

Abstract
Interaction between tumor cells and stromal cells plays an important role in the growth and metastasis of colon cancer. We previously found that carcinoma‐associated fibroblasts (CAFs) expressed platelet‐derived growth factor receptor‐β (PDGFR‐β) and that PDGFR targeted therapy using imatinib or nilotinib inhibited stromal reaction. Bone marrow‐derived mesenchymal stem cells (MSCs) migrate to tumor stroma and differentiate into CAFs. A novel oral multikinase inhibitor regorafenib inhibits receptor tyrosine kinases expressed on stromal cells (vascular endothelial growth factor receptor 1–3, TIE2, PDGFR‐β, and fibroblast growth factors) and tumor cells (c‐KIT, RET, and BRAF). These molecules are involved in tumor growth, angiogenesis, lymphangiogenesis, and stromal activation. Therefore, we examined whether regorafenib impaired the tumor‐promoting effect of CAFs/MSCs. KM12SM human colon cancer cells alone or KM12SM cells with MSCs were transplanted into the cecal wall of nude mice. Co‐implantation of KM12SM cells with MSCs into the cecal wall of nude mice produced tumors with abundant stromal component and promoted tumor growth and lymph node metastasis. Single treatment with regorafenib inhibited tumor growth and metastasis by inhibiting both tumor cells and stromal reaction. This tumor‐inhibitory effect of regorafenib was more obvious in tumors developed by co‐implanting KM12SM cells with MSCs. Our data suggested that targeting of the tumor microenvironment with regorafenib affected tumor cell–MSC interaction, which in turn inhibited the growth and metastasis of colon cancer.
Keywords: Colon cancer, mesenchymal stem cells, molecular target therapy, regorafenib, stroma
Colorectal cancer is the third most common cancer and a major cause of mortality worldwide.1 Recent improvements in surgery and chemotherapy have increased the survival of patients with CRC.2, 3 The overall 5‐year survival rate of patients with CRC is 64.3%; however, the 5‐year stage‐specific survival rate of patients with stage IV CRC is only 11.7%.4 Therefore, new strategies for treating CRC are urgently needed. Many studies have indicated that tumor growth and metastasis are determined by both tumor cells and stromal cells. Stroma constitutes a large part of most solid tumors, and tumor–stromal cell interaction contributes to tumor growth and metastasis.5, 6 Recent studies have shown that CAFs, which are the major components of tumor stroma, stimulate tumor progression by promoting tumor cell proliferation and invasion.7, 8, 9 We previously reported that the PDGF/PDGFR signaling pathway plays a critical role in the growth and metastasis of gastric and colon cancers in orthotopic transplantation models. Platelet‐derived growth factor‐B is expressed by cancer cells, whereas PDGFR‐β is mainly expressed by CAFs. Blockade of PDGFR signaling decreases stromal reaction, areas of vascular and lymphatic vessels, and number of pericytes.8, 9
Tumor stroma contains CAFs, smooth muscle cells, endothelial cells, pericytes, inflammatory cells, and ECM. Normal fibroblasts inhibit tumor progression, whereas CAFs promote tumor growth.10 Tumor stroma contains many growth factors, angiogenic factors, cytokines, and matrix‐remodeling proteins.11
Bone marrow‐derived MSCs are recruited from the bone marrow to inflamed or damaged tissues by local endocrine signals to induce tissue regeneration.12, 13 Mesenchymal stem cells have a strong capacity to migrate to various tumors14, 15, 16, 17, 18 and to promote tumor proliferation and metastasis.19 In addition, MSCs are involved in tumor angiogenesis and lymphangiogenesis,16, 20, 21, 22, 23 inhibition of apoptosis,24 and formation of a cancer stem cell niche.19, 25 We previously reported that MSCs migrate to tumor stroma and differentiate into CAFs.26 Treatment with PDGFR tyrosine kinase inhibitor imatinib impairs the migration of MSCs to tumor stroma, decreases the number of MSCs in the tumor microenvironment, and inhibits the progression of colon cancer in orthotopic transplantation models.27
Chemotherapy of CRC commonly includes combination regimens of 5‐fluorouracil, folinic acid, and oxaliplatin (FOLFOX) or irinotecan (FOLFIRI).28, 29 Recent studies have reported a combination of targeted therapies such as bevacizumab (anti‐VEGF antibody), cetuximab, and panitumumab (anti‐epidermal growth factor receptor antibody) with FOLFOX or FOLFIRI as the first‐line treatment for patients with CRC.28, 29, 30, 31 Regorafenib is a novel oral multikinase inhibitor that was recently approved by the FDA for treating metastatic CRC, which was previously treated using standard therapies, based on the results of a phase III study (CORRECT).32 Regorafenib inhibits various kinases such as c‐KIT, RET, BRAF, VEGFR1–3, TIE2, PDGFR‐β, and fibroblast growth factor receptor 1.33, 34, 35, 36 Although BRAF is involved in the RAS/RAF signaling pathway, it is unclear whether the mutation of KRAS/BRAF genes in tumor cells is associated with the effect of regorafenib. Vascular endothelial growth factor receptor 1–3, TIE2, and PDGFR‐β are mainly expressed by stromal cells; however, few studies have focused on the effect of regorafenib on tumor stroma.37 We examined the effect of regorafenib on several colorectal cell lines with diverse RAS/RAF gene mutation statuses. In addition, we analyzed molecular mechanisms underlying the inhibitory effect of regorafenib by co‐implanting colon cancer cells with MSCs to develop highly metastatic mixed‐cell tumors having abundant stroma. We found that regorafenib treatment inhibited tumor growth and lymph node metastasis, interrupted tumor cell–MSC interaction, and modified tumor‐supporting stroma.
Materials and Methods
Cell culture
Human MSCs were isolated from the iliac crest and were cultured according to a protocol described previously26, 38 and approved by the Ethics Committee of Hiroshima University Graduate School of Medicine (Hiroshima, Japan). Human colon cancer cell line KM12SM was kindly gifted by Dr. Isaiah J. Fidler (University of Texas, Austin, TX, USA).39 KM12SM cells did not have mutations in KRAS codons 12 and 13 or the BRAF V600E mutation. Mutations in KM12SM cells were determined by Sanger sequencing of KRAS exons 2 and 3 and BRAF exon 15, according to standard protocols. Human colon cancer cell lines Caco‐2, DLD‐1, LoVo, SW480, WiDr, HT‐29, and RKO were obtained from the Health Science Research Resources Bank (Osaka, Japan). Caco‐2 cells do not have mutations in KRAS or BRAF; DLD‐1 cells have a KRAS G13D mutation, LoVo cells have KRAS G13D and A14V mutations, SW480 cells have a KRAS G12V mutation, and WiDr, HT‐29, and RKO cells have a BRAF V600E mutation. Mutational status of key oncogenic CRC driver genes was obtained from the COSMIC database (http://www.sanger.ac.uk/genetics/CGP/cosmic/) of the Wellcome Trust Sanger Institute. All the cell lines were maintained in DMEM supplemented with 10% FBS and 1% penicillin–streptomycin. The cultures were maintained for not more than 12 weeks after recovery of the cells from frozen stocks.
Reagents
Regorafenib was kindly provided by Bayer Healthcare Pharmaceuticals (Leverkusen, Germany). The following primary antibodies were used: rat anti‐mouse CD31 antibody (BD Pharmingen, BD Biosciences, San Diego, CA, USA), monoclonal rat anti‐mouse LYVE‐1 antibody (R&D Systems, Minneapolis, MN, USA), rabbit anti‐αSMA antibody (Abcam, Cambridge, UK), Ki‐67 equivalent antibody (Novocastra; Leica Microsystems, Newcastle‐upon‐Tyne, UK), polyclonal rabbit anti‐mouse type I collagen antibody (Novotec, Saint Martin La Garenne, France), anti‐p44/42MAPK (anti‐ERK1/2) rabbit mAb (Cell Signaling Technology, Danvers, MA, USA), and anti‐phosphorylated p44/42MAPK (anti‐pERK1/2) rabbit mAb (Cell Signaling Technology).
Cell proliferation assay
The different colon cancer cell lines (cell density, 6 × 104 cells per well for all the cell lines) were seeded into 24‐well plates (Essen ImageLock; Essen Bioscience, Ann Arbor, MI, USA) containing DMEM supplemented with 10% FBS. The cells were treated with various concentrations of regorafenib (including 5 μM concentration, which is equivalent to the steady‐state plasma concentration of clinically effective doses of regorafenib).34, 35 Growth curves were generated from a bright field image obtained using a label‐free, high‐content time‐lapse assay system (Incucyte Zoom; Essen Bioscience) that automatically expresses cell confluence as a percentage over a 5‐day period. All experiments were carried out in triplicate.
Cell migration assay
Cell migration was assessed by scratch wound assay. Colon cancer cells (density, 1 × 105 cells per well) were seeded in 100 μg/L Matrigel‐coated (BD Biosciences, Bedford, MA, USA) 96‐well plates (Essen ImageLock) containing DMEM supplemented with 10% FBS. Use of ImageLock 96‐well plates allows the images of wounds to be taken automatically at the exact location by the Incucyte software. Confluent cell layers were scratched using a 96‐pin wound maker provided with Incucyte.40 After inducing the wound, the cells were washed twice with PBS to remove detached cells and were stimulated in the presence or absence of various doses of regorafenib. ImageLock 96‐well plates were then placed into Incucyte (Essen Bioscience), and wound images were acquired automatically every 2 h over a 5‐day period. Relative wound density was analyzed automatically by the Incucyte software. All experiments were carried out in triplicate.
Animals and transplantation of tumor cells
Female athymic BALB/c nude mice were obtained from Charles River Japan (Tokyo, Japan). The mice were maintained under specific pathogen‐free conditions and were used at 8 weeks of age. The study was carried out after obtaining the approval of the Committee on Animal Experimentation of Hiroshima University.
Cecal tumors were produced by injecting KM12SM cells in 50 μL Hanks’ balanced salt solution into the cecal wall of nude mice under a zoom stereomicroscope (Carl Zeiss, Gottingen, Germany).
Effect of regorafenib on tumor cell–MSC interaction in orthotopic colon tumors
Co‐implantation studies were carried out to examine the effect of regorafenib on tumor–MSC interaction in orthotopic colon tumors. Tumor xenograft models were generated by implanting KM12SM cells (1 × 106; KM12SM tumors) and KM12SM cells with MSCs (1:1 ratio; mixed‐cell tumors) into the cecal wall of nude mice. Next, these mice were divided into the following four groups: (i) mice with KM12SM tumors that were treated daily with water through oral gavage (1 × 106, n = 6); (ii) mice with mixed‐cell tumors that were treated daily with water through oral gavage (n = 10); (iii) mice with KM12SM tumors that were treated daily with 10 mg/kg regorafenib through oral gavage (n = 6); and (iv) mice with mixed‐cell tumors that were treated daily with 10 mg/kg regorafenib through oral gavage (n = 10). Regorafenib treatment was initiated 1 week after the intracecal transplantation of cells and was continued for 4 weeks.
Western blot analysis
Tumors in the cecal wall of nude mice were resected and homogenized in lysis buffer. Protein content of the tumor samples was quantified spectrophotometrically, as described previously.41
Necropsy and histological studies
Mice bearing orthotopic tumors were killed using diethyl ether, and their body weights were recorded. After necropsy, their tumors were excised and weighed. For immunohistochemical analysis, one part of the tumor tissue was fixed in formalin‐free immunohistochemistry zinc fixative (BD Pharmingen; BD Biosciences) and was embedded in paraffin. The other part of the tumor tissue was embedded in Tissue‐Tek OCT compound (Sakura Finetek, Torrance, CA, USA), rapidly frozen in liquid nitrogen, and stored at −80°C. All macroscopically enlarged regional (celiac and para‐aortal) lymph nodes were harvested, and presence of tumor metastasis was confirmed by histological analysis. Tumor volume was calculated using the following formula: V = 1/2 (length × width2).
Immunohistochemical staining
Formalin‐fixed, paraffin‐embedded tissues sliced into serial 4‐mm sections were used for immunohistochemical analyses of α‐SMA, CD31, LYVE‐1, Ki‐67, ERK1/2, and pERK1/2, as described previously.9 Apoptotic cells in the tissue sections were detected by TUNEL assay with ApopTag Plus Peroxidase in situ apoptosis detection kit (Millipore–Chemicon International, Temecula, CA, USA), according to the manufacturer's instructions.
Immunofluorescence staining
Frozen tissues were cut into 8‐μm sections for immunofluorescent analysis of CD31, LYVE‐1, Ki‐67, and apoptotic cells as described previously.26 Confocal fluorescence images were captured with a 20× or 40× objective lens on a Zeiss LSM 510 laser scanning microscopy system (Carl Zeiss Inc., Thornwood, NY, USA) equipped with a motorized Axioplan microscope, argon laser (458/477/488/514 nm, 30 mW), HeNe laser (543 nm, 1 mW), HeNe laser (633 nm, 5 mW), LSM 510 control and image acquisition software, and appropriate filters (Chroma Technology Corp., Brattleboro, VT, USA). Confocal images were exported to Adobe Photoshop software (Adobe, San Jose, CA, USA), and image montages were prepared.
Quantification of microvessel, CAF, and lymphatic vessel areas
Microvessel (CD31‐positive staining) and lymphatic vessel (LYVE‐1‐positive staining) areas were quantified to evaluate the angiogenic and lymphangiogenic activity of tumors. For this, 10 random fields at ×200 magnification were captured for each tumor, and vessels along with their lumen were counted manually. The CAF area (α‐SMA‐positive staining) was also determined from 10 optical fields (×200 magnification) of different sections. The areas were calculated using ImageJ software version 1.48v (NIH, Bethesda, MD, USA).
Determination of Ki‐67 labeling index and apoptotic index
Both Ki‐67 LI and AI were determined using the paraffin sections of orthotopic tumors, as described previously.42
Colocalization of TUNEL‐ and Ki67‐positive cells with CD31 and LYVE‐1
To assess the effect of MSCs and regorafenib on tumor microvessels and lymphatic vessels, fluorescent double‐staining for CD31/Ki67, CD31/TUNEL, LYVE‐1/Ki67, and LYVE‐1/TUNEL was carried out as previously described.8 Ki‐67‐positive vascular endothelial cells (CD31‐positive), TUNEL‐positive vascular endothelial cells (CD31‐positive), Ki‐67‐positive lymphatic endothelial cells (LYVE1‐positive), and TUNEL‐positive lymphatic endothelial cells (LYVE‐1‐positive) were quantified to evaluate the angiogenic and lymphangiogenic activity of tumors. For this, 10 optical fields at ×400 magnification were captured for each tumor. The ratios of proliferating and apoptotic cells to vascular and lymphatic endothelial cells, respectively, were expressed as percentages.
Statistical analysis
Between‐group differences in tumor volume and areas of α‐SMA‐positive, CD31‐positive, and LYVE‐1‐positive cells were analyzed using the Wilcoxon/Kruskal–Wallis test. Differences in the incidence of lymph node metastasis were analyzed using Fisher's exact test. Differences in the percentages of Ki‐67‐ and TUNEL‐positive cells were analyzed using unpaired Student's t‐test or Pearson's chi‐square‐test, as appropriate. The two‐sided t‐test was used to determine statistical differences between treatment and control groups with respect to the growth curves and scratch wound confluence analyzed by Incucyte at the end of the treatment period, as previously described.43 P‐values of <0.05 were considered statistically significant. All data are expressed as mean ± SE.
Results
Inhibitory effects of regorafenib on the proliferation and migration of colon cancer cells in vitro
We evaluated the effects of regorafenib on the proliferation and migration of colon cancer cells in vitro and correlated these effects with the mutation status of KRAS and BRAF. Proliferation and migration of colon cancer cells were analyzed in the presence or absence of regorafenib (2.5, 5, 10, and 20 μM) in real‐time by using Incucyte Zoom. Compared with control cells, proliferation and migration of all the tumor cell lines were inhibited by clinically effective concentrations of regorafenib (5–8 μM)33, 34, 44 irrespective of their KRAS and BRAF mutation status (Fig. 1).
Figure 1.
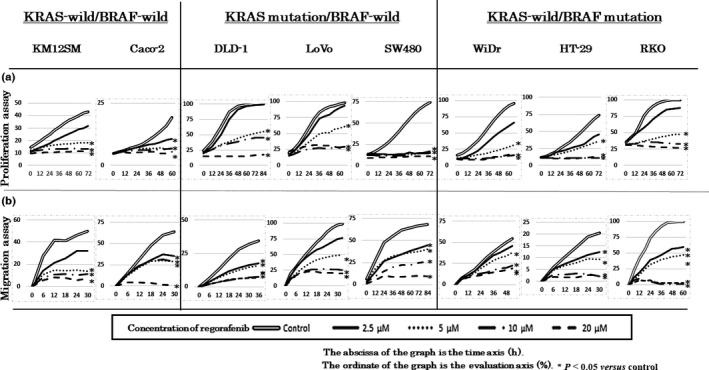
In vitro cell proliferation and migration assay. Growth (a) and migration (b) of tumor cells in the presence or absence of regorafenib was measured using Incucyte Zoom, which monitors cell confluency in real time. Regorafenib inhibited tumor proliferation and migration irrespective of KRAS and BRAF mutation status. *P < 0.05 versus control.
Production of rapidly growing and highly metastatic tumors by co‐implanting colon cancer cells with MSCs
We previously reported that co‐implantation of tumor cells with MSCs stimulated tumor growth and metastasis.26 To confirm this, we implanted KM12SM cells alone or along with MSCs into the cecal wall of the nude mice. Tumor volume and incidence of lymph node metastasis significantly increased in mice bearing mixed‐cell tumors compared with that in mice bearing KM12SM tumors (Table 1). Effects of MSCs on cell proliferation, stromal volume, angiogenesis, and lymphangiogenesis were evaluated using paraffinized tumor sections and proteins present in orthotopic implanted tumors. Ki‐67 LI, AI, α‐SMA expression, and lymphatic vessel and microvessel areas were significantly higher in mice bearing mixed‐cell tumors than in mice bearing KM12SM tumors (Table 1). Similarly, Western blot analysis showed that expression of collagen type I and α‐SMA was significantly higher in mice bearing mixed‐cell tumors than in mice bearing KM12SM tumors (Fig. 2a). Co‐implantation of KM12SM cells with MSCs produced tumors having abundant stromal component, which was consistent with our previous data.26
Table 1.
Effect of mesenchymal stem cells on tumor growth, stromal volume, angiogenesis, and lymphangiogenesis
| KM12SM mice | Mixed‐cell mice | |
|---|---|---|
| Tumor volume, mm3 | 144 ± 21 | 745 ± 135a |
| Lymph node metastasis | 0/6 | 9/10a |
| Ki67 labeling index | 10.2 ± 1.6 | 57.6 ± 2.4a |
| Apoptosis index | 2.88 ± 0.47 | 0.48 ± 0.19a |
| Area of α‐SMA‐positive staining, ×103 μm2 | 5.4 ± 1.1 | 15.8 ± 1.2a |
| Micro vessel area, ×103 μm2 | 44.6 ± 3.6 | 85.0 ± 3.0a |
| Lymphatic vessel area, ×103 μm2 | 3.6 ± 0.5 | 28.2 ± 2.3a |
P < 0.05 versus control group values. Α‐SMA, α‐smooth muscle actin.
Figure 2.
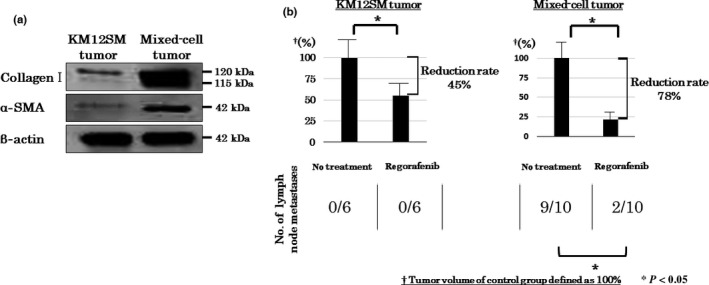
(a) Effect of mesenchymal stem cells on tumor stromal reaction. Expression of α‐smooth muscle actin (α‐SMA) and collagen I was higher in mixed‐cell tumors than in KM12SM tumors. (b) Effect of regorafenib on tumor growth and lymph node metastasis in orthotopic colon tumor models. Regorafenib inhibited the growth of both KM12SM and mixed‐cell tumors. Average tumor volume of mice not treated with regorafenib was set as 100% for both the tumor types. The rate of tumor reduction after regorafenib treatment was significantly higher (78% vs 45%; P < 0.05) in mixed‐cell tumors than in KM12SM tumors. Data are expressed as mean ± SE; *P < 0.05.
Effects of regorafenib on tumor growth and lymph node metastasis in the orthotopic colon tumor models
Regorafenib treatment significantly inhibited the growth of KM12SM and mixed‐cell tumors. However, the rate of reduction in tumor volume was significantly higher in mice bearing mixed‐cell tumors than in mice bearing KM12SM tumors (Fig. 2b). Co‐implantation of KM12SM cells with MSCs increased the potential of KM12SM tumors to metastasize toward lymph nodes. However, regorafenib treatment decreased the incidence of lymph node metastasis in mice bearing mixed‐cell tumors (Fig. 2b).
Effects of regorafenib on molecules involved in tumor signaling pathways in orthotopic colon tumor models
To assess the effects of regorafenib on molecules involved in tumor signaling pathways, we confirmed the expression of ERK1/2 and pERK1/2, which are downstream effecters of the RAS/RAF signaling pathway, by carrying out immunohistochemical and Western blot analyses. Regorafenib treatment did not influence the expression of ERK1/2, but inhibited the expression of pERK1/2 in both tumor types (Figs 3a,b,4). These results indicated that regorafenib treatment inhibited the RAS/RAF/ERK signaling pathway in KM12SM and mixed‐cell tumors.
Figure 3.
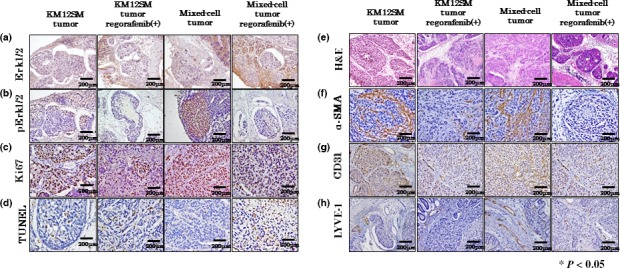
Effect of regorafenib on KM12SM and mixed‐cell tumors. Expression of ERK (a) and pERK (b), analysis of cell proliferation (Ki‐67 labeling index) (c), apoptosis index (TUNEL assay) (d), H&E staining (e), expression of α‐smooth muscle actin (α‐SMA) (carcinoma‐associated fibroblasts) (f), angiogenesis (CD31 staining) (g), and lymphangiogenesis (LYVE‐1 staining) (h). Scale bar = 200 μm.
Figure 4.
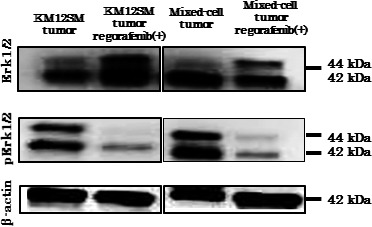
Western blot analysis showed that regorafenib inhibited the phosphorylation of ERK1/2.
Effects of regorafenib on tumor proliferation and apoptosis revealed by immunohistochemical analysis
To determine the mechanisms underlying the tumor‐inhibitory effect of regorafenib, we examined tumor proliferation (Ki‐67 LI) and apoptosis (AI) in xenografted mice using immunohistochemical analysis. Ki‐67 LI was significantly decreased after regorafenib treatment in mixed‐cell tumors but not in KM12SM tumors (Figs 3c,5a). The AI was significantly increased after regorafenib treatment in both KM12SM and mixed‐cell tumors (Figs 3d,5b).
Figure 5.
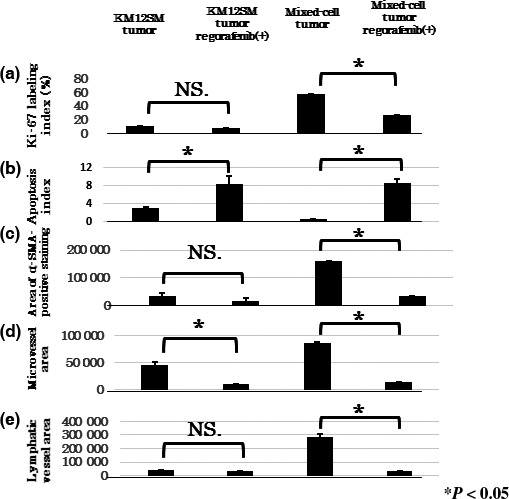
Quantification of the effects of regorafenib on KM12SM and mixed‐cell tumors. Regorafenib decreased the Ki‐67 labeling index (a), α‐smooth muscle actin (α‐SMA)‐positive area (c), and lymphatic vessel area (e) in mixed‐cell tumors. Furthermore, regorafenib increased the apoptosis index (b) and decreased microvessel area (d) in KM12SM tumors. Data are expressed as mean ± SE; *P < 0.05. NS., not significant.
Effects of regorafenib on stromal reaction, tumor angiogenesis, and lymphangiogenesis
We evaluated the effects of regorafenib treatment on stromal reaction, tumor angiogenesis, and lymphangiogenesis by immunohistochemical analysis. Angiogenesis and lymphangiogenesis were evaluated by determining the areas of CD31‐ and LYVE‐1‐positive staining, respectively. Stromal reaction was evaluated by determining the area of α‐SMA‐positive staining, which is a marker of CAFs. Regorafenib treatment significantly decreased the areas of α‐SMA‐positive staining and lymphatic vessels in mixed‐cell tumors but not in KM12SM tumors (Figs 3f,h,5c,e). Furthermore, regorafenib treatment significantly decreased the microvessel area in both the tumor types, especially in mixed‐cell tumors (Figs 3g,5d).
As shown in Figure 6, proliferating and apoptotic endothelial cells were analyzed by double fluorescence for CD31/TUNEL (Fig. 6a,e), LYVE‐1/TUNEL (Fig. 6b,f), CD31/KI‐67 (Fig. 6c,g), and LYVE‐1/TUNEL (Fig. 6d,h). Regorafenib treatment decreased the number of proliferating endothelial cells and increased the number of apoptotic endothelial cells (Fig. 6e–h).
Figure 6.
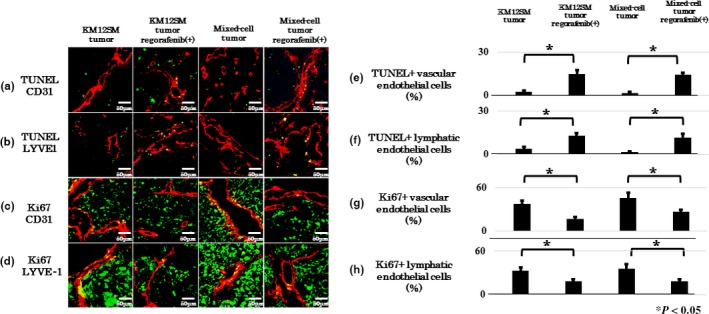
Effect of regorafenib on vascular and lymphatic endothelial cells in KM12SM and mixed‐cell tumors. Expression of CD31 and TUNEL in apoptotic vascular endothelial cells (a), LYVE‐1 and TUNEL in apoptotic lymphatic endothelial cells (b), CD31 and Ki‐67 in proliferating vascular endothelial cells (c), and LYVE‐1 and KI‐67 in proliferating lymphatic endothelial cells (d). Proliferating (Ki67‐positive) and apoptotic (TUNEL‐positive) cells are depicted in green. Vascular and lymphatic endothelial cells are depicted in red. Scale bar = 50 μm. (c) Average ratio of apoptotic vascular endothelial cells to the total number of vascular endothelial cells. (d) Average ratio of apoptotic lymphatic endothelial cells to the total number of lymphatic endothelial cells. (e) Average ratio of proliferating vascular endothelial cells to the total number of vascular endothelial cells. (f) Average ratio of proliferating lymphatic endothelial cells to the total number of lymphatic endothelial cells. Data are expressed as mean ± SE; *P < 0.05.
Discussion
The tumor microenvironment influences tumor growth and invasion and thus tumor metastasis.45 We previously reported that MSCs migrate to tumor stroma and differentiate into CAFs.26 CXCL12 (stromal cell derived factor‐1)/CXCR4,46 CCL2 (MCP‐1)/CCR2,18 transforming growth factor‐β/receptors,47 and PDGF/PDGFR,17 are involved in the tumor‐homing activity of MSCs. We previously reported that co‐implantation of tumor cells with MSCs into orthotopic (cecum) sites produced highly metastatic and fast‐growing tumors having abundant active stroma. The PDGFR tyrosine kinase inhibitor imatinib inhibits stromal reaction and metastasis.27 Target molecules of regorafenib are expressed by CAFs and pericytes (PDGFR), vascular endothelial cells (VEGFR2 and TIE2), lymphatic endothelial cells (PDGFR and VEGFR3), and tumor cells (BRAF). In this study, mixed‐cell tumors had higher Ki‐67 LI and lower AI than KM12SM tumors. Furthermore, mixed tumors had more active stroma than KM12SM tumors. Mesenchymal stem cells have been reported to express many anti‐apoptotic factors, such as VEGF, basic fibroblast growth factor, PDGF, stromal cell derived factor‐1α, IGF‐1,2, transforming growth factor‐β, and IGF binding protein‐2. 48 Therefore, MSCs may stimulate the biological aggressiveness of tumor cells and thus protect them. Mixed‐cell tumors showed highly metastatic properties, but mixed‐cell tumors were significantly larger than KM12SM tumors. Thus, in this study, it was unclear whether the highly metastatic properties were due to the significantly different tumor burden or different biological aggressiveness of the tumors. To further clarify this, a tumor size‐matched comparison of lymphatic metastasis frequency between KM12SM and mixed‐cell tumors is required, which may support a therapeutic strategy targeting tumor cells and MSCs.
Subsequently, we evaluated the effect of regorafenib on KM12SM and mixed‐cell tumors. Despite the presence of the same cell line, the inhibitory effect of regorafenib differed between KM12SM and KM12SM + MSC mixed‐cell tumors. Regorafenib treatment significantly decreased tumor cell growth, stromal reaction, angiogenesis, and lymphangiogenesis. In addition, regorafenib treatment completely inhibited the acquired ability of KM12SM cells co‐implanted with MSCs to metastasize to lymph nodes.
We also examined the inhibitory effect of regorafenib on the proliferation of several colon cancer cell lines, which show different abnormalities in the RAS/RAF/MAPK signaling pathway. We confirmed that regorafenib exerted inhibitory effects on all the cell lines irrespective of their KRAS and BRAF mutation status. The phase III COLLECT study showed that regorafenib exerted inhibitory effects on both wild‐type KRAS‐ and mutant KRAS‐containing CRCs.32 Results of biochemical assays have indicated that regorafenib treatment is effective irrespective of the presence of BRAF mutations.33, 49 However, the clinical effectiveness of RAF inhibitors such as vemurafenib and dabrafenib depends on BRAF mutation status. BRAF inhibitors suppress the growth of cells harboring BRAF mutations; however, these compounds paradoxically activate the RAS/RAF/ERK pathway in cells harboring oncogenic RAS or elevate upstream receptor signaling.50, 51 Therefore, it is unclear how KRAS and BRAF mutations are involved in the resistance of tumor cells to BRAF inhibitors. Regorafenib is a multikinase inhibitor. Therefore, the effect may depend on the inhibition effect not only against cancer cells by inhibiting the RAS/RAF/ERK signaling pathway but also against stroma and angiogenesis. However, the effect of regorafenib may be different depending on the mutation status of cancer cells, and the difference may be more apparent in vitro where stroma does not exist. We examined the tumor‐inhibitory effect of regorafenib in vitro by using cultured cell lines with different mutations. We confirmed the inhibitory effect of regorafenib on tumor cells and examined whether this effect was correlated with the mutation status of the cells. We observed that regorafenib exerted the same effect on all the cell lines with different mutation statuses, which was consistent with that reported in a previous study.52 Some studies have found no correlation between regorafenib‐induced antiproliferative effects and mutation status of cells and have suggested that resistance of tumor cells to regorafenib depends on the expression of pERK1/2 before regorafenib treatment.52, 53 However, a study showed that downregulation of the AKT/mTOR/S6 ribosomal protein axis, which is not the primary target of regorafenib, after regorafenib treatment was associated with longer progression‐free survival and metabolic responses.54 This suggests that the inhibitory effect of regorafenib does not strongly depend on the mutation status of the RAS/RAF/ERK axis.
We previously reported that blockade of mTOR and PDGFR pathways by inhibitors everolimus and imatinib inhibited the progression of CRC because of the modulation of the tumor microenvironment.42 Everolimus inhibited tumor cell proliferation but not stromal reaction. In contrast, imatinib inhibited stromal reaction but not tumor cell proliferation. Therefore, a combination therapy with these drugs should be used to disrupt tumor cell–stroma interactions. Regorafenib inhibits both tumor cells and stromal reactions by sole administration; therefore, regorafenib treatment is highly effective against tumors having abundant active stroma. In conclusion, regorafenib targets tumor cells as well as stromal cells, including MSC‐derived CAFs. Histopathological evaluation of tumor stroma may be important for the prognosis of patients with CRC.
Disclosure Statement
The authors have no conflict of interest.
Abbreviations
- α‐SMA
α‐smooth muscle actin
- AI
apoptosis index
- BRAF
B‐Raf proto‐oncogene
- CAF
cancer‐associated fibroblast
- CRC
colorectal carcinoma
- IGF
insulin‐like growth factor
- Ki‐67 LI
Ki‐67 labeling index
- LYVE‐1
lymphatic vessel endothelial hyaluronan receptor 1
- MSC
mesenchymal stem cell
- mTOR
mammalian target of rapamycin
- PDGF
platelet‐derived growth factor
- PDGFR
platelet‐derived growth factor receptor
- VEGF
vascular endothelial growth factor
Acknowledgments
This work was carried out with the kind cooperation of the Analysis Center of Life Science and Institute of Laboratory Animal Science, Hiroshima University. In addition, we thank Bayer Healthcare Pharmaceuticals for providing regorafenib.
Cancer Sci 107 (2016) 601–608
Funding Information
Supported in part by grants‐in‐aid for cancer research from the Ministry of Education, Culture, Science, Sports and Technology of Japan. Grant Number: 21590813.
References
- 1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin 2011; 61: 69–90. [DOI] [PubMed] [Google Scholar]
- 2. Kapse N, Goh V. Functional imaging of colorectal cancer: positron emission tomography, magnetic resonance imaging, and computed tomography. Clin Colorectal Cancer 2009; 8: 77–87. [PubMed] [Google Scholar]
- 3. Majek O, Gondos A, Jansen L et al Survival from colorectal cancer in Germany in the early 21st century. Br J Cancer 2012; 106: 1875–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Siegel R, DeSantis C, Virgo K et al Cancer treatment and survivorship statistics, 2012. CA Cancer J Clin 2012; 62: 220–41. [DOI] [PubMed] [Google Scholar]
- 5. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer‐related inflammation. Nature 2008; 454: 436–44. [DOI] [PubMed] [Google Scholar]
- 6. Whiteside TL. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008; 27: 5904–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liotta LA, Kohn EC. The microenvironment of the tumour‐host interface. Nature 2001; 411: 375–9. [DOI] [PubMed] [Google Scholar]
- 8. Kitadai Y, Sasaki T, Kuwai T, Nakamura T, Bucana CD, Fidler IJ. Targeting the expression of platelet‐derived growth factor receptor by reactive stroma inhibits growth and metastasis of human colon carcinoma. Am J Pathol 2006; 169: 2054–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sumida T, Kitadai Y, Shinagawa K et al Anti‐stromal therapy with imatinib inhibits growth and metastasis of gastric carcinoma in an orthotopic nude mouse model. Int J Cancer 2011; 128: 2050–62. [DOI] [PubMed] [Google Scholar]
- 10. De Wever O, Mareel M. Role of tissue stroma in cancer cell invasion. J Pathol 2003; 200: 429–47. [DOI] [PubMed] [Google Scholar]
- 11. Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med 1986; 315: 1650–9. [DOI] [PubMed] [Google Scholar]
- 12. Pittenger MF, Mackay AM, Beck SC et al Multilineage potential of adult human mesenchymal stem cells. Science 1999; 284: 143–7. [DOI] [PubMed] [Google Scholar]
- 13. Gregory CA, Prockop DJ, Spees JL. Non‐hematopoietic bone marrow stem cells: molecular control of expansion and differentiation. Exp Cell Res 2005; 306: 330–5. [DOI] [PubMed] [Google Scholar]
- 14. Studeny M, Marini FC, Champlin RE, Zompetta C, Fidler IJ, Andreeff M. Bone marrow‐derived mesenchymal stem cells as vehicles for interferon‐beta delivery into tumors. Cancer Res 2002; 62: 3603–8. [PubMed] [Google Scholar]
- 15. Nakamizo A, Marini F, Amano T et al Human bone marrow‐derived mesenchymal stem cells in the treatment of gliomas. Cancer Res 2005; 65: 3307–18. [DOI] [PubMed] [Google Scholar]
- 16. Hung SC, Deng WP, Yang WK et al Mesenchymal stem cell targeting of microscopic tumors and tumor stroma development monitored by noninvasive in vivo positron emission tomography imaging. Clin Cancer Res 2005; 11: 7749–56. [DOI] [PubMed] [Google Scholar]
- 17. Beckermann BM, Kallifatidis G, Groth A et al VEGF expression by mesenchymal stem cells contributes to angiogenesis in pancreatic carcinoma. Br J Cancer 2008; 99: 622–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dwyer RM, Potter‐Beirne SM, Harrington KA et al Monocyte chemotactic protein‐1 secreted by primary breast tumors stimulates migration of mesenchymal stem cells. Clin Cancer Res 2007; 13: 5020–7. [DOI] [PubMed] [Google Scholar]
- 19. Quante M, Tu SP, Tomita H et al Bone marrow‐derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell 2011; 19: 257–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Annabi B, Naud E, Lee YT, Eliopoulos N, Galipeau J. Vascular progenitors derived from murine bone marrow stromal cells are regulated by fibroblast growth factor and are avidly recruited by vascularizing tumors. J Cell Biochem 2004; 91: 1146–58. [DOI] [PubMed] [Google Scholar]
- 21. Sun B, Zhang S, Ni C et al Correlation between melanoma angiogenesis and the mesenchymal stem cells and endothelial progenitor cells derived from bone marrow. Stem Cells Dev 2005; 14: 292–8. [DOI] [PubMed] [Google Scholar]
- 22. Zhu W, Xu W, Jiang R et al Mesenchymal stem cells derived from bone marrow favor tumor cell growth in vivo. Exp Mol Pathol 2006; 80: 267–74. [DOI] [PubMed] [Google Scholar]
- 23. Koyama H, Kobayashi N, Harada M et al Significance of tumor‐associated stroma in promotion of intratumoral lymphangiogenesis: pivotal role of a hyaluronan‐rich tumor microenvironment. Am J Pathol 2008; 172(1): 179–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ramasamy R, Lam EW, Soeiro I, Tisato V, Bonnet D, Dazzi F. Mesenchymal stem cells inhibit proliferation and apoptosis of tumor cells: impact on in vivo tumor growth. Leukemia 2007; 21: 304–10. [DOI] [PubMed] [Google Scholar]
- 25. Liu S, Ginestier C, Ou SJ et al Breast cancer stem cells are regulated by mesenchymal stem cells through cytokine networks. Cancer Res 2011; 71: 614–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shinagawa K, Kitadai Y, Tanaka M et al Mesenchymal stem cells enhance growth and metastasis of colon cancer. Int J Cancer 2010; 127: 2323–33. [DOI] [PubMed] [Google Scholar]
- 27. Shinagawa K, Kitadai Y, Tanaka M et al Stroma‐directed imatinib therapy impairs the tumor‐promoting effect of bone marrow‐derived mesenchymal stem cells in an orthotopic transplantation model of colon cancer. Int J Cancer 2013; 132: 813–23. [DOI] [PubMed] [Google Scholar]
- 28. Van Cutsem E, Cervantes A, Nordlinger B, Arnold D. Metastatic colorectal cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow‐up. Ann Oncol 2014; 25(Suppl 3): iii1–9. [DOI] [PubMed] [Google Scholar]
- 29. Benson AB III, Bekaii‐Saab T, Chan E et al Metastatic colon cancer, version 3.2013: featured updates to the NCCN guidelines. J Natl Compr Canc Netw 2013; 11: 141–52; quiz 52. [DOI] [PubMed] [Google Scholar]
- 30. Hurwitz H, Fehrenbacher L, Novotny W et al Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004; 350: 2335–42. [DOI] [PubMed] [Google Scholar]
- 31. Ocvirk J, Brodowicz T, Wrba F et al Cetuximab plus FOLFOX6 or FOLFIRI in metastatic colorectal cancer: CECOG trial. World J Gastroenterol 2010; 16: 3133–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Grothey A, Cutsem EV, Sobrero A et al Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo‐controlled, phase 3 trial. Lancet 2013; 381: 303–12. [DOI] [PubMed] [Google Scholar]
- 33. Wilhelm SM, Dumas J, Adnane L et al Regorafenib (BAY 73‐4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int J Cancer 2011; 129(1): 245–55. [DOI] [PubMed] [Google Scholar]
- 34. Mross K, Frost A, Steinbild S et al A phase I dose‐escalation study of regorafenib (BAY 73‐4506), an inhibitor of oncogenic, angiogenic, and stromal kinases, in patients with advanced solid tumors. Clin Cancer Res 2012; 18: 2658–67. [DOI] [PubMed] [Google Scholar]
- 35. Strumberg D, Scheulen ME, Schultheis B et al Regorafenib (BAY 73‐4506) in advanced colorectal cancer: a phase I study. Br J Cancer 2012; 106: 1722–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. George S, Wang Q, Heinrich MC et al Efficacy and safety of regorafenib in patients with metastatic and/or unresectable GI stromal tumor after failure of imatinib and sunitinib: a multicenter phase II trial. J Clin Oncol 2012; 30: 2401–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Abou‐Elkacem L, Arns S, Brix G et al Regorafenib inhibits growth, angiogenesis, and metastasis in a highly aggressive, orthotopic colon cancer model. Mol Cancer Ther 2013; 12: 1322–31. [DOI] [PubMed] [Google Scholar]
- 38. Ishii M, Koike C, Igarashi A et al Molecular markers distinguish bone marrow mesenchymal stem cells from fibroblasts. Biochem Biophys Res Commun 2005; 332(1): 297–303. [DOI] [PubMed] [Google Scholar]
- 39. Morikawa K, Walker SM, Nakajima M, Pathak S, Jessup JM, Fidler IJ. Influence of organ environment on the growth, selection, and metastasis of human colon carcinoma cells in nude mice. Cancer Res 1988; 48: 6863–71. [PubMed] [Google Scholar]
- 40. Liu L, Wang YD, Wu J, Cui J, Chen T. Carnitine palmitoyltransferase 1A (CPT1A): a transcriptional target of PAX3‐FKHR and mediates PAX3‐FKHR‐dependent motility in alveolar rhabdomyosarcoma cells. BMC Cancer 2012; 12: 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kim SJ, Uehara H, Karashima T, Shepherd DL, Killion JJ, Fidler IJ. Blockade of epidermal growth factor receptor signaling in tumor cells and tumor‐associated endothelial cells for therapy of androgen‐independent human prostate cancer growing in the bone of nude mice. Clin Cancer Res 2003; 9: 1200–10. [PubMed] [Google Scholar]
- 42. Yuge R, Kitadai Y, Shinagawa K et al mTOR and PDGF pathway blockade inhibits liver metastasis of colorectal cancer by modulating the tumor microenvironment. Am J Pathol 2015; 185: 399–408. [DOI] [PubMed] [Google Scholar]
- 43. Green JM, Alvero AB, Kohen F, Mor G. 7‐(O)‐Carboxymethyl daidzein conjugated to N‐t‐Boc‐hexylenediamine: a novel compound capable of inducing cell death in epithelial ovarian cancer stem cells. Cancer Biol Ther 2009; 8: 1747–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rey JB, Launay‐Vacher V, Tournigand C. Regorafenib as a single‐agent in the treatment of patients with gastrointestinal tumors: an overview for pharmacists. Target Oncol 2015; 10: 199–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fidler IJ. Critical factors in the biology of human cancer metastasis: twenty‐eighth G.H.A. Clowes memorial award lecture. Cancer Res 1990; 50: 6130–8. [PubMed] [Google Scholar]
- 46. Menon LG, Picinich S, Koneru R et al Differential gene expression associated with migration of mesenchymal stem cells to conditioned medium from tumor cells or bone marrow cells. Stem Cells 2007; 25: 520–8. [DOI] [PubMed] [Google Scholar]
- 47. Goldstein RH, Reagan MR, Anderson K, Kaplan DL, Rosenblatt M. Human bone marrow‐derived MSCs can home to orthotopic breast cancer tumors and promote bone metastasis. Cancer Res 2010; 70: 10044–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yang X, Hou J, Han Z et al One cell, multiple roles: contribution of mesenchymal stem cells to tumor development in tumor microenvironment. Cell Biosci 2013; 3(1): 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wilhelm SM, Carter C, Tang L et al BAY 43‐9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res 2004; 64: 7099–109. [DOI] [PubMed] [Google Scholar]
- 50. Hatzivassiliou G, Song K, Yen I et al RAF inhibitors prime wild‐type RAF to activate the MAPK pathway and enhance growth. Nature 2010; 464: 431–5. [DOI] [PubMed] [Google Scholar]
- 51. Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild‐type BRAF. Nature 2010; 464: 427–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schmieder R, Hoffmann J, Becker M et al Regorafenib (BAY 73‐4506): antitumor and antimetastatic activities in preclinical models of colorectal cancer. Int J Cancer 2014; 135: 1487–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Koyama M, Matsuzaki Y, Yogosawa S, Hitomi T, Kawanaka M, Sakai T. ZD1839 induces p15INK4b and causes G1 arrest by inhibiting the mitogen‐activated protein kinase/extracellular signal‐regulated kinase pathway. Mol Cancer Ther 2007; 6: 1579–87. [DOI] [PubMed] [Google Scholar]
- 54. Wong AL, Lim JS, Sinha A et al Tumour pharmacodynamics and circulating cell free DNA in patients with refractory colorectal carcinoma treated with regorafenib. J Transl Med 2015; 13(1): 57. [DOI] [PMC free article] [PubMed] [Google Scholar]