


Abstract
High-throughput screening (HTS) is an indispensable tool for drug (target) discovery that currently lacks user-friendly software tools for the robust identification of putative hits from HTS experiments and for the interpretation of these findings in the context of systems biology. We developed HiTSeekR as a one-stop solution for chemical compound screens, siRNA knock-down and CRISPR/Cas9 knock-out screens, as well as microRNA inhibitor and -mimics screens. We chose three use cases that demonstrate the potential of HiTSeekR to fully exploit HTS screening data in quite heterogeneous contexts to generate novel hypotheses for follow-up experiments: (i) a genome-wide RNAi screen to uncover modulators of TNFα, (ii) a combined siRNA and miRNA mimics screen on vorinostat resistance and (iii) a small compound screen on KRAS synthetic lethality. HiTSeekR is publicly available at http://hitseekr.compbio.sdu.dk. It is the first approach to close the gap between raw data processing, network enrichment and wet lab target generation for various HTS screen types.
INTRODUCTION
High-throughput screening (HTS) is a versatile and powerful technique for systematic bio-medical research. Formerly used exclusively by pharmaceutical companies, more and more academic institutions seek to extend the application beyond screening for chemical compounds to screens concerned with the systematic manipulation of gene or microRNA (miRNA) expression. HTS is generally characterized by a high degree of assay miniaturization and robotic automation. This allows rapid screening of microtiter plates, each of which can hold up to 1536 experiments. The result of an HTS experiment is a numeric readout of cellular response, e.g. to determine cell viability or metabolic activity. It is acquired via absorbance, fluorescence, or luminescence, typically measured between two different conditions or for two different cell types (Figure 1A).
Figure 1.

(A) In robotic high-throughput screening, a large number of microtiter plates is screened successively to interrogate cells via suitable assays. This is often done in a differential setup. Our example shows siRNA screens between two cell lines (yellow and green). The majority of siRNAs is lethal to both cell lines, whereas some are specific and potentially of therapeutic interest. (B) Three types of high-throughput screens (HTS) are supported in HiTSeekR, namely gene centered, microRNA (multiple target genes) and small compound screens (targeting single or multiple proteins). (C) HiTSeekR complements the typical HTS work-flow for hit selection, where only the top fraction of hits are subjected to secondary screening (dashed green line), by proposing hits based on a multi-faceted systems biology analysis (solid green line). This is facilitated through integrated resources such as Bioconductor packages (BC) or external web services (WS) and allows, for example, to perform (D) de novo network enrichment as shown here for one of the application cases.
HTS data analysis is complicated and time-consuming. For example, a differential human genome-wide RNAi screen comparing two different cell-lines in triplicates results in ∼120 000 data points (experiments), which need to be processed to identify significant biological signals. HTS data suffers from experiment-specific signal variation caused by, e.g. plate, batch, library and positional bias (1,2). The robust identification of putative hits, i.e. samples with a desired phenotype, thus crucially depends on choosing appropriate data normalization routines. This poses a significant hurdle for the analysis of HTS data in general and for secondary and comparative analyses in particular (3). The few existing statistical tools do not provide support for all of the different kinds of HTS data, and they lack adaptive user interfaces (see Supplementary Material for a requirement analysis and an overview of the state of the art). In the standard HTS work-flow, a secondary screen is performed for hit confirmation, which, however, is limited to the most promising hits for economical reasons. In this step, false positive hits, including those caused by off target effects, are identified, and only fully validated hits are then subjected to in-depth functional characterization. Here, the major goal is the identification of the bio-molecular mechanisms underlying the emergence of the phenotype of interest. A considerable disadvantage of this approach is that the primary screening data are not fully utilized, since most of the moderate hits are neglected. Moreover, the development of complex diseases such as cancer is not centered around individual genes but on molecular pathways controlled by genes acting in concert.
MATERIALS AND METHODS
Normalization of raw signal
In addition to easily identifiable hits there are typically many hits with moderate yet significant effect. The specificity and sensitivity for identifying these hits depends largely on the ability to remove noise from the raw data. In general, two types of normalization methods exist, namely control based and plate based normalization.
Control based normalization
Control based normalization methods rely on a series of wells containing negative control samples that demonstrate little or no effect and positive control samples that exhibit a strong effect. Control samples can be used as a reference point to measure the relative effect observed in samples located on the same plate as the controls. This allows for inter-plate comparisons even if absolute values vary significantly between plates and batches, since the control normalized values express the signal of the various plates as a percentage of effect strength. The two most common control based normalization methods are implemented in HiTSeekR.
Percentage of control (POC)
Here, only a single control type is needed to establish a reference point, i.e.
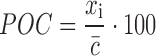 |
where 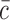 corresponds to the control mean and xi corresponds to the sample value.
corresponds to the control mean and xi corresponds to the sample value.
Normalized percentage inhibition (NPI)
Here, two controls establish an effect range that can be computed as a percentage, i.e.
 |
where  corresponds to the mean of the negative control and
corresponds to the mean of the negative control and 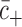 corresponds to the mean of the positive control, respectively.
corresponds to the mean of the positive control, respectively.
Plate based normalization
Control based normalization is often considered inappropriate, since the number of control wells is typically kept small to reserve more space for samples. Moreover, control wells are typically located on the outer wells, where the signal is often strongly biased by evaporation. Most importantly, control samples may occasionally not perform as expected, e.g. in cell viability assays, where negative controls may prove lethal for a particular cell line or where a cell line may be resistant toward a normally lethal positive control (4). A viable alternative is plate based normalization, which operates under the assumption that the majority of reagents do not demonstrate any significant effect. Consequently, most wells in a plate qualify as negative controls, which largely increases the number of wells contributing to a virtual reference point. A control reagent may show unexpected behavior in some scenarios, while the plate-based normalization is more robust due to the large number of contributing samples (5). Plate based normalization, however, is limited to primary screening. Follow-up screens are typically confirmation screens of hits found in the primary screen, where this assumption does not hold. Consequently, control wells are typically included in primary screening even when only plate based normalization methods are considered. The control wells will then be used at a later stage to compare the effect strength between primary and secondary screening results. Finally, screening libraries are sometimes not randomized but clustered. In this case, it is quite likely that groups of wells on a plate demonstrate comparable effects. Examples are small compound libraries, where structurally similar compounds are often grouped or siRNA libraries, where functionally related reagents may be located on the same plate. The most common plate based normalization methods are implemented in HiTSeekR:
(Robust) z-score
The z-score expresses effect strength as a function of the general variability of the data, i.e.
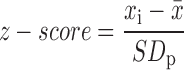 |
where xi corresponds to the sample value, 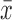 corresponds to the plate mean and SDp corresponds to the standard deviation (SD) of plate p. A disadvantage of this method is that it considers all samples for computing mean and SD. Alternatively, the robust z-score can be used, where mean and SD are exchanged with median and median absolute deviation (MAD), respectively. In z-score normalized data, the plate mean is 0 and the SD is 1. This type of normalization corrects for general differences in signal intensity and expresses effect strength in dependence of the general signal variation, thus allowing for inter-plate comparison.
corresponds to the plate mean and SDp corresponds to the standard deviation (SD) of plate p. A disadvantage of this method is that it considers all samples for computing mean and SD. Alternatively, the robust z-score can be used, where mean and SD are exchanged with median and median absolute deviation (MAD), respectively. In z-score normalized data, the plate mean is 0 and the SD is 1. This type of normalization corrects for general differences in signal intensity and expresses effect strength in dependence of the general signal variation, thus allowing for inter-plate comparison.
B-score
A common problem in HTS are positional effects. These can be caused by, e.g. increased evaporation of the outer wells or by a technical bias introduced in cell seeding, where each row is typically supplied by a different tube. The result of these effects is a signal bias that is in most cases row and column specific. To mitigate this, Brideau et al. suggest the B-score (6), which utilizes Tukey's two way median polish to obtain a signal estimate rijp that is corrected for position specific bias 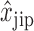 . It includes the estimated average of the plate p as
. It includes the estimated average of the plate p as 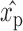 , the estimated offset of row i in plate p as
, the estimated offset of row i in plate p as 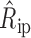 and the estimated offset of column j in plate p as
and the estimated offset of column j in plate p as 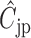 :
:
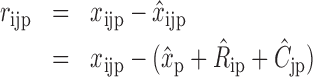 |
Similar to the robust z-score, the B-score can be obtained by dividing the corrected signal estimate by the MAD of the plate:
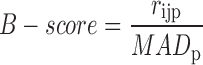 |
The B-score is ideally suited to deal with positional effects, but particularly in 96 well plates it may introduce an additional bias in case of rows or columns that contain many active samples.
Quality control
Quality control in HTS is imperative to identify batches or individual plates that did not perform as expected, e.g. if the transfection was inefficient or if the cell viability was affected. Moreover, quality control helps to monitor time dependent effects where the quality typically decreases with each following plate, thus limiting the maximal batch size. Several parameters of quality control can be investigated visually, such as row and column means to check for positional effects, or scatter plots to investigate the signal distribution. Ideally, wells with positive and negative controls are available on each plate. Two established measures that assess how well positive controls can be separated from negative controls are presented in the following.
Strictly standardized mean difference
Acknowledging that quality measures like the signal-to-noise and signal-to-background ratio fail to capture the variability of the data appropriately, Zhang et al. suggested a dimensionless parameter called Z-factor (7). It is defined as the ratio of the difference between sample and control mean, and the dynamic range of the signal. This measure called Z-factor, when comparing control values, is a widely used quality measure in HTS. However, Zhang et al. have criticized it for a lack of solid statistical interpretation and proposed an alternative score called strictly standardized mean difference (SSMD) (8), which is defined as:
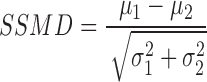 |
where μ1 and μ2 are the means and σ1 and σ2 the SDs of two populations 1 and 2. It should be noted that populations here can refer to the comparison of two controls or a sample and a control. Like the Z-factor, the SSMD captures not only the difference, but also the variability of both populations. In contrast to the Z-factor, however, the SSMD is easier to interpret. An SSMD > 3 indicates that the mean difference is at least three times the SD of the difference of the two populations. Moreover, an SSMD > 3 indicates that the probability that a value from the first population is larger than a value of the second population is close to 1 (0.99865), adding a probability interpretation. Following the three sigma-rule for significance, a SSMD of 3 is thus a suitable cutoff for a pass/fail test.
Hit detection
The goal of HTS is to identify active samples or reagents in a screen that show an effect. The threshold at which an observed effect is considered significant depends on the variability of the data. Therefore, a common and straight forward approach to detect hits is to define a threshold based on ±k SDs or, to increase robustness, on ±k MADs. The factor k is chosen by the user to control the number of hits to be selected. A small k results in less stringent filtering and is likely to include many false positives, while a large k will lead to stringent filtering with and lead to many false negatives. In reality, the choice of k mostly depends on economic considerations, where as many promising hits as possible are included in a secondary confirmation screen. Therefore, the false discovery rate (FDR) and its control via multiple testing correction are widely ignored in primary screening.
SSMD for hit detection
The intuitive approaches of defining a window based on SD or MAD are often criticized for being relative arbitrary. Moreover, these approaches are not suited to fully utilize replicates. An intuitive alternative that is often considered for HTS experiments with replicates is thus the t-test, which can be used to assign a P-value to the difference between sample and control replicates. However, (9) demonstrated the t-test is in fact an inappropriate measure of effect strength, due to its dependence on the number of available replicates. A suitable alternative for hit detection that separates the effect size of the impact of the number of replicate samples, is the SSMD score described above.
Bayesian hit detection
The hit discovery methods presented so far can be used to assess and rank the activity of all samples in a screen. A common disadvantage is that the FDR is unknown but expected to be inflated, since a large number of samples are tested independently. Moreover, variation between plates and batches is only taken into account on a per plate basis. While this strategy is generally appropriate, it might lead to misleading results if individual plates contain clusters of active samples (10), therefore propose an alternative hit discovery based on a Bayesian model. One of the main advantages of this method is that it calculates effect strength per plate while borrowing information from the entire experiment. Moreover, this model maintains a balance between contributions of sample wells and control wells. In practice, the experiment-wide information is used to calculate the priors for the model, while the actual likelihood is calculated per plate. Another major advantage of this method is that it allows one to effectively control the FDR via multiple testing correction, e.g. using the method of Benjamini–Hochberg. Bayesian hit detection thus offers a statistically motivated alternative for defining a threshold for hit detection. The method presented here is more robust to plate-specific signal bias due to its ability to utilize information of the entire experiment or batch, yet it does not account for positional effects. Similar to control based normalization methods, its effectiveness depends crucially on the reliability of controls. Pooling the variance of negative controls across plates, however, allows for this method to be used even if relatively few negative control wells are included. This is of particular importance for 96 well plates, where typically only very few controls are available. The result of hit discovery in RNAi or CRISPR/Cas9 screens is a list of genes that are associated with a particular phenotype. In contrast, miRNA inhibitor or mimics screens require a list of target genes.
microRNA target resources
The predictions of various miRNA tools are available through the Bioconductor package RmiR, including predicted targets from mirbase (11), targetScan (12), miRanda (13), miRDB (14) and PicTar (15), as well as experimentally validated targets from tarbase (16). In addition, we created a database of human miRNA targets using the tool RNAhybrid (17,18). RNAhybrid predicts miRNA target interactions via the free binding energy. The advantage of this database, named RNAhybrid_hsa, is that each miRNA target interaction is associated with a P-value. This allows for controlling the specificity of the target prediction and for keeping the number of false-positive predictions in a reasonable range. The target prediction was run against human 3’UTRs derived from human/mouse/rat multiz alignments from the UCSC Genome Browser (19). Target sites were required to have a seed match with the miRNA at miRNA positions 2 to 7, and no G:U base pairs in the seed were allowed. P-values were calculated from miRNA-specific binding energy distributions and normalized for target sequence length. Finally, we also integrated two miRNA target resources as web services, namely a more up-to-date version of tarbase providing experimentally validated targets and a prediction method called micro-T-CDS (20).
Drug target resources
HiTSeekR utilizes the STITCH databases (21) (v. 4.0 downloaded on 26/03/2015) to map small compounds to prospective target genes (proteins). The interactions in STITCH come with a score that can be used to filter for high-confidence target genes. STITCH is the ideal choice for HiTSeekR since it integrates relevant databases in the field. Similar to miRNA target resources introduced in the previous chapter, STITCH enables users to create a list of target genes that can be subjected to down-stream analysis as described in the following section.
Functional enrichment analysis
HiTSeekR offers three powerful methods for interpreting the results on the system biology level:
Gene Set Overrepresentation Analysis (GSOA): Databases such as Reactome (22) link sets of genes to functional categories and are used in GSOA. The hypergeometric test is used to calculate a probability for observing a given overlap between the genes in the hit list and pre-defined functional gene sets. The hypergeometric distribution in case of HTS data is based on the number of potential target genes in the screen (universe size) and the number of success states (hits). Overrepresentation analysis identifies gene sets for which significantly more genes are found to be hits than would be expected by chance.
Gene Set Enrichment Analysis (GSEA): In GSOA analysis, only the membership of a hit gene in a gene set is considered. In contrast, GSEA takes the rank of a all genes of a set into account and relies on permutations to assess significance (23).
Network Enrichment Analysis (NEA): The results of GSEA as well as GSOA depend crucially on the selection of the a priori defined gene sets. In contrast, network enrichment analysis utilizes biological networks such as BioGrid (24) or I2D (25) that consist of experimentally validated or predicted interactions between genes and/or proteins. This allows for functional pathways or functionally related gene sets to be discovered de novo through dedicated methods such as those provided by KeyPathwayMiner (26).
The first two are performed using the HTSAnalyzeR R package (27), which uses KEGG (28) and Gene Ontology (29) as sources of gene set annotations. However, since the bioconductor R package for KEGG is outdated, we have extended the HTSAnalyzeR package to also include ReactomeDB (22) via its bioconductor R package.
To provide de novo network enrichment, HiTSeekR integrates KeyPathwayMiner (26) through a RESTful web service API. Therefore, HiTSeekR creates an indicator matrix with one column (one case), and one row for each gene of the hit list. HiTSeekR now uses KeyPathwayMiner's INES function to query a maximal connected sub-network covering only genes from the hit list allowing for a user-given number (K) of exceptions (genes not in the hit list). As HiTSeekR utilizes the KeyPathwayMiner web service, pressing the start analysis button will trigger a KeyPathwayMiner run remotely. The user may either wait (up to 1 min) for the results or continue to work with HiTSeekR until the analysis is finished.
RESULTS
The lack of a user-friendly solution for HTS raw data analysis and the untapped potential of systems biology in the field motivated us to develop HiTSeekR, the first comprehensive tool to offer guided analyses for all major screen types, including small compound screens, RNAi knock-down and CRISPR/Cas9 knock-out screens, as well as miRNA inhibitor and mimics screens (Figure 1B). HiTSeekR, which is available at http://hitseekr.compbio.sdu.dk, complements the classical work-flow of HTS through integrating various systems biology methods for hypothesis generation (Figure 1C and D). The individual steps of this work-flow are described below.
Data import
HiTSeekR facilitates HTS data analysis in a user friendly and interactive web interface built with R shiny (http://shiny.rstudio.com/). The use of R in the back end provides us with powerful framework for statistical analysis as well as visualization. The general work-flow of HiTSeekR begins with the import of HTS raw data. Here, HiTSeekR is flexible with respect to the format of the input data and allows users to map columns to the properties required for analysis. Moreover, HiTSeekR supports mapping of the most common types of identifiers. Existing screening data already available through the PubChem assay repository https://pubchem.ncbi.nlm.nih.gov/ can be accessed directly through the corresponding identifier. Note that imported data are only saved transiently during the analysis and will be deleted when the session expires.
Quality control
Subsequent to the data import, various plots are generated to point the user to potential quality issues that need to be taken into account for further analysis (Figure 2). This includes plots concerned with the signal spread across different plates, the correlation of replicates, the performance of control samples, as well as row- or column-effect typically caused through evaporation or clogging. In this way, the user gains knowledge about the experiment-specific bias allowing an educated choice for the following data normalization strategy.
Figure 2.

HiTSeekR produces a series of plots for quality control. (A) Box plots of all plates and replicates. (B) Replicate correlation plot including a linear regression (black line) with corresponding R2 correlation factor. The grey line indicates the identity. (C) Signal spread of the various controls across all plates. The positive controls rela and tnfr1 are more robust than the negative controls sicon and lrp5. (D) Control separability measured by SSMD. Here, the separability between the negative control lrp5 and the other controls is shown. The red area indicates bad separability and the orange area good separability. An SSMD outside of the orange area indicates excellent separability. (E) Plate signal is centered by the mean and subsequently column means are plotted across all plates and replicates to indicate positional effects typically evident by a u-shape.
Hit discovery
Hit discovery is often the end-point of HTS analysis. Here, samples exhibiting the strongest effect after normalization are reported to the user. First, the user may select a number of readouts and/or experiments found in the data for the analysis. HiTSeekR supports various state-of-the-art normalization methods and is thus able to address experimental bias appropriately.
A normalization method should be selected based on the findings of the quality control step, i.e. a plate-based method for primary screens in which the majority of the samples do not show an effect or a control-based method otherwise. For the former, users can choose between the (robust) z-score normalization or, if positional effects are evident, the B-score normalization. For the latter, users should choose percentage of control if only one type of control is given (negative or positive control) or normalized percentage inhibition if both a robust negative and positive control are available. As is customary in the field, a fixed cutoff can be chosen based on the distribution of the (normalized) signal. Each of these normalization methods is applied to the individual plates and is thus not affected by batch effects or differences between experiments. If both robust negative controls and replicates are part of the screen, users are encouraged to apply the Bayesian normalization method (4). Here, the normalization is applied to each individual readout in an experiment. This method combines experiment-wide (negative controls) and plate-based information and is the only method implemented in HiTSeekR that allows controlling the false discovery rate.
Functional enrichment analysis
To better exploit primary screening data, computational systems biology may identify affected molecular mechanisms based on the molecular interplay of strong and/or moderate hits. Gene silencing screens like RNAi or CRISPR/Cas9 provide a list of hit genes that can be directly subjected to further analysis. HiTSeekR supports GSOA, GSEA (23), as well as de novo network enrichment analysis (26). While the former two are suitable for implicating known molecular and biological functions in the experimental results, the latter can be used to identify novel modulators and functional units from large biological interaction networks.
miRNA targets
A relatively new use case of HTS technology involves functional analysis of miRNAs, which operate as key regulators of biological processes by inhibiting the translation of up to hundreds of genes. Consequently, system-level effects of inhibiting or mimicking a miRNA can only be understood through interrogating the function of the affected genes. Interactions between miRNAs and their target genes are available through a number of experimental and predictive databases, many of which have been included in HiTSeekR. While lists of these target genes can be subjected to regular systems biology analysis, previous studies indicate that this may lead to erroneous results (30,31). We consider the issue that a gene might be (predicted to be) targeted by several miRNAs, possibly including a large number of miRNAs that did not have an effect in the assay used in the experiment. To mitigate this, we thus calculate, for each potential target gene, a P-value for the enrichment of miRNAs hits that (are predicted to) target a gene, compared to how many miRNAs in total (including effectors and non-effectors) are predicted to target the gene. More formally, we would like to calculate the probability that a target gene is targeted by k miRNAs, where k is the number of miRNAs in the hit list that target this gene. To compute this probability, we use a hypergeometric test. Consider an experiment where we draw k successes from a population of size N in n draws, given that there are K success states in the population:
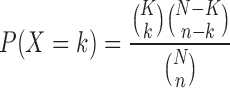 |
Similarly, we use this to calculate the probability that k out of n miRNAs target a particular gene given that this gene is predicted as a target for K out of N total miRNAs. Since we are interested in the probability of observing ≥k miRNAs that target a particular gene, we can calculate the cumulative probability as
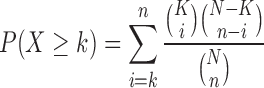 |
Since miRNAs from the same miRNA family will generally be predicted to target the same or largely overlapping sets of genes, we restrict the calculation of target gene effect specificity to one representative of each miRNA family. Genes with a low probability (P-value) are found more frequently as targets in the miRNA hit list than we would expect by chance and are thus important candidates after multiple testing correction with the method of Benjamini and Hochberg (32).
Drug targets
Small compound screening is the traditional use case of HTS and aims at identifying drugs that typically influence cellular function through targeting one or several proteins. Although several million drug target interactions have already been collected in public databases such as STITCH (21), a joint systems biology analysis of active compounds in a screen is not common. HiTSeekR mitigates this by integrating the STITCH database to perform functional analysis and network enrichment on the target genes. In this way, HiTSeekR enables researchers to identify drug sets addressing molecular functions or pathways responsible for a certain phenotype rather than focusing on individual genes.
Application cases
We demonstrate how HiTSeekR uniquely enables novel applications by studying three different kinds of publicly available HTS data sets that have been embedded in HiTSeekR and that are briefly described in the following. The full set of parameters are listed in Supplementary Tables S6–S9.
A genome-wide RNA interference screen identifies caspase 4 as a factor required for tumor necrosis factor alpha signaling
The first example is a genome-wide RNAi screen aimed at identifying modulators of tumor necrosis factor alpha (TNFα) (33), which is implicated in inflammatory diseases and cancer. Here, we compare different normalization strategies (Supplementary Figure S1) and go far beyond the results of the original study by applying de novo network enrichment analysis to evaluate the results of this screen in a wider biological context. The individual steps of this analysis are described in full detail in the Supplementary Material. As a result, HitSeekR implicates PHC2, a gene with unknown function, to be a TNFα modulator through its connection to the NF-κB pathway member CSNK2B (Figure 1D). In addition, we found several genes not directly hit in the screen, such as GRB2 or RNF2 (Supplementary Figure S2 and S3), which are surrounded by several hit genes in the interaction network. Consequently, these are interesting candidates for follow-up studies (guilt-by-association principle) that are neglected by existing HTS analysis pipelines.
Genome-wide functional genomics analysis for genes regulating sensitivity to vorinostat
Falkenberg et al. performed a genome-wide RNAi synthetic lethal drug screen to identify vorinostat sensitivity genes in human colon cancer cells (34). In addition, Falkenberg et al. included a genome-wide miRNA mimics screen under identical assay conditions. Vorinostat is a histone deacetylase inhibitor used in cancer treatment. It was observed that the drug is quite effective in some patients whereas other patients do not respond to therapy, raising the question what factors can explain the lack of a response against vorinostat in cancer. While we focus on the miRNA mimics screen as an example, the combination of a RNAi and miRNA screens offers a unique opportunity to study if the observed effect attributed to miRNA hits can partially be explained by individual target genes. Both screens were split into a plus and a minus drug arm, where the minus arm served to identify genes that were lethal by knockdown alone via DAPI staining. On the plus drug arm, a cell viability assay was used to assess rapid cell death, while a complementary caspase activity readout served to assess apoptosis, i.e. slower cell death. The raw data of the primary siRNA screen (PubChem AID 743454) and the miRNA mimics screen (PubChem AID 743456) can be downloaded directly into HiTSeekR and are also included as example data. Using the miRNA screen, we performed de novo network enrichment analysis based on 61 miRNA hits (5 caspase activity and 56 cell viability hits) and 442 putative target genes. In the union graph built from the top 20 solutions (Supplementary Figure S4), SF3A1 was a prominent gene with a node degree of eight. It is targeted directly by two different miRNAs, namely hsa-miR-497-5p and hsa-miR-15a-5p. Analysis of the RNAi screen reveals that SF3A1 as well as the related gene SF3A3 are indeed moderate suppressors (with a z-score of −3.3 and −3.6, respectively). They would not have been considered for secondary screening without the additional evidence uncovered by systems biology analysis, and illustrate the power of network enrichment integration with HTS data analysis. The individual steps of this analysis are also shown in a video screencast available through the HiTSeekR website.
KRAS synthetic lethal drug screen
A synthetic lethal drug screen was downloaded from ChemBank with the accession ID 1004158. KRAS is a major oncogene and synthetic lethal targets of KRAS have therefore been studied in the past. Here, we analyze a systematic screen of 3947 small compounds in the cell line HKE3, a model system for colon cancer. Since no quality issues were observed, we used z-score with standard deviation and a margin of 3 to extract 80 hits. Subsequently, we determined putative drug targets using the STITCH database and a STITCH score threshold of 500. The resulting 5128 compound–gene interactions were analyzed on the systems biology level. Non surprisingly, gene set analysis revealed “Pathways in Cancer” to be the most significantly enriched pathway (adjusted P-value 1.02e-29). Most interestingly, de novo network enrichment analysis yielded a single solution (Supplementary Figure S5) that revealed frequent interactions of hit compounds with cycline-dependent kinases such as CDK2 or CDK6 as well as glycogen synthase kinase 3 beta (GSK3B), both of which are synthetic lethal interactions previously established in the literature (35,36).
DISCUSSION
HiTSeekR is the first platform for integrated raw data and systems biomedicine analysis. It strives to follow the reactive design principle, which allows for a responsive and intuitive user interface, where changes to input parameters trigger an immediate update of the results. The user is guided in each step of the analysis and can consult additional documentation including a tutorial and video screencast. Each of the above results can be obtained from the raw data within 15–30 min using HiTSeekR. It is the first comprehensive tool that can be used to analyze HTS data in quite heterogeneous contexts down to the systems biology level. This enables users to generate suitable hypotheses for follow-up experiments efficiently.
The majority of screening data is often neglected in favor of a few of hits that are selected for in-depth functional characterization. We acknowledge that resources are limited but propose that hit selection should not be limited to the strongest effectors. Computational systems biology methods can be used to identify molecular mechanisms affected in the experiment more effectively on the basis of the entire data set. Here, key mechanisms may only be identifiable through moderate hits, which would be neglected in a classical analysis but are promoted for further studies through such a systematic analysis. The aim of HiTSeekR is thus to enable more widespread use of systems biology methodology in the HTS community by offering a unified and easily accessible processing platform. The results of extremely costly primary screening experiments may therefore be better utilized. In addition, the systematic analysis serves as an additional in silico filter for false positives. It is less likely that many genes of a pathway (compared to single genes) represent hits due to off target effects or measurement errors. Most importantly, pathways are more likely to unravel indications for systems biology events and alternative molecular signaling cascades behind complex diseases.
We demonstrated the potential of this strategy for the first application case, an RNAi screen aimed at identifying modulators of TNFalpha, which did not include a systems biology analysis in the original publication. With HiTSeekR we could perform GSOA and GSEA, which implicated RNA polymerase II and ribosomal activity. Both processes are related to NFκB signaling. These results are also supported by de novo network enrichment, which identified a subnetwork consisting of DNA polymerase II subunits (results not shown). Our findings are based on strong and moderate hits alike and allow for a hypothesis driven selection of samples for follow-up experiments.
HiTSeekR is the first HTS analysis tool to accommodate miRNA inhibitor or mimics screens. In order to perform systems biology analysis on this type of screen, we annotate miRNAs with corresponding target genes. The identification of target genes also enables comparison between classical RNAi and miRNA screens as we demonstrate for our second application case, in which both types of screen are included but were only analyzed separately. We used HiTSeekR to identify miRNA target genes such as SF3A1 (see online screencast), which can be confirmed in the RNAi screen where they are also hit.
Both, the experimental validation of miRNA target interactions and the computational prediction of miRNA targets are active fields of research and the accuracy of existing methods (both in terms of false positives and false negatives) is controversial. We included a variety of methods and resources in HiTSeekR to acknowledge this fact and plan to implement a mechanism that allows users to upload custom miRNA target annotation files. In general, we note that our selection of tools, methods and resources is not based on a comparative evaluation and does consequently not necessarily include the best performing options.
While powerful, the potential of the systems biology driven analysis in HiTSeekR is ultimately limited by the quality of database annotations and thus no substitute for confirmatory screens. In particular the prediction of miRNA and drug targets is challenging due to a presumably high false positive rate, where, for instance, genes are counted as targets even if they are not actually expressed in a particular tissue. The results of HTS analysis in general have to be considered carefully, since in vitro results can often not be confirmed in vivo (37).
In addition to proficient tools for data analysis, the HTS community also depends on sample management systems that can handle the large number of samples regularly created during the robotic screening. To this end, several tools exist, such as Screensaver (38), OpenBIS (39), Mscreen (40) and SAVANAH (http://nanocan.github.io/SAVANAH, manuscript in preparation). Currently, screening data have to be exported from these tools and uploaded to HiTSeekR or other HTS analysis tools for analysis. We thus plan implementing a mechanism that allows direct export from such applications to HiTSeekR in the future.
CONCLUSION
HiTSeekR paves the way for unique systems biomedicine analysis of miRNA as well as small compound screens through directly predicting target genes. Moreover, the seamless integration of additional resources, such as DIANA miRPath (41) or miRCancer (42), as well as functional enrichment analysis based on gene sets and de novo network enrichment is ideally suited to utilize HTS data more effectively in the research of complex diseases.
In conclusion, HiTSeekR closes the gap between raw data processing, functional enrichment and wet lab target generation for various HTS screen types. Most importantly, HiTSeekR may increase biological relevance of the selected HTS targets using systems biology methodology.
ENDNOTES
http://chembank.broadinstitute.org/assays/view-assay.htm?id=1004158 (last access 07/12/2015).
Supplementary Material
Acknowledgments
This work was supported by the Lundbeckfonden grant for the NanoCAN Center of Excellence in Nanomedicine, the Region Syddanmarks ph.d.-pulje and Forskningspulje, the Fonden Til Lægevidenskabens Fremme, by the DAWN-2020 project financed by Rektorspuljen SDU2020 program, and the MIO project of the OUH Frontlinjepuljen.
Footnotes
Present addresses:
Markus List, Max Planck Institute for Informatics, 66123 Saarbrücken, Germany.
Steffen Schmidt, Roche Innovation Center Copenhagen, 2970 Hørsholm, Denmark.
Helle Christiansen, Roche Innovation Center Copenhagen, 2970 Hørsholm, Denmark.
Marc Rehmsmeier, Integrated Research Institute (IRI) for the Life Sciences and Department of Biology, Humboldt-Universität zu Berlin, 10099 Berlin, Germany.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Lundbeckfonden grant for the NanoCAN Center of Excellence in Nanomedicine; Region Syddanmarks ph.d.-pulje and Forskningspulje, the Fonden Til Lægevidenskabens Fremme, by the DAWN-2020 project financed by Rektorspuljen SDU2020 program, and the MIO project of the OUH Frontlinjepuljen. Funding for open access charge: University/department budget.
Conflict of interest statement. None declared.
REFERENCES
- 1.Malo N., Hanley J.A., Cerquozzi S., Pelletier J., Nadon R. Statistical practice in high-throughput screening data analysis. Nat. Biotechnol. 2006;24:167–175. doi: 10.1038/nbt1186. [DOI] [PubMed] [Google Scholar]
- 2.Caraus I., Alsuwailem A.A., Nadon R., Makarenkov V. Detecting and overcoming systematic bias in high-throughput screening technologies: a comprehensive review of practical issues and methodological solutions. Brief. Bioinform. 2015;16:1–13. doi: 10.1093/bib/bbv004. [DOI] [PubMed] [Google Scholar]
- 3.Blucher A.S., McWeeney S.K. Challenges in secondary analysis of high throughput screening data. Pac. Symp. Biocomput. 2014;19:114–124. [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang X.D., Espeseth A.S., Johnson E.N., Chin J., Gates A., Mitnaul L.J., Marine S.D., Tian J., Stec E.M., Kunapuli P., et al. Integrating experimental and analytic approaches to improve data quality in genome-wide RNAi screens. J. Biomol. Screen. 2008;13:378–389. doi: 10.1177/1087057108317145. [DOI] [PubMed] [Google Scholar]
- 5.Birmingham A., Selfors L., Forster T., Wrobel D. Statistical methods for analysis of high-throughput RNA interference screens. Nat. Methods. 2009;6:569–575. doi: 10.1038/nmeth.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brideau C., Gunter B., Pikounis B., Liaw A. Improved statistical methods for hit selection in high-throughput screening. J. Biomol. Screen. 2003;8:634–647. doi: 10.1177/1087057103258285. [DOI] [PubMed] [Google Scholar]
- 7.Zhang J.-H. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 8.Zhang X.D. A pair of new statistical parameters for quality control in RNA interference high-throughput screening assays. Genomics. 2007;89:552–561. doi: 10.1016/j.ygeno.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 9.Zhang X.D. Illustration of SSMD, z Score, SSMD*, z* Score, and t Statistic for hit selection in RNAi high-throughput screens. J. Biomol. Screen. 2011;16:775–785. doi: 10.1177/1087057111405851. [DOI] [PubMed] [Google Scholar]
- 10.Zhang X.D., Kuan P.F., Ferrer M., Shu X., Liu Y.C., Gates A.T., Kunapuli P., Stec E.M., Xu M., Marine S.D., et al. Hit selection with false discovery rate control in genome-scale RNAi screens. Nucleic Acids Res. 2008;36:4667–4679. doi: 10.1093/nar/gkn435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Griffiths-Jones S., Grocock R.J., van Dongen S., Bateman A., Enright A.J. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34:D140–D144. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lewis B.P., Burge C.B., Bartel D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 13.John B., Enright A.J., Aravin A., Tuschl T., Sander C., Marks D.S. Human microRNA targets. PLoS Biol. 2004;2:11. doi: 10.1371/journal.pbio.0020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang X. miRDB: a microRNA target prediction and functional annotation database with a wiki interface. RNA. 2008;14:1012–1017. doi: 10.1261/rna.965408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krek A., Grün D., Poy M.N., Wolf R., Rosenberg L., Epstein E.J., MacMenamin P., da Piedade I., Gunsalus K.C., Stoffel M., et al. Combinatorial microRNA target predictions. Nat. Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 16.Papadopoulos G.L., Reczko M., Simossis V.A., Sethupathy P., Hatzigeorgiou A.G. The database of experimentally supported targets: A functional update of TarBase. Nucleic Acids Res. 2009;37:D155–D158. doi: 10.1093/nar/gkn809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rehmsmeier M., Steffen P., Höchsmann M., Ho M. Fast and effective prediction of microRNA/target duplexes. RNA. 2004:1507–1517. doi: 10.1261/rna.5248604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krueger J., Rehmsmeier M. RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res. 2006;34:W451–W454. doi: 10.1093/nar/gkl243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosenbloom K.R., Armstrong J., Barber G.P., Casper J., Clawson H., Diekhans M., Dreszer T.R., Fujita P.A., Guruvadoo L., Haeussler M., et al. The UCSC Genome Browser database: 2015 update. Nucleic Acids Res. 2015;43:D670–D681. doi: 10.1093/nar/gku1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paraskevopoulou M.D., Georgakilas G., Kostoulas N., Vlachos I.S., Vergoulis T., Reczko M., Filippidis C., Dalamagas T., Hatzigeorgiou A.G. DIANA-microT web server v5.0: service integration into miRNA functional analysis workflows. Nucleic Acids Res. 2013;41:W169–W173. doi: 10.1093/nar/gkt393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuhn M., Szklarczyk D., Pletscher-Frankild S., Blicher T.H., Von Mering C., Jensen L.J., Bork P. STITCH 4: Integration of protein-chemical interactions with user data. Nucleic Acids Res. 2014;42:D401–D407. doi: 10.1093/nar/gkt1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Joshi-Tope G., Gillespie M., Vastrik I., D'Eustachio P., Schmidt E., de Bono B., Jassal B., Gopinath G.R., Wu G.R., Matthews L., et al. Reactome: a knowledgebase of biological pathways. Nucleic Acids Res. 2005;33:D428–D432. doi: 10.1093/nar/gki072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chatr-aryamontri A., Breitkreutz B.-J., Oughtred R., Boucher L., Heinicke S., Chen D., Stark C., Breitkreutz A., Kolas N., O'Donnell L., et al. The BioGRID interaction database: 2015 update. Nucleic Acids Res. 2015;43:D470–D478. doi: 10.1093/nar/gku1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brown K.R., Jurisica I. Unequal evolutionary conservation of human protein interactions in interologous networks. Genome Biol. 2007;8:R95. doi: 10.1186/gb-2007-8-5-r95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alcaraz N., Pauling J., Batra R., Barbosa E., Junge A., Christensen A., Azevedo V., Ditzel H.J., Baumbach J. KeyPathwayMiner 4.0: condition-specific pathway analysis by combining multiple omics studies and networks with Cytoscape. BMC Syst. Biol. 2014;8:99. doi: 10.1186/s12918-014-0099-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang X., Terfve C., Rose J.C., Markowetz F. HTSanalyzeR: an R/Bioconductor package for integrated network analysis of high-throughput screens. Bioinformatics. 2011;27:879–880. doi: 10.1093/bioinformatics/btr028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kanehisa M., Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Godard P., van Eyll J. Pathway analysis from lists of microRNAs: common pitfalls and alternative strategy. Nucleic Acids Res. 2015;43:3490–3497. doi: 10.1093/nar/gkv249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bleazard T., Lamb J.A., Griffiths-Jones S. Bias in microRNA functional enrichment analysis. Bioinformatics. 2015;31:1592–1598. doi: 10.1093/bioinformatics/btv023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. 1995;57:289–300. [Google Scholar]
- 33.Nickles D., Falschlehner C., Metzig M., Boutros M. A genome-wide RNA interference screen identifies Caspase 4 as a factor required for tumor necrosis factor alpha signaling. Mol. Cell. Biol. 2012;32:3372–3381. doi: 10.1128/MCB.06739-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Falkenberg K.J., Gould C.M., Johnstone R.W., Simpson K.J. Genome-wide functional genomic and transcriptomic analyses for genes regulating sensitivity to vorinostat. Sci. Data. 2014;1:1–13. doi: 10.1038/sdata.2014.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Puyol M., Martín A., Dubus P., Mulero F., Pizcueta P., Khan G., Guerra C., Santamaría D., Barbacid M. A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell. 2010;18:63–73. doi: 10.1016/j.ccr.2010.05.025. [DOI] [PubMed] [Google Scholar]
- 36.Remsing Rix L.L., Kuenzi B.M., Luo Y., Remily-Wood E., Kinose F., Wright G., Li J., Koomen J.M., Haura E.B., Lawrence H.R., et al. GSK3 alpha and beta are new functionally relevant targets of tivantinib in lung cancer cells. ACS Chem. Biol. 2014;9:353–358. doi: 10.1021/cb400660a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wünsch D., Fetz V., Heider D., Tenzer S., Bier C., Kunst L., Knauer S., Stauber R. Chemico-genetic strategies to inhibit the leukemic potential of threonine aspartase-1. Blood Cancer J. 2012;2:e77. doi: 10.1038/bcj.2012.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tolopko A.N., Sullivan J.P., Erickson S.D., Wrobel D., Chiang S.L., Rudnicki K., Rudnicki S., Nale J., Selfors L.M., Greenhouse D., et al. Screensaver: an open source lab information management system (LIMS) for high throughput screening facilities. BMC Bioinformatics. 2010;11:260. doi: 10.1186/1471-2105-11-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bauch A., Adamczyk I., Buczek P., Elmer F.-J., Enimanev K., Glyzewski P., Kohler M., Pylak T., Quandt A., Ramakrishnan C., et al. openBIS: a flexible framework for managing and analyzing complex data in biology research. BMC Bioinformatics. 2011;12:468. doi: 10.1186/1471-2105-12-468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jacob R.T., Larsen M.J., Larsen S.D., Kirchhoff P.D., Sherman D.H., Neubig R.R. MScreen: an integrated compound management and high-throughput screening data storage and analysis system. J. Biomol. Screen. 2012;17:1080–1087. doi: 10.1177/1087057112450186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vlachos I.S., Kostoulas N., Vergoulis T., Georgakilas G., Reczko M., Maragkakis M., Paraskevopoulou M.D., Prionidis K., Dalamagas T., Hatzigeorgiou A.G. DIANA miRPath v.2.0: investigating the combinatorial effect of microRNAs in pathways. Nucleic Acids Res. 2012;40:W498–W504. doi: 10.1093/nar/gks494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xie B., Ding Q., Han H., Wu D. miRCancer: a microRNA-cancer association database constructed by text mining on literature. Bioinformatics. 2013;29:638–644. doi: 10.1093/bioinformatics/btt014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.