


Abstract
DNA nanoassemblies have demonstrated wide applications in various fields including nanomaterials, drug delivery and biosensing. In DNA origami, single-stranded DNA template is shaped into desired nanostructure by DNA staples that form Holliday junctions with the template. Limited by current methodologies, however, mechanical properties of DNA origami structures have not been adequately characterized, which hinders further applications of these materials. Using laser tweezers, here, we have described two mechanical properties of DNA nanoassemblies represented by DNA nanotubes, DNA nanopyramids and DNA nanotiles. First, mechanical stability of DNA origami structures is determined by the effective density of Holliday junctions along a particular stress direction. Second, mechanical isomerization observed between two conformations of DNA nanotubes at 10–35 pN has been ascribed to the collective actions of individual Holliday junctions, which are only possible in DNA origami with rotational symmetric arrangements of Holliday junctions, such as those in DNA nanotubes. Our results indicate that Holliday junctions control mechanical behaviors of DNA nanoassemblies. Therefore, they can be considered as ‘mechanophores’ that sustain mechanical properties of origami nanoassemblies. The mechanical properties observed here provide insights for designing better DNA nanostructures. In addition, the unprecedented mechanical isomerization process brings new strategies for the development of nano-sensors and actuators.
INTRODUCTION
Because of the highly stable and specific recognition between two complementary DNA strands, DNA has been used as an attractive component in nanoassembly. In the DNA origami nanoassembly, a long ssDNA serves as a template to fold into nanostructures through hundreds of Holliday junctions formed between short DNA staples complementary to the template sequences at particular locations (1). The simple, robust and highly efficient synthesis strategy of DNA origami has established this structure as a highly potent nanomaterial (2–10) yet to be fully characterized for its properties. Among these unknown territories, mechanical property is certainly a notable missing link with high significance. Mechanical stability of the connecting regions in DNA nanoassemblies is essential to sustain robust interactions between biomolecules (11) and nanoassemblies. Likewise, mechanical rigidity of DNA nanocavities is critical to define the morphology of inorganic nanoparticles contained within (12,13). In the sensing applications using DNA nanoassemblies, (14–18), the mechanical rigidity and stability of the DNA nanoassemblies can directly affect the accuracy in the signal output and the sensitivity in the analyte recognition (5).
So far, only a handful of investigations have been reported with a main focus on the mechanical functionalities of the DNA nanoassemblies (14,19–22). The insufficient information on the mechanical rigidity and stability of DNA nanoassemblies in general and DNA origami in particular hinders rational design of DNA nanomaterials for applications that exploit their mechanical properties. One reason for this lack of knowledge lies in the difficulty in the characterization of individual nano-objects for their mechanical properties. Special tools with high resolution for mechanical force measurement, such as AFM and optical tweezers, must be employed to carry out the characterization (22,23) after immobilization of nanoparticles with different shapes, which is another challenging practice.
We reasoned that due to the flexible nature in the design of a DNA origami with the single-nucleotide precision, it is possible to introduce two duplex DNA handles to tether a DNA origami. These handles are then linked to the two optically trapped polystyrene beads in laser tweezers, which allow the quantification of mechanical properties of DNA nanoassembly. Previously, with the aim to develop origami based nanomechanical devices, Sugiyama and coworkers have found that tubular designs of origami structures can have two stable conformations: a short and a long tubular forms that may stem from different isomers of Holliday junctions contained inside the origami devices (10). Given the potential applications of controlling conformations of DNA origami structures by mechanical factors as discussed above, here we wish to understand whether it is a unique feature for the tubular-shaped origami to demonstrate different structural isomers. To this purpose, we applied two duplex DNA handles to the tubular DNA origami structures for the comparison of their mechanical properties with those from other nanoassemblies, DNA nanopyramids and DNA nanotiles. We found mechanical stabilities of DNA origami structures are correlated with the effective density of Holliday junctions in a particular nanoassembly. The mechanical stability is anisotropic in nature with the short axis of a DNA nanotube resisting higher external stress than the long axis. Interestingly, mechanical isomerization between two conformations of a DNA nanoassemby at 10–35 pN external force was observed only in DNA nanotubes, which have unique symmetric arrangements of Holliday junctions. Similar to the mechanical stability, the mechanical isomerization also showed anisotropic behavior. Given that individual Holliday junctions have isomerization force in the sub-picoNewton range (24) and they are anisotropically arranged in DNA nanotubes, we have attributed the mechanical isomerization of the DNA nanotubes to the collective actions of many Holliday junctions that experience similar microenvironment. All these results indicate that Holliday junctions in DNA origami structures serve as mechanophores (25,26), which determine the mechanical property of DNA nanoassemblies, similar to chromophores and fluorophores that carry spectroscopic information in a molecule. We anticipate these new findings are instrumental to optimize the mechanical strength of DNA nanoassemblies.
RESULTS AND DISCUSSION
Mechanical isomerization and mechanical disassembly of DNA tubes
Optical tweezers have been used to apply and measure forces in picoNewtons (27–29). Compared to AFM, it has better force resolution and therefore is particularly suitable to characterize mechanical stability of macromolecules such as proteins and nucleic acids. Here, we used a home-built laser tweezers instrument (30,31) to characterize the mechanical properties of individual DNA origami nanoassemblies. To this end, we introduced two double-stranded (ds) DNA handles to DNA origami structures (see Figure 1A, Supplementary Figure S1A and B and Materials and Methods) (14). DNA handles were inserted into the origami structure via overhang single-stranded staple sequence, which contains 40 nucleotides complementary to the single-stranded M13mp18 template (it has been reported that the shearing force of 30 bp duplex DNA is >60 pN) (32). The other end of the handle was labeled with a digoxigenin or biotin molecule. These DNA handles were mixed and incubated with rest of the DNA components for the origami construction (see Materials and Methods and Supplementary Figures S1–S7). AFM images have revealed successful incorporation of the handles (see Figure 1B and C, Supplementary Figures S8 and S9 for the handles attached to the DNA nanotubes; see Supplementary Figures S6 and S7 for handle incorporation of other origami structures).
Figure 1.

Mechanoanalytical characterization of DNA origami nanotubes. (A) Schematic of the experimental set up for the mechanical isomerization of DNA origami tubes. DNA origami tube is sandwiched between two long dsDNA handles and the whole construct is tethered between two optically trapped beads with affinity linkages. The mechanochemical property of DNA tubes is revealed by moving one of the beads away from another at a loading rate of ∼5.5 pN/s. AFM image of the eight-tube DNA for longitudinal (B) and horizontal stretching (C). Schematic pictures are shown to the right of corresponding AFM images.
Using the digoxigenin and the biotin molecules labeled at the free end of the DNA handles, the modified DNA origami construct was tethered to the two optically trapped particles coated with digoxigenin antibody and streptavidin through respective affinity interactions (14). The mechanical stability of the origami was probed by force ramping experiments in which one of the optically trapped particles was moved away from the other at a loading rate of ∼5.5 pN/s in laser tweezers.
Due to the unique behavior of isomerization displayed in DNA origami tubes (10), we chose these origami structures as our first samples to evaluate mechanical properties of DNA origami structures. Using the reported procedures (10), we prepared eight-tube and six-tube DNA origami nanoassemblies that respectively consist of eight and six Holliday junctions in each of the circular layers that are spirally arranged into desired tubes. Previously, AFM images (10) have revealed that each type of tube has two equilibrated isomers, long and short tubes (similar to those shown in Figure 1B and C, Supplementary Figures S8 and S9). Due to the limited time resolution in AFM, the isomerization between the two conformers has not been observed in real time. The dynamic nature of mechanical unfolding at the single nanostructure level may provide an unprecedented opportunity to investigate this isomerization process. To this end, by using the procedures reported earlier (14), either eight-tube (short and long) or six-tube (short and long) DNA origami structures were obtained and tethered to two optically trapped polystyrene beads as described above.
In the first set of experiments, we mechanically stretched DNA origami samples from longitudinal direction of the tube (see the eight-tube DNA in Figure 1 as an example). We observed two types of rupture transitions (Figure 2A and B), which were assigned to mechanical isomerization and disassembly of origami tubes on the basis of the following aspects. First, the two types of transitions can be separated according to the low (10–35 pN) and high (35–60 pN) force of transitions (Figure 2C, two left panels). The hierarchical cluster analysis (HCA) (33), which is a non-supervised method (34) to classify groups using the distance between two data points, confirmed the presence of these two populations (Figure 2C, two right panels). Here, we used rupture force as a distance variable in the HCA calculation (see Materials and Methods for details). When we scrutinized the properties of the transition in these two groups, we found that the single or multiple transitions in the lower force region (10–35 pN) were reproducibly observed after incubating the construct for 90 s before each force–extension cycle (Supplementary Figure S10). On the other hand, we observed successive transition events in the higher force region (35–60 pN) that were not reversible after the same incubation time. These high-force transitions, all centered in a similar force region (35–60 pN) even for different DNA tubes (Supplementary Figures S11 and S12), suggesting the disassembly of the structures with similar mechanical stabilities. Since the force range we observed here fell into that for the slide-opening of DNA duplex (35), we surmised these transitions may be due to the force-induced disassembly of Holliday junctions (shearing breakage), whose re-assembly is slow. Consistent with this scenario, when the tension for the origami in the higher-force region was relaxed to 0 pN, we did not observe the same trajectory in subsequent force-ramping cycles. Instead, the contour length of the origami became longer even after incubation up to 90 s at 0 pN (see curves in the disassembly region in Figure 2B). Similar events were observed during the mechanical stretching of DNA nanotiles, which have been ascribed to the disassembly of DNA origami structures (14). Therefore, these high-force transitions likely represent the disintegration of a DNA origami structure through the irreversible breakage in the Holliday junctions during the time scale of the experiment.
Figure 2.

Mechanochemical properties of DNA origami tubes. (A) A typical force–extension (F–X) trace for the longitudinal stretching of a eight-tube DNA in optical tweezers experiment. Dotted circles depict the isomerization and disassembly processes. (B) A set of F–X traces for another eight-tube DNA origami. The first transition event in the red curve depicts mechanical isomerization. The relaxing curve after mechanical isomerization is shown in black. After incubation, the next stretching curve is identical with the first red curve. After mechanical disassembly, the traces do not overlap with the first red curve after incubation at 0 pN. Instead, they depict longer contour lengths (blue). (C) Distribution plots for the transition forces (left two panels) and the hierarchical cluster analyses (right two panels) clearly indicate the presence of two populations. The red population with lower transition force is due to isomerization of the nanotube while the blue population with higher transition force is due to nanotube disassembly. Solid curves in the left panel represent Gaussian fittings. The green data points are identified as stand-alone groups in HCA. We assigned these points to each of the populations based on force. (D) Schematic of mechanical isomerization of the short to long eight-tube DNA origami structures followed by disassembly. Magnified sections show the rearrangement of Holliday junctions before and after mechanical isomerization. (E) Schematic of the molecular rearrangements of Holliday junctions in a DNA tube during mechanical isomerization and disassembly processes. Numbers in black and green respectively indicate the duplex layers and the Holliday junctions in DNA origami tubes.
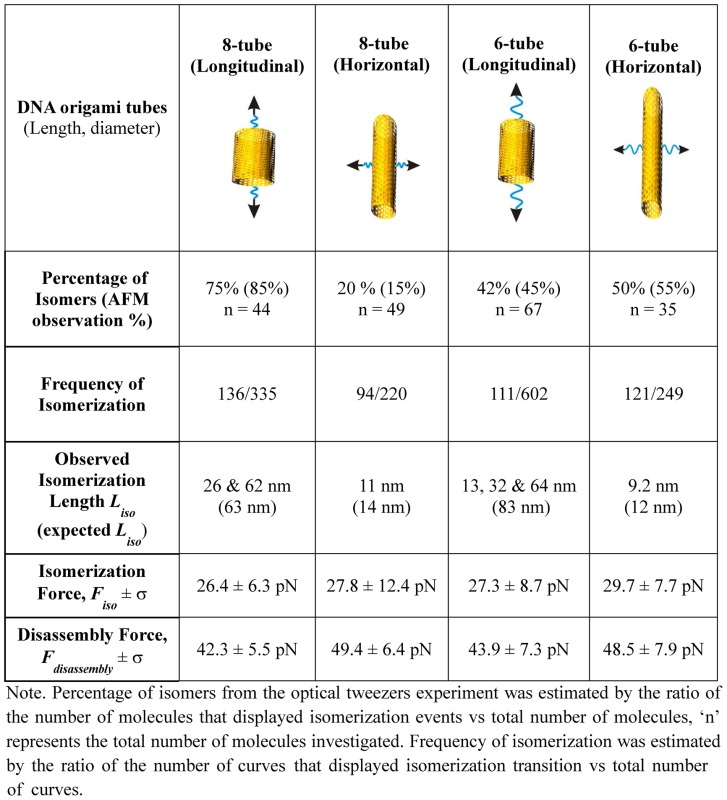
To explain the nature of the low-force transitions, we analyzed the population of the origami that yielded reversible transitions in the lower force region. We found that 42% (Supplementary Figure S13B) and 75% (Supplementary Figure S10) of the origami showed these transitions in the six-tube and eight-tube DNA stretched from the longitudinal direction, respectively. During longitudinal stretching, only short tubes can isomerize into the long tubes (Figure 2D), therefore, 42% and 75% populations represent short tube isomers. These values are close to that obtained from AFM images, which showed 45% and 85% (Table 1) for the short isomers in the six-tube and eight-tube DNA, respectively. The striking agreement between the mechanical unfolding and AFM analyses strongly suggests that these low-force transitions are likely force-induced isomerizations, which are expected to be reversible after incubation as the origami structures remain intact during the process (Figure 2D and E). To confirm this scenario, we measured the changes-in-extension due to the rupture transitions in the lower force region. We found that the measured values were closely matched with the calculated difference in length between the short and long isomers in each type of the DNA tube (see next section for details). It is noteworthy that in the force range (10–35 pN) we observed for the isomerization, the changes-in-extension for different DNA nanotubes do not vary with force significantly (calculations see SI).
Table 1. Mechanochemical properties of tubular DNA origami structures.
|
Based on these facts, we assigned the lower-force transition as mechanical isomerization while the higher-force transition as disintegration of origami structures (see schematic models in Figure 2D (nanostructure level) and Figure 2E (molecular level)).
Given that each Holliday junction consists of Watson–Crick base pairing that can be taken apart by different mechanical force from varying directions, (24,36) it is interesting to see whether or not the same force is required to disintegrate the DNA origami tube from different directions. To test this scenario, we stretched the same type of tube from horizontal direction by using two dsDNA handles facing each other in the middle of the eight-tube DNA (Figure 1C and Supplementary Figure S3). Similar to what was observed for the longitudinal stretching, two regions were found for the horizontal stretching. However, the disassembly process was abrupt. While the lower force region (10-35 pN) showed reproducible features after incubation at 0 pN, the higher force region (35–60 pN) again revealed irreversible disassembly, likely due to disintegration of staples. Similar characteristics were observed during the longitudinal and horizontal stretchings of the six-tube DNA (see below for detailed discussion).
Mechanical disassembly (or stability) of a DNA origami structure is correlated with the effective density of Holliday junctions
When we scrutinized the higher-force mechanical disassembly region that reflects the mechanical stability of a DNA nanoassembly, we found that the mechanical stability for horizontal stretching of the eight-tube DNA was higher (49 pN, see Supplementary Figure S13A for F–X curves and Figure 3B for histogram) than longitudinal stretching (42 pN, Figure 2A and B for F–X curves and Figure 3A for histogram). Similar anisotropy in mechanical stability was observed for the six-tube DNA origami (see Supplementary Figures S4 and S5 for its structure and preparation and Supplementary Figure S9 for AFM images). While the horizontal mechanical stability was 49 pN, that of longitudinal was lower (44 pN, summarized in Table 1 and Figure 3C, see representative F–X curves in Supplementary Figure S13 B&C). During disassembly, gradual deviation in the extension that corresponds to the sequential rupture of Holliday junctions was not observed in the F–X curves for either horizontal or longitudinal pulling direction. Instead, sudden and big (80–100 nm, see Figure 2A and B) rupture events that were consistent with the layer-by-layer disintegration during longitudinal pulling were observed (Figure 3E). Peeling off each layer during layer-by-layer disassociation in the eight-tube DNA requires the breakage of eight Holliday junctions (see black arrows in Supplementary Figure S2), while it requires breaking six Holliday junctions for the same effect in the six-tube DNA (see black arrows in Supplementary Figure S4). On the other hand, with horizontal stretching, the layer-by-layer peeling is rather difficult (Supplementary Figures S3 and S5) and the disassembly of the structure is likely due to force induced melting (35,37) of Holliday junctions (Figure 3F). As a result, complete disassembly of many more Holliday junctions is necessary to break the tube. During stretching experiments, it is more likely that the loss of the Holliday junction connected to the DNA handles would cause the sudden breakage of the tether (Supplementary Figure S13A and C). These breakage forces are used to represent the lower end of the disassembly forces of the origami tubes at the horizontal direction (Figure 3C). This geometry difference also predicts that during longitudinal stretching, more intermediates can be accommodated compared to those from horizontal direction. In fact, this has been confirmed during experiments, in which on average 6 intermediates were observed in longitudinal disassembly while it contained only one intermediate in horizontal direction (Figure 3D).
Figure 3.

Disassembly processes of DNA origami structures. (A and B) Histograms of the mechanical disassembly forces measured along longitudinal and horizontal stretching directions of DNA eight-tubes, respectively. The disassembly force was determined at the first disassembly event (the red arrow in Figure 2B). The insets depict the stretching directions. N and n respectively indicate the number of F–X traces and the number of molecules. (C) Comparison of the disassembly force of DNA nanotiles (Tiles), DNA nanopyramid (Py), eight-tube DNA (8T) and six-tube DNA (6T). (D) Average number of the disassembly events observed during the mechanical disintegration of the 8-tube DNA (8T) in longitudinal (L) or horizontal (H) stretching. (E and F) Schematics of the disassembly with respect to Holliday junctions during longitudinal and horizontal stretching directions, respectively. Dotted yellow lines depict Holliday junction layers in different stretching orientations. For clarity, only one complete Holliday junction is shown in the white circle in each scheme.
To further characterize the mechanical stability of DNA origami structures, we performed stretching experiments of origami nanotiles (14) (see Supplementary Figure S6 for design and AFM image, S13D for F–X curves) and pyramid-shaped origami (see Materials and Methods and Supplementary Figure S7 for the design and AFM image, Supplementary Table S1 for the staple sequences and Supplementary Figure S13 E for F–X curves). As shown in Figure 3C, DNA nanotiles have shown the weakest mechanical stability (disassembly force 28 pN), which was followed by a 3D structure, DNA nanopyramid (35 pN). The most compact 3D structures, horizontal eight-tube and six-tube DNA, demonstrated the strongest stabilities (49.4 and 48.5 pN respectively). Due to the open geometry in the 2D origami nanotiles (14), disassembly of the structure via stretching only involves the breakage of Holliday junctions located close to the axis of the stress. In contrast, in the 3D DNA assemblies, the stability of a structure can be contributed from distally located Holliday junctions that come close to the stress axis through the long-range, 3D arrangement. In fact, the density of Holliday junctions along the longitudinal pulling for the 3D tubular structure increases significantly as the Holliday junctions in the same layer becomes more compact after isomerization (see Supplementary Figure S15). Therefore, the effective density of the Holliday junctions along the stretching direction becomes a decisive factor for the mechanical stability of an origami structure along that direction. Here, we defined the density of Holliday junctions as the number of contributing Holliday junctions per nanometer of the force axis in the nanostructure. For nanotiles and nanopyramids, the effective Holliday junction densities are 0.17 and 0.18 HJ/nm, respectively. For the eight-tube and the six-tube DNA along the longitudinal pulling direction, due to the symmetric geometries, all the Holliday junctions contribute equally to the mechanical property of the origami. Therefore, they are all counted in the density calculation, which leads to much higher densities (1.43 HJ/nm for the eight-tube and 1.09 HJ/nm for the six-tube). Corresponding to this trend, the disassembly forces of the isomerizable structures along the longitudinal direction for both six- and eight-tube DNA origami are higher (35–60 pN) than those of the non-isomerizable structures such as nanotiles and nanopyramids (28–40 pN). This clearly shows that the mechanical stability of DNA origami structures may depend on the effective density of the Holliday junctions. Therefore, the observed data of mechanical stability suggest that Holliday junctions serve as mechanophores (26,38,39) to impart the mechanical stability to the entire DNA origami nanoassemblies.
Holliday junctions in DNA origami tubes are responsible for mechanical isomerization between nanotube isomers
Next, we investigated the mechanical property of the DNA origami at the lower force region (Figures 2A–C and 4), in which transition events were reproducibly observed during consecutive stretching of the DNA nanotubes (see Supplementary Figure S10). F–X curves obtained during the stretching of the 8-tube DNA showed characteristic changes-in-extension along the longitudinal (Figure 4C) or horizontal direction (Figure 4D). This confirms that the low-force transition events represent isomerization of the DNA tubes. To determine the nature of these rupture transitions, we measured the change-in-extension during each event of isomerization transition. The change-in-extension is described as the isomerization length (Liso) in Figures 4 and 5. For the eight-tube DNA, we found two major transitions of ∼62 and ∼26 nm during the longitudinal stretching (Figure 4E) and one transition of ∼11 nm during the horizontal stretching (Figure 4F). These change-in-extension histograms show broad distributions, which is expected as the transition events are likely mediated by one or more intermediates presented in DNA origami structures with large sizes (40,41). It is also possible that some reversible disassembly or melting processes may contribute to this broad feature. For example, it has been observed that under mechanical stress, force induced melting can weaken Watson-Crick base pairing (32), which may lead to transitions with longer-than-expected sizes. To clearly determine the most likely change-in-extension values, we used Population Deconvolution at Nanometer resolution (PoDNano), a statistical tool based on bootstrapping analysis as described in literature (42,43). With a total of 5000 random resampling processes, we confirmed the presence of the ∼62 and ∼26 nm transitions in the change-in-extension histogram with the broadest distribution (Figure 4E inset). In AFM imaging, we have observed two different DNA tube isomers, long and short tubular structures (Supplementary Figure S8) (10). Compared to the expected change-in-extension between these two isomers (63 and 14 nm for longitudinal and horizontal stretching directions, respectively, see Supplementary Figure S16 for detailed calculation), the most probable values obtained from the histograms of the change-in-extension (Table 1) strongly suggest that the transitions are due to the force induced isomerization between the long and short DNA eight-tubes (Figure 4A and B). Stretching of the six-tube DNA origami confirmed that the transitions in the low force region are indeed due to the mechanical isomerization between the long and short DNA tubes (80 and 9.2 nm in sum of all transition events vs expected values of 83 and 12 nm for the longitudinal and horizontal stretching directions, respectively, see Figure 5, Supplementary Figures S16 and S17). It is noteworthy that multiple transitions were observed in every longitudinal stretching experiment. This suggests the presence of intermediates during isomerization. For example, a stable intermediate at ∼46 nm (Figure 5E) can be attributed to a conformation trapped during incomplete isomerization (Supplementary Figure S18).
Figure 4.

Mechanical isomerization of the eight-tube DNA origami. Mechanical isomerizations between the long and short DNA tubes are explained from the perspective of Holliday junctions in the origami structure during longitudinal (A) or horizontal (B) stretching. For clarity, only one complete Holliday junction is shown in the middle of each structure. Dotted yellow lines depict Holliday junction layers in different stretching orientations. Representative F-X curves were obtained during longitudinal (C) or horizontal (D) stretching of the eight-tube DNA. Arrow heads in both F-X curves indicate the isomerization events (see insets for respective schematic diagrams). The blue/red and green traces depict stretching and relaxing curves, respectively. Due to the nature of the mechanical isomerization, only the transitions from the short tube to the long tube in the longitudinal pulling, and the long tube to the short tube in the horizontal pulling can be observed. Histograms of the isomerization length (Liso) of the eight-tube DNA are plotted for longitudinal (E) or horizontal (F) stretching (inset in E depicts the most probable populations using PoDNano approach, see text). Histograms of the isomerization force (Fiso) of the same DNA tube measured during longitudinal (G) or horizontal (H) stretching. Solid curves represent Gaussian fittings. N and n respectively depict the number of F–X traces and number of molecules in experiments.
Figure 5.

Mechanical isomerization of the six-tube DNA origami. AFM images of the DNA nanoassembly with longitudinal (A) or horizontal (B) dsDNA handles. Representative F-X traces were obtained by longitudinal (C) or horizontal (D) stretching of the six-tube DNA. The arrowheads in both F-X curves indicate isomerization events (see insets for respective schematic diagrams). The blue/red and green traces depict stretching and relaxing curves, respectively. (E) Histogram of the isomerization length (Liso) measured from the longitudinal stretching shows individual transitions that involve different intermediates. Inset depicts the histogram of the sum of all transitions observed in individual F-X curves. While the 46.5 nm population suggests transitions involving intermediates, the 80 nm population is consistent with the full transition from the short to long DNA tubes (expected value: 83 nm). (F) Histogram of the isomerization length (Liso) of the six-tube DNA during horizontal stretching. Isomerization force (Fiso) histograms of the six-tube DNA during longitudinal (G) or horizontal (H) stretching. Solid curves represent Gaussian fittings. N and n respectively depict the number of F–X traces and number of molecules in experiments.
Interestingly, during horizontal stretching, the mechanical isomerization involves fewer intermediate states compared to the longitudinal stretching (compare between Figure 4E and F; and between Figure 5E and F). This reflects different origami arrangements along various stretching orientations. During longitudinal stretching, it travels longer (62 nm for the eight-tube and 80 nm for the six-tube) from the shorter DNA tube to the longer tube with respect to the horizontal stretching, in which the distance is much shorter between the two isomers (11 nm for the eight-tube and 9.2 nm for the six-tube). The extra transition distance in the former case (longitudinal) renders a more elastic energetic profile that allows more intermediates to populate. In the latter case (horizontal), however, the entire process becomes more cooperative due to the shorter and therefore more rigid isomerization pathways that cannot host as many intermediate populations. Such an observation is consistent with the disassembly of the origami structures at the high force region discussed above. While the DNA tube experiences more intermediates during the longitudinal, layer-by-layer disintegration, it has fewer intermediates during the horizontal disassembly with increased cooperativity (Figure 3D). Similar observations have been previously obtained in the mechanical unfolding of short and long DNA duplexes. While the short DNA duplexes such as hairpins almost do not present intermediates, (44) the saw-teeth features observed in force–extension traces demonstrate the existence of many intermediates in long DNA duplexes (41).
During the longitudinal isomerization of the eight-tube DNA origami, the isomerization force (Fiso = 26.4 pN, Figure 4G and Table 1) is comparable to that of the horizontal Fiso (27.8 pN, Figure 4H and Table 1). Similar forces between the two stretching orientations have been observed in the isomerization of the six-tube DNA origami (Fiso = 27.3 pN for longitudinal and 29.7 pN for horizontal, see Figure 5G and H, respectively). We reasoned that the isomerization of the DNA nanotubes can be a result of collective isomerization of individual Holliday junctions at the microscopic level. Based on this assumption, we calculated the isomerization force for individual Holliday junctions as 0.12–0.14 pN (see SI). This value is in agreement with the isomerization force of a single Holliday junction (0.1–0.3 pN) predicted by Hohng et al. (24).
It is noteworthy that mechanical isomerization was not observed in DNA nanotiles or nanopyramid structures. Close inspection on all three types of origami nanoassemblies shows that the latter two structures are rather different from the tubular structures. While the DNA nanotiles have a planar geometry, nanopyramids consisting of four triangular tiles fold into an object in which the perimeter of each layer gradually reduces from the base to the apex of the pyramid. In DNA nanotubes, the circular geometry with the same tube diameter gives rise to less hierarchical contributions between the distal and proximal regions with respect to the force axis, which bring a similar microenvironment to all Holliday junctions. However, due to the loss of symmetry in either DNA nanotiles or nanopyramids, individual Holliday junctions experience different environment and therefore, behave differently. As a result, the mechanical isomerization between two origami isomers can only be carried out by many identical Holliday junctions as a collective action in DNA nanotubes, but not in other structures.
CONCLUSIONS
Serving as essential components inside DNA origami nanoassemblies, DNA staples offer stability to DNA nanoassemblies by forming Holliday junctions with the single-stranded DNA template. Using optical tweezers, we have quantified and explained two emergent properties of DNA nanoassemblies not seen in their individual components from mechanical perspective. First, we have found the mechanical stability of DNA origami structures can be geometry dependent, which is determined by the effective density of Holliday junctions along a particular stretching direction. Second, we have quantitatively ascribed the cooperative transition between short and long DNA nanotubes to the collective mechanical isomerizations of many individual Holliday junctions. These observations indicate that the Holliday junctions serve as mechanophores in DNA origami nanoassemblies investigated here. Further testing is required to validate this point for more complex DNA nanoassemblies such as DNA helix bundles (2,21,22,45). We anticipate our new findings can provide unprecedented guidelines to design DNA nanoassemblies with better mechanical properties in both thermodynamic and kinetic aspects.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
NSF [CHE-1026532, CHE-1415883 to H.M.]; JSPS KAKENHI [15H03837, 24104002, 24225005, 26620133 to M.E. and H.S.]. Funding for open access charge: NSF [CHE-1026532, CHE-1415883 to H.M.]; JSPS KAKENHI [15H03837, 24104002, 24225005, 26620133 to M.E. and H.S.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Rothemund P.W.K. Folding DNA to create nanoscale shapes and patterns. Nature. 2006;440:297–302. doi: 10.1038/nature04586. [DOI] [PubMed] [Google Scholar]
- 2.Douglas S.M., Dietz H., Liedl T., Högberg B., Graf F., Shih W.M. Self-assembly of DNA into nanoscale three-dimensional shapes. Nature. 2009;459:414–418. doi: 10.1038/nature08016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Han D., Pal S., Nangreave J., Deng Z., Liu Y., Yan H. DNA origami with complex curvatures in three-dimensional space. Science. 2011;332:342–346. doi: 10.1126/science.1202998. [DOI] [PubMed] [Google Scholar]
- 4.Douglas S.M., Marblestone A.H., Teerapittayanon S., Vazquez A., Church G.M., Shih W.M. Rapid prototyping of 3D DNA-origami shapes with caDNAno. Nucleic Acids Res. 2009;37:5001–5006. doi: 10.1093/nar/gkp436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gerling T., Wagenbauer K.F., Neuner A.M., Dietz H. Dynamic DNA devices and assemblies formed by shape-complementary, non–base pairing 3D components. Science. 2015;347:1446–1452. doi: 10.1126/science.aaa5372. [DOI] [PubMed] [Google Scholar]
- 6.Maune H.T., Han S.-p., Barish R.D., Bockrath M., Goddard I.I.A., RothemundPaul W.K., Winfree E. Self-assembly of carbon nanotubes into two-dimensional geometries using DNA origami templates. Nat. Nano. 2010;5:61–66. doi: 10.1038/nnano.2009.311. [DOI] [PubMed] [Google Scholar]
- 7.Rajendran A., Endo M., Katsuda Y., Hidaka K., Sugiyama H. Programmed two-dimensional self-assembly of multiple DNA origami jigsaw pieces. ACS Nano. 2010;5:665–671. doi: 10.1021/nn1031627. [DOI] [PubMed] [Google Scholar]
- 8.Andersen E.S., Dong M., Nielsen M.M., Jahn K., Subramani R., Mamdouh W., Golas M.M., Sander B., Stark H., Oliveira C.L.P., et al. Self-assembly of a nanoscale DNA box with a controllable lid. Nature. 2009;459:73–76. doi: 10.1038/nature07971. [DOI] [PubMed] [Google Scholar]
- 9.Marras A.E., Zhou L., Su H.-J., Castro C.E. Programmable motion of DNA origami mechanisms. Proc. Natl. Acad. Sci. U.S.A. 2015;112:713–718. doi: 10.1073/pnas.1408869112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Endo M., Yamamoto S., Emura T., Hidaka K., Morone N., Heuser J.E., Sugiyama H. Helical DNA origami tubular structures with various sizes and arrangements. Angew. Chem. Int. Ed. 2014;53:7484–7490. doi: 10.1002/anie.201402973. [DOI] [PubMed] [Google Scholar]
- 11.Fu J., Yang Y.R., Johnson-Buck A., Liu M., Liu Y., Walter N.G., Woodbury N.W., Yan H. Multi-enzyme complexes on DNA scaffolds capable of substrate channelling with an artificial swinging arm. Nat. Nano. 2014;9:531–536. doi: 10.1038/nnano.2014.100. [DOI] [PubMed] [Google Scholar]
- 12.Sun W., Boulais E., Hakobyan Y., Wang W.L., Guan A., Bathe M., Yin P. Casting inorganic structures with DNA molds. Science. 2014;346:1258361. doi: 10.1126/science.1258361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Helmi S., Ziegler C., Kauert D.J., Seidel R. Shape-controlled synthesis of gold nanostructures using DNA origami molds. Nano Lett. 2014;14:6693–6698. doi: 10.1021/nl503441v. [DOI] [PubMed] [Google Scholar]
- 14.Koirala D., Shrestha P., Emura T., Hidaka K., Mandal S., Endo M., Sugiyama H., Mao H. Single-molecule mechanochemical sensing using DNA origami nanostructures. Angew. Chem. Int. Ed. Engl. 2014;53:8137–8141. doi: 10.1002/anie.201404043. [DOI] [PubMed] [Google Scholar]
- 15.Goodman R.P., Heilemann M., Doose S., Erben C.M., Kapanidis A.N., Turberfield A.J. Reconfigurable, braced, three-dimensional DNA nanostructures. Nat. Nano. 2008;3:93–96. doi: 10.1038/nnano.2008.3. [DOI] [PubMed] [Google Scholar]
- 16.Pei H., Liang L., Yao G., Li J., Huang Q., Fan C. Reconfigurable three-dimensional DNA nanostructures for the construction of intracellular logic sensors. Angew. Chem. Int. Ed. 2012;124:9154–9158. doi: 10.1002/anie.201202356. [DOI] [PubMed] [Google Scholar]
- 17.Wen Y., Liu G., Pei H., Li L., Xu Q., Liang W., Li Y., Xu L., Ren S., Fan C. DNA nanostructure-based ultrasensitive electrochemical microRNA biosensor. Methods. 2013;64:276–282. doi: 10.1016/j.ymeth.2013.07.035. [DOI] [PubMed] [Google Scholar]
- 18.Ge Z., Lin M., Wang P., Pei H., Yan J., Shi J., Huang Q., He D., Fan C., Zuo X. Hybridization chain reaction amplification of microRNA detection with a tetrahedral DNA nanostructure-based electrochemical biosensor. Z. Anal. Chem. 2014;86:2124–2130. doi: 10.1021/ac4037262. [DOI] [PubMed] [Google Scholar]
- 19.Zhou L., Marras A.E., Su H.-J., Castro C.E. DNA origami compliant nanostructures with tunable mechanical properties. ACS Nano. 2013;8:27–34. doi: 10.1021/nn405408g. [DOI] [PubMed] [Google Scholar]
- 20.Chen H., Weng T.-W., Riccitelli M.M., Cui Y., Irudayaraj J., Choi J.H. Understanding the mechanical properties of DNA origami tiles and controlling the kinetics of their folding and unfolding reconfiguration. J. Am. Chem. Soc. 2014;136:6995–7005. doi: 10.1021/ja500612d. [DOI] [PubMed] [Google Scholar]
- 21.Kauert D.J., Kurth T., Liedl T., Seidel R. Direct mechanical measurements reveal the material properties of three-dimensional DNA origami. Nano Lett. 2011;11:5558–5563. doi: 10.1021/nl203503s. [DOI] [PubMed] [Google Scholar]
- 22.Pfitzner E., Wachauf C., Kilchherr F., Pelz B., Shih W.M., Rief M., Dietz H. Rigid DNA beams for high-resolution single-molecule mechanics. Angew. Chem. Int. Ed. 2013;52:7766–7771. doi: 10.1002/anie.201302727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhipeng M., Young-Joo K., Seongsu P., Hirai Y., Tsuchiya T., Kim D.N., Tabata O. Nano/Micro Engineered and Molecular Systems (NEMS) IEEE. 2015:581–584. [Google Scholar]
- 24.Hohng S., Zhou R., Nahas M.K., Yu J., Schulten K., Lilley D.M.J., Ha T. Fluorescence-force spectroscopy maps two-dimensional reaction landscape of the Holliday junction. Science. 2007;318:279–283. doi: 10.1126/science.1146113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Caruso M.M., Davis D.A., Shen Q., Odom S.A., Sottos N.R., White S.R., Moore J.S. Mechanically-induced chemical changes in polymeric materials. Chem. Rev. 2009;109:5755–5798. doi: 10.1021/cr9001353. [DOI] [PubMed] [Google Scholar]
- 26.Mandal S., Koirala D., Selvam S., Ghimire C., Mao H. A molecular tuning fork in single-molecule mechanochemical sensing. Angew. Chem. Int. Ed. 2015;54:7607–7611. doi: 10.1002/anie.201502580. [DOI] [PubMed] [Google Scholar]
- 27.Moffitt J.R., Chemla Y.R., Smith S.B., Bustamante C. Recent advances in optical tweezers. Annu. Rev. Biochem. 2008;77:205–228. doi: 10.1146/annurev.biochem.77.043007.090225. [DOI] [PubMed] [Google Scholar]
- 28.Wang M.D., Yin H., Landick R., Gelles J., Block S.M. Stretching DNA with optical tweezers. Biophys. J. 1997;72:1335–1346. doi: 10.1016/S0006-3495(97)78780-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mehta A.D., Rief M., Spudich J.A., Smith D.A., Simmons R.M. Single-molecule biomechanics with optical methods. Science. 1999;283:1689–1695. doi: 10.1126/science.283.5408.1689. [DOI] [PubMed] [Google Scholar]
- 30.Luchette P., Abiy N., Mao H. Microanalysis of clouding process at the single droplet level. Sens. Actuators B: Chem. 2007;128:154–160. [Google Scholar]
- 31.Mao H., Luchette P. An integrated laser-tweezers instrument for microanalysis of individual protein aggregates. Sens. Actuators B. 2008;129:764–771. [Google Scholar]
- 32.Morfill J., Kühner F., Blank K., Lugmaier R.A., Sedlmair J., Gaub H.E. B-S transition in short oligonucleotides. Biophys. J. 2007;93:2400–2409. doi: 10.1529/biophysj.107.106112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abraham Punnoose J., Cui Y., Koirala D., Yangyuoru P.M., Ghimire C., Shrestha P., Mao H. Interaction of G-quadruplexes in the full-length 3′ human telomeric overhang. J. Am. Chem. Soc. 2014;136:18062–18069. doi: 10.1021/ja510079u. [DOI] [PubMed] [Google Scholar]
- 34.Zhao Y., Karypis G., Fayyad U. Hierarchical clustering algorithms for document datasets. Data Min. Knowl. Disc. 2005;10:141–168. [Google Scholar]
- 35.Williams M.C., Rouzina I., Bloomfield V.A. Thermodynamics of DNA interactions from single molecule stretching experiments. Accounts Chem. Res. 2002;35:159–166. doi: 10.1021/ar010045k. [DOI] [PubMed] [Google Scholar]
- 36.Amit R., Gileadi O., Stavans J. Direct observation of RuvAB-catalyzed branch migration of single Holliday junctions. Proc. Natl. Acad. Sci. U.S.A. 2004;101:11605–11610. doi: 10.1073/pnas.0404332101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koirala D., Yu Z., Dhakal S., Mao H. Detection of single nucleotide polymorphism using tension-dependent stochastic behavior of a single-molecule template. J. Am. Chem. Soc. 2011;133:9988–9991. doi: 10.1021/ja201976r. [DOI] [PubMed] [Google Scholar]
- 38.May P.A., Moore J.S. Polymer mechanochemistry: techniques to generate molecular force via elongational flows. Chem. Soc. Rev. 2013;42:7497–7506. doi: 10.1039/c2cs35463b. [DOI] [PubMed] [Google Scholar]
- 39.Brantley J.N., Wiggins K.M., Bielawski C.W. Polymer mechanochemistry: the design and study of mechanophores. Polym. Int. 2013;62:2–12. [Google Scholar]
- 40.Bae W., Kim K., Min D., Ryu J.-K., Hyeon C., Yoon T.-Y. Programmed folding of DNA origami structures through single-molecule force control. Nat. Commun. 2014;5:5654. doi: 10.1038/ncomms6654. [DOI] [PubMed] [Google Scholar]
- 41.Bockelmann U., Thomen P., Essevaz-Roulet B., Viasnoff V., Heslot F. Unzipping DNA with optical tweezers: high sequence sensitivity and force flips. Biophys. J. 2002;82:1537–1553. doi: 10.1016/S0006-3495(02)75506-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu Z., Gaerig V., Cui Y., Kang H., Gokhale V., Zhao Y., Hurley L.H., Mao H. Tertiary DNA structure in the single-stranded hTERT promoter fragment unfolds and refolds by parallel pathways via cooperative or sequential events. J. Am. Chem. Soc. 2012;134:5157–5164. doi: 10.1021/ja210399h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu Z., Mao H. Non-B DNA structures show diverse conformations and complex transition kinetics comparable to RNA or proteins ― a perspective from mechanical unfolding and refolding experiments. Chem. Rec. 2013;13:102–116. doi: 10.1002/tcr.201200021. [DOI] [PubMed] [Google Scholar]
- 44.Woodside M.T., Anthony P.C., Behnke-Parks W.M., Larizadeh K., Herschlag D., Block S.M. Direct measurement of the full, sequence-dependent folding landscape of a nucleic acid. Science. 2006;314:1001–1004. doi: 10.1126/science.1133601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mathieu F., Liao S., Kopatsch J., Wang T., Mao C., Seeman N.C. Six-helix bundles designed from DNA. Nano Lett. 2005;5:661–665. doi: 10.1021/nl050084f. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.