


Abstract
Integrase strand transfer inhibitors (INSTIs) are highly effective against HIV infections. Co-crystal structures of the prototype foamy virus intasome have shown that all three FDA-approved drugs, raltegravir (RAL), elvitegravir and dolutegravir (DTG), act as interfacial inhibitors during the strand transfer (ST) integration step. However, these structures give only a partial sense for the limited inhibition of the 3′-processing reaction by INSTIs and how INSTIs can be modified to overcome drug resistance, notably against the G140S-Q148H double mutation. Based on biochemical experiments with modified oligonucleotides, we demonstrate that both the viral DNA +1 and −1 bases, which flank the 3′-processing site, play a critical role for 3′-processing efficiency and inhibition by RAL and DTG. In addition, the G140S-Q148H (SH) mutant integrase, which has a reduced 3′-processing activity, becomes more active and more resistant to inhibition of 3′-processing by RAL and DTG in the absence of the −1 and +1 bases. Molecular modeling of HIV-1 integrase, together with biochemical data, indicate that the conserved residue Q146 in the flexible loop of HIV-1 integrase is critical for productive viral DNA binding through specific contacts with the virus DNA ends in the 3′-processing and ST reactions. The potency of integrase inhibitors against 3′-processing and their ability to overcome resistance is discussed.
INTRODUCTION
Diketo acid derivatives (DKAs) were the first selective and potent integrase (IN) inhibitors (1–3). These molecules preferentially inhibit the strand transfer (ST) reaction and, to a lesser extent, the 3′-processing (3′-P) activity of IN [For a review on HIV-1 IN and its activities, see (4,5)]. For this reason, DKAs have been referred to as integrase strand transfer inhibitors or INSTIs. Among this family of molecules, raltegravir (RAL, Merck & Co.) was the first IN inhibitor approved by the U.S. Food and Drug Administration (FDA) for the treatment of acquired immune deficiency syndrome (AIDS) (6). More recently, two additional INSTIs have been approved by the FDA: elvitegravir (EVG, Gilead Sciences) and dolutegravir (DTG, ViiV Healthcare) (7,8). Crystal structures of the prototype foamy virus (PFV) IN nucleoprotein complexes (intasomes) have been solved showing the detailed organization of four IN PFV subunits and two viral DNA ends (9). PFV IN co-crystal structures bound to the INSTIs developed against HIV-1 IN show that INSTIs act as interfacial inhibitors, interacting simultaneously with IN, the viral DNA and the two active site cations [reviewed in (10)].
Using recombinant enzymes and oligonucleotides mimicking the ends of the retroviral DNA, IN mutants have been extensively characterized and RAL resistance can be recapitulated with three primary mutants: Y143R, N155H and G140S-Q148H (SH) (11–14). PFV IN co-crystal structures showed that residue Y212 (equivalent to Y143 in HIV-1 IN) was the only amino acid directly in contact with RAL (9). The interaction between RAL and Y212 involves π–π stacking between the oxadiazole moiety of RAL and the tyrosine side chain of IN, which is critical for the tight binding of RAL in the IN catalytic site. The crystal structures also suggested that the N155H and SH mutations indirectly impact the binding of INSTIs in the intasome by altering the conformation of the IN active site (15).
EVG was the second INSTI approved by the FDA (7,8). It is three to five time more potent than RAL both in its ability to inhibit the ST reaction in biochemical assays and HIV replication in cell-based assays (12). In a previous study, we compared EVG to RAL and showed that EVG retains potency against the RAL-specific mutant Y143R (12). However, the N155H and SH mutants, which emerge in patients treated with RAL, confer cross-resistance to EVG (11,12,16). Notably, the effect of these mutations on resistance is more profound in the case of EVG than RAL. Indeed, the SH mutant is ∼1000-fold resistant to EVG versus 150-fold for RAL (12). Relative to RAL, EVG also shows a higher selectivity for the ST reaction over 3′-P (12,17). Several groups have developed new molecules with improved resistance profiles and some of the best compounds are effective 3′-P inhibitors (18–21). Based on this observation, we asked whether the 3′-P/ST selectivity of a drug could be linked to its ability to retain potency against the clinically important SH mutant.
DTG is the latest FDA-approved drug for clinical use, and because of its reduced resistance profile, it has been considered a second generation IN inhibitor. While DTG effectively inhibits the Y143R and N155H mutants, the SH double mutant still reduces DTG potency by about 5- to 6-fold (22,23). Despite its enhanced potency and resistance profile, some patients are failing DTG therapy and IN resistance mutations can be selected in vitro at positions surrounding the active site (24). Until the present study, DTG had not yet been characterized for its ability to inhibit the 3′-P reaction and the reason for relatively broad efficacy against many of the common INST-resistant mutants is not clearly established.
Published PFV intasome crystal structures show that the active site conformations before and after 3′-P are quite similar (25). The binding of an INSTIs to the cleaved donor complex involves the displacement of the terminal nucleotide (−1 position, see Figure 1A for numbering convention) of the viral DNA from its normal binding in the IN active site (9). In contrast, the molecular mechanism by which INSTIs inhibit 3′-P (at higher drug concentrations) remains unknown.
Figure 1.

Differential 3′-processing of modified DNA substrates by the wild-type IN (WT) and the drug-resistant IN mutant G140S-Q148H (SH), and inhibition of 3-processing by raltegravir (RAL) and dolutegravir (DTG). (A) Schematic representation of the 21 base pair duplex oligonucleotide used as IN substrate. The canonical CAGT terminal sequence is numbered according to the 3′-P cleavage site (red arrow). Modified substrates have a tetrahydrofuran in place of a nucleobase at the locations, which is indicated by an underscore. 3′-P activity was monitored with the indicated substrates using WT (B) or the RAL-resistant SH mutant (E). Using the 120-min time point, the inhibition of 3′-P by increasing concentration of RAL or DTG for the various substrates was measured in reactions with WT IN (C and D) or the SH mutant (F and G). Fit curves and standard deviation (SD) were derived from at least 4 and up to 14 independent experiments. For comparison, the vertical blue dashed lines in panels C, D, F and G represent the IC50 value for RAL and DTG for ST inhibition (STI) for WT IN and the SH mutant.
In the present study, we tested the effects of modified viral DNA substrates on the 3′-P reaction in the absence and presence of RAL and DTG to investigate the links between the molecular mechanisms involved in the 3′-P/ST selectivity of INSTIs and the effects of resistance mutations. We also investigated potential protein–nucleic acid interactions using our HIV-1 IN molecular model (26,27) pointing to a previously unreported role for residue Q146 in viral DNA end recognition. Finally, we describe a correlation between the ability of INSTIs to overcome drug resistance and their potency in the 3′-P reaction.
MATERIALS AND METHODS
Drugs
RAL, EVG and DTG were purchased from Selleck chemicals. Dihydro-1H-isoindole integrase inhibitors (XZ-89, XZ-90, XZ-115, XZ-116 and XZ-259) were prepared according to previous reports (28–32). Stock solutions (10 mM) were prepared in 100 % dimethyl sulfoxide (DMSO).
DNA substrate generation
Oligonucleotides 21t (GTGTGGAAAATCTCTAGCAGT), 21t-2AP-1 (GTGTGGAAAATCTCTAGC-2AP-GT), 21t-iG-1 (GTGTGGAAAATCTCTAGC-iG-GT), 21t-iG+1 (GTGTGGAAAATCTCTAGCA-iG-T), 21b (ACTGCTAGAGATTTTCCACAC), 21b-iC-1 (AC-iC-GCTAGAGATTTTCCACAC), 21b-iC+1 (A-iC-TGCTAGAGATTTTCCACAC) and 19t (GTGTGGAAAATCTCTAGCA) were purchased from Integrated DNA Technologies, Inc. (Coralville, IA, USA) and gel purified with electro-elution. Oligonucleotides harboring a tetrahydrofuran group in place of the nucleobase at position −1 (21Ab-1, GTGTGGAAAATCTCTAGC_GT), at position +1 (21Ab+1, GTGTGGAAAATCTCTAGCA_T), position +2 (21Ab+2, GTGTGGAAAATCTCTAGCAG_) and in place of the last 2 bases (21-2Ab, GTGTGGAAAATCTCTAGCA__) were purchased from Eurogentec with PAGE purification. 5’-Labeling was performed using T4 polynucleotide kinase (New England Biolabs, Ipswich, MA, USA) with [γ-32P]-ATP (Perkin-Elmer Life and Analytical Sciences, Boston, MA, USA) according to the manufacturers’ instructions. Unincorporated isotopes were removed using the Mini Quick spin oligo columns (Roche). Duplex substrates were obtained by annealing ‘top’ (21t or 19t) and ‘bottom’ (21b) oligonucleotides at equimolar ratio.
IN activity
WT and mutant INs were purified and their enzymatic activities measured as previously reported (11). Briefly, enzymes were present in the reactions at a concentration of 400 nM and the substrate was present at 20 nM in the activity buffer which contained 50 mM 3-(N-morpholino)propanesulfonic acid (MOPS) pH 7.2, 7.5 mM MgCl2, 14 mM 2-mercaptoethanol, any of the compounds in DMSO or DMSO to give a final concentration in DMSO of 10 %. 3′-P and ST reactions were performed at 37°C for 120 min unless otherwise indicated. Products were separated in denaturing 16% polyacrylamide sequencing gels, visualized using a Typhoon 8600 and analyzed using ImageQuant 5.1 (GE Healthcare, Piscataway, NJ, USA).
Data analysis
Linear regressions, fit curves, IC50 determinations, standard deviations, standard error of the mean and statistical analyses (P-values) were generated using GraphPad Prism software 5.0c. Pearson and Spearman correlations were determined using the following equation:
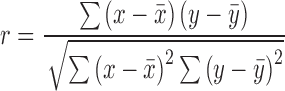 |
Molecular modeling
The full-length HIV-1 IN model used in these studies has been described previously (26,27). To generate models of HIV-1 IN bound to frayed CA_T and C_GT DNA substrates, the atoms of the appropriate base were deleted from the model bound to the CAGT substrate, leaving the phosphor-sugar backbone intact. The resulting model was energy minimized using the AMBER99 forcefield and Born solvation (33). Following minimization, a sphere of explicit water molecules was generated with an 8 Å radius around the four terminal bases of the transferred DNA strand and subjected to another round of energy minimization. Molecular dynamics simulations were then run with 2.5 ns equilibration periods followed by 10 ns simulations. Active site structures at the end of the 10 ns simulation were used to generate the images shown in Figure 4.
Figure 4.

Molecular model of the interaction of the HIV IN monomer with viral DNA. (A) Overall shape of an IN monomer interacting with the double-stranded DNA corresponding to the full-length viral DNA after reverse transcription and prior to ST (substrate for 3′-P). The catalytic core of IN is represented as gray surface. The catalytic amino acid triad DDE is colored as red sticks. Residue Q146 is colored as gray sticks. The two catalytic Mg2+ are shown as green spheres and the water molecules are presented as red balls. The viral DNA is shown with a ribbon backbone, with the bases shown as sticks. The strand cleaved during 3′-P is colored in yellow and its complementary strand in cyan. The dashed rectangle corresponds to the region highlighted in the close-up views of the active site of WT IN (B) and of the SH mutant (C). The DNA backbones are represented as ribbon with the −1 and +1 bases shown as sticks and balls colored according to their constituent elements. IN is represented as gray cartoon with residue Q146 shown as gray sticks and balls. Interatomic distances (in Å) between the Q146 side chain amine and oxy-substituent and the surrounding nucleobases are shown in green.
RESULTS
Probing the interactions of the viral DNA ends with WT HIV-1 integrase
To determine the role of the three terminal bases of the viral DNA in the binding of RAL to the IN active site and 3′-P inhibition, we designed oligonucleotides with tetrahydrofuran substitution at these positions (Figure 1A). First, we compared the 3′-P activity of WT IN on a DNA substrate with the canonical CAGT sequence to the 3′-P activity on a substrate containing an abasic site at the −1 position relative to the cleavage site on the scissile strand (C_GT or 21Ab-1, Figure 1A). The conserved adenosine at the -1 position is known to be important for the activities of IN (34–37). Accordingly, 3′-P of this 21Ab-1 DNA was markedly reduced (Figure 1B).
Under conditions where RAL inhibited ST with an IC50 of about 30 nM (11), its IC50 for 3′-P was about 7 μM (Figure 1C and Table 1). Notably, the ability of RAL to inhibit 3′-P of the 21Ab-1 substrate was enhanced by ∼50-fold relative to the normal substrate (Figure 1C). Under these conditions, RAL inhibited 3′-P in the 21Ab-1 substrate at about the same concentration as it inhibited ST with a normal pre-cleaved CA-OH substrate (IC50 33 ± 7 nM and 34 ± 2 nM, respectively, Figure 1C and Table 1). Similar results were obtained with DTG, the best in-class INSTI with considerable potency against several resistance mutants (21,23,38,39). The introduction of an abasic site in the IN substrate also enhanced 3′-P inhibition by DTG with a resulting IC50 close to that obtained for the inhibition of ST (Figure 1D).
Table 1. Potency of RAL against WT and SH mutant HIV-1 IN depends on the substrate used.
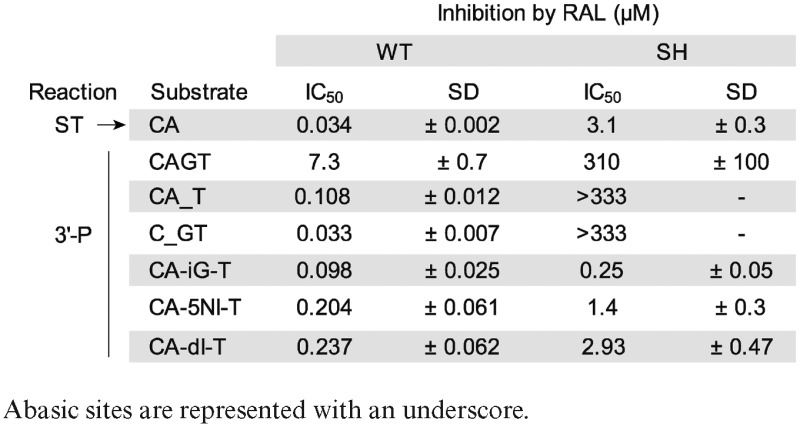
|
Next, we investigated the role of the terminal GT dinucleotide of the viral DNA. Replacing both nucleotides with abasic sites (keeping the DNA backbone intact) did not significantly affect the 3′-P activity of WT IN (data not shown). However, 3′-P inhibition by RAL was greatly enhanced, with a shift in IC50 similar to the reduction seen with the 21Ab-1 substrate (Supplementary Figure S1). Next, we tested substrates with only one abasic site, either at the +1 (21Ab+1, penultimate position) or +2 position (21Ab+2, terminal position). While the ability of RAL to inhibit 3′-P with the 21Ab+2 substrate was similar to its ability to inhibit 3′-P with a normal substrate (CAGT, Supplementary Figure S1), removing only the +1 base from the substrate recapitulated the effects seen with the 21Ab-1 substrate.
When the +1 base was absent (21Ab+1 substrate; Figure 1A), IN catalyzed 3′-P with reduced kinetics and efficiency compared to the normal substrate (Figure 1B). Also, the ability of RAL to inhibit 3′-P was enhanced ∼30-fold with the 21Ab+1 substrate compared to the normal substrate (Figure 1C and Table 1). Similar results were obtained with DTG. DTG inhibited 3′-P by WT IN with an IC50 of 1.64 μM ± 0.11 (Figure 1D). By contrast, using the modified substrates missing the +1 base (Figure 1D), the 3′-P inhibition by DTG was also enhanced and the observed IC50 values were close to the IC50 of DTG for ST (Figure 1D). Together, these results suggest that both the canonical A-1 and the penultimate G+1 play a critical role in stabilizing the DNA in the IN active site for 3′-P. Furthermore, removing either of these two bases allows more potent inhibition of 3′-P by INSTIs.
Abasic substrates differentially affect the 3′-P activity of the G140S-Q148H (SH) IN mutant and RAL resistance
Different 3′-P activity and inhibition profiles were observed when the modified DNA substrates described above were used in assays preformed with the SH mutant. With the canonical CAGT substrate, the 3′-P activity of the SH mutant was markedly slower and less effective than the 3′-P activity of WT IN (Figure 1B and E), which is consistent with prior studies (11,14). Yet, removing either the −1 or +1 base of the viral DNA tended to restore 3′-P activity (Figure 1E).
In addition, removing either base dramatically decreased RAL inhibition of 3′-P reaction catalyzed by the SH mutant compared to the normal DNA substrate (Figure 1F). Similarly, removing either the −1 or +1 base of the viral DNA increased the IC50 values for DTG for 3′-P carried out by the SH mutant by about 100-fold (Figure 1G). These results suggest that the SH mutant alters both the DNA–protein and the drug-complex interactions in the active site.
Role of the 2-amino group of the penultimate G of the viral DNA end
To further examine the structural constraints on the G+1 base of the viral scissile DNA strand, we tested the 3′-P activity of WT and SH mutant integrases after replacing the G+1 guanine with one of the following non-natural bases: inosine, isoguanine or 5-nitroindole (Figure 2A). None of these modifications markedly affected the 3′-P activity of the WT enzyme (Figure 2B). However, those alterations caused a marked increase in RAL potency (Figure 2C and Table 1) with a shift from an IC50 of 7300 ± 700 nM for the inhibition of 3′-P with the normal CAGT substrate to 237 ± 62, 204 ± 61 and 98 ± 25 nM for the inosine-, 5-nitroindole- and isoguanine-containing substrates, respectively. These IC50 values are comparable to the IC50 of RAL for the inhibition of ST (vertical dashed blue line in Figure 2C). Similar results were obtained with DTG (Figure 2D). Given that the common structural change for those modified bases is the absence of the exocyclic NH2 substituent (2-amino group), these results suggest that the guanine amine N2 residue reduces the binding of INSTIs for the 3′-P reaction.
Figure 2.

Effect of +1 nucleobase modifications on IN activity and RAL inhibition. (A) Chemical structure of the modified bases tested. Inosine (dI) and isoguanine (iG) lack the 2-amino group of the guanine base (dG) and iG has flipped substitutions at positions 2 and 6 compared to dG. 5-NitroIndole (5NI) has a nitro substitution at position 5. Time course experiments were used to monitor the 3′-P activity of WT IN (B) or of the SH mutant (D) with the indicated substrates. Using the 120-min time point, inhibition of 3′-P by RAL was determined with the indicated substrates for WT IN (C) and the SH mutant (E). Fit curves and SD were derived from three to five independent experiments. For comparison, the vertical dashed vertical lines represent the IC50 value for RAL and DTG for ST inhibition (STI) for WT IN and the SH mutant.
The G+1 base modifications markedly decreased the 3′-P activity of the SH mutant (Figure 2E). However, these substrates increased the ability of RAL to inhibit 3′-P (Figure 2F), consistent with the results obtained with WT IN (Figure 2C). The observed IC50 values were 2.93 ± 0.47, 1.4 ± 0.3 and 0.25 ± 0.05 μM for the inosine-, 5-nitroindole- and isoguanine-containing substrates, respectively (Figure 2F and Table 1). This trend was also observed with DTG; 3′-P inhibition was enhanced both in the context of WT IN or the SH mutant (Figure 2D and G). Together, these results indicate that removing the 2-amino group at the G+1 position of the scissile strand alters a significant contact between DNA and IN, and that this contact is conserved between the WT enzyme and the SH mutant.
Possible role of Mg2+ ions in altered 3′-P activity
Using our HIV-1 homology model (26,27), we examined the interactions of the WT and SH mutant active sites with the abasic DNA substrates 21Ab-1 and 21Ab+1 (Figure 3). In the context of the WT enzyme, removing either the A-1 (cyan) or G+1 (pink) bases induced a marked shift in the position of the Mg2+ ions with displacements of cation B by 2.5 Å and 0.8 Å, respectively. These displacements are in agreement with the observed reduction in IN activity (see Figure 1B). Comparing models of the WT and SH integrases in the presence of the native substrate (green and gray, respectively) also highlighted a conformational change in the active site with a shift of 1.6 Å for the Mg2+ ion B. However, removing the A-1 or G+1 base in the model of the SH mutant (blue and orange, respectively) placed the Mg2+ ions closer to their position in the model of WT IN with the canonical CAGT substrate (green, Figure 3). Removing the G+1 base in the mutant model generated an active site that was structurally the closest to a WT active site with the normal (WT) DNA bound; the shift of the Mg2+ ion B was only 0.8 Å. Notably, this combination also showed a level of biochemical activity that was closest to WT IN with the canonical substrate (Figure 1E and F).
Figure 3.

Modeling of the WT and G140S-Q148H (SH) IN active sites in complexes with the modified substrates. Models were generated for the WT and SH mutant intasomes carrying the native viral DNA sequence CAGT or the −1/+1 abasic derivatives. The positions of the two catalytic amino acids D64 and E152 are shown as sticks, and the positions of the two magnesium ions by spheres. Color coding and cation displacements (Mg B Shift, Å) compared to WT are tabulated.
Interaction of IN residue Q146 with the viral DNA end
Earlier studies showed that the RAL-resistant mutant Q148H and the double mutant G140S-Q148H (SH mutant), in which the amino acids that are changed flank the flexible loop of IN, both exhibit an impaired catalytic activity and have a kinetic defect (11,14). As expected, there were differences in the 3′-P activity between the WT and the SH mutant enzymes (see Figure 1B and E). Using the canonical substrate (CAGT), WT activity was biphasic (Figure 1B). This could be explained by either: (i) a slow IN–DNA complex formation and/or (ii) a slow step involving DNA end fraying. Under the same conditions, the SH mutant was slower in both the first and second phases of the reaction (Figure 1E). However, the SH mutant was significantly more efficient at processing the modified 21Ab+1 and 21Ab-1 substrates compared to the canonical substrate, and the initial step was reduced (Figure 1E). The absence of a nucleobase would destabilize the end of the double helix, making it easier to open the end of the viral DNA substrate, which would allow the SH mutant to regain some activity. Thus, it is plausible that the limiting step in the catalysis of 3′-P by the SH double mutant could be the opening of the ends of the viral DNA.
To better understand this phenomenon, we used in silico approaches to model the interactions between the WT and the SH mutated catalytic core domains with double-stranded viral DNA ends derived from the NMR structure of the terminal 17 base pairs of the HIV LTR DNA (PDB ID: 1TQR, Figure 4) (40). This model incorporated several contacts between the flexible loop (residues 140–149) and the end of the viral DNA. The model shows that the side chain of residue Q146 is near the G:C base pair at the position +1 (Figure 4). Molecular dynamics simulations were run using the WT and SH mutant models with 2.5 ns equilibration times followed by 10 ns simulations. The distances between the nitrogen and oxygen atoms involved in base pairing were monitored throughout the simulations (hydrogen atoms were not monitored because of the rotation of NH2 groups). In both the WT and SH models, the side chain of residue Q146 was close enough to potentially form hydrogen bonds with the DNA, either with the exocyclic carbonyl moiety of the C+1 or with the carbonyl at the 2-position of the T-1 bases of the non-cleaved strand (Figure 4B and C). At the end of the equilibration (2.5 ns), there were differences in the positioning of the side chain of residue Q146 in the WT and the mutant active sites. Q146 was about 5.6 Å from the G+1 base of the cleaved strand in the WT model (Figure 4B), while the same residue was only 3.1 Å away in the SH mutant model (Figure 4C). This enabled a potential H-bond between the carboxamide moiety of the Q146 side chain and the 2-amino group of the penultimate G+1 base of the cleaved strand (Figure 4C). These results suggest that residue Q146 interacts with the viral DNA end during 3′-P.
Role of residue Q146 in HIV IN catalysis of 3′-P
To investigate the role of residue Q146, we generated four mutants at this position: Q146A, Q146E, Q146T and Q146K. First, we evaluated these mutants for their ability to affect the fraying of the viral DNA ends using a FRET assay (41,42). However, we did not observe any significant difference in FRET efficiency between WT and the mutant INs at various IN/DNA ratios (Supplementary Figure S2).
Next, the 3′-P and ST catalytic activities of the Q146A, E, T and K IN mutants were tested using the full-length canonical duplex 21 bp substrate (Figure 5A). In addition, we tested those mutants with the pre-cleaved 19/21 bp substrate to measure ST only (Figure 5B). The catalytic activities of all of the mutants were reduced, except Q146K which had 3′-P and ST levels similar to WT IN (Figure 5A and B). The mutation from Q to E, which results in the replacement of a carboxamide by a carboxylic group on the side chain, led to a 2-fold reduction in 3′-P (Figure 5A). The most deleterious mutation for 3′-P activity was Q146A (Figure 5A). The fact that the Q146K mutant had full catalytic activity could be explained by the fact that the flexible amino side chain of the lysine residue can probably still interact with the T-1 and C+1 of the non-cleaved strand (Figure 4). Taken together, these results suggest that the NH2 group of the Q146 side chain interacts with the bases at the end of the viral DNA.
Figure 5.

3′-Processing and strand transfer (ST) activity of the HIV IN WT and Q146A, Q146E, Q146T and Q146K mutants. (A) 3′-Processing of the canonical 21 bp substrate by the WT, Q146A, Q146E, Q146T and Q146K IN mutants. Percent substrate conversion versus enzyme concentration is quantified at left; WT IN is plotted as dotted gray line. A representative experiment is shown on the right. (B) ST activity of the WT, Q146A, Q146E, Q146T and Q146K IN mutants with the precleaved 19/21 bp substrate. Percent substrate conversion versus enzyme concentration is shown on the left; WT IN is plotted as dotted gray line. A representative experiment is shown on the right.
3′-P/ST selectivity of INSTIs is correlated with their potency against the SH resistant mutant
We recently developed a series of INSTIs based on the dihydro-1H-isoindole scaffold. These compounds exhibited a range of activity against WT IN and the three main RAL resistance mutants (28–32). Among the five compounds shown in Figure 6, those with the highest selectivity for ST over 3′-P were also the most susceptible to the SH and N155H mutants (Figures 6 and Supplementary Figure S3) (30). XZ-259, which was the most potent ST inhibitor against WT IN with an IC50 of 77 nM (30) had a selectivity index (SI) for 3′-P/ST of 112 was also the most susceptible to the SH double-mutant with a 57-fold increase in IC50. In contrast, XZ-115, which is less selective for ST (SI of 32) had the lowest fold increase in IC50 for ST (<9-fold in the context of the SH mutant). Figure 6 summarizes the relationship between SI and the relative activity toward the SH mutant for our 5 inhibitors (XZ-89, XZ-90, XZ-115, XZ-116 and XZ-259) and for RAL and EVG. Both parameters were significantly correlated with a Spearman coefficient of 0.98 and a Pearson coefficient >0.9995 (P-values < 0.0001).
Figure 6.

Correlation between the selectivity of different INSTIs for ST over 3′-P and the change in susceptibility induced by the SH resistance mutant. The selectivity index was calculated as the IC50 for 3′-P for each compound divided by the corresponding IC50 for ST in the context of WT IN. Change in susceptibility was calculated as the IC50 for ST for each compound in the context of the SH mutant divided by the corresponding IC50 in the context of WT IN. Linear regression and statistical analyses including P-values and Pearson correlation (r) are indicated.
Our present data showing that DTG is one of the most potent 3′-P inhibitor among the 3 clinical INSTIs, with an IC50 of 1.6 μM toward WT IN (Figure 1), strengthen the correlation (Figure 6). As DTG also inhibited ST with an IC50 of 33 nM (38), its SI was 50. This is substantially lower than RAL and EVG, which have SIs of 430 and 2600, respectively. Thus, the correlation presented in Figure 6 is consistent with the fact that DTG retains potency against the RAL-resistant SH mutant (2,23,27).
We also tested the ST selectivity and resistance index relationships for the N155H mutant. As for the SH mutant, the correlation was significant with a Pearson correlation of 0.96 and a P-value of 0.0005 (Supplementary Figure S3). No correlation was found for the Y143R mutant (Supplementary Figure S4). Together, these results reveal a significant correlation between the relative activity of a drug as a 3′-P inhibitor and its ability to overcome resistance to the SH mutant, and to a lesser extent, to the N155H mutant. The lack of correlation for the Y143R mutant is consistent with the specific interaction of this residue with RAL but not with the other INSTIs (6). These results imply a relationship between the structural changes resulting from the SH mutations and the ability of drugs to bind to the 3′-P site of IN. They also suggest that 3′-P inhibition is an indicator of the ability of compounds to retain efficacy against the SH (and N155H) mutants.
DISCUSSION
Biochemical studies using recombinant IN and various DNA oligonucleotide substrates have established that IN recognizes the conserved CA dinucleotide (−2 and −1 positions, see Figure 1A) located near the 3′-end of the viral DNA for 3′-P (34,35,37,43–46). Here, we performed biochemical experiments and molecular modeling with the WT and the double-mutant (G140S-Q148H [SH]) that causes significant resistance to RAL and EVG and to a lesser extent to DTG (2,11,12,22,26). Viral DNA substrates with modifications at positions +1 and −1 were tested to probe the 3′-P activity of WT and SH IN enzymes in the absence and presence of RAL and DTG.
Our results highlight the critical role of the +1 and −1 bases for optimum 3′-P activity of WT and SH mutant IN enzymes. In the case of the WT IN, removing either of these two bases flanking the 3′-P site (positions +1 and −1) markedly reduced 3′-P while enabling RAL and DTG to inhibit 3′-P almost as efficiently as ST. An important difference between the SH and the WT enzymes, with respect to the viral DNA interactions (Figures 1 and 2) is our finding that the +1 guanine base (immediately 3′ from the conserved CA; see Figure 1A) is critical for optimum 3′-P activity in the case of the WT IN (34,45,46). By contrast removing this +1 guanine enables the SH mutant to regain 3′-P activity. Based on our biochemical and molecular modeling analyses, we propose that the distorted active site of the SH mutant appears able to accommodate a nucleobase at position +1 only when an amine is present at position 2 of the purine ring system (as in the guanine that is present in the normal viral DNA substrates; see Figure 2). The crucial role of the exocyclic amine of the G+1 base is consistent with the mechanism of the RNase P. In this system, the presence of the G+1 is not essential but participates in the correct positioning of the cleavage site and improves the efficiency of catalysis (47). This exocyclic amino group was important for both the WT and SH enzymes. Therefore, we surmise that the Q146 residue in the flexible loop of HIV-1 IN (140–149) plays an important role in positioning the viral DNA end.
Residue Q146 is among the most conserved amino acid residues of HIV-1 integrase; it is also conserved in all other retroviruses except for deltaretroviruses. In the PFV IN crystal structures, residue Q215 was shown to displace the +1 base of the pre-cleaved viral DNA substrate (9). However, there were no data, either structural or biochemical, regarding the status of the complex before end fraying. We propose that glutamine (Q146 for HIV-1 IN) has a critical role in binding and positioning the double-stranded end(s) of the viral DNA. Our molecular modeling simulations highlight a potential interaction of Q146 (through its amine substituent) with the carbonyl located at position 2 of the pyrimidine ring on the non-reactive +1 and/or −1 bases. This is compatible with the presence of either a cytosine or a thymidine at position +1 of the non-reactive DNA strand, allowing the following two combinations for the cleaved strand: CAGT (e.g. HIV), CAAT (e.g. PFV). Deltaretroviruses possess a threonine in place of this conserved glutamine, thus lacking an amine substituent. The sequence of the corresponding viral DNA substrate is CAGT for HTLV-I and CAAG for HTLV-II. Accordingly, the hydroxyl group present on the side chain of threonine could play a similar role in opening the viral DNA at with the −1 positions through the exocyclic amine of the A-1 and A/G+1 bases of the cleaved strand. This hypothesis is supported by the observation that the HIV-1 IN mutant Q146E has reduced 3′P activity (Figure 5). From the structural data obtained with the PFV intasome, it has been proposed that the SH double-mutation in HIV-1 IN alters the active site conformation to one that is less favorable for binding the available drugs (15). Based on the data presented here, we propose that the active site of the SH mutant constrains the full-length viral DNA in a more stable conformation, but one which is suboptimal for cleavage during 3′-P (i.e. there is a loss of activity compared to WT protein). In this model, the 21Ab-1 and 21Ab+1 substrates would destabilize the double helical character of the viral DNA end, which would allow more efficient 3′-P compared to the canonical substrate.
This proposal is consistent with our predictions for the WT and SH mutant active sites after 3′-P (27). We predicted that the 3′ hydroxyl at the end of the viral DNA (CA-OH) was more tightly bound to the magnesium ion (Mg2+ B) in the context of the SH mutant as compared to the WT. In such a case, it would appear that the INSTI-resistant SH mutant has an enhanced affinity for the 3′-end of the viral DNA both before and after 3′-P, compared to WT. While a recent study by Hightower et al. showed that the dissociative half-life (t1/2) of INSTIs from the IN active site was decreased by RAL resistant mutants (22), our data indicate that resistance to INSTIs could also arise from a decrease in accessibility of the active site, thus increasing the on-rate constant kon. Thus, we propose that resistance to INSTIs might result from a combination of an increased stability of the viral DNA end when the viral DNA is bound to IN, coupled with a reduced ability of the distorted active site to process its substrate, both of which could help to reduce the ability of the drug to bind.
Among the three clinically approved INSTIs, DTG is the best in class in terms of the development of resistance in patients (2). Hightower et al. showed that DTG was bound to the WT HIV-1 intasome was 8- and 26-fold longer than RAL and EVG, respectively (22). However, this measurement cannot be used in the screening of a large number of compounds. Most of the compounds that have been tested for their potency in a ST assay have also been tested for their ability to inhibit 3′-P. Because we demonstrated a clear correlation between the 3′-P potency and the ability of compounds to retain potency against RAL-associated resistant mutants (Figure 6 and Supplementary Figure S3), we propose that evaluation of 3′-P inhibition could be used as a surrogate for the potential activity of drugs against resistant mutants. This parameter could be used to identify compounds exhibiting high potency for inhibition of both 3′-P and ST (i.e. reduced 3′-P/ST selectivity), such as DTG.
In conclusion, our study sheds light on aspects of the 3′-P reaction, the first step of the integration process, for WT and SH-mutant IN enzymes. It suggests that HIV-1 IN recognizes its substrate through specific interactions involving the Q146 residue and the penultimate G of the conserved CAGT sequence at the end of the viral DNA.
Supplementary Material
Footnotes
Present address: Mathieu Métifiot, Laboratoire de Microbiologie Fondamentale et Pathogénicité, CNRS—UMR 5234, Université de Bordeaux, 146 rue Léo Saignat 33076 Bordeaux cedex.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Intramural Program of the National Cancer Institute; Center for Cancer Research [Z01 BC 007333, ZIA BC 007363]; Intramural AIDS Targeted Antiviral Program (IATAP); National Institutes of Health. Funding for open access charge: Developmental Therapeutics Branch; Laboratory of Molecular Pharmacology; CCR/NCI/NIH.
Conflict of interest statement. None declared.
REFERENCES
- 1.Marchand C., Maddali K., Metifiot M., Pommier Y. HIV-1 IN inhibitors: 2010 update and perspectives. Curr. Top. Med. Chem. 2009;9:1016–1037. doi: 10.2174/156802609789630910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Metifiot M., Marchand C., Pommier Y. HIV integrase inhibitors: 20-year landmark and challenges. Adv. Pharmacol. 2013;67:75–105. doi: 10.1016/B978-0-12-405880-4.00003-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liao C., Marchand C., Burke T.R., Jr, Pommier Y., Nicklaus M.C. Authentic HIV-1 integrase inhibitors. Future Med. Chem. 2010;2:1107–1122. doi: 10.4155/fmc.10.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pommier Y., Johnson A.A., Marchand C. Integrase inhibitors to treat HIV/AIDS. Nat. Rev. Drug Discov. 2005;4:236–248. doi: 10.1038/nrd1660. [DOI] [PubMed] [Google Scholar]
- 5.Lesbats P., Engelman A.N., Cherepanov P. Retroviral DNA Integration. Chem. Rev. 2016 doi: 10.1021/acs.chemrev.6b00125. doi:10.1021/acs.chemrev.6b00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Metifiot M., Marchand C., Maddali K., Pommier Y. Resistance to integrase inhibitors. Viruses. 2010;2:1347–1366. doi: 10.3390/v2071347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marchand C. The elvitegravir Quad pill: the first once-daily dual-target anti-HIV tablet. Expert Opin. Investig. Drugs. 2012;21:901–904. doi: 10.1517/13543784.2012.685653. [DOI] [PubMed] [Google Scholar]
- 8.Wills T., Vega V. Elvitegravir : a once-daily inhibitor of HIV-1 integrase. Expert Opin. Investig. Drugs. 2012;21:395–401. doi: 10.1517/13543784.2012.658914. [DOI] [PubMed] [Google Scholar]
- 9.Hare S., Gupta S.S., Valkov E., Engelman A., Cherepanov P. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature. 2010;464:232–236. doi: 10.1038/nature08784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pommier Y., Marchand C. Interfacial inhibitors: targeting macromolecular complexes. Nat. Rev. Drug Discov. 2012;11:25–36. doi: 10.1038/nrd3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Metifiot M., Maddali K., Naumova A., Zhang X., Marchand C., Pommier Y. Biochemical and pharmacological analyses of HIV-1 integrase flexible loop mutants resistant to raltegravir. Biochemistry. 2010;49:3715–3722. doi: 10.1021/bi100130f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Metifiot M., Vandegraaff N., Maddali K., Naumova A., Zhang X., Rhodes D., Marchand C., Pommier Y. Elvitegravir overcomes resistance to raltegravir induced by integrase mutation Y143. AIDS. 2011;25:1175–1178. doi: 10.1097/QAD.0b013e3283473599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Delelis O., Thierry S., Subra F., Simon F., Malet I., Alloui C., Sayon S., Calvez V., Deprez E., Marcelin A.G., et al. Impact of Y143 HIV-1 integrase mutations on resistance to raltegravir in vitro and in vivo. Antimicrob. Agents Chemother. 2010;54:491–501. doi: 10.1128/AAC.01075-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Delelis O., Malet I., Na L., Tchertanov L., Calvez V., Marcelin A.G., Subra F., Deprez E., Mouscadet J.F. The G140S mutation in HIV integrases from raltegravir-resistant patients rescues catalytic defect due to the resistance Q148H mutation. Nucleic Acids Res. 2009;37:1193–1201. doi: 10.1093/nar/gkn1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hare S., Vos A.M., Clayton R.F., Thuring J.W., Cummings M.D., Cherepanov P. Molecular mechanisms of retroviral integrase inhibition and the evolution of viral resistance. Proc. Natl. Acad. Sci. U.S.A. 2010;107:20057–20062. doi: 10.1073/pnas.1010246107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garrido C., Villacian J., Zahonero N., Pattery T., Garcia F., Gutierrez F., Caballero E., Van Houtte M., Soriano V., de Mendoza C., et al. Broad phenotypic cross-resistance to elvitegravir in HIV-infected patients failing on raltegravir-containing regimens. Antimicrob. Agents Chemother. 2012;56:2873–2878. doi: 10.1128/AAC.06170-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marinello J., Marchand C., Mott B.T., Bain A., Thomas C.J., Pommier Y. Comparison of raltegravir and elvitegravir on HIV-1 integrase catalytic reactions and on a series of drug-resistant integrase mutants. Biochemistry. 2008;47:9345–9354. doi: 10.1021/bi800791q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Desimmie B.A., Demeulemeester J., Suchaud V., Taltynov O., Billamboz M., Lion C., Bailly F., Strelkov S.V., Debyser Z., Cotelle P., et al. 2-Hydroxyisoquinoline-1,3(2H,4H)-diones (HIDs), novel inhibitors of HIV integrase with a high barrier to resistance. ACS Chem. Biol. 2013;8:1187–1194. doi: 10.1021/cb4000426. [DOI] [PubMed] [Google Scholar]
- 19.Zhao X.Z., Smith S.J., Metifiot M., Johnson B.C., Marchand C., Pommier Y., Hughes S.H., Burke T.R., Jr Bicyclic 1-hydroxy-2-oxo-1,2-dihydropyridine-3-carboxamide-containing HIV-1 integrase inhibitors having high antiviral potency against cells harboring raltegravir-resistant integrase mutants. J. Med. Chem. 2014;57:1573–1582. doi: 10.1021/jm401902n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao X.Z., Smith S.J., Metifiot M., Marchand C., Boyer P.L., Pommier Y., Hughes S.H., Burke T.R., Jr 4-amino-1-hydroxy-2-oxo-1,8-naphthyridine-containing compounds having high potency against raltegravir-resistant integrase mutants of HIV-1. J. Med. Chem. 2014;57:5190–5202. doi: 10.1021/jm5001908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao X.Z., Smith S.J., Maskell D.P., Metifiot M., Pye V.E., Fesen K., Marchand C., Pommier Y., Cherepanov P., Hughes S.H., et al. HIV-1 Integrase Strand Transfer Inhibitors with Reduced Susceptibility to Drug Resistant Mutant Integrases. ACS Chem. Biol. 2016;11:1074–1081. doi: 10.1021/acschembio.5b00948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hightower K.E., Wang R., Deanda F., Johns B.A., Weaver K., Shen Y., Tomberlin G.H., Carter H.L., 3rd, Broderick T., Sigethy S., et al. Dolutegravir (S/GSK1349572) exhibits significantly slower dissociation than raltegravir and elvitegravir from wild-type and integrase inhibitor-resistant HIV-1 integrase-DNA complexes. Antimicrobial. Agents Chemother. 2011;55:4552–4559. doi: 10.1128/AAC.00157-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Underwood M.R., Johns B.A., Sato A., Martin J.N., Deeks S.G., Fujiwara T. The activity of the integrase inhibitor dolutegravir against HIV-1 variants isolated from raltegravir-treated adults. J. Acquir. Immune Defic. Syndr. 2012;61:297–301. doi: 10.1097/QAI.0b013e31826bfd02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mesplede T., Wainberg M.A. Resistance against Integrase Strand Transfer Inhibitors and Relevance to HIV Persistence. Viruses. 2015;7:3703–3718. doi: 10.3390/v7072790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hare S., Maertens G.N., Cherepanov P. 3′-processing and strand transfer catalysed by retroviral integrase in crystallo. EMBO J. 2012;31:3020–3028. doi: 10.1038/emboj.2012.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnson B.C., Metifiot M., Ferris A., Pommier Y., Hughes S.H. A homology model of HIV-1 integrase and analysis of mutations designed to test the model. J. Mol. Biol. 2013;425:2133–2146. doi: 10.1016/j.jmb.2013.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson B.C., Metifiot M., Pommier Y., Hughes S.H. Molecular dynamics approaches estimate the binding energy of HIV-1 integrase inhibitors and correlate with in vitro activity. Antimicrob. Agents Chemother. 2012;56:411–419. doi: 10.1128/AAC.05292-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao X.Z., Maddali K., Metifiot M., Smith S.J., Vu B.C., Marchand C., Hughes S.H., Pommier Y., Burke T.R., Jr Bicyclic hydroxy-1H-pyrrolopyridine-trione containing HIV-1 integrase inhibitors. Chem. Biol. Drug Des. 2012;79:157–165. doi: 10.1111/j.1747-0285.2011.01270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao X.Z., Maddali K., Smith S.J., Metifiot M., Johnson B.C., Marchand C., Hughes S.H., Pommier Y., Burke T.R., Jr 6,7-Dihydroxy-1-oxoisoindoline-4-sulfonamide-containing HIV-1 integrase inhibitors. Bioorg. Med. Chem. Lett. 2012;22:7309–7313. doi: 10.1016/j.bmcl.2012.10.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Metifiot M., Maddali K., Johnson B.C., Hare S., Smith S.J., Zhao X.Z., Marchand C., Burke T.R., Jr, Hughes S.H., Cherepanov P., et al. Activities, crystal structures, and molecular dynamics of dihydro-1H-isoindole derivatives, inhibitors of HIV-1 integrase. ACS Chem. Biol. 2013;8:209–217. doi: 10.1021/cb300471n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao X.Z., Maddali K., Vu B.C., Marchand C., Hughes S.H., Pommier Y., Burke T.R., Jr Examination of halogen substituent effects on HIV-1 integrase inhibitors derived from 2,3-dihydro-6,7-dihydroxy-1H-isoindol-1-ones and 4,5-dihydroxy-1H-isoindole-1,3(2H)-diones. Bioorg. Med. Chem. Lett. 2009;19:2714–2717. doi: 10.1016/j.bmcl.2009.03.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao X.Z., Semenova E.A., Vu B.C., Maddali K., Marchand C., Hughes S.H., Pommier Y., Burke T.R., Jr 2,3-Dihydro-6,7-dihydroxy-1H-isoindol-1-one-based HIV-1 integrase inhibitors. J. Med. Chem. 2008;51:251–259. doi: 10.1021/jm070715d. [DOI] [PubMed] [Google Scholar]
- 33.Brooks B.R., Brooks C.L., 3rd, Mackerell A.D., Jr, Nilsson L., Petrella R.J., Roux B., Won Y., Archontis G., Bartels C., Boresch S., et al. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johnson A.A., Sayer J.M., Yagi H., Patil S.S., Debart F., Maier M.A., Corey D.R., Vasseur J.J., Burke T.R., Jr, Marquez V.E., et al. Effect of DNA modifications on DNA processing by HIV-1 integrase and inhibitor binding: role of DNA backbone flexibility and an open catalytic site. J. Biol. Chem. 2006;281:32428–32438. doi: 10.1074/jbc.M605101200. [DOI] [PubMed] [Google Scholar]
- 35.Bushman F.D., Craigie R. Activities of human immunodeficiency virus (HIV) integration protein in vitro: specific cleavage and integration of HIV DNA. Proc. Natl. Acad. Sci. U.S.A. 1991;88:1339–1343. doi: 10.1073/pnas.88.4.1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sherman P.A., Fyfe J.A. Human immunodeficiency virus integration protein expressed in Escherichia coli possesses selective DNA cleaving activity. Proc. Natl. Acad. Sci. U.S.A. 1990;87:5119–5123. doi: 10.1073/pnas.87.13.5119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vink C., van Gent D.C., Elgersma Y., Plasterk R.H. Human immunodeficiency virus integrase protein requires a subterminal position of its viral DNA recognition sequence for efficient cleavage. J. Virol. 1991;65:4636–4644. doi: 10.1128/jvi.65.9.4636-4644.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hare S., Smith S.J., Metifiot M., Jaxa-Chamiec A., Pommier Y., Hughes S.H., Cherepanov P. Structural and functional analyses of the second-generation integrase strand transfer inhibitor dolutegravir (S/GSK1349572) Mol. Pharmacol. 2011;80:565–572. doi: 10.1124/mol.111.073189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Canducci F., Ceresola E.R., Boeri E., Spagnuolo V., Cossarini F., Castagna A., Lazzarin A., Clementi M. Cross-resistance profile of the novel integrase inhibitor Dolutegravir (S/GSK1349572) using clonal viral variants selected in patients failing raltegravir. J. Infect. Dis. 2011;204:1811–1815. doi: 10.1093/infdis/jir636. [DOI] [PubMed] [Google Scholar]
- 40.Renisio J.G., Cosquer S., Cherrak I., El Antri S., Mauffret O., Fermandjian S. Pre-organized structure of viral DNA at the binding-processing site of HIV-1 integrase. Nucleic Acids Res. 2005;33:1970–1981. doi: 10.1093/nar/gki346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Katz R.A., Merkel G., Andrake M.D., Roder H., Skalka A.M. Retroviral integrases promote fraying of viral DNA ends. J. Biol. Chem. 2011;286:25710–25718. doi: 10.1074/jbc.M111.229179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Merkel G., Andrake M.D., Ramcharan J., Skalka A.M. Oligonucleotide-based assays for integrase activity. Methods. 2009;47:243–248. doi: 10.1016/j.ymeth.2008.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chow S.A., Brown P.O. Substrate features important for recognition and catalysis by human immunodeficiency virus type 1 integrase identified by using novel DNA substrates. J. Virol. 1994;68:3896–3907. doi: 10.1128/jvi.68.6.3896-3907.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Esposito D., Craigie R. Sequence specificity of viral end DNA binding by HIV-1 integrase reveals critical regions for protein-DNA interaction. EMBO J. 1998;17:5832–5843. doi: 10.1093/emboj/17.19.5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johnson A.A., Marchand C., Patil S.S., Costi R., Di Santo R., Burke T.R., Jr, Pommier Y. Probing HIV-1 Integrase Inhibitor Binding Sites with Position-Specific Integrase-DNA Cross-Linking Assays. Mol. Pharmacol. 2007;71:893–901. doi: 10.1124/mol.106.030817. [DOI] [PubMed] [Google Scholar]
- 46.Johnson A.A., Santos W., Pais G.C., Marchand C., Amin R., Burke T.R., Jr, Verdine G., Pommier Y. Integration requires a specific interaction of the donor DNA terminal 5′-cytosine with glutamine 148 of the HIV-1 integrase flexible loop. J. Biol. Chem. 2006;281:461–467. doi: 10.1074/jbc.M511348200. [DOI] [PubMed] [Google Scholar]
- 47.Kikovska E., Brannvall M., Kirsebom L.A. The exocyclic amine at the RNase P cleavage site contributes to substrate binding and catalysis. J. Mol. Biol. 2006;359:572–584. doi: 10.1016/j.jmb.2006.03.040. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.