death | 13 | 15.5 | 9.10E-07 | 2.60E-04 |
| GOTERM_BP_FAT | negative regulation of cell death | 13 | 15.5 | 9.40E-07 | 2.20E-04 |
| GOTERM_BP_FAT | membrane organization | 13 | 15.5 | 1.70E-06 | 3.50E-04 |
| GOTERM_BP_FAT | vesicle-mediated transport | 15 | 17.9 | 4.50E-06 | 8.10E-04 |
| GOTERM_BP_FAT | membrane invagination | 10 | 11.9 | 4.70E-06 | 7.40E-04 |
| GOTERM_BP_FAT | endocytosis | 10 | 11.9 | 4.70E-06 | 7.40E-04 |
| GOTERM_BP_FAT | response to hypoxia | 8 | 9.5 | 1.20E-05 | 1.70E-03 |
| GOTERM_BP_FAT | response to oxygen levels | 8 | 9.5 | 1.60E-05 | 2.10E-03 |
| GOTERM_BP_FAT | response to inorganic substance | 9 | 10.7 | 2.30E-05 | 2.70E-03 |
| GOTERM_BP_FAT | anti-apoptosis | 9 | 10.7 | 2.40E-05 | 2.60E-03 |
| GOTERM_BP_FAT | positive regulation of multicellular organismal process | 9 | 10.7 | 7.90E-05 | 8.00E-03 |
| GOTERM_BP_FAT | drug metabolic process | 4 | 4.8 | 9.80E-05 | 9.20E-03 |
| GOTERM_BP_FAT | response to metal ion | 7 | 8.3 | 9.80E-05 | 8.70E-03 |
| GOTERM_BP_FAT | phagocytosis | 5 | 6 | 1.60E-04 | 1.40E-02 |
| GOTERM_BP_FAT | response to organic substance | 14 | 16.7 | 2.20E-04 | 1.80E-02 |
| GOTERM_BP_FAT | regulation of tube size | 5 | 6 | 2.40E-04 | 1.80E-02 |
| KEGG_PATHWAY | Metabolism of xenobiotics by cytochrome P450 | 6 | 7.1 | 2.00E-04 | 1.70E-02 |
| KEGG_PATHWAY | Retinol metabolism | 5 | 6 | 1.40E-03 | 6.00E-02 |
| KEGG_PATHWAY | Drug metabolism | 5 | 6 | 2.40E-03 | 6.70E-02 |
| KEGG_PATHWAY | Linoleic acid metabolism | 3 | 3.6 | 2.70E-02 | 4.50E-01 |
| KEGG_PATHWAY | Pathways in cancer | 8 | 9.5 | 2.90E-02 | 4.00E-01 |
| KEGG_PATHWAY | Focal adhesion | 6 | 7.1 | 3.70E-02 | 4.10E-01 |
| KEGG_PATHWAY | Porphyrin and chlorophyll metabolism | 3 | 3.6 | 3.70E-02 | 3.70E-01 |
| KEGG_PATHWAY | Small cell lung cancer | 4 | 4.8 | 4.20E-02 | 3.70E-01 |
| KEGG_PATHWAY | ECM-receptor interaction | 4 | 4.8 | 4.20E-02 | 3.70E-01 |
| KEGG_PATHWAY | TGF-beta signaling pathway | 4 | 4.8 | 4.60E-02 | 3.60E-01 |
Table 2.
Enrichment analysis of the maximally functional module in rifampin
| DAVID (Term) | Genes | P-value |
|---|---|---|
| GO: Response to drug | ABCB1,UGT1A4,CAV1,CAV2 | 3.6E-2 |
| KEGG: Metabolism of xenobiotics by cytochrome P450 | UGT1A4,ADH6,CYP1A1,CYP2C19,CYP2C9,CYP2E1 | 2.0E-4 |
| KEGG: Retinol metabolism | UGT1A4,ADH6,CYP1A1,CYP2C19,CYP2C9 | 1.4E-3 |
| KEGG: Drug metabolism | UGT1A4,ADH6, CYP2C19,CYP2C9,CYP2E1 | 2.4E-3 |
| KEGG: Linoleic acid metabolism | CYP2C19,CYP2C9,CYP2E1 | 2.7E-2 |
| KEGG: Pathways in cancer | CEBPA,CREBBP,SMAD3,TRAF2,BIRC3,EGLN2,FN1,IKBKG | 2.9E-2 |
| Focal adhesion | BIRC3,CAV1,CAV2,FN1,ITGA1,THBS1 | 3.7E-2 |
The results show that the maximally functional module is relevant with seven functional enrichment terms: response to drug, metabolism of xenobiotics by cytochrome P450, retinol metabolism, drug metabolism, linoleic acid metabolism, cancer pathways, and focal adhesion. Among these terms, retinol metabolism, drug metabolism and linoleic acid metabolism contained many similarities in genes, since these three function terms were functionally correlated and clustered in functional annotation clustering in DAVID. In particular, the function pathways coincided with previously reported rifampin-induced biological functions. For example, rifampin affected the hepatic drug disposition and metabolism [28, 29] and it was a potent inducer of drug-metabolizing enzymes [6, 29–31]. Rifampin is also an inhibitor which rapidly downregulates angiogenesis and mitogenesis-related genes to target cancer cells [12, 32, 33].
In addition, we define the percentage of identified SDGs in each pathway relative to the total number of SDGs as pathway relative abundance. Assume that there are N SDGs-associated biological pathways, for i–th pathway, S(i) is number of identified SDGs, and N(i) is enriched number of the total SDGs. The pathway relative abundance E(x) is defined as:
| 1 |
Figure 4 shows the pathway relative abundance of maximally functional module’s genes and all SDGs on the associated seven functional enrichment terms. It can be seen that the SDGs of maximally functional module are more enriched and representative on each terms than the total SDGs. This suggests that our strategy in integrating PPI network with the differential expression analysis helped us in capturing more biologically relevant signals.
Fig. 4.
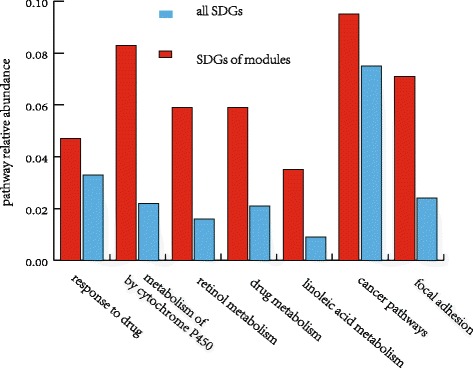
The pathway relative abundance of maximally functional module’s genes and all SDGs on the associated seven functional enrichment terms
To analyze each functional enrichment term, we focus on the analysis of their genes. There were 19 key genes associated with 7 functions that we extracted using DAVID. Then we extend the protein interaction network of 19 genes based on the RrPIN with one level interaction. The RrPIN extension network of 19 genes consists of 50 nodes and 53 interactions. The RrPIN extension network of 19 genes and associated 7 functions are shown as Fig. 5.
Fig. 5.
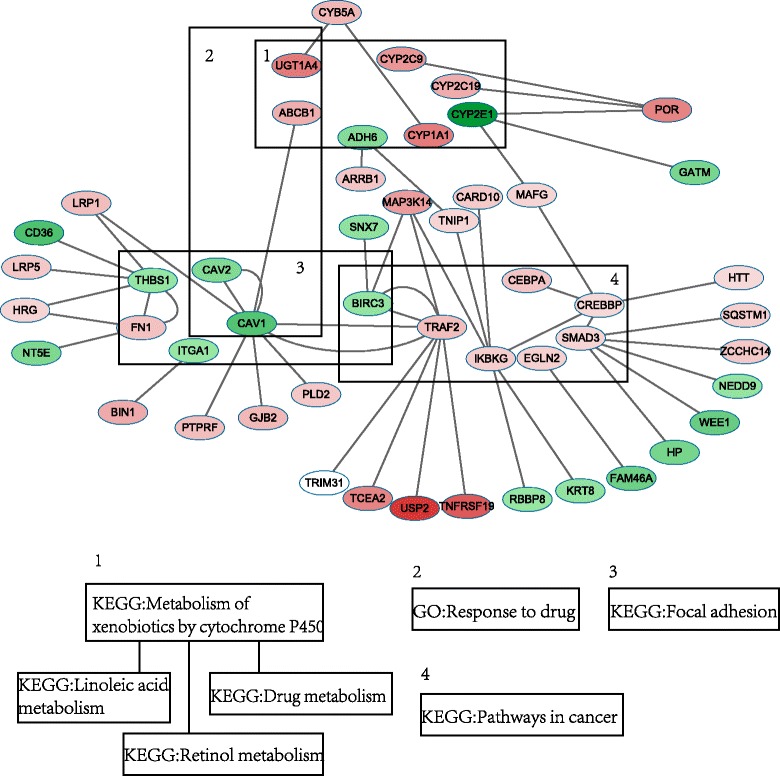
The RrPIN extension network of 19 genes and associated 7 functions
It is worth noting that UGT1A4, ADH6, CYP1A1, CYP2C19, CYP2C9 and CYP2E1 are all associated with metabolism of xenobiotics, drug metabolism, retinol metabolism, and linoleic acid metabolism. BIRC3, CAV1, CAV2, FN1, ITGA1 and THBS1 were functionally enriched to focal adhesion, which contributes to antiangiogenic and anti-tumour effects. These results indicate that rifampin induced drug metabolism, partially, by regulating UGT1A4, ADH6, CYP1A1, CYP2C19, CYP2C9 and CYP2E1. These results also signify that rifampin can influence the anti-angiogenesis and anti-tumour effects of drugs by regulating BIRC3, CAV1, CAV2, FN1, ITGA1 and THBS1. Previous reports support these findings, stating that UGT1A4 CYP1A1, CYP2C19, CYP2C9 and CYP2E1 are drug-metabolizing enzymes [34, 35], and ADH6 modulates the risk for drug dependence [35]. BIRC3 contains anti-apoptotic genes, which can be suppressed to counteract cancerous activity [36]. CAV1 and CAV2 were correlated with tumour growth and metastasis [37–39], and FN1 was a potential biomarker for some cancers [40, 41], while ITGA1 and THBS1 were also associated with cancer risk [42, 43].
In addition, some of the 19 key genes were hub proteins that interacted with multiple proteins. For example, CAV1, CREBBP, SMAD3, TRAF2, KBKG and THBS1 had at least four interactions with other proteins. These results suggest that these six genes are important components in biological pathways regulated by rifampin.
Joint analysis of key genes and associated miRNAs
To identify miRNAs that may regulate these key genes within the functional modules, we correlated the alterations in the miRNA and gene expression. In this process, we extracted the significant differentially expressed miRNAs (p < 0.05), and identified 20 miRNAs. The significant differentially expressed miRNAs are shown in Table 3.
Table 3.
The significant differentially expressed miRNAs
| miRNA | P-value | miRNA | P-value |
|---|---|---|---|
| Upregulated | Upregulated | ||
| miR-886-3p | 0.0002 | miR-660 | 0.0297 |
| miR-766 | 0.0075 | miR-638 | 0.0302 |
| miR-92a | 0.0169 | miR-25 | 0.0338 |
| miR-107 | 0.0177 | miR-616 | 0.0446 |
| miR-30d# | 0.0195 | miR-576-3p | 0.0453 |
| miR-335 | 0.0241 | miR-218 | 0.0499 |
| Downregulated | Downregulated | ||
| miR-186 | 0.0018 | miR-320 | 0.0376 |
| miR-361 | 0.0111 | miR-202 | 0.0396 |
| miR-95 | 0.0219 | miR-200b# | 0.0426 |
| miR-345 | 0.0239 | let-7 g | 0.0435 |
In order to identify the target genes of significant differentially expressed miRNAs, three databases (Targetscan, miRanda and PicTar) were used [20–23]. The miRNA-mRNA pairs were extracted for each significant differentially expressed miRNA. In order to include verified miRNA-mRNA pairs, we also extracted the miRNA-mRNA pairs from the miRTarBase [25, 26]. And the miRNA-mRNA pairs of which mRNA is a gene of maximally functional module were chosen. Then, we established the miRNA-regulated PIN, which showed a negative correlation between the miRNA and the mRNA. The miRNA-regulated PIN, which is constructed of the genes in the functional modules, is shown in Table 4 and Fig. 6.
Table 4.
The miRNA-regulated PIN which constructed by the genes of functional modules
| Gene list | logFC | miRNA | P-value | Fold change |
|---|---|---|---|---|
| CYP2E1 | −1.4341 | miR-335 | 0.0242 | 1.3300 |
| CAV1 | −0.8518 | miR-34b | 0.1753 | 185.3764 |
| miR-886-3p | 0.0001 | 1.5645 | ||
| miR-218 | 0.0499 | 1.9012 | ||
| miR-576-3p | 0.0453 | 2.1916 | ||
| CAV2 | −0.5386 | miR-200c | 0.0913 | 4.8313 |
| miR-576-3p | 0.0453 | 2.1916 | ||
| CEBPA | 0.5812 | miR-186 | 0.0017 | 0.8356 |
| CREBBP | 0.3821 | miR-186 | 0.0017 | 0.8356 |
| miR-95 | 0.0216 | 0.6320 | ||
| miR-769 | 0.1249 | 0.8388 | ||
| EGLN2 | 0.4574 | miR-202 | 0.0396 | 0.5988 |
| let-7 g | 0.0435 | 0.8402 | ||
| ITGA1 | −0.3754 | miR-616 | 0.0446 | 1.3337 |
| miR-660 | 0.0297 | 1.2642 | ||
| miR-576-3p | 0.0453 | 2.1916 | ||
| miR-335 | 0.0242 | 1.3300 | ||
| THBS1 | −0.3951 | miR-886-3p | 0.0001 | 1.5645 |
| miR-335 | 0.0242 | 1.3300 | ||
| miR-616 | 0.0446 | 1.3337 | ||
| miR-92a | 0.0169 | 1.1319 |
Fig. 6.
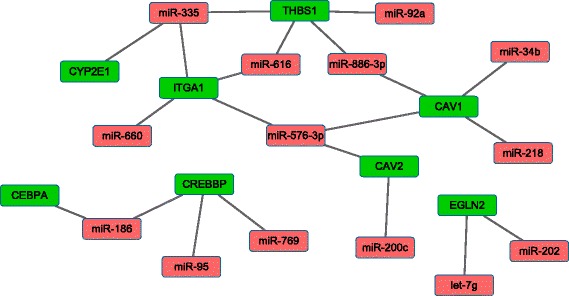
The miRNA-regulated PIN which is constructed by the genes of functional modules
Eight genes and 14 miRNAs were identified to have significant differential expression changes. Each of these genes was regulated by multiple miRNAs. Due to the miRNAs control of gene expression, either by degradation of the target mRNAs or by inhibition of protein translation, miRNA-regulated PPI networks can uncover new rules of miRNA regulation or protein interaction. Thus, we predicted the potential functions of miRNAs based on the function of their target genes as shown in Table 5.
Table 5.
The potential functions of miRNAs
| DAVID (Term) | miRNA |
|---|---|
| GO: Response to drug | miR-34b, miR-886-3p, miR-218, miR-576-3p, miR-200c |
| KEGG: Metabolism of xenobiotics by cytochrome P450 | miR-335 |
| KEGG: Drug metabolism | miR-335 |
| KEGG: Linoleic acid metabolism | miR-335 |
| KEGG: Pathways in cancer | miR-186, miR-95, miR-769 |
| Focal adhesion | miR-34b, miR-886-3p, miR-218, miR-576-3p, miR-200c, miR-616, miR-660, miR-335, miR-92a |
Twelve miRNAs were extracted which associated with 6 biological pathways including response to drug, metabolism of xenobiotics by cytochrome P450, drug metabolism, linoleic acid metabolism, cancer pathways, and focal adhesion through regulation of 8 target genes. The results suggest that miR-335 influences drug metabolism through negative regulation of CYP2E1, which is a drug metabolizing enzyme that is affected by rifampin treatment. Therefore, it is possible that rifampin may alter miRNA expression, which in turn affects the expression of the drug metabolizing enzyme gene CYP2E1. MiR-186 was found to regulate two genes (CEBPA, CREBBP), which were associated with cancer pathways. MiR-186, miR-769, miR-95, miR-202 and let-7 g were also relevant to cancer pathways, but did not serve other functions. Previous studies have demonstrated that rifampin also inhibited anti-angiogenesis by regulating the expression of multiple miRNAs (miR-34b, miR-886-3p, miR-218, miR-576-3p, miR-200c, miR-616, miR-660, miR-335, miR-92a), and further induced the gene expression of BIRC3, CAV1, CAV2, FN1, ITGA1 and THBS1.
Conclusions
In conclusion, a novel integrative network-based method was used to identify the functional modules and discover the potential functions of miRNAs based on human protein network, mRNA and miRNA expression profile in rifampin treated hepatocytes. Furthermore, this method identifies 19 genes and 7 crucial biological pathways. By analysing the miRNA-regulated PIN, we suggested that 12 miRNAs were associated with 6 biological pathways through regulation of 8 target genes. Our results suggest that rifampin contributes to changes in the expression of genes and miRNAs, and induces multiple biological pathways. This study not only provides an insight into functional modules that are associated with rifampin-treated human hepatocytes in human protein interaction network, it also shows that the integrated analysis of mRNA, miRNA expression profile and PIN can be used to study the molecular mechanism of rifampin-induced drug disposition.
Acknowledgments
Declarations
Publication charges for this article have been funded by the National Key Scientific Instrument and Equipment Development Projects of China (2012YQ04014001 and 2012YQ04014010), National Natural Science Foundation of China (61471139), Fundamental Research Funds for the Central Universities (HEUCF160412), Natural Science Fund of Heilongjiang Province (F201331, F201241).
This article has been published as part of BMC Genomics Volume 17 Supplement 7, 2016: Selected articles from the International Conference on Intelligent Biology and Medicine (ICIBM) 2015: genomics. The full contents of the supplement are available online at http://bmcgenomics.biomedcentral.com/articles/supplements/volume-17-supplement-7.
Availability of data and materials
The complete RNA-seq data used in this paper can be downloaded from the GEO database with the accession number: GSE79933; The complete microRNA OpenArray data used in this paper can be downloaded from http://compbio.iupui.edu/group/6/pages/rifampin.
Authors’ contributions
JL and WY developed the programs and workflows, analysed the data, and wrote the manuscript. LW, XFD and YW contributed to the data analysis. WC provided some advice on analysis and contributed partly to writing of the manuscript. CZX and WXF contributed to the computational analyses. YLD, TCS was responsible for sample collection and processing for analysis. HL and YLL conceived and directed the project, arranged the sampling, provided advice on analysis, and contributed to writing of the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Additional files
P-values, Fold Change and false discovery rates (FDR) for the genes of the maximally functional module. (PDF 161 kb)
P-values, Fold Change and false discovery rates (FDR) for the genes of the five functional modules. (PDF 105 kb)
Top 10 GO terms and KEGG terms for the genes in functional modules from the DAVID were provided as Additional file 3. (PDF 264 kb)
Contributor Information
Jin Li, Email: lijin@hrbeu.edu.cn.
Ying Wang, Email: wangying0129@126.com.
Lei Wang, Email: wl@hrbeu.edu.cn.
Xuefeng Dai, Email: daixuefeng203@163.com.
Wang Cong, Email: cwcy9156@163.com.
Weixing Feng, Email: fengweixing@hrbeu.edu.cn.
Chengzhen Xu, Email: xu5115@163.com.
Yulin Deng, Email: deng@bit.edu.cn.
Yue Wang, Email: yuewang@iu.edu.
Todd C. Skaar, Email: tskaar@iu.edu
Hong Liang, Email: lh@hrbeu.edu.cn.
Yunlong Liu, Email: yunliu@iupui.edu.
References
- 1.Koyuturk M. Algorithmic and analytical methods in network biology. Wiley Interdiscip Rev Syst Biol Med. 2010;2(3):277–292. doi: 10.1002/wsbm.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lin CC, Chen YJ, Chen CY, Oyang YJ, Juan HF, Huang HC. Crosstalk between transcription factors and microRNAs in human protein interaction network. BMC Syst Biol. 2012;6:18. doi: 10.1186/1752-0509-6-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gennarino VA, D’Angelo G, Dharmalingam G, Fernandez S, Russolillo G, Sanges R, Mutarelli M, Belcastro V, Ballabio A, Verde P, et al. Identification of microRNA-regulated gene networks by expression analysis of target genes. Genome Res. 2012;22(6):1163–1172. doi: 10.1101/gr.130435.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Egelund EF, Isaza R, Brock AP, Alsultan A, An G, Peloquin CA. Population pharmacokinetics of rifampin in the treatment of Mycobacterium tuberculosis in Asian elephants. J Vet Pharmacol Ther. 2015;38(2):137–143. doi: 10.1111/jvp.12156. [DOI] [PubMed] [Google Scholar]
- 5.Liu CL, Lim YP, Hu ML. Fucoxanthin attenuates rifampin-induced cytochrome P450 3A4 (CYP3A4) and multiple drug resistance 1 (MDR1) gene expression through pregnane X receptor (PXR)-mediated pathways in human hepatoma HepG2 and colon adenocarcinoma LS174T cells. Marine drugs. 2012;10(1):242–257. doi: 10.3390/md10010242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mahatthanatrakul W, Nontaput T, Ridtitid W, Wongnawa M, Sunbhanich M. Rifampin, a cytochrome P450 3A inducer, decreases plasma concentrations of antipsychotic risperidone in healthy volunteers. J Clin Pharm Ther. 2007;32(2):161–167. doi: 10.1111/j.1365-2710.2007.00811.x. [DOI] [PubMed] [Google Scholar]
- 7.Takahashi K, Tatsumi N, Fukami T, Yokoi T, Nakajima M. Integrated Analysis of Rifampicin-induced MicroRNA and Gene Expression Changes in Human Hepatocytes. Drug Metab Pharmacokinet. 2014;29(4):333–340. doi: 10.2133/dmpk.DMPK-13-RG-114. [DOI] [PubMed] [Google Scholar]
- 8.Ramamoorthy A, Liu Y, Philips S, Desta Z, Lin H, Goswami C, Gaedigk A, Li L, Flockhart DA, Skaar TC. Regulation of microRNA expression by rifampin in human hepatocytes. Drug Metab Dispos. 2013;41(10):1763–1768. doi: 10.1124/dmd.113.052886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim B, Moon JY, Choi MH, Yang HH, Lee S, Lim KS, Yoon SH, Yu KS, Jang IJ, Cho JY. Global metabolomics and targeted steroid profiling reveal that rifampin, a strong human PXR activator, alters endogenous urinary steroid markers. J Proteome Res. 2013;12(3):1359–1368. doi: 10.1021/pr301021p. [DOI] [PubMed] [Google Scholar]
- 10.Kliewer SA, Goodwin B, Willson TM. The nuclear pregnane X receptor: a key regulator of xenobiotic metabolism. Endocr Rev. 2002;23(5):687–702. doi: 10.1210/er.2001-0038. [DOI] [PubMed] [Google Scholar]
- 11.Benson EA, Eadon MT, Desta Z, Liu Y, Lin H, Burgess KS, Segar MW, Gaedigk A, Skaar TC. Rifampin Regulation of Drug Transporters Gene Expression and the Association of MicroRNAs in Human Hepatocytes. Front. Pharmacol. 2016;7:111. doi: 10.3389/fphar.2016.00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shichiri M, Fukai N, Kono Y, Tanaka Y. Rifampicin as an oral angiogenesis inhibitor targeting hepatic cancers. Cancer Res. 2009;69(11):4760–4768. doi: 10.1158/0008-5472.CAN-08-3417. [DOI] [PubMed] [Google Scholar]
- 13.Keshava Prasad TS, Goel R, Kandasamy K, Keerthikumar S, Kumar S, Mathivanan S, Telikicherla D, Raju R, Shafreen B, Venugopal A, et al. Human Protein Reference Database--2009 update. Nucleic Acids Res. 2009;37(Database issue):D767–D772. doi: 10.1093/nar/gkn892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang J, Huo K, Ma L, Tang L, Li D, Huang X, Yuan Y, Li C, Wang W, Guan W, et al. Toward an understanding of the protein interaction network of the human liver. Mol Syst Biol. 2011;7:536. doi: 10.1038/msb.2011.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kohl M, Wiese S, Warscheid B. Cytoscape: software for visualization and analysis of biological networks. Methods Mol Biol. 2011;696:291–303. doi: 10.1007/978-1-60761-987-1_18. [DOI] [PubMed] [Google Scholar]
- 16.Ideker T, Ozier O, Schwikowski B, Siegel AF. Discovering regulatory and signalling circuits in molecular interaction networks. Bioinformatics. 2002;18(Suppl 1):S233–S240. doi: 10.1093/bioinformatics/18.suppl_1.S233. [DOI] [PubMed] [Google Scholar]
- 17.Dittrich MT, Klau GW, Rosenwald A, Dandekar T, Muller T. Identifying functional modules in protein-protein interaction networks: an integrated exact approach. Bioinformatics. 2008;24(13):i223–i231. doi: 10.1093/bioinformatics/btn161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beisser D, Klau GW, Dandekar T, Muller T, Dittrich MT. BioNet: an R-Package for the functional analysis of biological networks. Bioinformatics. 2010;26(8):1129–1130. doi: 10.1093/bioinformatics/btq089. [DOI] [PubMed] [Google Scholar]
- 19.Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40(Database issue):D109–D114. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Helvik SA, Snove O, Jr, Saetrom P. Reliable prediction of Drosha processing sites improves microRNA gene prediction. Bioinformatics. 2007;23(2):142–149. doi: 10.1093/bioinformatics/btl570. [DOI] [PubMed] [Google Scholar]
- 21.Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19(1):92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garcia DM, Baek D, Shin C, Bell GW, Grimson A, Bartel DP. Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs. Nat Struct Mol Biol. 2011;18(10):1139–1146. doi: 10.1038/nsmb.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Betel D, Wilson M, Gabow A, Marks DS, Sander C. The microRNA.org resource: targets and expression. Nucleic Acids Res. 2008;36(Database issue):D149–D153. doi: 10.1093/nar/gkm995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M, et al. Combinatorial microRNA target predictions. Nat Genet. 2005;37(5):495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 25.Hsu SD, Lin FM, Wu WY, Liang C, Huang WC, Chan WL, Tsai WT, Chen GZ, Lee CJ, Chiu CM, et al. miRTarBase: a database curates experimentally validated microRNA-target interactions. Nucleic Acids Res. 2011;39(Database issue):D163–D169. doi: 10.1093/nar/gkq1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hsu SD, Tseng YT, Shrestha S, Lin YL, Khaleel A, Chou CH, Chu CF, Huang HY, Lin CM, Ho SY, et al. miRTarBase update 2014: an information resource for experimentally validated miRNA-target interactions. Nucleic Acids Res. 2014;42(Database issue):D78–D85. doi: 10.1093/nar/gkt1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Assefa SA, Preston MD, Campino S, Ocholla H, Sutherland CJ, Clark TG. estMOI: estimating multiplicity of infection using parasite deep sequencing data. Bioinformatics. 2014;30(9):1292–1294. doi: 10.1093/bioinformatics/btu005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lam JL, Shugarts SB, Okochi H, Benet LZ. Elucidating the effect of final-day dosing of rifampin in induction studies on hepatic drug disposition and metabolism. J Pharmacol Exp Ther. 2006;319(2):864–870. doi: 10.1124/jpet.106.108282. [DOI] [PubMed] [Google Scholar]
- 29.Anderson MS, Cote J, Liu Y, Stypinski D, Auger P, Hohnstein A, Rasmussen S, Johnson-Levonas AO, Gutstein DE. Effects of Rifampin, a potent inducer of drug-metabolizing enzymes and an inhibitor of OATP1B1/3 transport, on the single dose pharmacokinetics of anacetrapib. J Clin Pharmacol. 2013;53(7):746–752. doi: 10.1002/jcph.97. [DOI] [PubMed] [Google Scholar]
- 30.Hamilton M, Wolf JL, Drolet DW, Fettner SH, Rakhit AK, Witt K, Lum BL. The effect of rifampicin, a prototypical CYP3A4 inducer, on erlotinib pharmacokinetics in healthy subjects. Cancer Chemother Pharmacol. 2014;73(3):613–621. doi: 10.1007/s00280-014-2390-3. [DOI] [PubMed] [Google Scholar]
- 31.Loboz KK, Gross AS, Williams KM, Liauw WS, Day RO, Blievernicht JK, Zanger UM, McLachlan AJ. Cytochrome P450 2B6 activity as measured by bupropion hydroxylation: effect of induction by rifampin and ethnicity. Clin Pharmacol Ther. 2006;80(1):75–84. doi: 10.1016/j.clpt.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 32.Shichiri M, Tanaka Y. Inhibition of cancer progression by rifampicin: involvement of antiangiogenic and anti-tumor effects. Cell Cycle. 2010;9(1):64–68. doi: 10.4161/cc.9.1.10354. [DOI] [PubMed] [Google Scholar]
- 33.Fardel O, Lecureur V, Loyer P, Guillouzo A. Rifampicin enhances anti-cancer drug accumulation and activity in multidrug-resistant cells. Biochem Pharmacol. 1995;49(9):1255–1260. doi: 10.1016/0006-2952(95)00045-2. [DOI] [PubMed] [Google Scholar]
- 34.Cho YY, Jeong HU, Kim JH, Lee HS. Effect of honokiol on the induction of drug-metabolizing enzymes in human hepatocytes. Drug Des Devel Ther. 2014;8:2137–2145. doi: 10.2147/DDDT.S72305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luo X, Kranzler HR, Zuo L, Wang S, Schork NJ, Gelernter J. Multiple ADH genes modulate risk for drug dependence in both African- and European-Americans. Hum Mol Genet. 2007;16(4):380–390. doi: 10.1093/hmg/ddl460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kumar S, Ingle H, Mishra S, Mahla RS, Kumar A, Kawai T, Akira S, Takaoka A, Raut AA, Kumar H. IPS-1 differentially induces TRAIL, BCL2, BIRC3 and PRKCE in type I interferons-dependent and -independent anticancer activity. Cell Death Dis. 2015;6:e1758. doi: 10.1038/cddis.2015.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ando T, Ishiguro H, Kimura M, Mitsui A, Mori Y, Sugito N, Tomoda K, Mori R, Harada K, Katada T, et al. The overexpression of caveolin-1 and caveolin-2 correlates with a poor prognosis and tumor progression in esophageal squamous cell carcinoma. Oncol Rep. 2007;18(3):601–609. [PubMed] [Google Scholar]
- 38.Capozza F, Trimmer C, Castello-Cros R, Katiyar S, Whitaker-Menezes D, Follenzi A, Crosariol M, Llaverias G, Sotgia F, Pestell RG, et al. Genetic ablation of Cav1 differentially affects melanoma tumor growth and metastasis in mice: role of Cav1 in Shh heterotypic signaling and transendothelial migration. Cancer Res. 2012;72(9):2262–2274. doi: 10.1158/0008-5472.CAN-11-2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Elsheikh SE, Green AR, Rakha EA, Samaka RM, Ammar AA, Powe D, Reis-Filho JS, Ellis IO. Caveolin 1 and Caveolin 2 are associated with breast cancer basal-like and triple-negative immunophenotype. Br J Cancer. 2008;99(2):327–334. doi: 10.1038/sj.bjc.6604463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Amundson SA, Smilenov LB. Integration of biological knowledge and gene expression data for biomarker selection: FN1 as a potential predictor of radiation resistance in head and neck cancer. Cancer Biol Ther. 2010;10(12):1252–1255. doi: 10.4161/cbt.10.12.13731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Viana Lde S, Affonso RJ, Jr, Silva SR, Denadai MV, Matos D, Salinas de Souza C, Waisberg J. Relationship between the expression of the extracellular matrix genes SPARC, SPP1, FN1, ITGA5 and ITGAV and clinicopathological parameters of tumor progression and colorectal cancer dissemination. Oncology. 2013;84(2):81–91. doi: 10.1159/000343436. [DOI] [PubMed] [Google Scholar]
- 42.Yim DH, Zhang YW, Eom SY, Moon SI, Yun HY, Song YJ, Youn SJ, Hyun T, Park JS, Kim BS, et al. ITGA1 polymorphisms and haplotypes are associated with gastric cancer risk in a Korean population. World J Gastroenterol. 2013;19(35):5870–5876. doi: 10.3748/wjg.v19.i35.5870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin XD, Chen SQ, Qi YL, Zhu JW, Tang Y, Lin JY. Polymorphism of THBS1 rs1478604 A > G in 5-untranslated region is associated with lymph node metastasis of gastric cancer in a Southeast Chinese population. DNA Cell Biol. 2012;31(4):511–519. doi: 10.1089/dna.2011.1344. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The complete RNA-seq data used in this paper can be downloaded from the GEO database with the accession number: GSE79933; The complete microRNA OpenArray data used in this paper can be downloaded from http://compbio.iupui.edu/group/6/pages/rifampin.