

Abstract
Background
Most existing tools for detecting next-generation sequencing-based splicing events focus on generic splicing events. Consequently, special types of non-canonical splicing events of short mRNA regions (IRE1α targeted) have not yet been thoroughly addressed at a genome-wide level using bioinformatics approaches in conjunction with next-generation technologies. During endoplasmic reticulum (ER) stress, the gene encoding the RNase Ire1α is known to splice out a short 26 nt region from the mRNA of the transcription factor Xbp1 non-canonically within the cytosol. This causes an open reading frame-shift that induces expression of many downstream genes in reaction to ER stress as part of the unfolded protein response (UPR). We previously published an algorithm termed “Read-Split-Walk” (RSW) to identify non-canonical splicing regions using RNA-Seq data and applied it to ER stress-induced Ire1α heterozygote and knockout mouse embryonic fibroblast cell lines. In this study, we have developed an improved algorithm “Read-Split-Run” (RSR) for detecting genome-wide Ire1α-targeted genes with non-canonical spliced regions at a faster speed. We applied the RSR algorithm using different combinations of several parameters to the previously RSW tested mouse embryonic fibroblast cells (MEF) and the human Encyclopedia of DNA Elements (ENCODE) RNA-Seq data. We also compared the performance of RSR with two other alternative splicing events identification tools (TopHat (Trapnell et al., Bioinformatics 25:1105–1111, 2009) and Alt Event Finder (Zhou et al., BMC Genomics 13:S10, 2012)) utilizing the context of the spliced Xbp1 mRNA as a positive control in the data sets we identified it to be the top cleavage target present in Ire1α+/− but absent in Ire1α−/− MEF samples and this comparison was also extended to human ENCODE RNA-Seq data.
Results
Proof of principle came in our results by the fact that the 26 nt non-conventional splice site in Xbp1 was detected as the top hit by our new RSR algorithm in heterozygote (Het) samples from both Thapsigargin (Tg) and Dithiothreitol (Dtt) treated experiments but absent in the negative control Ire1α knock-out (KO) samples. Applying different combinations of parameters to the mouse MEF RNA-Seq data, we suggest a General Linear Model (GLM) for both Tg and Dtt treated experiments. We also ran RSR for a human ENCODE RNA-Seq dataset and identified 32,597 spliced regions for regular chromosomes. TopHat (Trapnell et al., Bioinformatics 25:1105–1111, 2009) and Alt Event Finder (Zhou et al., BMC Genomics 13:S10, 2012) identified 237,155 spliced junctions and 9,129 exon skipping events (excluding chr14), respectively. Our Read-Split-Run algorithm also outperformed others in the context of ranking Xbp1 gene as the top cleavage target present in Ire1α+/− but absent in Ire1α−/− MEF samples. The RSR package including source codes is available at http://bioinf1.indstate.edu/RSR and its pipeline source codes are also freely available at https://github.com/xuric/read-split-run for academic use.
Conclusions
Our new RSR algorithm has the capability of processing massive amounts of human ENCODE RNA-Seq data for identifying novel splice junction sites at a genome-wide level in a much more efficient manner when compared to the previous RSW algorithm. Our proposed model can also predict the number of spliced regions under any combinations of parameters. Our pipeline can detect novel spliced sites for other species using RNA-Seq data generated under similar conditions.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-2896-7) contains supplementary material, which is available to authorized users.
Keywords: Alternative splicing, Non-canonical, RNA-Seq, Xbp1, ENCODE
Background
In metazoans, during endoplasmic reticulum (ER) stress, the endoribonuclease (RNase) Inositol Requiring Enzyme 1a (Ire1α) initiates removal of a 26 nt region from the mRNA encoding the transcription factor Xbp1 via an non-canonical mechanism (atypically within the cytosol). This causes a transitional open reading frame-shift to produce a potent transcription factor, Xbp1s, that induces expression of numerous downstream genes in response to ER stress as part of the unfolded protein response (UPR) [1, 2]. In addition, spliceosome-independent cytoplasmic splicing, as a part of the unfolded protein response pathway, has been described in yeast [3] where HAC1p was found to be the sole splicing substrate of Ire1. The mechanism of Ire1α-mediated RNA-splicing is likely conserved in all eukaryotes as well [4].
In recent years, many popular methods have been developed to identify novel splice sites in RNA-Seq data, including TopHat [5] and Alt Event Finder [6]. A detailed review on the limitations of several other tools for identification of alternative splicing events (TrueSight [7], Splicing-Compass [8], and PASTA [9]) was described previously [10]. In short, indeed none of these existing tools were suitably designed for detecting the type of non-canonical sometimes called non-canonical splice sites generated by Ire1α-targeted Xbp1 mRNA splicing. Given that non-canonical splicing events of short mRNA regions occurring within the cytosol have not yet been investigated using next-generation technologies at a genome-wide level, cutting-edge bioinformatics methods of detecting such targets are needed to quickly discover such splicing events in a patient-specific manner in order to derive future therapeutic value.
In order to supply the medical and scientific fields with such a tool we previously developed a novel bioinformatics pipeline method, named Read-Split-Walk [10] for detecting non-canonical, short, splicing regions using RNA-Seq data. We applied the method to ER stress-induced Ire1α heterozygous and knockout mouse embryonic fibroblast (MEF) cell lines to identify Ire1α targets of which the 26 nt non-canonical splice site in Xbp1 was detected as the most prominent splice target by our initial RSW pipeline in heterozygous (Het) samples, not mapped in the negative control Ire1α knockout (KO) samples for both Thapsigargin (Tg) and Dithiothreitol (Dtt) treated experiments. In our previous study, we also compared the Xbp1 results from our approach with results using the alignment program BWA [11], Bowtie2 [12], STAR [13], Exonerate [14] and the Unix “grep” command. Although our previous RSW method gave better results overall than the above-mentioned approaches, we realized that RSW’s running speed needed to be further improved in order to handle the massive amount of data in other experiments (human ENCODE project: https://www.encodeproject.org). In addition, we wanted to test, under different combinations of parameters, how and where reported spliced regions would differ. Therefore, we have designed a newer algorithm which we call “Read-Split-Run” (RSR) that can process RNA-Seq data in a more efficient manner with flexible parameters. We also proposed a linear regression equation under the assumption of the Generalized Linear Model for RSR parameters that can automatically predict the number of spliced regions given any parameter settings for a particular experiment.
We compared our RSR algorithm with the above-mentioned alternative splicing events detection tools using metrics of how each tool ranks Xbp1 as the top cleavage target and its presence and absence in Ire1α+/− and Ire1α−/− MEF samples. We have also compared our RSR pipeline and other tools to process a human ENCODE dataset and reported their statistics of running performance and sensitivity (the number of spliced junctions identified).
Results
The web interface features of RSR
In addition to providing the source code for download, the current web site of RSR (http://bioinf1.indstate.edu/RSR) allows users to use RSR by using a web form to upload data to the RSR server. After a job is submitted the server runs the pipeline and sends an email with a download link when the results are ready. The web form allows selection of a flexible combination of parameters. For example, users can select “Mode (Comparative or Non-comparative)”, “Reads Type (Single or Paired-end reads)”, “Experimental Replicates (1, 2, 3, …)”. The input files must be in FASTQ format. Based on the user’s initial selection, the pre-processing step will automatically reflect the number of input files needed. Users also have the options of checking the quality encoding and read length for short read input sequence files. The pipeline moves to the next step only if the read lengths for all input files are confirmed to be equal. A screenshot for the RSR web interface is shown in Fig. 1.
Fig. 1.
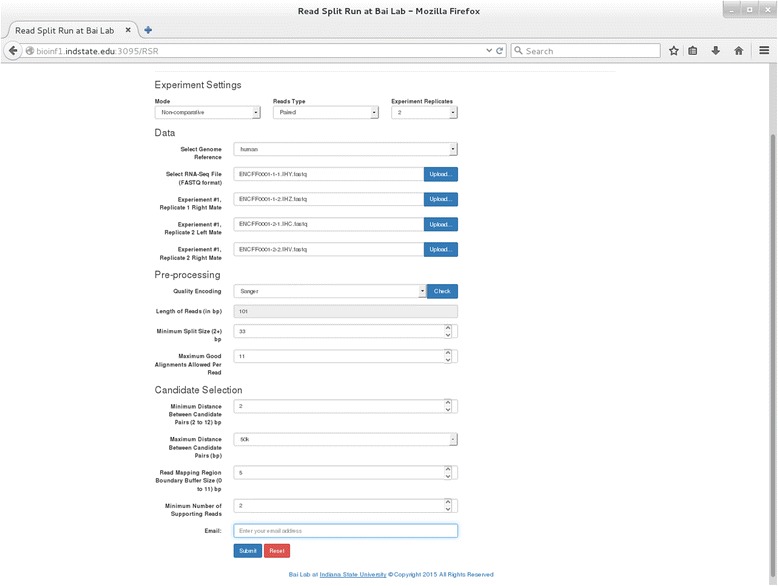
A screenshot for the Read-Split-Run web interface
The spliced regions detected by the RSR pipeline for mouse MEF RNA-Seq data
The identified spliced regions by the RSR pipeline for five cases with different combinations of parameters when processing Ire1α(+/−) and Ire1α(−/−) samples in both experiments (Tg and Dtt treated) are shown in Table 1. The detailed information of spliced regions identified by RSR for Tg and Dtt treated samples under different parameter settings are reported in Additional files 1 and 2. The attached Additional files 3 and 4 give statistics from running the new pipeline on mouse Tg and Dtt data with various parameter settings for MS, MD, and BB on a system with two Intel Xeon E5-2650 2.6GHz processors (a total of 16 cores and 32 hyperthreads).
Table 1.
Comparison of total number of junctions identified by RSR for five cases from Tg and Dtt treated samples
| Parameter | Case 1 | Case 2 | Case 3 | Case 4 | Case 5 | |
|---|---|---|---|---|---|---|
| Variable Name | Minimum split size | 8 | 11 | 11 | 15 | 18 |
| Maximum candidate distance | 40,000 | 40,000 | 50,000 | 50,000 | 50,000 | |
| Read mapping region boundary buffer size | 5 | 5 | 5 | 8 | 8 | |
| Minimum candidate distance | 2 | 2 | 2 | 2 | 2 | |
| Minimum number of supporting reads | 2 | 2 | 2 | 2 | 2 | |
| Maximum good alignment allowed per read | 8 | 11 | 11 | 15 | 18 | |
| Tg Het | Total number of junctions identified | 122 | 140 | 143 | 135 | 141 |
| Tg KO | Total number of junctions identified | 153 | 177 | 177 | 170 | 177 |
| Dtt Het | Total number of junctions identified | 6496 | 6614 | 6661 | 6673 | 5683 |
| Dtt KO | Total number of junctions identified (Novel/Known) | 6135 | 6247 | 6285 | 6308 | 5687 |
The bolded numbers show parameters with different test settings
The spliced regions detected by the RSR pipeline for human ENCODE data
The total number of spliced regions and running time of RSR on each chromosome of human ENCODE RNA-Seq data are shown in Fig. 2. The analysis for Chr14 was not performed due to memory constraints on the machine running the RSR pipeline. Additional file 5 shows the number of spliced regions identified by our RSR algorithm for each chromosome of the human ENCODE RNA-Seq dataset.
Fig. 2.
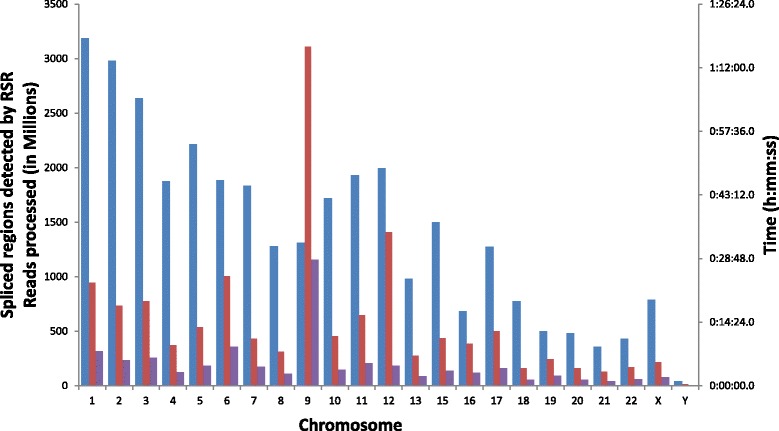
Number of junctions and clock time reported by RSR for the human ENCODE RNA-Seq sample (Separated by chromosomes). Blue bar: junctions; red bar: clock time; purple bar: reads processed (million)
Comparison of detected spliced regions between RSR and other tools
We also compared our RSR algorithm with other NGS based alternative splicing events detection tools (TopHat and Alt Event Finder). Our Read-Split-Run algorithm outperformed the other two software in the context of ranking Xbp1 gene as the top cleavage target present in Ire1α+/− but absent in Ire1α−/− MEF samples. In particular, we have ran TopHat and Alt Event Finder on both Tg and Dtt samples of MEF cell line RNA-Seq data. TopHat identified 23 reads supporting Xbp1 in Tg Het and 86 reads for Tg KO samples. For Dtt samples, TopHat reported a total of 59 (Het) and 289 (KO). Although the number of reads supporting Xbp1 reported by TopHat in Tg Het sample is slightly higher than our RSR method (23 vs 21), our RSR accomplished a better turnaround (173) when compared to TopHat (59) in the Dtt dataset. Surprisingly, TopHat also reported reads supporting Xbp1 in KO samples (86 in Tg and 289 in Dtt), which are false positive reads. Alt Event Finder did not identify any reads in supporting 26 nt Xbp1 spliced regions. The comparison results between RSR and other tools in running RNA-Seq data from MEF cell line is listed in Table 2.
Table 2.
Number of reads for supporting Xbp1 26 nt spliced regions reported by RSR and other tools
| 500 nM Thapsigargin (Tg) | 1 mM Dithiothreitol (Dtt) | |||
|---|---|---|---|---|
| Software | Het (Ire1α +/−) | KO (Ire1α −/−) | Het (Ire1α +/−) | KO (Ire1α −/−) |
| Read-Split-Run (RSR) | 21 | 0 | 173 | 0 |
| TopHat | 23 | 86 | 59 | 289 |
| BWA | 0 | 0 | 67 | 0 |
| Bowtie2 | 0 | 0 | 171 | 0 |
| STAR | 0 | 0 | 0 | 0 |
| Alt Event Finder | 0 | 0 | 0 | 0 |
When applying these tools on human ENCODE RNA-Seq data, we found that the running speed and splicing regions detected by these tools are different. Due to the memory constraint of our server, we ran RSR by splitting the genome alignment files into individual chromosomes. It took RSR less than half an hour to run most chromosomes except for chromosome 9 and 12. In contrast, it took TopHat roughly 20 h to run the ENCODE RNA-Seq dataset. Alt Event Finder only required 3 h time to process the dataset, but the result was not informative for identifying short non-canonical spliced regions. Indeed, we have designed the web-based interface to and results reporting of our RSR pipeline to be as user-friendly as possible. Figure 3 shows comparison results between RSR and other tools in identifying number of spliced regions for the human ENCODE RNA-Seq dataset that we have tested.
Fig. 3.
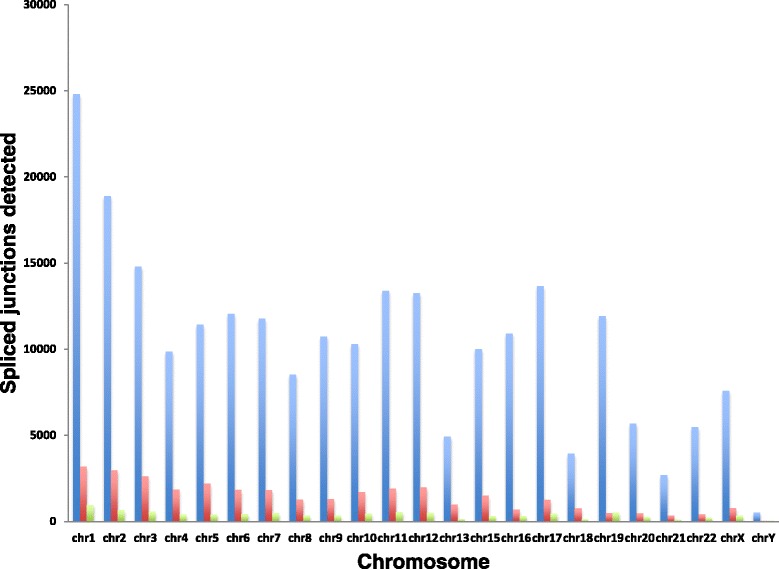
Comparison results in identifying number of spliced regions between RSR, Alt Event Finder, TopHat tools. Blue bar: TopHat; red bar: RSR; green bar: AltEventFinder
The general linear model for RSR algorithm
We generated a General Linear Model for both Tg and Dtt samples in the context of the total number of unique spliced regions (i.e. present in Het and not in KO samples) identified by the RSR algorithm (Fig. 4). The model is derived from the results based on many pairs of Het and KO test cases (100 for Tg and 64 for Dtt samples) with different combinations of three RSR algorithm parameters (minimum split size (MS), maximum candidate distance (MD), and read mapping region boundary buffer size (BB)). We therefore obtained two linear regression equations (one for Tg and the other for Dtt) as shown in Fig. 4. It is clear that the numbers of spliced regions identified by RSR decreases as MS values increase. This is true because the numbers of split pairs fed into the second step of bowtie decrease when MS values increase. The parameter of MD plays less critical roles as we expected. The BB parameter seems to follow the correlation (the numbers of spliced regions increase as BB values increase) for the Tg dataset, but not for the Dtt dataset. We would like to increase test cases for the Dtt sample to see whether the trend will change.
Fig. 4.

Linear regression equations for mouse MEF Tg and Dtt experiments
Discussion
Parameters consideration in RSR pipeline
Our proposed model of taking different parameter combinations to run the bowtie aligner and RSR algorithm could be applied to other species. However, different parameter combinations would predict different outcomes (i.e. number of spliced regions) for different species, even for different experiments. In our pipeline, we chose three parameters (MS, MD, and BB) to test how different combinations affect the prediction outcome. Specifically, we have run a number of test cases (100 for Tg and 64 for Dtt) with different combinations and used the results to generate linear regression equations. To increase the robustness of our RSR algorithm, it is ideal to perform a large-scale simulation study in order to look for the optimal combination. Typical questions remain to be answered: What would be the trade-off between lower/upper bound split size vs alignment sensitivity? What is the optimal consolidation slip/buffer size? More or fewer supporting reads will be reported if different cut-off criteria are applied, and these can be adjusted to achieve the desired balance between sensitivity and specificity in specific applications.
We chose three parameters (minimum split size (MS), maximum candidate distance (MD), and read mapping region boundary buffer size (BB)) for the RSR algorithm because the number of reads supporting spliced regions could be different given different combination values of these parameters. Our GLM was generated by running a number of test cases. A smaller MS and bigger MD and BB could increase the number of junctions reported. Empirically MS could be set to approximately 1/3 of the read length, and should also be larger than 8 bp to ensure the split half is not too short to be mapped accurately. An estimated MD value could be determined according to the average gene length of species tested. Therefore, users could choose their customized MD value according to the species on which their experiments were performed. This criterion is determined according to the assumption that the split pairs supporting the junction are often mapped onto the same gene. Finally, BB could be set to a number that should not reflect a large boundary variation (5 or less would be reasonable).
The file deletion of our RSR algorithm
We noticed, by splitting reads into multiple read half pairs, the size of the result files substantially increased when the human ENCODE dataset was processed. To reduce the hard drive storage of large data files, we automatically delete files throughout the pipeline as they are no longer needed. For example, if the split step only needs to utilize the unmapped read datasets, the alignment files generated from the first step of bowtie are deleted. After the second step of bowtie is finished, all unmapped files will be deleted and only the alignment file will be kept for the next RSR step. The RSR pipeline can also compare spliced regions between samples and output reported regions side-by-side.
RSR running speed, sensitivity, and specificity
In this study, we have developed a newer pipeline (RSR) of RSW with an improved running speed and proposed a General Linear Model for the algorithm. We used RSR to process different combinations of running parameters for the MEF and human ENCODE RNA-Seq data. We have compared our RSR algorithm with two popular NGS based alternative splicing events detection tools (TopHat and Alt Event Finder) and reported the spliced regions detection results. Neither of these two tools achieved better sensitivity (Number of junctions identified) than our RSR algorithm in identifying reads supporting the Xbp1 26 bp spliced region. This can be explained in part due to the fact that Alt Event Finder processes the mapped reads to report splice regions yet does not consider unmapped reads in the analysis input. Moreover, the current version of Alt Event Finder focuses on identifying exon skipping events only. TopHat reported canonical exon-exon splice sites as well. The spliced junctions identified by TopHat and our RSR are reported in Table 3. TopHat reported more junctions than our RSR. But many of them were known junctions or false positive ones. It is clear that the common junctions detected by both tools or overlapping rate is low. Indeed, the overlapping rate is even smaller for results from the human ENCODE dataset.
Table 3.
The overlapping spliced junctions identified by TopHat and our RSR
| mouse-Het | mouse-KO | mouse-Het | mouse-KO | human | |
|---|---|---|---|---|---|
| Software | 500 nM Thapsigargin (Tg) | 1 mM Dithiothreitol (Dtt) | ENCODE | ||
| TopHat | 956 | 923 | 8897 | 7847 | 237,155 |
| RSR | 144 | 183 | 6727 | 6343 | 32,597 |
| Common | 38 | 41 | 2398 | 2128 | 314 |
| Common/TopHat | 3.97 % | 4.44 % | 26.95 % | 27.12 % | 0.13 % |
| Common/RSR | 26.39 % | 22.40 % | 35.65 % | 33.55 % | 0.96 % |
In our original RSW paper, we also compared the Xbp1 results from our approach with results using the alignment program BWA [11], Bowtie2 [12], STAR [13], Exonerate [14] and the Unix “grep” command. Although our RSW method gave better results overall than the above-mentioned approaches, comparison results also suggested that reads supporting removal of the 26 nt intron from Xbp1 mRNA were not fully acknowledged. A study using in vitro cleavage assay combined with microarray analysis reported 13 additional mRNAs as Ire1α cleavage targets [15]. The discovery shed light on the existence of other possible targets. A future version of the algorithm will focus on rescuing these false negative reads in order to achieve a better sensitivity.
Applying RSR on human ENCODE RNA-Seq data
The discovery of a new set of non-canonical splicing events in humans is important not only because of the obvious potential for novel alteration of targeted transcript function, but also the potential for the resulting excised sequences to function as silencing RNAs associated with particular disease states. In addition, the frequency of these novel splicing events could be subject to altered regulation in some individuals, resulting in identifiable splicing profiles associated with the risk of certain diseases. We used ENCODE RNA-Seq datasets to train our RSR algorithm and hope to identify additional targets and elucidate their splicing patterns. Results should eventually provide unique insight of elucidating how short non-canonical spliced sequences act their biological functions in the context of relevant biological processes and diseases.
Conclusions
The positive control for our application, the Xbp1 26 nt non-canonical splice site, was clearly detected in Het samples but not in the KO control samples from Tg and Dtt treated MEF experiments, and was reported again as the top cleavage target for an Ire1α target splice site. Although we have tested the RSR using human ENCODE datasets, our algorithm could also be easily extended for prediction of spliced regions for other species under any given parameter settings.
Methods
The RNA-Seq read sequence data
The mouse test data were downloaded from NCBI Gene Expression Omnibus (GEO) under the accession number GSE54631. Mouse embryonic fibroblast cells (MEF) that were heterozygous for Ire1α (Ire1α(+/−)) and cells which had Ire1α knocked out (Ire1α(−/−)) treated for 4 h with either 500nM Thapsigargin (Tg) or 1 mM Dithiothreitol (Dtt). Both RNA-Seq experiments are single end reads and had no experimental replicates performed.
The human test data are ENCSR000CUR which were downloaded from the ENCODE project (https://www.encodeproject.org/experiments/ENCSR000CUR/). The data were paired-end RNA-Seq experiments performed on human skin melanocytes primary whole cells (NHEM-M2) and sequenced using Illumina HiSeq 2000. There were two biological replicates (adult 52 years old and adult 55 years old) and no technical replicates used in this experiment.
The reference genome for Read-Split-Run
We downloaded the mouse (mm9) and human (hg19) genome reference sequences from the University of California Santa Cruz (UCSC) genome browser (http://genome.ucsc.edu). We also downloaded respective UCSC gene files (knownGene.txt) from the UCSC genome browser. The splice junction file was created by setting the sequence entry on each side of the junction site to 4 bp shorter than the read length using a RNA-Seq software python script (getsplicefa.py) from ERANGE version 3.1 (http://woldlab.caltech.edu/~alim/RNAseq/). The original reference genome and splice junction site file were merged together to form an expanded genome.
The algorithm of Read-Split-Run
We first recall the basic pipeline of the earlier work [10] before highlighting areas of improvement. Pseudocode is given for the pipeline in Fig. 5. The pipeline takes as input an RNA-seq file containing many short reads. The bowtie sequence aligner, version 1.0.1, [16] is first invoked, and unmapped reads are passed to the next stage of the pipeline as possible candidates resulting from the splicing. If a given non-aligned read sequence S did result from the splicing, the splice point could be at any position within S. The next stage of the algorithm splits each non-aligned sequence S into pairs (S1, S2) in all ways so that both parts are at least some minimum size (a parameter we denote MS, with a typical value between 8 and 1/3 of length of the original read sequences). Bowtie is invoked again, this time on each sub-sequence that resulted from splitting a non-aligned sequence from the first invocation of bowtie. Alignments of the sub-sequences are scanned for sub-sequences that were (i) derived as split pairs from the same original non-aligned read sequence, and (ii) aligned at positions on the same chromosome that are not too far apart (a parameter we denote MD, with a typical value of around 40,000). These conditions are consistent with a splicing event, and we save all pairs of alignments that satisfy the conditions, which we call “matched pairs”. The final stage of the pipeline scans all matched pairs to determine for each matched pair how many other matched pairs are likely a result of the same splice location; one matched pair “supports” another if the spliced region between the two ends is the same length and at a position on the same chromosome that is very close (a parameter we denote BB, with a typical value of between 2 and 5). The most interesting splice junctions are those with the highest number of matched pairs that support them.
Fig. 5.

Pseudocode for Read-Split-Run algorithm. The junctions output by step 6 of the algorithm can optionally be restricted to those which are supported by some minimum number of sequences
The present work began by porting the previous pipeline from being written in Perl to C++ (compiled with g++ 4.8.3 using optimization parameter –O4 on Linux). Porting to C++ resulted in a speedup by a factor of roughly two to three. Other than the sequence alignment using bowtie, most of the running time in the pipeline is in comparing aligned sub-sequences to determine the set of matched pairs, and comparing matched pairs to determine which support each other. The previous pipeline compared all pairs in each step, resulting in a running time that is quadratic in the number of sub-sequences coming out of the second bowtie step. We improve this step drastically so that the running time is quadratic only in the number of sub-sequences that were derived from the same initial non-aligned read sequence (typically less than a few hundred, whereas there may be millions of sub-sequences coming out of the second bowtie step). We obtain a similar improvement in the step that scans matched pairs to determine which support each other.
Methods of running Read-Split-Run on mouse MEF RNA-Seq data
The running parameter values employed for both Tg and Dtt samples are shown in Table 4. The steps of finding matched pairs and scanning matched pairs for supporting reads for Dtt samples were performed on a separate system running two Intel Xeon E5-2680 2.8Gz processors (a total of 20 cores and 40 hyperthreads). The highest running times are for tests with lower values of MS – this increases the number of sub-sequences that must be considered. A larger value for the length of initial read sequences also increases running time; the Dtt tests had higher running times because they consist of 77 bp (as opposed to 33 bp for Tg tests) and because the Dtt tests had roughly three times as many read sequences to begin with.
Table 4.
The running parameter values employed for both Tg and Dtt samples
| Sample | Minimum split size (MS) | Maximum candidate distance (MD) | Read mapping region boundary buffer size (BB) |
|---|---|---|---|
| Tg | 8, 11, 12, 16 | 10000, 20000, 30000, 40000, 50000 | 1, 3, 5, 7, 9 |
| DTT | 11, 16, 20, 24 | 10000, 20000, 40000, 50000 | 3, 5, 7, 9 |
Methods of running Read-Split-Run on human ENCODE RNA-Seq dataset
The phases: bowtie, splitting, and second step of bowtie were performed on same hardware mentioned above as the Dtt and Tg sets, whereas the RSR program was run on the “compute node,” described above. The parameters for this run were: MS - 33, MD - 50,000, and BB - 5. Before we could run the split-pairs portion of the pipeline, the output from bowtie (phase 2) had to be split into individual chromosomes so that they could fit into memory. Even in doing so, chromosome 14 had so much data (611Gb) that it could not be run. No junctions were identified on chromosome M.
Comparison with other tools
We compared our RSR algorithm with a couple of other NGS based alternative splicing events detection tools (TopHat [5] and Alt Event Finder [6]). We applied these tools on RNA-Seq data from a mouse embryonic fibroblast (MEF) cell line to check which of these tools can identify and rank Xbp1 as the top cleavage target and its presence and absence in Ire1+/− and Ire1−/− MEF samples and extended the analysis to the ENCODE RNA-Seq datasets. We ran TopHat v2.0.13 using options: −I 3000000, −g 10, --coverage-search, −microexon-search, to generate the “accepted_hits.bam” file for RNA-Seq data for each experiment condition from MEF cell line. Alt Event Finder v0.1 was ran by taking the “transcript.gtf” file generated from Cufflinks [17–20] and the “accepted_hits.bam” file generated by TopHat. Other metrics (i.e. running speed and usability) of these tools were also examined.
A general linear model for RSR
We proposed a modified General Linear Model (Fig. 6) for RSR. The variables (parameters) considered in the model are: minimum split size, maximum candidate distance, and read mapping region boundary buffer size. 80 test cases for Het and KO samples of both Tg and Dtt experiments were run on mouse MEF datasets to produce the General Linear Model equation.
Fig. 6.

A modified General Liner Model for the Read-Split-Run algorithm
Abbreviations
BB, boundary buffer size; Dtt, Dithiothreitol; ENCODE, Encyclopedia of DNA Elements; ER, endoplasmic reticulum; GLM, General Linear Model; Het, heterozygote; KO, knock-out; MD, maximum candidate distance; MEF, mouse embryonic fibroblast; MS, minimum split size; RSR, Read-Split-Run; RSW, Read-Split-Walk; Tg, Thapsigargin; UPR, unfolded protein response
Acknowledgements
This research was supported by ISU start-up funds to YB. RJK acknowledges support from NIH grants DK088227, DK042394, DK103183 and CA128814 and the Hevery Foundation. The authors thank The Center for Genomic Advocacy (TCGA) and Department of Mathematics and Computer Science at Indiana State University for computing servers. Authors also thank reviewers for comments and Gary Stuart for helping biological interpretation.
Declarations
The publication charges for this article have been funded by the corresponding author.
This article has been published as part of BMC Genomics Volume 17 Supplement 7, 2016: Selected articles from the International Conference on Intelligent Biology and Medicine (ICIBM) 2015: genomics. The full contents of the supplement are available online at http://bmcgenomics.biomedcentral.com/articles/supplements/volume-17-supplement-7.
Availability of data and material
The datasets supporting the conclusions of this article are included within the article and its additional files. The RSR source and accompanying examples are freely available for academic use at https://github.com/xuric/read-split-run under the Apache License, Version 2.0 license.
Authors’ contributions
YB designed and supervised the project, performed the analysis, provided biological interpretation, and wrote the manuscript. JK wrote the software code, performed the analysis, and wrote the manuscript. BD wrote the software code, ran the pipeline, and provided the human data results. FJ and LD participated in result comparisons between RSR and other tools. JRH performed the experiments and provided biological interpretation. RJK designed the experiments and wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Additional files
Spliced regions identified by RSR for the Tg sample. This file contains all supplementary results for five test cases for the Tg sample. (XLSX 105 kb)
Spliced regions identified by RSR for the Dtt sample. This file contains all supplementary results for five test cases for the Dtt sample. (XLSX 2828 kb)
Statistics reported by RSR for 100 test cases for the Tg sample. This file contains all supplementary results for 100 test cases for the Tg sample. (XLSX 92 kb)
Statistics reported by RSR for 64 test cases for the Dtt sample. This file contains all supplementary results for 64 test cases for the Dtt sample. (XLSX 79 kb)
Total number of spliced regions identified by RSR in the human ENCODE RNA-Seq dataset. This file includes all supplementary results for number of supporting reads, splice length, range of supporting reads (spliced regions) identified by RSR in the human ENCODE RNA-Seq dataset. (XLSX 1411 kb)
Contributor Information
Yongsheng Bai, Email: Yongsheng.Bai@indstate.edu.
Jeff Kinne, Email: jkinne@cs.indstate.edu.
Brandon Donham, Email: bdonham@cs.indstate.edu.
Feng Jiang, Email: fjiang@sycamores.indstate.edu.
Lizhong Ding, Email: lding@sycamores.indstate.edu.
Justin R. Hassler, Email: jhassler@sbpdiscovery.org
Randal J. Kaufman, Email: rkaufman@sbpdiscovery.org
References
- 1.Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415(6867):92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- 2.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107(7):881–891. doi: 10.1016/S0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 3.Ruegsegger U, Leber JH, Walter P. Block of HAC1 mRNA translation by long-range base pairing is released by cytoplasmic splicing upon induction of the unfolded protein response. Cell. 2001;107(1):103–114. doi: 10.1016/S0092-8674(01)00505-0. [DOI] [PubMed] [Google Scholar]
- 4.Back SH, Lee K, Vink E, Kaufman RJ. Cytoplasmic IRE1alpha-mediated XBP1 mRNA splicing in the absence of nuclear processing and endoplasmic reticulum stress. J Biol Chem. 2006;281(27):18691–18706. doi: 10.1074/jbc.M602030200. [DOI] [PubMed] [Google Scholar]
- 5.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou A, Breese MR, Hao Y, Edenberg HJ, Li L, Skaar TC, Liu Y. Alt Event Finder: a tool for extracting alternative splicing events from RNA-seq data. BMC Genomics. 2012;13(Suppl 8):S10. doi: 10.1186/1471-2164-13-S8-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li Y, Li-Byarlay H, Burns P, Borodovsky M, Robinson GE, Ma J. TrueSight: a new algorithm for splice junction detection using RNA-seq. Nucleic Acids Res. 2013;41(4):e51. doi: 10.1093/nar/gks1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aschoff M, Hotz-Wagenblatt A, Glatting KH, Fischer M, Eils R, Konig R. SplicingCompass: differential splicing detection using RNA-seq data. Bioinformatics. 2013;29(9):1141–1148. doi: 10.1093/bioinformatics/btt101. [DOI] [PubMed] [Google Scholar]
- 9.Tang S, Riva A. PASTA: splice junction identification from RNA-sequencing data. BMC Bioinformatics. 2013;14:116. doi: 10.1186/1471-2105-14-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bai Y, Hassler J, Ziyar A, Li P, Wright Z, Menon R, Omenn GS, Cavalcoli JD, Kaufman RJ, Sartor MA. Novel bioinformatics method for identification of genome-wide non-canonical spliced regions using RNA-Seq data. PLoS One. 2014;9(7):e100864. doi: 10.1371/journal.pone.0100864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Slater GS, Birney E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics. 2005;6:31. doi: 10.1186/1471-2105-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oikawa D, Tokuda M, Hosoda A, Iwawaki T. Identification of a consensus element recognized and cleaved by IRE1 alpha. Nucleic Acids Res. 2010;38(18):6265–6273. doi: 10.1093/nar/gkq452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roberts A, Pimentel H, Trapnell C, Pachter L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics. 2011;27(17):2325–2329. doi: 10.1093/bioinformatics/btr355. [DOI] [PubMed] [Google Scholar]
- 18.Roberts A, Trapnell C, Donaghey J, Rinn JL, Pachter L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 2011;12(3):R22. doi: 10.1186/gb-2011-12-3-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31(1):46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]