

Abstract
In the two decades since the discovery of the nociceptin opioid receptor (NOP) and its ligand, nociceptin/orphaninFQ (N/OFQ), steady progress has been achieved in understanding the pharmacology of this fourth opioid receptor/peptide system, aided by genetic and pharmacologic approaches. This research spawned an explosion of small-molecule NOP receptor ligands from discovery programs in major pharmaceutical companies. NOP agonists have been investigated for their efficacy in preclinical models of anxiety, cough, substance abuse, pain (spinal and peripheral) and urinary incontinence, whereas NOP antagonists have been investigated for treatment of pain, depression and motor symptoms in Parkinson’s disease. Translation of preclinical findings into the clinic is guided by PET and receptor occupancy studies, particularly for NOP antagonists. Recent progress in preclinical NOP research suggests that NOP agonists may have clinical utility for pain treatment and substance abuse pharmacotherapy. This review discusses the progress towards validating the NOP-N/OFQ system as a therapeutic target.
INTRODUCTION
The nociceptin opioid peptide receptor (NOP) was discovered in 1994, after cloning of the other three opioid receptors mu, delta and kappa (MOP, DOP and KOP in IUPHAR nomenclature).1-5 The characterization that rapidly followed clearly placed the NOP receptor (previously named the Opioid Receptor-like receptor-1, ORL-1) in the opioid receptor family of GPCRs, which couples to Gi/Go and inhibits adenylate cyclase activity. These properties informed the quest for an endogenous ligand for this ‘then orphan’ opioid receptor, and a year later, resulted in the successful identification, by two independent groups, of a heptadecapeptide from rat and porcine brains, which was named nociceptin and orphanin FQ, respectively.6,7 This neuropeptide, henceforth referred to as N/OFQ, has a high degree of similarity with the heptadecapeptide dynorphin, an endogenous neuropeptide that binds the kappa opioid receptor. It is therefore remarkable that N/OFQ has a 1000-fold lower affinity for the kappa opioid receptor than for NOP.8 Further, N/OFQ has no measurable affinity for the mu or delta opioid receptors. While both the NOP receptor and its endogenous ligand N/OFQ have structural and functional similarity to the other three opioid receptors and their endogenous ligands respectively, the NOP receptor does not bind classical opioid ligands, whereas the endogenous NOP ligand N/OFQ does not bind to the other opioid receptors, making the NOP–N/OFQ receptor-ligand system a class in itself, distinct from the opioid family in several important ways.
The NOP–N/OFQ Receptor–Ligand System
Over the two decades since its discovery, the study of NOP–N/OFQ pharmacology revealed similarities and differences of this system from the opioid receptor family, and continues to be actively pursued, in the hope that the NOP receptor can prove to be a valuable therapeutic target for pharmacotherapy of various human conditions, provided that suitable NOP receptor-targeted compounds can be developed. The importance of the NOP–N/OFQ system in physiological processes is suggested by its widespread distribution in the brain, spinal cord and peripheral organs. Under normal homeostatic conditions, the endogenous peptide N/OFQ functions primarily as an inhibitory neurotransmitter, acting via the NOP receptor, to suppress cellular neuronal activity and release of other neurotransmitters.9 This functional activity may provide a balance with other excitatory systems, to maintain a homeostatic state of a physiological process. At the molecular level, N/OFQ actions at the NOP receptor results in inhibition of adenylate cyclase, increase in K+ conductance (which leads to hyperpolarization in neuronal cells) and inhibition of Ca+2 conductance.10 However, dysregulation of the NOP-N/OFQ system has been shown for many pathophysiological states, justifying the interest in this system as a potential therapeutic target in these disease states. Examples of disease states where the NOP-N/OFQ system is upregulated is the development of chronic pain, Parkinson’s disease and depression. For instance, NOP receptor mRNA is upregulated in various regions of the brain and spinal cord in rats after induction of chronic constriction injury, a model of neuropathic pain.11-14 Interestingly, both NOP receptor protein and N/OFQ immunoreactivity were increased in rats with inflammatory pain induced by Complete Freund’s adjuvant (CFA) and intrathecal administration of N/OFQ decreased inflammatory pain markers.15, 16 The significance of the NOP receptor as a target for pain therapeutics is on the verge of clinical validation (as discussed below), but target validation thus far has been riddled with complexities arising from species differences and site-specific pharmacology. N/OFQ levels were found to be elevated in the cerebrospinal fluid of parkinsonian patients,17 providing the first clinical evidence implicating N/OFQ upregulation in the pathophysiology of Parkinson's disease in humans. These findings suggest a rationale for developing NOP receptor antagonists as antiparkinsonian drugs.18-20
Alternately, the NOP-N/OFQ system may be activated (using NOP agonists) to provide inhibitory control over physiological processes errantly upregulated during disease states. For example, most drugs of abuse (such as opioids, nicotine, cocaine, and even alcohol) cause an increase in dopamine levels in the nucleus accumbens, an area of the brain that is the nexus of the brain reward circuitry.21 Exogenous administration of N/OFQ suppresses basal and drug-induced dopamine release in the nucleus accumbens,22-25 a phenomenon known to decrease the rewarding effects of abused drugs.26-30 NOP agonists, therefore, would be potentially useful as medications for drug abuse.
The widespread distribution of the NOP receptor and N/OFQ throughout the central nervous system (CNS) and in many peripheral organs is now well documented from studies in rodents, nonhuman primates and humans. However, the involvement of the NOP system in disease states has been mainly obtained from rodent studies. Further, the majority of studies to understand the pharmacology of NOP receptor have been carried out in rodents. A caveat that is unfolding as the NOP receptor is being vetted as a human clinical target is that there are significant differences in the pharmacological effects of NOP ligands in rodents, compared to that in nonhuman primates and possibly humans, that need to be carefully considered and which impact the success of the preclinical advancement of suitable drug candidates and selection of clinical endpoints. Studies on localization of the NOP system in the rodent,31, 32 primate 33, 34 and human brain35, 36 suggests that there are important differences in the distribution and localization of the NOP-N/OFQ system between species, that may play a significant role in translating the validation of this system as a potential target for different pathophysiological states.
This perspective attempts to correlate and compare NOP pharmacology from rodent and primate studies to what is being revealed about NOP ligands in the clinic from PET and human occupancy studies, in order to highlight challenges and promises of this target for clinical translation. It is not the intent of this review to recapitulate the extensive literature on NOP pharmacology. For that the reader is referred to several excellent in-depth reviews published recently.37-40 Previous studies on NOP-N/OFQ localization in the rodent, primate and human brain can be correlated to understand the relevance and face validity of rodent or primate models used in preclinical development to inform efficacy in human clinical trials. This review highlights the issues and challenges revealed in the translation of preclinical rodent data to efficacy in human. Given that the NOP receptor is primarily a CNS target, the development of NOP-targeted drug candidates is also faced with the usual challenges of CNS drug development. The discussion of preclinical data presented here will hopefully provide a broad perspective on the issues encountered during development of NOP-targeted drug candidates and encourage new preclinical research to ease the challenges of translating preclinical findings into human therapeutic potential.
NOP Receptor-targeted Compounds in the Pipeline from Research to the Clinic
Fast forwarding to the present, the current state of progress in translation of NOP-N/OFQ research into clinical therapeutics development is, at the time of writing this review, limited to just one compound, a dual-targeted NOP agonist-MOP agonist Cebranopadol41 (1, GRT 6005, Table 1) from Grunenthal, for which Phase III clinical trials are currently ongoing, for treatment of various pain conditions.42, 43 Thus far, this is the most advanced into clinical development that any NOP-targeted compound has reached (Table 1). A NOP antagonist from Eli Lilly, 2 (LY2940094),44 is also in Phase II clinical trials for major depressive disorder45 and for alcohol dependence,46 but no further information is available regarding the outcome of these trials or future progress of this compound (Table 1). MK-5757 (3, Table 1), a NOP antagonist identified as a clinical candidate after extensive optimization by Banyu47, was in Phase II clinical trials, for the treatment of cognitive impairment in men with schizophrenia. Although the trial has been completed, no results have been yet disclosed.48
Table 1.
Status of NOP-targeted compounds that entered clinical development
| Compound Name |
Structure | NOP profile | Clinical Phase (Ref) | Indication |
|---|---|---|---|---|
|
1 Cebranopadol |

|
NOP full agonist-MOP full agonist |
Ongoing Phase III42, 43 |
Various Pain modalities |
|
2 LY2940094 |
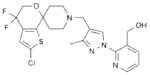
|
NOP antagonist | Completed Phase II45, 46 |
Major
Depressive Disorder Alcohol Dependence |
|
3 MK-5757 |
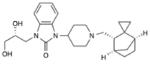
|
NOP antagonist | Completed Phase II48 |
Cognitive Impairment in Schizophrenia |
|
4 SCH486757 |
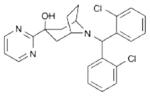
|
NOP full agonist | Completed Phase I (Discontinued)57 |
Cough |
|
5 JNJ-19385899 |
Not available | NOP agonist | Completed Phase I (No information)51 |
Anxiety and Depression |
|
6 Ser100, (ZP120) |
Ac-RYYRWKKKKKKK-NH2 | NOP partial agonist |
Completed Phase IIa56 |
Treatment resistant systolic hypertension |
|
7 JTC-801 |
|
NOP antagonist | Completed Phase II59 |
Neuropathic
Pain Postoperative Pain |
| N/OFQ | FGGFTGARKSARKLANQ | Natural ligand NOP agonist |
Completed Exploratory Phase II60 |
Neurogenic urinary incontinence |
One of the first investigations into the therapeutic applications of targeting the NOP-N/OFQ system suggested that NOP agonists, including the natural peptide N/OFQ, had anxiolytic activity in several animal models.49,50 However, the only small-molecule NOP agonist that appears to have entered human clinical trials is 5 (JNJ-19385899)51, for which no structural or biological information is available. A Phase I pharmacodynamics/pharmacokinetics (PKPD) and safety study suggested an inverted U-shaped dose-response curve for this NOP agonist,51 however, no further information is available on the status of its development. Among peptidic NOP agonists, compound 6 (ZP-120, Table 1), a dodecapeptide NOP partial agonist,52-55 now called SER100 (licensed from Zealand Pharmaceuticals by Serodus ASA, Norway), completed a Phase IIa clinical trial for systolic hypertension. However, there is no further information on the future clinical progression of 6. Zealand Pharma previously advanced 6 up to Phase II clinical trials for urinary incontinence, but its development was terminated because of a blood pressure-lowering side effect. This sodium-sparing peripherally-targeted peptide was resurrected by Serodus, who capitalized on the blood pressure-lowering side effect as a potential approach in patients with treatment-resistant isolated systolic hypertension and advanced it to Phase IIa, with plans for further development with an oral formulation.56
There have been NOP-targeted compounds that were advanced into clinical development, but later terminated due to various reasons. The NOP full agonist 4 (SCH486757, Figure 1) was advanced into Phase I clinical trials as a potential treatment for cough. 57,58 However, its development was terminated due to lack of efficacy and therapeutic window separating the efficacy and the somnolence effect in patients. NOP antagonist 7 (JTC-801, from Japan Tobacco, Table 1) was also taken into Phase II clinical trials in Japan and the UK, as an injectable and oral formulation for the treatment of neuropathic and postoperative pain,59 but was dropped from further development due to unknown reasons. The natural peptide N/OFQ itself has also been investigated in patients, as an intravesical injection for the control of neurogenic detrusor overactivity in a placebo controlled, randomized exploratory trial in Italy, and demonstrated clinical efficacy during 10 days of treatment.60-62 While these findings support the use of NOP agonists as a potential therapy for urinary incontinence, no investigations of this therapeutic approach have been reported with small-molecule NOP agonists.
Figure 1.

Structures and in vitro pharmacological profiles of known NOP agonists. Note that the in vitro data from the cited references are from different laboratories and use different radioligands and protocols for the assays, and therefore cannot be directly compared.
The discovery of the endogenous nociceptin/orphanin FQ peptide and investigations into the pharmacology of the NOP-N/OFQ system spawned a large effort to discover small-molecule NOP ligands in almost every major pharmaceutical company.63-67 From this effort, several small-molecule NOP agonists and antagonists emerged as being suitable for in vivo pharmacological evaluation in animal models using systemic routes of administration. These NOP ligands have been invaluable as tools to investigate the modulation of NOP pharmacology for therapeutic benefit and characterize other target-related pharmacology that may impact a useful therapeutic profile, and as lead compounds for drug development. Presented below is the collection of small-molecule NOP agonists and antagonists that have been well characterized in the literature. The pharmacological characterization of these ‘literature’ NOP compounds in various preclinical disease models (Table 2) reveals several issues and challenges that have plagued NOP-targeted drug development and slowed the clinical validation of the NOP receptor as a pharmacological target. Rather than being an exhaustive compilation of every study published with these known NOP ligands, the discussion below presents the interesting observations from these studies that point to the therapeutic potential of NOP ligands and highlights the issues related to animal models, on-target/off-target effects and other challenges that must be addressed on the path to translation of the NOP receptor as a viable clinical target for various CNS disorders.
Table 2.
NOP-selective Agonists and Antagonists: Preclinical testing and outcomes
| Indication | Compound | Preclinical Species |
Preclinical Model (Dose, route) |
Outcomes (Side-effects) | Ref. |
|---|---|---|---|---|---|
| NOP Agonists | |||||
| Anxiety * | 8 | Rats | Elevated plus-maze (1.0, 3.2 mg/kg, i.p.) |
Decreased open arm avoidance (no
effect on locomotion at these doses) |
50 |
| Fear-potentiated auditory
startle (3.2, 10 mg/kg, i.p.) |
Attenuated startle responses (no
effect on locomotion at 3.2 mg/kg dose, but impaired motor activity at 10 mg/kg dose) |
50 | |||
| Modified Geller-Seifter
conflict procedure (0.3, 1 and 3.2 mg/kg, i.p.) |
Bell-shaped dose-response,
highest response at 1 mg/kg (no effect on locomotion at these doses) |
50 | |||
| Conditioned lick suppression
(punished responding) (1, 3, 10 mg/kg, i.p.) |
3 and 10 mg/kg doses increased
punished licking (impaired motor activity at 10 mg/kg and above) |
71 | |||
| Panic-like anxiety test (1, 3.2, 10 mg/kg, i.p.) |
No effect at 1 and 3.2 mg/kg
(impaired motor activity at 10 mg/kg dose, but not lower doses) |
50 | |||
| Separation-induced rat pup
vocalization (0.3, 1 and 3 mg/kg, i.p.) |
1 and 3 mg/kg doses decreased
vocalization (no effect on locomotion at these doses) |
71 | |||
| Vogel conflict drinking
(punished responding) (0.1, 0.3, 1 and 3 mg/kg, i.p.) |
1 and 3 mg/kg doses increased
punished drinking (no effect on locomotion at these doses) |
76 | |||
| Social approach avoidance (0.3, 1 and 3 mg/kg, i.p.) |
All three doses increased time in
social compartment |
76 | |||
| Mice (C57BL/6– 129X1Sv mixed) |
Mouse Geller-Seifter conflict
procedure (1 and 3 mg/kg, i.p.) |
Increase in punished responding at 3
mg/kg dose (decreased time on rotarod and locomotor activity at 3 and 10 mg/kg) |
71 | ||
| Mice (C57Bl/6) |
Novelty-induced hypophagia (0.3, 1 and 3 mg/kg, i.p.) |
In novel cage, decrease in latency to
drink at 1 mg/kg and increase in milk intake at 0.3 and 1 mg/kg. (In home cage, increase in latency to drink and decrease in intake at 1 and 3 mg/kg, due to sedative effects) |
76 | ||
| Mice (C57BL/6) |
Stress-induced hyperthermia (0.3, 1 and 3 mg/kg, i.p.) |
All three doses reversed
stress-induced hyperthermia (no effect on baseline temperature measures at these doses) |
76 | ||
| 9 | Rats | Elevated plus-maze (0.32, 1 and 3.2 mg/kg, i.p.) |
Increased time in open arms at 1 and
3.2 mg/kg doses (no effect on locomotion at these doses) |
102 | |
| 12 | Rats | Elevated plus-maze (1, 3 and 10 mg/kg, p.o.) |
Increased open arm time and entries at
3 and 10 mg/kg doses (no effect on locomotion at these doses) |
108 | |
| Vogel conflict drinking
(punished responding) (1, 3, 10 and 30 mg/kg, p.o.) |
3, 10 and 30 mg/kg doses
increased punished drinking (no effect on locomotion at these doses) |
108 | |||
| Conditioned lick suppression
(punished responding) (1, 3, 10 mg/kg, p.o.) |
Only the 10 mg/kg dose increased
punished licking (no effect on locomotion at this dose) |
108 | |||
| Fear-potentiated auditory
startle (3, 6 and 10 mg/kg, p.o.) |
Attenuated startle responses at 10
mg/kg (no effect on locomotion at these doses) |
108 | |||
| 13 | Rats | Conditioned lick suppression
(punished responding) (1, 3, 10 mg/kg, p.o.) |
3 and 10 mg/kg doses increased
punished licking response (no effect on locomotion at these doses) |
73 | |
| Fear-potentiated startle (1, 3 and 10 mg/kg, p.o.) |
Decreased startle response at 3 and
10 mg/kg (no effect on locomotion at these doses) |
73 | |||
| Mice (C57BL/6) |
Mouse Geller-Seifter conflict
procedure (3, 10 and 30 mg/kg, p.o.) |
Increase in punished responding at
30 mg/kg dose only (decreased time on rotarod and locomotor activity at 30 mg/kg) |
73 | ||
| Marble burying activity (3, 10 and 30 mg/kg, p.o.) |
Decreased in number of marbles buried
at 30 mg/kg dose only (decreased time on rotarod and locomotor activity at 30 mg/kg) |
73 | |||
| 10 | Mice (ddY strain) |
Mouse Vogel conflict test (3, 10 and 30 mg/kg, p.o.) |
Only the 10 mg/kg dose increased
the number of shocks accepted, bell-shaped dose-response (no decrease in locomotion observed at these doses) |
75 | |
| Pain (Acute) | 8 | Mice (Swiss) | Thermal Tail immersion test (1 and 3 mg/kg, i.p.) |
No effect on latency (ataxia
observed at both doses in mice for first 30 mins) |
84 |
| Mice (C57BL/6N) |
Tail-flick test (0.3, 1 and 3 mg/kg, i.p.) |
Increases in pain sensitivity, decrease
in tail- flick latency at 1 and 3 mg/kg doses (decrease in locomotion observed at 3 mg/kg dose) |
85 | ||
| Hot-plate test (forepaw licking and
jump latency) (0.3, 1 and 3 mg/kg, i.p.) |
Decreases in pain sensitivity, increase
in latency of forepaw licking at 3 mg/kg and jump latency at all three doses (decrease in locomotion observed at 3 mg/kg dose) |
85 | |||
| Shock threshold test (flinch
and vocalization latency and jump threshold) (0.3, 1 and 3 mg/kg, i.p.) |
No effect on flinch latency, increase
in vocalization latency at all doses, trend to increase in jump threshold (decrease in locomotion observed at 3 mg/kg dose) |
85 | |||
| Nonhuman primate (rhesus) |
Warm water (50°C) tail
withdrawal (0.001–0.06 mg/kg, s.c.) |
Increase in withdrawal latency at all
doses (no side-effect on respiration, itch response or activity observed) |
78 | ||
| Capsaicin-induced allodynia
(46°C warm water withdrawal latency) (0.001–0.06 mg/kg, s.c.) |
Increase in withdrawal latency at all
doses (no side-effect on respiration, itch response or activity observed) |
78 | |||
| 12 | Nonhuman primate (squirrel monkeys) |
Operant antinociception
assay (Increase in thermal threshold and pull duration of thermode for a food reward) (0.003, 0.01 and 0.03 mg/kg, i.m.) |
Produced larger antinociceptive effects
than morphine, increased thermal threshold and pull duration of a thermode linked to a food reward in the operant paradigm. (no disruptions in activity noted) |
112 | |
| 16 | Mice (ICR) | Tail-flick assay (3, 10 and 30 mg/kg, s.c.) |
No effect on tail-flick latency
(no effect on locomotor activity at these doses) |
74 | |
| 17 | Mice (ICR) | Tail-flick assay (3, 10 and 30 mg/kg, s.c.) |
Increase in tail-flick latency at the 10
and 30 mg/kg doses (30 mg/kg dose causes decrease in locomotor activity) |
74 | |
| UFP-112 | Rats | Paw pressure/vocalization or
withdrawal threshold (0.03, 0.1 and 0.3 nmol i.t.) |
Antinociceptive response at the 0.1 and
0.3 nmol doses, lower doses than morphine (no effect on activity) |
163 | |
| Nonhuman primate (rhesus) |
Warm water (50°C) tail
withdrawal (1, 3 and 10 nmol, i.t.) |
Increase in withdrawal latency at 3 and
10 nmol doses (no side-effect on itch response observed) |
90 | ||
| Capsaicin-induced allodynia
(46°C warm water withdrawal latency) (10 nmol, i.t.) |
Increase in withdrawal latency up to 4
hours (no side-effect on itch response observed) |
90 | |||
|
Pain (Chronic,
neuropathic, inflammatory) |
8 | Rats | Sciatic nerve injury (0.2, 2.3, 11.5, 23 nmol, i.t.) |
Anti-allodynic effects (Von Frey)
and (thermal cold water allodynia) at doses above 2.3 nmol (no locomotor effects reported) |
91 |
| Sciatic nerve injury (23, 46, 69 nmol, intraplantar) |
Anti-allodynic effects (Von Frey)
and (thermal cold water allodynia) at the 46 and 69 nmol doses |
91 | |||
| Sciatic nerve injury (262 nmol/kg, s.c.) |
No anti-allodynic effects observed | 91 | |||
| Nonhuman Primates |
Carrageenan-induced
thermal hyperalgesia ((46°C warm water withdrawal latency) (0.0003, 0.001 and 0.003 mg/kg, s.c.) |
Dose-dependent increase in %
maxiumum possible antihyperalgesic effect (no side- effects on respiration, itch or activity) |
86 | ||
| 9 | Rats | Streptozotocin-induced
diabetic polyneuropathy (mechanical paw withdrawal) (7.1, 23.8 and 71.4 nmol, intraplantar) |
Dose-dependent increase in %
maxiumum possible antihyperalgesic effect (no confounding effects via this route at these doses) |
104 | |
| Mice (ICR) | Formalin paw withdrawal
(formalin- induced licking, flinching, paw-lifting) (0.03, 0.1, 0.3 and 1 micromol/kg, i.v.) |
Dose-dependent inhibition of
formalin- induced hyperalgesic effects (highest dose caused loss of righting reflex) |
106 | ||
| 12 | Mice (BalbC) |
Mustard oil-induced abdominal pain
in mice with colitis (3 mg/kg p.o.) |
Significant antinociceptive activity in
an acutely inflamed intestinal colitis model |
114 | |
| 16 | Mice (ICR) | Spinal nerve ligation model
of neuropathic pain (10 and 30 mg/kg, s.c.) |
Significant mechanical anti-allodynic
effects (Von Frey) but not thermal antinociception in tail-flick (no effect on locomotor activity) |
128 | |
| 17 | Mice (ICR) | Spinal nerve ligation model
of neuropathic pain (3 and 10 mg/kg, s.c.) |
Significant mechanical anti-allodynic
effects (Von Frey) and thermal antinociception in tail-flick assay (no decrease in locomotor activity at these doses) |
128 | |
| 11 | Rats | Chronic constriction injury model
of neuropathic pain (1.0, 10 and 30 mg/kg, s.c.) |
Significant mechanical anti-allodynic
effects (Von Frey) (no effect on locomotor activity at these doses) |
127 | |
| Chronic constriction injury model
of neuropathic pain (5.5, 55 and 170 nmol, i.t.) |
Significant mechanical anti-allodynic
effects (Von Frey) at 55 and 170 nmol doses |
127 | |||
| Chronic constriction injury model
of neuropathic pain (5.5, 55 and 170 nmol, i.c.v.) |
No anti-allodynic effects (Von Frey) at
any dose |
127 | |||
|
Substance
Abuse Disorders * |
8 | Mice (C57BL/6) |
Morphine conditioned place
preference (0.3 and 1 mg/kg, i.p.) |
1 mg/kg blocked acquisition of
morphine CPP and morphine prime-induced reinstatement of CPP, but not the expression of morphine CPP or the rate of extinction of morphine CPP (no effect on locomotor) |
96 |
| Mice (NMRI) | Alcohol conditioned place
preference (0.1, 0.3, and 1 mg/kg, i.p.) |
0.3 and 1 mg/kg blocked
acquisition, expression as well as alcohol prime-induced reinstatement of alcohol CPP (decrease in locomotor activity at this dose) |
97 | ||
| Rats (Wistar) |
Alcohol self-administration
and deprivation-induced relapse (0.1, 0.3 and 1 mg/kg, i.p.) |
Dose-dependent decrease in alcohol
self- administration. Decreased deprivation- induced increase in alcohol intake |
98 | ||
| Rats (alcohol- preferring msP rats) |
Alcohol self-administration (0.3 and 1 mg/kg, i.p.) |
Increase in alcohol consumption at the
1 mg/kg dose, reversible by naloxone |
99 | ||
| 12 | Rats | Remifentanil (opioid)
self-administration (1, 3, 10, 30 mg/kg s.c.) |
Only the 30 mg/kg dose
decreased remifentanil self-administration (at this dose, rats were unresponsive and showed loss of righting reflex) |
110 | |
| Remifentanil (opioid)
self-administration (0.3, 1, 3 micrograms intracisternally) |
All three doses decrease remifentanil
self- administration (at 3 μg, sedation observed) |
110 | |||
| 15 | Rats (msP rats) |
Alcohol self-administration, and
cue- and stress-induced reinstatement (0.3, 1 and 3 mg/kg p.o.) |
Decrease in alcohol self-administration
and reinstatement (no effect on locomotion observed) |
100 | |
| Rats (Wistar) |
Alcohol withdrawal in dependent
rats (1, 3 and 10 mg/kg, p.o.) |
Decreased withdrawal signs at 3 and
10 mg/kg doses |
100 | ||
| Alcohol self-administration
and reinstatement in post-dependent rats (0.3 and 1 mg/kg p.o.) |
Decrease in alcohol self-administration
and reinstatement |
123 | |||
| 16 | Mice (ICR) | Morphine CPP (3, 10 and 30 mg/kg, s.c.) |
Decrease in morphine CPP (no
effect on locomotor activity at these doses) |
74 | |
| Mice (ICR) | Cocaine CPP and prime- and
stress- induced reinstatement (3, 10 and 30 mg/kg, s.c.) |
No effect on acquisition of cocaine CPP.
But 30 mg/kg blocked cocaine prime- and stress- induced reinstatement of CPP (no effect on locomotor activity at this dose) |
134 | ||
|
Inflammatory
bowel disease |
12 | Mice (BalbC) |
2,4,6-trinitrobenzesulfonic acid
(TNBS)- induced colitis (Twice daily 0.1 and 1 mg/kg i.p. or 3 mg/kg p.o.) |
Attenuation of TNBS colitis via both
routes, but not intracolonic route |
114 |
| NOP Antagonists | |||||
| Depression | 18 | Mice (CD-1) | Forced swim test (20 mg/kg, i.p.) |
Decreased immobility time (no
effect on general locomotor activity) |
144 |
| 19 | Mice (Swiss) | Forced swim test (1, 3, and 10 mg/kg, i.p.) |
10 mg/kg dose decreased immobility time | 141 | |
| 2 | Rats | Forced swim test (10 and 30 mg/kg, p.o.) |
30 mg/kg decreased immobility time | 44 | |
|
Parkinson’s
disease |
18 | Mice | MPTP-induced Parkinson’s
model (0.01 mg/kg, i.p.) |
Improved motor performance in
several motor tests (higher doses 10 mg/kg worsened hypokinesia) |
20 |
| Nonhuman primate (macaques) |
MPTP-induced Parkinson’s
model (0.01 mg/kg, i.m.) |
Improvement in Parkinsonian scores
(but less than that with L-DOPA) |
20 | ||
| Rats | 6-hydroxydopamine hemilesioned
rat model (0.3, 1 and 3 mg/kg, i.p.) |
Relieved akinesia and attenuated
motor asymmetry in hemiparkinsonian rats |
148 | ||
| 19 | Rats | 6-hydroxydopamine hemilesioned
rat model (0.01, 0.1 and 1 mg/kg, i.p.) |
Relieved akinesia and improved
motor performance in hemiparkinsonian rats |
18 | |
|
Pain (Chronic,
inflammatory) |
19 | Rats | Carrageenan-induced inflammatory
pain model (0.1, 0.3, 1, 3, 5 mg/kg, i.v.) |
Increase in paw withdrawal latencies at
3 and 5 mg/kg doses |
139 |
| 7 | Rats | Formalin-induced inflammatory
pain model (3 mg/kg, p.o.) |
Decreased licking response up to
45 minutes after administration |
152 | |
| Rats | Hyperalgesia in the single
prolonged stress model of post-traumatic stress disorder (6 mg/kg, i.p. once daily, 21d) |
Decreased mechanical allodynia,
thermal hyperalgesia |
164 | ||
Preclinical assays in nonhuman primates not reported
NOP agonists
Ro64-6198 (8) (Figure 1) was among the first small-molecule NOP agonists reported, from a series of 1,3,8-triazaspirodecanones disclosed by Hoffman La-Roche,68,69 and has been the most widely studied nonpeptide NOP agonist. It has subnanomolar binding affinity for NOP, 100-fold binding selectivity over MOP and negligible affinity for KOP or DOP (Figure 1).69,70 Its potency as a full agonist at NOP is comparable to that of the endogenous agonist N/OFQ.69,70 Compound 8 has been invaluable as a pharmacological tool to validate NOP receptor agonists as a potential treatment of several conditions such as anxiety, substance abuse, pain, and cough. Importantly, however, characterization of the in vivo pharmacology of 8, after systemic administration, in several pharmacological tests and in different species, has been very informative for identifying the issues and challenges for translational development of NOP agonists as therapeutics. One of the first indications of the potential clinical utility of NOP agonists came from the demonstration that 8 and its congeners (e.g. 9, Ro65-6570,68 Figure 1, also see below) showed significant anxiolytic activity in many different assay paradigms,50,69 later independently confirmed in a report from Schering-Plough researchers, which characterized the anxiolytic properties of 8 across three different rodent species and a variety of conditioned and unconditioned tests of anxiety.71
While these studies clearly established the anxiolytic potential of 8 after systemic administration, they also revealed several issues regarding systemic effects of NOP agonists that confound behavioral assays, particularly in rodents–
(i) For example, in mice, compound 8 showed significant anxiolytic activity only at doses at which it adversely affected general neurological functions such as rotarod performance, locomotor activity and body temperature.71 A detailed study of the effect of 8 on rodent neurological functions previously showed that at doses 3 mg/kg and above, in mice, compound 8, given intraperitoneally, produced marked suppressive effects on body posture, rotarod performance, grip strength and righting reflex.72 Together, these studies showed that in mice, there is no dose separation between anxiolytic doses and doses that disrupt general behavior. This is also observed with other selective NOP agonists studied in mouse anxiety assays such as 13 (SCH655842)73 (Figure 1). Interestingly, there seems to be some influence of the mouse strain on the severity of these adverse behavioral effects, as no decrease in locomotor or other activity was observed in ICR mice, using a different selective NOP agonist 16 (SR16835)126 (Figure 1), at doses at which it inhibited morphine-conditioned place preference (see below)74. Similarly, no adverse effects on activity were observed at anxiolytic doses with the Pfizer NOP agonist 10 (MCOPPB)75 (Figure 1) (Table 2) in ddY mice, a general purpose inbred mouse strain used in Japan.75 The anxiety studies with 8 were all conducted in mice with a C57Bl/6 background and consistently showed dose-dependent disruptive effects on mouse behavior (Table 2).71,72,76 It appears that the C57Bl/6 mice are sensitive to the adverse neurological effects of NOP agonists. Interestingly, in studies in isolated tissue preparations, 8 mimicked the effect of N/OFQ in Swiss mouse vas deferens, but this effect was not reversed by NOP antagonists or by naloxone, suggesting that in this mouse strain also, 8 appears to have effects mediated by an uncharacterized biological target.77
(ii) In rats, however, compound 8 and other NOP agonists mentioned above show distinct and consistent dose separation of at least >3-fold between the doses at which anxiolytic effects are observed in a variety of assay paradigms, and those at which decreased activity is observed. Compound 8 not only showed significant anxiolytic activity, comparable to benzodiazepines in several rat anxiety assays, but also showed a better side-effect profile than benzodiazepines at efficacious doses, pointing to potential therapeutic utility as a novel approach for anxiolytics.50,71,76 Moreover, unlike the case with C57Bl/6 mice, the disruptive neurological effects of 8 in rats were only observed at doses of 10 mg/kg and above.72
Another dimension to species differences in the pharmacological profile of 8 was revealed in the studies by Ko et al. in nonhuman primates, which showed that 8, given systemically (s.c. or i.v), did not cause sedation or other adverse effects at doses at which it exhibited significant antinociceptive efficacy.78,79 However, the anxiolytic potential of 8 (or any other NOP agonist) has not been reported in nonhuman primates.
Compound 8 has also been useful in highlighting the complexities of the role of the NOP-N/OFQ system in modulation of pain pathways. It is well documented that the effect of NOP agonists in pain modulation in animal models varies with the assay, route of administration and species.80-82 Compound 8 has been investigated in animal models of various pain modalities, and shows differential effects depending on the route of administration and the type of pain assay. However, recent developments in the characterization of the NOP ligands in nonhuman primates reveals important information suggesting that NOP agonists may have some unique advantages over classical opioid therapy, offering opioid-type analgesia without the classical opioid liabilities such as itch, constipation, sedation and dependence.82,83
As expected of a NOP agonist, compound 8, given i.p., suppressed the antinociceptive effect of an acute injection of morphine in the mouse warm water tail immersion test, but did not, by itself, increase the latency of response to this thermal stimuli.84 These results were in line with the observations by the Hoffman La-Roche researchers who first reported the pharmacological characterization of 8 in rat anxiolytic assays, but found no effect of this compound on pain thresholds in rat tail-flick assay or on tactile allodynia measured with Von Frey hairs.50 Further characterization of 8 in other acute pain paradigms in C57BL/6 mice showed that it increased pain sensitivity in the tail-flick test at the 1 and 3 mg/kg doses given i.p., but showed antinociceptive effects in the hot-plate test and shock threshold tests.85
In contrast to these effects in rodent acute pain assays, 8, administered systemically (s.c.) to nonhuman primates, showed significant antinociceptive efficacy against an acute noxious stimulus (50° C water) tail-immersion test78,86 and also inhibited capsaicin-induced allodynia manifested as reduced warm water tail-flick latency when capsaicin was injected into the monkey tail.78 Clearly, the effect of systemic administration of NOP agonist 8 was different between rodents and nonhuman primates. Since the effect of systemic administration is a net effect of central (supraspinal and spinal) and peripheral action, it is possible that in rodents, the supraspinal NOP-receptor mediated hyperalgesia is countered by spinal and peripheral antinociceptive effects of NOP agonists (such as 8), resulting in a net outcome of ‘no effect’ on antinociception in rodents after systemic administration, as was observed with 8. The fact that in primates, systemic 8 consistently shows significant antinociceptive activity, points to a differential role of the NOP-N/OFQ in modulation of pain pathways in rodents and primates, and at the different physiological sites (supraspinal, spinal and peripheral) in the two species. Indeed, a recent report by the Ko group shows that, unlike the hyperalgesic effect of icv N/OFQ in rodents, supraspinal administration of N/OFQ to nonhuman primates via an intracisternal injection produced robust antinociceptive effects without increasing scratching response.87 These provocative results suggests that supraspinal actions of N/OFQ and its involvement in regulating pain may be different in rodents versus nonhuman primates, and possibly humans, and offers the possibility of obtaining antinociceptive efficacies in humans, using NOP agonists, without the confounding side-effects such as itch, that accompanies the traditional opioids like morphine.
On the other hand, the intrathecal (i.t.) route of administration has been explored in larger animals such as NHPs, with peptides such as N/OFQ and a synthetic peptide agonist UFP-112,90 which showed significant acute thermal antinociception in the warm water test and capsaicin-induced allodynia.88-90 However, small-molecule NOP agonists such as 8 have not yet been reported to be administered intrathecally to nonhuman primates.
In chronic pain models in rodents, such as the rat sciatic nerve injury model, 8 showed anti-allodynic and antinociceptive effects only upon intrathecal and intraplantar (local peripheral) administration, but not systemic (s.c.) administration.91 In nonhuman primates however, systemic administration (s.c) of 8 shows significant antinociceptive effects against carrageenan-induced hyperalgesia (a model of inflammatory pain), with potencies similar to mu opioid agonists, but without producing the itch and respiratory depression effects typically associated with mu opioid-type analgesics.86 Taken together, the results of the extensive characterization of 8 in acute and chronic pain models in rodents and primates via various routes of administration, suggest that selective NOP agonists may have clinical utility as analgesics in humans, providing efficacies similar to opioids, but without opioid-associated side-effects such as pruritis, respiratory depression, and dependence (see below).83
Given that the NOP-N/OFQ system is in the opioid family, and present in areas of the brain associated with reward processing, NOP ligands have been investigated for their rewarding and motivational properties. Early studies with 8 showed that it did not induce a place preference in rats, up to doses of 3.2 mg/kg, when administered systemically.92 However, a more recent study in rats showed that 8, at doses 2.5 mg/kg and 3.16 mg/kg, produced a significant place preference, reversible by the D2 antagonist haloperidol, but not by naloxone or the NOP antagonist J-113397 (18, Figure 2)137, suggesting a dopaminergic effect of 8.93 The reasons for this discrepancy are not known, but could be attributed to several factors, including the chemical supply of 8 itself, given that these two studies were conducted in different laboratories. Compound 8 (0.3, 1, and 3.2 mg/kg) was shown not to significantly modify the threshold for intracranial self-stimulation (ICSS) in animals implanted with electrodes into the ventral tegmental area, in contrast to cocaine, which, expectedly, rapidly decreased the ICSS threshold.50 These studies further confirmed that compound 8 was devoid of reward-stimulating effects characteristic of pyschostimulants such as cocaine.
Figure 2.

Structures and in vitro pharmacological profiles of NOP antagonists widely used as tool compounds. Note that the in vitro data from the cited references are from different laboratories and use different radioligands and protocols for the assays, and therefore cannot be directly compared.
On the other hand, 8 was shown to produce a discriminative stimulus effect in rats, reversible by the NOP antagonist 18 (Figure 2), and weakly generalized to morphine, but not kappa or delta agonists.94 This discriminative stimulus was not as strong as that produced by mu opioid agonists, suggesting that NOP agonists are likely to be devoid of reinforcing or euphorigenic effects. In nonhuman primates, 8 did not produce reinforcing effects at doses at which it showed significant antinociceptive effects.79 For potential clinical utility as analgesics, as discussed above, NOP agonists may provide opioid-like efficacies without the abuse liabilities of classical opioids.
The natural peptide agonist N/OFQ reduces basal and drug-induced dopamine release in the nucleus accumbens,22-25 pointing to the potential of NOP agonists in reducing drug-induced dopamine release and consequently the rewarding effects of abused drugs.95 Indeed, 8 (and other small-molecule NOP agonists, see below) was shown to decrease the acute rewarding effects of morphine in the conditioned place preference paradigm in C57BL/6 mice at a dose of 1 mg/kg, at which it does not produce nonspecific effects on locomotion or activity of the animals.96 Compound 8 has, however, not been tested for its effect on morphine drug reward in rats, nor has it been tested for effects on drug reward induced by psychostimulants such as cocaine. However, i.c.v., N/OFQ itself has been shown to block the conditioned place preference induced by cocaine, amphetamine and methamphetamine in mice and rats.27-30 On the other hand, 8 however, has been shown to block the acquisition and reinstatement of alcohol place preference in NMRI mice at the 1 mg/kg dose s.c. without nonspecific effects on activity.97 The inhibitory effect of 8 on alcohol-induced rewarding effects were further confirmed in rats, where it was shown to reduce alcohol self-administration and deprivation-induced relapse-like alcohol drinking in Wistar rats, when administered i.p. at doses up to 1 mg/kg.98 However, in genetically modified alcohol-preferring rats, 8, at 1 mg/kg administered i.p., actually increased alcohol consumption.99 This effect was not observed with other NOP agonists, and was inhibited by naloxone, hence attributed to a residual mu opioid agonist activity of 8. Notably, recently, another nonpeptidic NOP agonist 15 (MT-7716, Figure 1)100 was shown to decrease ethanol-seeking in alcohol-preferring rats.100 Therefore, the potential of NOP agonists to reduce alcohol reward needs further investigation.
Although 8 has been extensively characterized in vivo and shown to have good brain penetration,50,71 it was not further developed for any of the above indications, likely because its oral bioavailability in multiple preclinical species, was poor (1% in cynomolgous monkeys and 4% in rodent species).101
Ro65-6570 (9) was another congener from the same family of 1,3,8-triazaspirodecanones as 8, reported by Hoffman La-Roche, but not as extensively characterized. Compound 9 differs from 8 only in the structure of the piperidine nitrogen substituent (Figure 1).68,102 Interestingly, this small structural change results in a significant difference in binding affinity for the NOP and MOP receptor (Figure 1), compared to 8, such that its selectivity for NOP is lower than that of 8 (~10-fold, compared to 100-fold for 8).103 Nevertheless, both compounds are full agonists at the NOP receptor. While it has not been directly compared with 8 in any study, compound 9 was reported to show dose-dependent anxiolytic effects in the elevated plus maze test in rats in doses (1 and 3.2 mg/kg i.p.) at which it did not affect motor performance.102 However, its low selectivity versus the mu opioid receptor may be responsible for the ‘increase’ in locomotor activity observed with 3 and 6 mg/kg doses in rats, as well as increase in cocaine-induced hyperactivity, when 9 was administered prior to cocaine in the conditioned place preference paradigm.27 In the same study, 9 was also shown to produce a significant place preference alone, at doses of 3 mg/kg and 6 mg/kg, at which it produced an increase in locomotor activity, while having no effect on the expression of cocaine place preference. While this study did not report the effect of 9 on the acquisition (development) of cocaine CPP, a later study showed that 9 increased the minimal effective doses of morphine, heroin, oxycodone and cocaine, but not amphetamine, to produce a place preference, when it was administered as a 15-min pretreatment, but not when administered 30-sec before these drugs.93 These results show that while NOP agonists reduce the rewarding effects of opioids and psychostimulants, the attenuation of rewarding effects is more consistent with abused drugs of the opioid class. The discrepancy between the effect of 9 on CPP induced by psychostimulants cocaine versus amphetamine is not entirely clear, and points to the need to study the efficacy of well-characterized selective NOP agonists for their effects on psychostimulant reward.
Unlike 8, compound 9 has not been extensively evaluated in antinociceptive assays, possibly because of its lower selectivity profile. However, consistent with observations with 8,91 compound 9 showed robust peripheral anti-hyperalgesic activity when administered intraplantarly to rats with diabetic polyneuropathy, showing efficacy similar to that of morphine, administered by the same route.104 Similarly to 8, compound 9 also did not show significant antihyperalgesic efficacy, with a good dose separation from confounding side effects, when administered systemically to rats.105, 106 Nevertheless, these results with both NOP agonists 8 and 9 further attest that NOP agonists may be useful for relief of peripheral pain associated with chronic pain conditions such as diabetic neuropathy, and opens up possibilities for development of peripherally targeted NOP agonists which may be devoid of centrally-mediated side-effects such as suppression of locomotor activity at higher doses.
SCH 221510 (12) and SCH 655842 (13) belong to a class of N-benzhydryl nortropane-based NOP agonists (Figure 1) developed by Schering-Plough Corporation.107 These two NOP agonists were characterized extensively by Schering-Plough in rodent models of anxiety,73,108 both in rat and mouse, and found to have robust anxiolytic profiles, in a variety of anxiety paradigms, similar to that observed with 8. Notably, the anxiolytic efficacy of 12 and 13 was observed at doses at which there was no nonspecific disruption of activity, as was observed with classical benzodiazepines. As seen with agonist 8,71 there appeared to be a better dose separation between efficacy and reduction in activity in the rat, compared to mouse.73 Although both SCH agonists were reported to have good oral bioavailability and modest brain penetration (average rat brain-to-plasma ratio for 13 = 0.43–0.59), neither of these compounds were advanced into clinical trials. While it is not expressly discussed or reported, it is possible that its pharmacokinetic profile (e.g. modest brain penetration observed with 13) preclude an effective therapeutic window between anxiolytic effects and target-based side effects.72 As discussed below, others have investigated different NOP agonists (e.g. 10,109 by Pfizer, Figure 1) as anxiolytics, but have not progressed these NOP agonists into clinical trials for this indication. The clinical activity of the only NOP agonist that was advanced into Phase I trials for anxiety (5) (Table 1) has not been reported thus far.
Compound 12, however, is now commercially available as a selective NOP agonist research tool, and its availability has spawned investigations into the utility of NOP agonists for several indications, including those investigated with compound 8 (such as drug abuse, pain) and others such as gastrointestinal disorders. Sukhtankar et al. have shown that, in rats, 12 is not reinforcing on its own, but attenuates self-administration of the opioid remifentanil. Interestingly, this effect of decreasing opioid reinforcing effects was only observed when 12 was administered intracisternally (directly into the brain) to rats, but not when administered systemically (s.c.).110 Following s.c. administration, 12 was found to cause adverse behavioral abnormalities in rats at doses of 30 mg/kg and above. Even though 12 was shown to have oral efficacy in anxiety assays, it seems likely that its further development was negatively impacted by its poor distribution into the brain.108
Unlike agonist 8, compound 12 has not been evaluated for efficacy as an antinociceptive in rodents. However, a recent study in primates showed that NOP agonists 8 and 12 enhanced opioid buprenorphine antinociception synergistically, without causing a concomitant increase in respiratory depression or itch, adverse effects typically observed with opioid-type antinociceptives.111 A further confirmation of the antinociceptive efficacy of 12 in primates comes from a recent study by Kangas and Bergman, who utilized a novel operant paradigm to evaluate antinociceptive efficacies of compounds, and showed that compound 12, injected intramuscularly, showed antinociceptive efficacy exceeding that of morphine, as a significant increase in the thermal threshold and pull duration of a thermode-driven operant food reward in a novel thermal threshold antinociceptive assay (Table 2).112 These results with 12 further confirm the notion that NOP agonists have the potential to have potent antinociceptive activity in nonhuman primates and therefore likely in humans.
Among recent therapeutic applications explored in animal models with compound 12 as a NOP agonist tool compound, are reports that NOP agonists show significant antinociceptive and anti-inflammatory efficacy in mouse models of intestinal disorders such as inflammatory bowel disease (IBD)113 and diarrhea-predominant IBD.114 Interestingly, only the i.p. and oral routes of administration were effective for the antinociceptive effects of 12; direct intracolonic administration did not have an effect on the intestinal swelling observed in these animals.114 It has been speculated that N/OFQ and therefore NOP agonists may play a role in neurogenic inflammation and pain, such as that in IBD.115 These interesting findings, however, warrant further investigation before these applications can be translated to the clinic.
SCH486757 (4)116 a NOP agonist, also belongs to the N-benzhydryl-nortropane series of NOP ligands developed by Schering-Plough (Figure 1). This was the first small-molecule NOP agonist to enter human clinical testing.57 This compound and a close analog 14 (SCH225288, Figure 1)117 are high affinity NOP full agonists, and were extensively characterized in experimental models of cough (guinea pig capsaicin-induced cough, and cat mechanical-induced cough) and shown to be more potent than commonly used antitussives codeine and dextromethorphan.116,117 Indeed, the natural peptide N/OFQ, given intravenously, has also been shown to inhibit cough in experimental models at doses that do not affect general activity in the animal.118-120 Interestingly, compound 8 was also tested in the same models of experimental cough, and found to be effective, when administered i.p. up to 3 mg/kg.121 The authors hypothesized that the antitussive action of N/OFQ and 8 could be mediated by a peripheral mechanism, and proposed that oral NOP agonists that do not penetrate the blood-brain barrier may provide a novel approach for the treatment of cough, without central effects on activity. Further support for this hypothesis came from the work of Lee et al.,122 which showed that the inhibition of acid-induced cough effect by nociceptin results from the inhibition of acid-induced transient receptor potential vanilloid-1 (TRPV1) activation in peripheral C-fibers.
Compound 4 was advanced into Phase Ib/II clinical trials in patients with subacute cough;57 however, the development of this NOP agonist was terminated due to a lack of significant efficacy and therapeutic window. The high doses at which cough suppression was observed also resulted in significant somnolence in patients. It is possible that 4 was not sufficiently excluded from the blood brain barrier at the high dose at which antitussive efficacy was observed, resulting in an unwanted effect of sleepiness.
MT-7716 (15)100 is a NOP agonist belonging to the benzimidazolone class, similar to the well-known NOP antagonist 18 (Figure 2). Compound 15 is a high-affinity and potent NOP full agonist, with greater affinity and selectivity than compound 8. It was recently shown to decrease alcohol drinking in the genetically alcohol-preferring Marchigian Sardinian P rats (msP rats) after chronic treatment (p.o, twice daily dosing) for 14 days, an effect that lasted after the drug was discontinued.100 Compound 15 also significantly inhibited stress- and cue-induced reinstatement of alcohol drinking in these rats.100 When administered to Wistar rats made dependent on ethanol and withdrawn from ethanol, compound 15 (3 and 10 mg/kg p.o.) decreased the expression of withdrawal signs up to 4 h after administration.100 Interestingly, 15 appeared to preferentially decrease alcohol drinking and reinstatement in post-dependent Wistar rats, but was ineffective in non-dependent rats.123 On the other hand, compound 8 was found to inhibit reinstatement of deprivation-induced alcohol seeking in Wistar rats that were non-dependent.98 The reason for this difference between the two NOP agonists is unclear, but likely has to do with their overall profiles of NOP efficacy and selectivity, their pharmacokinetics and brain penetration. Notably, compound 15, previously characterized as W212393,124 was shown to have significant affinity at SERT, albeit with a 20-fold lower affinity than for NOP.124 However, the differences between the effect of 15 in post-dependent versus non-dependent rats suggest that alcohol dependence leads to neuroadaptations in the N/OFQ-NOP system, and that NOP agonists offer a potential approach to treat alcohol dependence, particularly relapse.123
SR16835 (16, now named AT-202) and SR14150 (17, now named AT-200) (Figure 1) are NOP agonists from the dihydroindolinone class of NOP ligands.125,126 The NOP binding affinities for 16 and 17, while not in the subnanomolar range as for compounds 8 and 15, are comparable to those of the SCH series (compounds 4, 12–14) and the Pfizer agonists 10 and 11127 (Figure 1). Compound 16 is a NOP full agonist whereas 17 shows partial agonist activity at NOP (Figure 1).125 Interestingly, both these compounds, administered systemically, show significant antinociceptive and anti-allodynic activity in a mouse spinal nerve ligation model of neuropathic pain, which is reversed by a NOP antagonist but not an opioid antagonist.128 Notably, even though both these NOP agonists have modest selectivity versus the MOP receptor, their antinociceptive efficacy in the chronic pain model is mediated only via their NOP agonist efficacy, since an opioid antagonist did not affect the overall efficacy, when co-administered.128 A further confirmation of the potential antinociceptive efficacy of NOP agonists in chronic pain comes from a recent study in which 17 was shown to have antinociceptive and anti-allodynic activity superior to morphine, in a transgenic mouse model of sickle-cell disease pain, in which the transgenic sickle mice develop spontaneous hyperalgesia akin to the pain observed in patients with sickle cell disease.129 The potent antinociceptive efficacy of 17 in this animal model was reversed only by the NOP antagonist 19 (SB-612111, Figure 2)139 but not by the opioid antagonist naloxone. Moreover, the antinociceptive efficacy of 17 did not diminish over a 7-day daily treatment, in contrast to that of morphine. As expected of NOP agonists, both 17 and 16 show no abuse liabilities, as determined in mouse conditioned place preference assays.74
The antinociceptive action of 16 and 17 mediated by their NOP agonist efficacy in chronic pain is consistent with the efficacy observed with NOP agonists 8 and 11 (Table 2) in rodent peripheral neuropathy models, and suggests that spinal and peripheral NOP receptors provide inhibitory control in pain pathways activated in these conditions. Further mechanistic studies are needed to shed light on this, however. In the light of the efficacies observed with NOP agonists in nonhuman primates, it is clear that NOP agonists may have the potential of antinociceptive efficacies in human chronic pain conditions, offering therapeutic efficacy equal to or superior to that of traditional opioids.
Perspective on the Therapeutic Potential of NOP agonists
From the extensive pharmacological characterization of a variety of NOP agonists from different chemical classes, a few common themes emerge that suggest that NOP agonists still hold significant promise as therapeutics in several clinical indications, where the NOP agonist pharmacological profile offers unique advantages over current approaches. Two such applications are (i) Treatment of neuropathic, inflammatory and other chronic pain conditions (ii) substance abuse medications, particularly to prevent stress-induced relapse and maintain abstinence.
Treatment of chronic, neuropathic and inflammatory pain
As discussed above, studies in rodent and nonhuman primate models of pain show that NOP agonists, administered systemically or spinally, show significant antinociceptive activity in neuropathic and inflammatory pain, with efficacies similar to that of opioids, but without the usual opioid-induced side effects such as itch, respiratory depression and abuse liability. One might argue that the locomotor-inhibiting activity of NOP agonism may lead to undesired sedative effects; however, NOP agonists 8 and 12 showed no sedative effects in nonhuman primates at antinociceptive doses.78, 111 Given that the antinociceptive activity of NOP agonists and N/OFQ is likely through a spinally-mediated mechanism and the sedative properties are mediated via a supraspinal mechanism, it may be possible to design NOP agonists with limited penetration into the brain. Such NOP agonists may also be useful for peripheral neuropathic pain as well as pain conditions such as IBD.
Treatment of substance abuse
Studies with NOP agonists discussed above indicate that they reduce the rewarding effects of drugs of abuse. From the different classes of abused substances, the data for alcohol dependence appears to be the most compelling.37, 100, 123, 130, 131 Notwithstanding the inhibitory effect of NOP agonism on dopamine release in the nucleus accumbens and dopamine neurotransmission, the anti-CRF effect of NOP agonists in the central amygdala132, 133 may be particularly relevant for the therapeutic use of NOP agonists in preventing relapse and maintaining abstinence. Further, NOP agonism was also shown to reduce alcohol withdrawal symptoms in rats.100,131 Even for psychostimulants, studies with NOP agonist 16 show that it blocks stress- and drug prime-induced reinstatement of cocaine conditioned place preference in mice, at doses at which there is no effect on locomotor activity.134 Clearly, NOP agonism appears to be a promising approach for substance abuse treatment that should be evaluated in the clinic. Studies in nonhuman primates may also provide a suitable translational model to evaluate NOP agonists for cocaine abuse treatment, although such studies have yet to be reported.
NOP antagonists
There has been a significantly higher interest in pharmaceutical companies for developing NOP antagonists than has been for NOP agonists. The potential therapeutic indications explored are depression, cognitive impairment, obesity and Parkinson’s disease, to name a few.38,66,135 As seen in Table 1, NOP antagonists from Lilly and Merck were advanced to Phase II clinical trials for major depressive disorder and schizophrenia-associated cognitive impairment, respectively, although no further information on the development of these compounds is available. After the discovery of the NOP-N/OFQ receptor/endogenous peptide system, both peptide (e.g. UFP-101) and small-molecule (18, 19) NOP antagonist ligands (Figure 2) were reported in the literature, and significantly aided the characterization of NOP-mediated pharmacology.136-141 These NOP antagonist tool compounds continue to be used today, are commercially available and serve as invaluable tools to study NOP pharmacology and therapeutic potential. The intense effort in pharmaceutical companies around developing small-molecule NOP antagonists also culminated in the development of radioligand and PET tracers (Figure 3), to aid clinical trials and human receptor occupany studies for NOP antagonists that entered the clinic (Table 1). The NOP antagonist tool compounds and radiotracers are briefly described below. It is interesting to note that in contrast to NOP agonists, which have been explored in many pharmacological paradigms for uncovering their therapeutic potential (anxiolytic, drug abuse treatment, pain, cough, GI disorders), there are very few explorations of NOP antagonists in pathophysiological states that indicate their therapeutic potential. As discussed below, NOP antagonist tool compounds have primarily been used as ‘pharmacological knock-outs’ to confirm the involvement of the NOP-N/OFQ system in disease states or to confirm the mechanism of action of NOP agonists. The exceptions to this are reports of the effect of NOP antagonists in inhibiting parkinsonian symptoms in rodent and primate models of Parkinson’s disease,19,20 and the antidepressant effects of NOP antagonists in rodent models.38,142-145 As noted in the discussion above and seen in Table 1, small-molecule NOP antagonists have had a much higher success rate of entering the clinic, compared to small-molecule NOP agonists, albeit the outcome of the clinical trials with NOP antagonists are not disseminated or published yet.
Figure 3.

Structures and in vitro pharmacological profiles of NOP antagonist-based PET tracers and radioligands recently developed for the NOP receptor.
J-113397 (18) (Figure 2) was the very first small-molecule NOP ligand and antagonist to be reported, by researchers at Banyu Japan.137 Since, unlike peptidic NOP antagonists available at that time, 18 could be administered systemically, there were several reports independently characterizing 18 in vitro and in vivo.103,138,146 These consistently reported that 18 was a high affinity, competitive antagonist at NOP but had modest selectivity (~15-fold, see Figure 2) versus the mu opioid receptor. Nevertheless, it was active when administered systemically, and reversed the hyperalgesic effect of icv N/OFQ quite effectively.138 Interestingly, when administered s.c., 18 was found to be rewarding in mice and stimulated accumbal dopamine release by a non-NOP receptor-mediated mechanism,147 suggesting that its in vivo effects may be a composite of actions at the NOP receptor and another undetermined target. Nevertheless, compound 18 has been a very popular NOP antagonist that is still used in pharmacological studies to block actions mediated by the NOP receptor.
Compound 18 was also very instrumental in the seminal results from the Morari group that showed NOP receptor blockade attenuates Parkinson-like symptoms in rodent and in nonhuman primate models of Parkinson’s disease.20,148,149 These results were recapitulated, at least in rodent models, with other NOP antagonists, such as 1-[1-cyclooctylmethyl-5-(1-hydroxy-1-methyl-ethyl)-1,2,3,6-tetrahydropyridin-4-yl]-3-ethyl-1,3-dihydro-benzoimidazol-2-one (GF-4),150 2-[3-[4-(2-Chloro-6-fluoro-phenyl)-piperidin-1-ylmethyl]-2-(morpholine-4-carbonyl)-indol-1-yl]-acetamide (Nik-21273),18 and 1-(1-Cyclooctylmethyl-5-hydroxymethyl-1,2,3,6-tetrahydro-pyridin-4-yl)-3-ethyl-1,3-dihydro-benzoimidazol-2-one (Trap-101),151 suggesting that NOP antagonists may represent a novel approach to treat symptoms in Parkinson's disease. Although NOP antagonists such as 18 were determined to enhance the effect of levodopa, findings which were validated in nonhuman primate models of Parkinson’s disease,19 no NOP antagonist has been developed to progress into human clinical trials for Parkinson’s disease symptom management.
Compound 18 was also the first small-molecule NOP antagonist to be tested in the mouse forced swim test, an assay usually employed to investigate an anti-depressant-like profile.144 Compound 18, administered i.p. at 20 mg/kg, showed a significant reduction in immobility time,144 an effect that was absent in NOP −/− mice.143 In the same study, 18 was shown to have no effect on general locomotor activity of mice in an open field test, indicating that the reduction in immobility time was not due to increase in locomotion.144 This anti-depressant-like activity of 18 was later confirmed with other NOP antagonists such as 19 and peptide UFP-101.141, a142 Interestingly, NOP−/− mice have also been characterized to possess an anti-depressant-like phenotype.142 These studies have provided an important basis for the translation of NOP-targeted pharmacology into the clinical setting, since NOP antagonist 2 (LY2940094) was advanced to Phase II clinical trials for major depressive disorder (see Table 1 and discussion above).45
SB-612111 (19) (Figure 2) is a high affinity, highly selective NOP antagonist reported by GSK.139 It is significantly more selective for NOP versus the other opioid receptors than is 18, when compared in the same study (Figure 2)139. Compound 19 has been widely used to reverse the effects of N/OFQ. When administered systemically (i.p. or s.c.), 19 effectively reverses hyperalgesia produced by an icv injection of N/OFQ139 as well as antinociception produced by an intrathecal injection of N/OFQ.141 Compound 19 was also shown to have antihyperalgesic effects to thermal hyperalgesia in the rat carageenan inflammatory pain model and reverse the expression of morphine tolerance.139 Interestingly, the aminoquinoline NOP antagonist JTC-801 (7, Table 1) also showed an antihyperalgesic effect in the rat formalin model.152 However, the evaluation of NOP antagonist 18 has not been reported in models of nociception. None of the above-mentioned antagonists or their congeners have successfully been investigated for their clinical potential as antinociceptives or anti-hyperalgesic agents for chronic or inflammatory pain. The clinical trials of 7, on the other hand, were discontinued, and its antinociceptive activity was speculated to be due to a non-NOP-mediated effect.
Compound 19 was also shown to ameliorate parkinsonian symptoms in the 6-hydroxydopamine hemilesioned rat model of Parkinson’s disease, and synergized with L-DOPA to provide motor benefit in parkinsonian rats.18 Rizzi et al. also showed that 19 decreased immobility time in the forced swim test for anti-depressant-like activity, an effect that was abolished in NOP−/− mice,141 confirming the anti-depressant potential of NOP antagonists.38 Compound 19 was not further developed, and is commercially available as a NOP antagonist tool compound.
The NOP antagonist 20 (C-24)153 was reported by researchers at Banyu, and later fully characterized by Fischetti et al.154 As expected of a small-molecule NOP antagonist, systemic administration of up to 10 mg/kg of 20 inhibited the effect of N/OFQ in analgesia assays.154 Compound 20 was also used as the bound ligand for crystallization of the NOP receptor GPCR.155
NOP antagonists PET and Radioligands
Perhaps one of the more useful developments in the NOP field, signaling the translation of NOP-targeted ligands into clinical trials, has been the development of PET tracers, which have been used to guide clinical trials of NOP antagonists from Lilly and Merck (Table 1). PET tracers are valuable for clinical development because they can enable quantitation of the target, receptor occupancy by the test compound, and pharmacodynamic assessments, particularly during Phase I clinical trials. Development of a suitable CNS PET tracer involves a significant effort in medicinal chemistry and optimization due to the demanding criteria of a CNS-penetrant PET tracer. Multiple parameters have to be simultaneously optimized, including the choice of the PET isotope and the synthesis route. Previous attempts to make PET radiotracers out of high-affinity NOP ligands 8 (viz. [11C]-Methyl-Ro 64-6198), and 18 (viz. [11C]-CPEB) were not successful due to problems of high nonspecific binding and low brain uptake.156,157
Pike and coworkers reported the development of [11C] NOP-1A (21, Figure 3), a NOP antagonist-based PET tracer, which has subnanomolar affinity for NOP and excellent selectivity versus the opioid receptors and a majority of other CNS targets.158, 159 Compound 21 was used to label NOP receptor sites in rhesus monkey34 and human brain.36, 160 A 18F-containing PET tracer, [18F]-MK0911 (22, Figure 3) was recently reported by Merck scientists, which was developed to support the clinical development of NOP antagonist 3 (Table 1).161 Compound 22 was also investigated in both rhesus monkeys and humans, and showed high specific binding in monkey and human brains, dose-dependently displaced by test NOP antagonists being developed in their studies.161
Pfizer scientists recently reported the development of a tritiated NOP antagonist radioligand, for use in rodent in vivo imaging and receptor occupancy studies, to support lead optimization and clinical candidate selection. [3H]PF-7191 (23, Figure 3) was developed after significant SAR and optimization of its physicochemical properties.162 Robust labeling of rat binding sites with high NOP receptor expression was observed, which were displaceable with a test NOP antagonist in a dose-dependent manner, indicating the suitability of this radioligand for in vivo receptor occupancy studies.
CONCLUSIONS
In the twenty years following the discovery of this fourth opioid receptor and its endogenous ligand, the NOP-N/OFQ research field has been actively engaged, using state-of-the-art approaches on all fronts–pharmacologic, genetic, drug discovery, and tracer discovery, to understand the role of this system in normal function and in pathophysiological states. Given the widespread distribution of the NOP-N/OFQ system in areas of the brain and periphery, it is clear that this system plays an important role in several physiological processes. At the same time, this also poses a challenge in targeting the NOP receptor for therapeutic applications. Nevertheless, the discussion above shows that the NOP-targeted therapies may have key advantages over existing approaches in several therapeutic areas, notably, chronic pain and substance abuse for NOP agonists, and psychiatric disorders for NOP antagonists. Other therapeutic areas under active investigation include treatment of gastrointestinal, inflammatory disorders and possibly Parkinson’s disease symptom management.
The NOP-N/OFQ field is at the cusp of an exciting time, because of the entry and successful progression of NOP-targeted compounds into the clinic (Table 1). It is hoped that the findings from these clinical investigations will be disseminated to benefit the ongoing research into the validating the NOP receptor system as a viable clinical target.
ACKNOWLEDGEMENTS
Support from the National Institutes of Health through grants R01DA014026, R01DA027811, R43NS070664, and R43HL115984 is gratefully acknowledged.
LIST OF ABBREVIATIONS
- CNS
Central nervous system
- CPP
Conditioned place preference
- DOP
delta opioid receptor
- GPCR
G-protein coupled receptor
- IBD
Inflammatory bowel disease
- ICSS
Intracranial self stimulation
- i.c.v.
intracerebroventricular
- i.m.
intramuscular
- i.p.
intraperitoneal
- i.t.
intrathecal
- IUPHAR
International Union of Basic and Clinical Pharmacology
- i.v.
intravenous
- KOP
kappa opioid receptor
- MOP
mu opioid receptor
- MPTP
1-methyl-4-phenyl 1,2,3,6 tetrahydropyridine
- N/OFQ
nociceptin/orphaninFQ peptide
- NOP
nociceptin/orphaninFQ peptide receptor
- PET
Positron emission tomography
- p.o.
peroral
- s.c.
subcutaneous
- SERT
Serotonin transporter
AUTHOR BIOGRAPHY
Nurulain T. Zaveri, Ph.D. is founder, President and Chief Scientific Officer of Astraea Therapeutics, a preclinical-stage company founded in 2009, focused on the development of small-molecule therapeutics for the treatment of CNS disorders. She has a Bachelor’s degree in Pharmacy and obtained her PhD in Medicinal Chemistry in 1994 from Duquesne University. Prior to Astraea, she was Principal Investigator and Drug Discovery Program Director at a nonprofit research institute for 15 years. Her drug discovery research has spanned a variety of molecular targets in oncology and CNS disorders, such as nuclear receptors, GPCRs, enzyme modulators, ion channels and natural products. Her current work is focused on the discovery, optimization and development of small-molecule therapeutics for two major molecular targets important in CNS and neurodegenerative diseases.
Footnotes
DECLARATION OF INTERESTS
The author has no competing interests to declare.
REFERENCES
- 1.Mollereau C, Parmentier M, Mailleux P, Butour JL, Moisand C, Chalon P, Caput D, Vassart G, Meunier JC. ORL1, a novel member of the opioid receptor family. Cloning, functional expression and localization. FEBS Lett. 1994;341:33–38. doi: 10.1016/0014-5793(94)80235-1. [DOI] [PubMed] [Google Scholar]
- 2.Bunzow JR, Saez C, Mortrud M, Bouvier C, Williams JT, Low M, Grandy DK. Molecular cloning and tissue distribution of a putative member of the rat opioid receptor gene family that is not a mu, delta or kappa opioid receptor type. FEBS Lett. 1994;347:284–288. doi: 10.1016/0014-5793(94)00561-3. [DOI] [PubMed] [Google Scholar]
- 3.Fukuda K, Kato S, Mori K, Nishi M, Takeshima H, Iwabe N, Miyata T, Houtani T, Sugimoto T. cDNA cloning and regional distribution of a novel member of the opioid receptor family. FEBS Lett. 1994;343:42–46. doi: 10.1016/0014-5793(94)80603-9. [DOI] [PubMed] [Google Scholar]
- 4.Wang JB, Johnson PS, Imai Y, Persico AM, Ozenberger BA, Eppler CM, Uhl GR. cDNA cloning of an orphan opiate receptor gene family member and its splice variant. FEBS Lett. 1994;348:75–79. doi: 10.1016/0014-5793(94)00557-5. [DOI] [PubMed] [Google Scholar]
- 5.Pan YX, Cheng J, Xu J, Rossi G, Jacobson E, Ryan-Moro J, Brooks AI, Dean GE, Standifer KM, Pasternak GW. Cloning and functional characterization through antisense mapping of a kappa 3-related opioid receptor. Mol. Pharmacol. 1995;47:1180–1188. [PubMed] [Google Scholar]
- 6.Meunier JC, Mollereau C, Toll L, Suaudeau C, Moisand C, Alvinerie P, Butour JL, Guillemot JC, Ferrara P, Monsarrat B, Mazarguil H, Vassart G, Parmentier M, Costentiná J. Isolation and structure of the endogenous agonist of opioid receptor-like ORL1 receptor. Nature. 1995;377:532–535. doi: 10.1038/377532a0. [DOI] [PubMed] [Google Scholar]
- 7.Reinscheid RK, Nothacker HP, Bourson A, Ardati A, Henningsen RA, Bunzow JR, Grandy DK, Langen H, Monsma FJ, Jr., Civelli O. Orphanin FQ: a neuropeptide that activates an opioidlike G protein-coupled receptor. Science. 1995;270:792–794. doi: 10.1126/science.270.5237.792. [DOI] [PubMed] [Google Scholar]
- 8.Mollereau C, Mouledous L, Lapalu S, Cambois G, Moisand C, Butour JL, Meunier JC. Distinct mechanisms for activation of the opioid receptor-like 1 and kappa-opioid receptors by nociceptin and dynorphin A. Mol. Pharmacol. 1999;55:324–331. doi: 10.1124/mol.55.2.324. [DOI] [PubMed] [Google Scholar]
- 9.Yu TP, Fein J, Phan T, Evans CJ, Xie CW. Orphanin FQ inhibits synaptic transmission and long-term potentiation in rat hippocampus. Hippocampus. 1997;7:88–94. doi: 10.1002/(SICI)1098-1063(1997)7:1<88::AID-HIPO9>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 10.Hawes BE, Graziano MP, Lambert DG. Cellular actions of nociceptin: transduction mechanisms. Peptides. 2000;21:961–967. doi: 10.1016/s0196-9781(00)00232-1. [DOI] [PubMed] [Google Scholar]
- 11.Briscini L, Corradini L, Ongini E, Bertorelli R. Up-regulation of ORL-1 receptors in spinal tissue of allodynic rats after sciatic nerve injury. Eur. J. Pharmacol. 2002;447:59–65. doi: 10.1016/s0014-2999(02)01833-2. [DOI] [PubMed] [Google Scholar]
- 12.Ma W, Eisenach JC. Chronic constriction injury of sciatic nerve induces the upregulation of descending inhibitory noradrenergic innervation to the lumbar dorsal horn of mice. Brain Res. 2003;970:110–118. doi: 10.1016/s0006-8993(03)02293-5. [DOI] [PubMed] [Google Scholar]
- 13.Mika J, Li Y, Weihe E, Schafer MK. Relationship of pronociceptin/orphanin FQ and the nociceptin receptor ORL1 with Substance P and calcitonin gene-related peptide expression in dorsal root ganglion of the rat. Neurosci. Lett. 2003;348:190–194. doi: 10.1016/s0304-3940(03)00786-9. [DOI] [PubMed] [Google Scholar]
- 14.Chen Y, Sommer C. Nociceptin and its receptor in rat dorsal root ganglion neurons in neuropathic and inflammatory pain models: implications on pain processing. J. Peripher. Nerv. Syst. 2006;11:232–240. doi: 10.1111/j.1529-8027.2006.0093.x. [DOI] [PubMed] [Google Scholar]
- 15.Chen Y, Sommer C. Activation of the nociceptin opioid system in rat sensory neurons produces antinociceptive effects in inflammatory pain: involvement of inflammatory mediators. J. Neurosci. Res. 2007;85:1478–1488. doi: 10.1002/jnr.21272. [DOI] [PubMed] [Google Scholar]
- 16.Jia Y, Linden DR, Serie JR, Seybold VS. Nociceptin/orphanin FQ binding increases in superficial laminae of the rat spinal cord during persistent peripheral inflammation. Neurosci. Lett. 1998;250:21–24. doi: 10.1016/s0304-3940(98)00430-3. [DOI] [PubMed] [Google Scholar]
- 17.Marti M, Sarubbo S, Latini F, Cavallo M, Eleopra R, Biguzzi S, Lettieri C, Conti C, Simonato M, Zucchini S, Quatrale R, Sensi M, Candeletti S, Romualdi P, Morari M. Brain interstitial nociceptin/orphanin FQ levels are elevated in Parkinson's disease. Mov. Disord. 2010;25:1723–1732. doi: 10.1002/mds.23271. [DOI] [PubMed] [Google Scholar]
- 18.Marti M, Mela F, Budri M, Volta M, Malfacini D, Molinari S, Zaveri NT, Ronzoni S, Petrillo P, Calò G, Morari M. Acute and chronic antiparkinsonian effects of the novel nociceptin/orphanin FQ receptor antagonist NiK-21273 in comparison with SB-612111. Br. J. Pharmacol. 2013;168:863–879. doi: 10.1111/j.1476-5381.2012.02219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Visanji NP, de Bie RMA, Johnston TH, McCreary AC, Brotchie JM, Fox SH. The nociceptin/orphanin FQ (NOP) receptor antagonist J-113397 enhances the effects of levodopa in the MPTP-lesioned nonhuman primate model of Parkinson's disease. Mov. Disord. 2008;23:1922–1925. doi: 10.1002/mds.22086. [DOI] [PubMed] [Google Scholar]
- 20.Viaro R, Sanchez-Pernaute R, Marti M, Trapella C, Isacson O, Morari M. Nociceptin/orphanin FQ receptor blockade attenuates MPTP-induced parkinsonism. Neurobiol. Dis. 2008;30:430–438. doi: 10.1016/j.nbd.2008.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chau DT, Roth RM, Green AI. The neural circuitry of reward and its relevance to psychiatric disorders. Curr. Psychiatry Rep. 2004;6:391–399. doi: 10.1007/s11920-004-0026-8. [DOI] [PubMed] [Google Scholar]
- 22.Di Giannuario A, Pieretti S. Nociceptin differentially affects morphine-induced dopamine release from the nucleus accumbens and nucleus caudate in rats. Peptides. 2000;21:1125–1130. doi: 10.1016/s0196-9781(00)00250-3. [DOI] [PubMed] [Google Scholar]
- 23.Di Giannuario A, Pieretti S, Catalani A, Loizzo A. Orphanin FQ reduces morphine-induced dopamine release in the nucleus accumbens: a microdialysis study in rats. Neurosci. Lett. 1999;272:183–186. doi: 10.1016/s0304-3940(99)00579-0. [DOI] [PubMed] [Google Scholar]
- 24.Murphy NP, Ly HT, Maidment NT. Intracerebroventricular orphanin FQ/nociceptin suppresses dopamine release in the nucleus accumbens of anaesthetized rats. Neuroscience. 1996;75:1–4. doi: 10.1016/0306-4522(96)00322-3. [DOI] [PubMed] [Google Scholar]
- 25.Murphy NP, Maidment NT. Orphanin FQ/nociceptin modulation of mesolimbic dopamine transmission determined by microdialysis. J. Neurochem. 1999;73:179–186. doi: 10.1046/j.1471-4159.1999.0730179.x. [DOI] [PubMed] [Google Scholar]
- 26.Murphy NP, Lee Y, Maidment NT. Orphanin FQ/nociceptin blocks acquisition of morphine place preference. Brain Res. 1999;832:168–170. doi: 10.1016/s0006-8993(99)01425-0. [DOI] [PubMed] [Google Scholar]
- 27.Kotlinska J, Wichmann J, Legowska A, Rolka K, Silberring J. Orphanin FQ/nociceptin but not Ro 65-6570 inhibits the expression of cocaine-induced conditioned place preference. Behav. Pharmacol. 2002;13:229–235. doi: 10.1097/00008877-200205000-00006. [DOI] [PubMed] [Google Scholar]
- 28.Sakoori K, Murphy NP. Central administration of nociceptin/orphanin FQ blocks the acquisition of conditioned place preference to morphine and cocaine, but not conditioned place aversion to naloxone in mice. Psychopharmacology (Berl) 2004;172:129–136. doi: 10.1007/s00213-003-1643-3. [DOI] [PubMed] [Google Scholar]
- 29.Zhao RJ, Woo RS, Jeong MS, Shin BS, Kim DG, Kim KW. Orphanin FQ/nociceptin blocks methamphetamine place preference in rats. Neuroreport. 2003;14:2383–2385. doi: 10.1097/00001756-200312190-00019. [DOI] [PubMed] [Google Scholar]
- 30.Kotlinska J, Rafalski P, Biala G, Dylag T, Rolka K, Silberring J. Nociceptin inhibits acquisition of amphetamine-induced place preference and sensitization to stereotypy in rats. Eur. J. Pharmacol. 2003;474:233–239. doi: 10.1016/s0014-2999(03)02081-8. [DOI] [PubMed] [Google Scholar]
- 31.Florin S, Leroux-Nicollet I, Meunier JC, Costentin J. Autoradiographic localization of [3H]nociceptin binding sites from telencephalic to mesencephalic regions of the mouse brain. Neurosci. Lett. 1997;230:33–36. doi: 10.1016/s0304-3940(97)00470-9. [DOI] [PubMed] [Google Scholar]
- 32.Florin S, Meunier J-C, Costentin J. Autoradiographic localization of [3H]nociceptin binding sites in the rat brain. Brain Res. 2000;880:11–16. doi: 10.1016/s0006-8993(00)02669-x. [DOI] [PubMed] [Google Scholar]
- 33.Bridge KE, Wainwright A, Reilly K, Oliver KR. Autoradiographic localization of (125)i[Tyr(14)] nociceptin/orphanin FQ binding sites in macaque primate CNS. Neuroscience. 2003;118:513–523. doi: 10.1016/s0306-4522(02)00927-2. [DOI] [PubMed] [Google Scholar]
- 34.Kimura Y, Fujita M, Hong J, Lohith TG, Gladding RL, Zoghbi SS, Tauscher JA, Goebl N, Rash KS, Chen Z, Pedregal C, Barth VN, Pike VW, Innis RB. Brain and whole-body imaging in rhesus monkeys of 11C-NOP-1A, a promising PET radioligand for nociceptin/orphanin FQ peptide receptors. J. Nucl. Med. 2011;52:1638–1645. doi: 10.2967/jnumed.111.091181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berthele A, Platzer S, Dworzak D, Schadrack J, Mahal B, Buttner A, Assmus HP, Wurster K, Zieglgansberger W, Conrad B, Tolle TR. [3H]-nociceptin ligand-binding and nociceptin opioid receptor mRNA expression in the human brain. Neuroscience. 2003;121:629–640. doi: 10.1016/s0306-4522(03)00484-6. [DOI] [PubMed] [Google Scholar]
- 36.Lohith TG, Zoghbi SS, Morse CL, Araneta MF, Barth VN, Goebl NA, Tauscher JT, Pike VW, Innis RB, Fujita M. Brain and whole-body imaging of nociceptin/orphanin FQ peptide receptor in humans using the PET ligand 11C-NOP-1A. J. Nucl. Med. 2012;53:385–392. doi: 10.2967/jnumed.111.097162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Witkin JM, Statnick MA, Rorick-Kehn LM, Pintar JE, Ansonoff M, Chen Y, Tucker RC, Ciccocioppo R. The biology of nociceptin/orphanin FQ (N/OFQ) related to obesity, stress, anxiety, mood, and drug dependence. Pharmacol. Ther. 2014;141:283–299. doi: 10.1016/j.pharmthera.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gavioli EC, Calo G. Nociceptin/orphanin FQ receptor antagonists as innovative antidepressant drugs. Pharmacol. Ther. 2013;140:10–25. doi: 10.1016/j.pharmthera.2013.05.008. [DOI] [PubMed] [Google Scholar]
- 39.Schröder W, Lambert DG, Ko MC, Koch T. Functional plasticity of the N/OFQ-NOP receptor system determines analgesic properties of NOP receptor agonists. Br. J. Pharmacol. 2014;171:3777–3800. doi: 10.1111/bph.12744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mogil JS, Pasternak GW. The molecular and behavioral pharmacology of the orphanin FQ/nociceptin peptide and receptor family. Pharmacol. Rev. 2001;53:381–415. [PubMed] [Google Scholar]
- 41.Linz K, Christoph T, Tzschentke TM, Koch T, Schiene K, Gautrois M, Schröder W, Kögel BY, Beier H, Englberger W, Schunk S, De Vry J, Jahnel U, Frosch S. Cebranopadol: A novel potent analgesic nociceptin/orphanin FQ peptide and opioid receptor agonist. J. Pharmacol. Exp. Ther. 2014;349:535–548. doi: 10.1124/jpet.114.213694. [DOI] [PubMed] [Google Scholar]
- 42.Lambert DG, Bird MF, Rowbotham DJ. Cebranopadol: a first in-class example of a nociceptin/orphanin FQ receptor and opioid receptor agonist. Br. J. Anaesthesia. 2015;114:364–366. doi: 10.1093/bja/aeu332. [DOI] [PubMed] [Google Scholar]
- 43.Sałat K, Jakubowska A, Kulig K. Cebranopadol: a first-in-class potent analgesic agent with agonistic activity at nociceptin/orphanin FQ and opioid receptors. Expert Opin. Invest. Drugs. 2015;24:837–844. doi: 10.1517/13543784.2015.1036985. [DOI] [PubMed] [Google Scholar]
- 44.Post A, Smart TS, Krikke-Workel J, Dawson GR, Harmer CJ, Browning M, Jackson K, Kakar R, Mohs R, Statnick M, Wafford K, McCarthy A, Barth V, Witkin JM. A selective nociceptin receptor antagonist to treat depression: Evidence from preclinical and clinical studies. Neuropsychopharmacology. 2015 doi: 10.1038/npp.2015.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eli Lilly & Co Study of the efficacy and safety of LY2940094 in participants with major depressive disorder (MDD) 2014 In https://clinicaltrials.gov/ct2/show/NCT01724112. NCT01724112, Phase 2.
- 46.Eli Lilly & Co A Study of LY2940094 in participants with alcohol dependence. 2014 In https://clinicaltrials.gov/ct2/show/NCT01798303. NCT0198303, Phase 2.
- 47.Satoh A, Sagara T, Sakoh H, Hashimoto M, Nakashima H, Kato T, Goto Y, Mizutani S, Azuma-Kanoh T, Tani T, Okuda S, Okamoto O, Ozaki S, Iwasawa Y, Ohta H, Kawamoto H. Identification of an orally active opioid receptor-like 1 (ORL1) receptor antagonist 4-{3-[(2R)-2,3-dihydroxypropyl]-2-oxo-2,3-dihydro-1H-Page benzimidazol-1-yl}-1-[(1S,3S ,4R)-spiro[bicyclo[2.2.1]heptane-2,1'-cyclopropan]-3-ylmethyl]piperidine as clinical candidate. J. Med. Chem. 2009;52:4091–4094. doi: 10.1021/jm900581g. [DOI] [PubMed] [Google Scholar]
- 48.Merck, Sharp and Dohme Corp Treatment of cognitive impairment in men with schizophrenia (MK5757-005)(COMPLETED) 2015 https://clinicaltrials.gov/ct2/show/results/NCT00848484. NCT00848484, Phase 2.
- 49.Jenck F, Moreau JL, Martin JR, Kilpatrick GJ, Reinscheid RK, Monsma FJ, Jr., Nothacker HP, Civelli O. Orphanin FQ acts as an anxiolytic to attenuate behavioral responses to stress. Proc. Natl. Acad. Sci. U. S. A. 1997;94:14854–14858. doi: 10.1073/pnas.94.26.14854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jenck F, Wichmann J, Dautzenberg FM, Moreau JL, Ouagazzal AM, Martin JR, Lundstrom K, Cesura AM, Poli SM, Roever S, Kolczewski S, Adam G, Kilpatrick G. A synthetic agonist at the orphanin FQ/nociceptin receptor ORL1: anxiolytic profile in the rat. Proc. Natl. Acad. Sci. U. S. A. 2000;97:4938–4943. doi: 10.1073/pnas.090514397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Canuso CM, Hutchison J, Manitpisitkul P, Moyer JA. NOP agonism: A Novel mechanism for the treatment of anxiety and depression; American Society of Clinical Psychopharmacology NCDEU Meeting; Phoenix, AZ. May 29, 2012; Phoenix, AZ: American Society of Clinical Psychopharmacology; 2012. p. 19. 2012. [Google Scholar]
- 52.Rizzi A, Rizzi D, Marzola G, Regoli D, Larsen BD, Petersen JS, Calo' G. Pharmacological characterization of the novel nociceptin/orphanin FQ receptor ligand, ZP120: in vitro and in vivo studies in mice. Br. J. Pharmacol. 2002;137:369–374. doi: 10.1038/sj.bjp.0704894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kapusta DR, Thorkildsen C, Kenigs VA, Meier E, Vinge MM, Quist C, Petersen JS. Pharmacodynamic characterization of ZP120 (Ac-RYYRWKKKKKKK-NH2), a novel, functionally selective nociceptin/orphanin FQ peptide receptor partial agonist with sodium-potassium-sparing aquaretic activity. J. Pharmacol. Exp. Ther. 2005;314:652–660. doi: 10.1124/jpet.105.083436. [DOI] [PubMed] [Google Scholar]
- 54.Fischetti C, Rizzi A, Gavioli EC, Marzola G, Trapella C, Guerrini R, Petersen JS, Calo G. Further studies on the pharmacological features of the nociceptin/orphanin FQ receptor ligand ZP120. Peptides. 2009;30:248–255. doi: 10.1016/j.peptides.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 55.van Deurs US, Hadrup N, Petersen JS, Christensen S, Jonassen TE. Nociceptin/orphanin FQ peptide receptor agonist Ac-RYYRWKKKKKKK-NH2 (ZP120) induces antinatriuresis in rats by stimulation of amiloride-sensitive sodium reabsorption. J. Pharmacol. Exp. Ther. 2009;328:533–539. doi: 10.1124/jpet.108.144774. [DOI] [PubMed] [Google Scholar]
- 56.SERODUS PHARMA Pipeline [Access date: September 20, 2015]; SER100TRISH. http://www.serodus.com/Pipeline/SER100TRISH.
- 57.Woodcock A, McLeod RL, Sadeh J, Smith JA. The efficacy of a NOP1 agonist (SCH486757) in subacute cough. Lung. 2010;188(Suppl 1):S47–52. doi: 10.1007/s00408-009-9197-8. [DOI] [PubMed] [Google Scholar]
- 58.McLeod RL, Tulshian DB, Sadeh J. Where are the new cough treatments: a debriefing of recent clinical proof-of-concept trials with the NOP agonist SCH 486757. Pharmacology. 2011;88:50–54. doi: 10.1159/000328782. [DOI] [PubMed] [Google Scholar]
- 59. [Access date: June 30, 2015];JTC 801 Japan Tobacco discontinued, Japan, UK (pain) http://business.highbeam.com/436989/article-1G1-109477099/jtc-801-japan-tobacco-discontinued-japan-uk.
- 60.Lazzeri M, Calo G, Spinelli M, Malaguti S, Guerrini R, Salvadori S, Beneforti P, Regoli D, Turini D. Daily intravesical instillation of 1 mg nociceptin/orphanin FQ for the control of neurogenic detrusor overactivity: a multicenter, placebo controlled, randomized exploratory study. J. Urol. 2006;176:2098–2102. doi: 10.1016/j.juro.2006.07.025. [DOI] [PubMed] [Google Scholar]
- 61.Lazzeri M, Calo G, Spinelli M, Guerrini R, Beneforti P, Sandri S, Zanollo A, Regoli D, Turini D. Urodynamic and clinical evidence of acute inhibitory effects of intravesical nociceptin/orphanin FQ on detrusor overactivity in humans: a pilot study. J. Urol. 2001;166:2237–2240. [PubMed] [Google Scholar]
- 62.Lazzeri M, Calo G, Spinelli M, Guerrini R, Salvadori S, Beneforti P, Sandri S, Regoli D, Turini D. Urodynamic effects of intravesical nociceptin/orphanin FQ in neurogenic detrusor overactivity: a randomized, placebo-controlled, double-blind study. Urology. 2003;61:946–950. doi: 10.1016/s0090-4295(02)02587-6. [DOI] [PubMed] [Google Scholar]
- 63.Zaveri N. Peptide and nonpeptide ligands for the nociceptin/orphanin FQ receptor ORL1: research tools and potential therapeutic agents. Life Sci. 2003;73:663–678. doi: 10.1016/s0024-3205(03)00387-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ronzoni S, Peretto I, Giardina GAM. Lead generation and lead optimisation approaches in the discovery of selective, non-peptide ORL-1 receptor agonists and antagonists. Expert Opin. Ther. Patents. 2001;11:525–546. [Google Scholar]
- 65.Bignan GC, Connolly PJ, Middleton SA. Recent advances towards the discovery of ORL-1 receptor agonists and antagonists. Expert Opin. Ther. Patents. 2005;15:357–388. [Google Scholar]
- 66.Lambert DG. The nociceptin/orphanin FQ receptor: a target with broad therapeutic potential. Nat. Rev. Drug Discov. 2008;7:694–710. doi: 10.1038/nrd2572. [DOI] [PubMed] [Google Scholar]
- 67.Largent-Milnes TM, Vanderah TW. Recently patented and promising ORL-1 ligands: where have we been and where are we going? Expert Opin. Ther. Patents. 2010;20:291–305. doi: 10.1517/13543771003602004. [DOI] [PubMed] [Google Scholar]
- 68.Rover S, Adam G, Cesura AM, Galley G, Jenck F, Monsma FJ, Jr., Wichmann J, Dautzenberg FM. High-affinity, non-peptide agonists for the ORL1 (orphanin FQ/nociceptin) receptor. J. Med. Chem. 2000;43:1329–1338. doi: 10.1021/jm991129q. [DOI] [PubMed] [Google Scholar]
- 69.Wichmann J, Adam G, Rover S, Hennig M, Scalone M, Cesura AM, Dautzenberg FM, Jenck F. Synthesis of (1S,3aS)-8-(2,3,3a,4,5, 6-hexahydro-1H-phenalen-1-yl)-1-phenyl-1,3,8-triaza-spiro[4. 5]decan-4-one, a potent and selective orphanin FQ (OFQ) receptor agonist with anxiolytic-like properties. Eur. J. Med. Chem. 2000;35:839–851. doi: 10.1016/s0223-5234(00)00171-9. [DOI] [PubMed] [Google Scholar]
- 70.Dautzenberg FM, Wichmann J, Higelin J, Py-Lang G, Kratzeisen C, Malherbe P, Kilpatrick GJ, Jenck F. Pharmacological characterization of the novel nonpeptide orphanin FQ/nociceptin receptor agonist Ro 64-6198: rapid and reversible desensitization of the ORL1 receptor in vitro and lack of tolerance in vivo. J. Pharmacol. Exp. Ther. 2001;298:812–819. [PubMed] [Google Scholar]
- 71.Varty GB, Hyde LA, Hodgson RA, Lu SX, McCool MF, Kazdoba TM, Del Vecchio RA, Guthrie DH, Pond AJ, Grzelak ME, Xu X, Korfmacher WA, Tulshian D, Parker EM, Higgins GA. Characterization of the nociceptin receptor (ORL-1) agonist, Ro64-6198, in tests of anxiety across multiple species. Psychopharmacology (Berl) 2005;182:132–143. doi: 10.1007/s00213-005-0041-4. [DOI] [PubMed] [Google Scholar]
- 72.Higgins GA, Grottick AJ, Ballard TM, Richards JG, Messer J, Takeshima H, Pauly-Evers M, Jenck F, Adam G, Wichmann J. Influence of the selective ORL1 receptor agonist, Ro64-6198, on rodent neurological function. Neuropharmacology. 2001;41:97–107. doi: 10.1016/s0028-3908(01)00048-x. [DOI] [PubMed] [Google Scholar]
- 73.Lu SX, Higgins GA, Hodgson RA, Hyde LA, Del Vecchio RA, Guthrie DH, Kazdoba T, McCool MF, Morgan CA, Bercovici A, Ho GD, Tulshian D, Parker EM, Hunter JC, Varty GB. The anxiolytic-like profile of the nociceptin receptor agonist, endo-8-[bis(2-chlorophenyl)methyl]-3-phenyl-8-azabicyclo[3.2.1]octane-3-carboxami de (SCH 655842): comparison of efficacy and side effects across rodent species. Eur. J. Pharmacol. 2011;661:63–71. doi: 10.1016/j.ejphar.2011.04.034. [DOI] [PubMed] [Google Scholar]
- 74.Toll L, Khroyan TV, Polgar WE, Jiang F, Olsen C, Zaveri NT. Comparison of the anti-nociceptive and anti-rewarding profiles of novel bifunctional nociceptin/orphanin FQ receptor (NOPr)-mu opioid receptor (MOPr) ligands: Implications for therapeutic applications. J. Pharmacol. Exp. Ther. 2009;331:954–964. doi: 10.1124/jpet.109.157446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hirao A, Imai A, Sugie Y, Yamada Y, Hayashi S, Toide K. Pharmacological characterization of the newly synthesized nociceptin/orphanin FQ-receptor agonist 1-[1-(1-methylcyclooctyl)-4-piperidinyl]-2-[(3R)-3-piperidinyl]-1H-benzimidazole as an anxiolytic agent. J. Pharmacol. Sci. 2008;106:361–368. doi: 10.1254/jphs.fp0071742. [DOI] [PubMed] [Google Scholar]
- 76.Goeldner C, Spooren W, Wichmann J, Prinssen EP. Further characterization of the prototypical nociceptin/orphanin FQ peptide receptor agonist Ro 64-6198 in rodent models of conflict anxiety and despair. Psychopharmacology (Berl) 2012;222:203–214. doi: 10.1007/s00213-012-2636-x. [DOI] [PubMed] [Google Scholar]
- 77.Rizzi D, Bigoni R, Rizzi A, Jenck F, Wichmann J, Guerrini R, Regoli D, Calo G. Effects of Ro 64-6198 in nociceptin/orphanin FQ-sensitive isolated tissues. Naunyn-Schmiedebergs Arch. Pharmacol. 2001;363:551–555. doi: 10.1007/s002100100399. [DOI] [PubMed] [Google Scholar]
- 78.Ko MC, Woods JH, Fantegrossi WE, Galuska CM, Wichmann J, Prinssen EP. Behavioral effects of a synthetic agonist selective for nociceptin/orphanin FQ peptide receptors in monkeys. Neuropsychopharmacology. 2009;34:2088–2096. doi: 10.1038/npp.2009.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Podlesnik CA, Ko MC, Winger G, Wichmann J, Prinssen EP, Woods JH. The effects of nociceptin/orphanin FQ receptor agonist Ro 64-6198 and diazepam on antinociception and remifentanil self-administration in rhesus monkeys. Psychopharmacology (Berl) 2011;213:53–60. doi: 10.1007/s00213-010-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zeilhofer HU, Calo G. Nociceptin/orphanin FQ and its receptor-potential targets for pain therapy? J. Pharmacol. Exp. Ther. 2003;306:423–429. doi: 10.1124/jpet.102.046979. [DOI] [PubMed] [Google Scholar]
- 81.Whiteside GT, Kyle DJ. A Review of the NOP (ORL-1)-Nociceptin/orphanin FQ system covering receptor structure, distribution, role in analgesia and reward and interactions with other receptors. Research and Development of Opioid-Related Ligands; American Chemical Society Symposium Series; Washington DC: Oxford University Press; 2013. pp. 327–368. [Google Scholar]
- 82.Sukhtankar D, Ko M-C. Pharmacological investigation of NOP-related ligands as analgesics without abuse liability. Research and Development of Opioid-Related Ligands; American Chemical Society Symposium Series; Washington DC: Oxford University Press; 2013. pp. 393–416. [Google Scholar]
- 83.Lin AP, Ko MC. The therapeutic potential of nociceptin/orphanin FQ receptor agonists as analgesics without abuse liability. ACS Chem. Neurosci. 2013;4:214–224. doi: 10.1021/cn300124f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kotlinska J, Wichmann J, Rafalski P, Talarek S, Dylag T, Silberring J. Non-peptidergic OP4 receptor agonist inhibits morphine antinociception but does not influence morphine dependence. Neuroreport. 2003;14:601–604. doi: 10.1097/00001756-200303240-00015. [DOI] [PubMed] [Google Scholar]
- 85.Reiss D, Wichmann J, Tekeshima H, Kieffer BL, Ouagazzal AM. Effects of nociceptin/orphanin FQ receptor (NOP) agonist, Ro64-6198, on reactivity to acute pain in mice: comparison to morphine. Eur. J. Pharmacol. 2008;579:141–148. doi: 10.1016/j.ejphar.2007.10.031. [DOI] [PubMed] [Google Scholar]
- 86.Sukhtankar DD, Lee H, Rice KC, Ko MC. Differential effects of opioid-related ligands and NSAIDs in nonhuman primate models of acute and inflammatory pain. Psychopharmacology (Berl) 2014;231:1377–1387. doi: 10.1007/s00213-013-3341-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ding H, Hayashida K, Suto T, Sukhtankar DD, Kimura M, Mendenhall V, Ko MC. Supraspinal actions of nociceptin/orphanin FQ, morphine and substance P in regulating pain and itch in non-human primates. Br. J. Pharmacol. 2015;172:3302–3312. doi: 10.1111/bph.13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ko MC, Naughton NN. Antinociceptive effects of nociceptin/orphanin FQ administered intrathecally in monkeys. J. Pain. 2009;10:509–514. doi: 10.1016/j.jpain.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ko MC, Wei H, Woods JH, Kennedy RT. Effects of intrathecally administered nociceptin/orphanin FQ in monkeys: behavioral and mass spectrometric studies. J. Pharmacol. Exp. Ther. 2006;318:1257–1264. doi: 10.1124/jpet.106.106120. [DOI] [PubMed] [Google Scholar]
- 90.Hu E, Calo G, Guerrini R, Ko MC. Long-lasting antinociceptive spinal effects in primates of the novel nociceptin/orphanin FQ receptor agonist UFP-112. Pain. 2010;148:107–113. doi: 10.1016/j.pain.2009.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Obara I, Przewlocki R, Przewlocka B. Spinal and local peripheral antiallodynic activity of Ro64-6198 in neuropathic pain in the rat. Pain. 2005;116:17–25. doi: 10.1016/j.pain.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 92.Le Pen G, Wichmann J, Moreau JL, Jenck F. The orphanin receptor agonist RO 64-6198 does not induce place conditioning in rats. Neuroreport. 2002;13:451–454. doi: 10.1097/00001756-200203250-00018. [DOI] [PubMed] [Google Scholar]
- 93.Rutten K, De Vry J, Bruckmann W, Tzschentke TM. Effects of the NOP receptor agonist Ro65-6570 on the acquisition of opiate- and psychostimulant-induced conditioned place preference in rats. Eur. J. Pharmacol. 2010;645:119–126. doi: 10.1016/j.ejphar.2010.07.036. [DOI] [PubMed] [Google Scholar]
- 94.Recker MD, Higgins GA. The opioid receptor like-1 receptor agonist Ro 64-6198 (1S,3aS-8-2,3,3a,4,5,6-hexahydro-1H-phenalen-1-yl-1-phenyl-1,3,8-triaza-spiro[4.5]decan-4-one) produces a discriminative stimulus in rats distinct from that of a μ, κ, and δ opioid receptor agonist cue. J. Pharmacol. Exp. Ther. 2004;311:652–658. doi: 10.1124/jpet.104.071423. [DOI] [PubMed] [Google Scholar]
- 95.Zaveri NT. The Nociceptin/orphanin FQ receptor (NOP) as a target for drug abuse medications. Curr. Topics in Med. Chem. 2011;11:1151–1156. doi: 10.2174/156802611795371341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shoblock JR, Wichmann J, Maidment NT. The effect of a systemically active ORL-1 agonist, Ro 64-6198, on the acquisition, expression, extinction, and reinstatement of morphine conditioned place preference. Neuropharmacology. 2005;49:439–446. doi: 10.1016/j.neuropharm.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 97.Kuzmin A, Sandin J, Terenius L, Ogren SO. Acquisition, expression, and reinstatement of ethanol-induced conditioned place preference in mice: effects of opioid receptor-like 1 receptor agonists and naloxone. J. Pharmacol. Exp. Ther. 2003;304:310–318. doi: 10.1124/jpet.102.041350. [DOI] [PubMed] [Google Scholar]
- 98.Kuzmin A, Kreek MJ, Bakalkin G, Liljequist S. The nociceptin/orphanin FQ receptor agonist Ro 64-6198 reduces alcohol self-administration and prevents relapse-like alcohol drinking. Neuropsychopharmacology. 2007;32:902–910. doi: 10.1038/sj.npp.1301169. [DOI] [PubMed] [Google Scholar]
- 99.Economidou D, Fedeli A, Fardon RM, Weiss F, Massi M, Ciccocioppo R. Effect of novel nociceptin/orphanin FQ-NOP receptor ligands on ethanol drinking in alcohol-preferring msP rats. Peptides. 2006;27:3299–3306. doi: 10.1016/j.peptides.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ciccocioppo R, Stopponi S, Economidou D, Kuriyama M, Kinoshita H, Heilig M, Roberto M, Weiss F, Teshima K. Chronic treatment with novel brain-penetrating selective NOP receptor agonist MT-7716 reduces alcohol drinking and seeking in the rat. Neuropsychopharmacology. 2014;39:2601–2610. doi: 10.1038/npp.2014.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Heinig K, Kratochwil N, Bucheli F, Thomae A. Bioanalytics and pharmacokinetics of the nociceptin/orphanin FQ peptide receptor agonist RO0646198 in Wistar rats and cynomolgus monkeys. J. Chromatogr. B. 2010;878:2101–2105. doi: 10.1016/j.jchromb.2010.06.013. [DOI] [PubMed] [Google Scholar]
- 102.Wichmann J, Adam G, Rover S, Cesura AM, Dautzenberg FM, Jenck F. 8-acenaphthen-1-yl-1-phenyl-1,3,8-triaza-spiro[4.5]decan-4-one derivatives as orphanin FQ receptor agonists. Bioorg. Med. Chem. Lett. 1999;9:2343–2348. doi: 10.1016/s0960-894x(99)00385-6. [DOI] [PubMed] [Google Scholar]
- 103.Hashiba E, Harrison C, Galo G, Guerrini R, Rowbotham DJ, Smith G, Lambert DG. Characterisation and comparison of novel ligands for the nociceptin/orphanin FQ receptor. Naunyn-Schmiedebergs Arch. Pharmacol. 2001;363:28–33. doi: 10.1007/s002100000327. [DOI] [PubMed] [Google Scholar]
- 104.Schiene K, Tzschentke TM, Schröder W, Christoph T. Mechanical hyperalgesia in rats with diabetic polyneuropathy is selectively inhibited by local peripheral nociceptin/orphanin FQ receptor and μ-opioid receptor agonism. Eur. J. Pharmacol. 2015;754:61–65. doi: 10.1016/j.ejphar.2015.01.049. [DOI] [PubMed] [Google Scholar]
- 105.Schiene K, Christoph T, Kögel B, Tzschentke T. Antinociceptive, antihyperalgesic and antiallodynic activity of the NOP receptor agonist Ro65-6570 in rodent models of pain; EFIC–8th "Pain in Europe" Congress, 2013; 2013.pp. 9–12. [Google Scholar]
- 106.Byford AJ, Anderson A, Jones PS, Palin R, Houghton AK. The hypnotic, electroencephalographic, and antinociceptive properties of nonpeptide ORL1 receptor agonists after intravenous injection in rodents. Anesthesia & Analgesia. 2007;104:174–179. doi: 10.1213/01.ane.0000250403.88649.51. [DOI] [PubMed] [Google Scholar]
- 107.Ho GD, Anthes J, Bercovici A, Caldwell JP, Cheng KC, Cui X, Fawzi A, Fernandez X, Greenlee WJ, Hey J, Korfmacher W, Lu SX, McLeod RL, Ng F, Torhan AS, Tan Z, Tulshian D, Varty GB, Xu X, Zhang H. The discovery of tropane derivatives as nociceptin receptor ligands for the management of cough and anxiety. Bioorg. Med. Chem. Lett. 2009;19:2519–2523. doi: 10.1016/j.bmcl.2009.03.031. [DOI] [PubMed] [Google Scholar]
- 108.Varty GB, Lu SX, Morgan CA, Cohen-Williams ME, Hodgson RA, Smith-Torhan A, Zhang H, Fawzi AB, Graziano MP, Ho GD, Matasi J, Tulshian D, Coffin VL, Carey GJ. The anxiolytic-like effects of the novel, orally active nociceptin opioid receptor agonist 8-[bis(2-methylphenyl)methyl]-3-phenyl-8-azabicyclo[3.2.1]octan-3-ol (SCH 221510) J. Pharmacol. Exp. Ther. 2008;326:672–682. doi: 10.1124/jpet.108.136937. [DOI] [PubMed] [Google Scholar]
- 109.Hayashi S, Hirao A, Imai A, Nakamura H, Murata Y, Ohashi K, Nakata E. Novel non-peptide nociceptin/orphanin FQ receptor agonist, 1-[1-(1-methylcyclooctyl)-4-piperidinyl]-2-[(3R)-3-piperidinyl]-1H-benzimidazole: design, synthesis, and structure-activity relationship of oral receptor occupancy in the brain for orally potent antianxiety drug. J. Med. Chem. 2009;52:610–625. doi: 10.1021/jm7012979. [DOI] [PubMed] [Google Scholar]
- 110.Sukhtankar DD, Lagorio CH, Ko M-C. Effects of the NOP agonist SCH221510 on producing and attenuating reinforcing effects as measured by drug self-administration in rats. Eur. J. Pharmacol. 2014;745:182–189. doi: 10.1016/j.ejphar.2014.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cremeans CM, Gruley E, Kyle DJ, Ko MC. Roles of mu-opioid receptors and nociceptin/orphanin FQ peptide receptors in buprenorphine-induced physiological responses in primates. J. Pharmacol. Exp. Ther. 2012;343:72–81. doi: 10.1124/jpet.112.194308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kangas BD, Bergman J. Operant nociception in nonhuman primates. Pain. 2014;155:1821–1828. doi: 10.1016/j.pain.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fichna J, Sobczak M, Mokrowiecka A, Cygankiewicz A, Zakrzewski P, Cenac N, Sałaga M, Timmermans JP, Vergnolle N, Małecka-Panas E. Activation of the endogenous nociceptin system by selective nociceptin receptor agonist SCH 221510 produces antitransit and antinociceptive effect: a novel strategy for treatment of diarrhea-predominant IBS. Neurogastroenterol. Motil. 2014;26:1539–1550. doi: 10.1111/nmo.12390. [DOI] [PubMed] [Google Scholar]
- 114.Sobczak M, Mokrowiecka A, Cygankiewicz AI, Zakrzewski PK, Salaga M, Storr M, Kordek R, Malecka-Panas E, Krajewska WM, Fichna J. Anti-inflammatory and antinociceptive action of an orally available nociceptin receptor agonist SCH 221510 in a mouse model of inflammatory bowel diseases. J. Pharmacol. Exp. Ther. 2014;348:401–409. doi: 10.1124/jpet.113.209825. [DOI] [PubMed] [Google Scholar]
- 115.Agostini S, Petrella C. The endogenous nociceptin/orphanin FQ-NOP receptor system as a potential therapeutic target for intestinal disorders. Neurogastroenterol. Motil. 2014;26:1519–1526. doi: 10.1111/nmo.12460. [DOI] [PubMed] [Google Scholar]
- 116.McLeod RL, Tulshian DB, Bolser DC, Varty GB, Baptista M, Fernandez X, Parra LE, Zimmer JC, Erickson CH, Ho GD, Jia Y, Ng FW, Korfmacher W, Xu X, Veals J, Smith-Torhan A, Wainhaus S, Fawzi AB, Austin TM, van Heek M, Hey JA. Pharmacological profile of the NOP agonist and cough suppressing agent SCH 486757 (8-[bis(2-chlorophenyl)methyl]-3-(2-pyrimidinyl)-8-azabicyclo[3.2.1]octan-3-ol) in preclinical models. Eur. J. Pharmacol. 2010;630:112–120. doi: 10.1016/j.ejphar.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.McLeod RL, Tulshian DB, Ho GD, Fernandez X, Bolser DC, Parra LE, Zimmer JC, Erickson CH, Fawzi AB, Jayappa H, Lehr C, Erskine J, Smith-Torhan A, Zhang H, Hey JA. Effect of a novel NOP receptor agonist (SCH 225288) on guinea pig irritant-evoked, feline mechanically induced and canine infectious tracheobronchitis cough. Pharmacology. 2009;84:153–161. doi: 10.1159/000235601. [DOI] [PubMed] [Google Scholar]
- 118.McLeod RL, Parra LE, Mutter JC, Erickson CH, Carey GJ, Tulshian DB, Fawzi AB, Smith-Torhan A, Egan RW, Cuss FM, Hey JA. Nociceptin inhibits cough in the guinea-pig by activation of ORL(1) receptors. Br. J. Pharmacol. 2001;132:1175–1178. doi: 10.1038/sj.bjp.0703954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bolser DC, McLeod RL, Tulshian DB, Hey JA. Antitussive action of nociceptin in the cat. Eur. J. Pharmacol. 2001;430:107–111. doi: 10.1016/s0014-2999(01)01244-4. [DOI] [PubMed] [Google Scholar]
- 120.McLeod RL, Bolser DC, Jia Y, Parra LE, Mutter JC, Wang X, Tulshian DB, Egan RW, Hey JA. Antitussive effect of nociceptin/orphanin FQ in experimental cough models. Pulm. Pharmacol. Ther. 2002;15:213–216. doi: 10.1006/pupt.2002.0357. [DOI] [PubMed] [Google Scholar]
- 121.McLeod RL, Jia Y, Fernandez X, Parra LE, Wang X, Tulshian DB, Kiselgof EJ, Tan Z, Fawzi AB, Smith-Torhan A, Zhang H, Hey JA. Antitussive profile of the NOP agonist Ro-64-6198 in the guinea pig. Pharmacology. 2004;71:143–149. doi: 10.1159/000077448. [DOI] [PubMed] [Google Scholar]
- 122.Lee MG, Undem BJ, Brown C, Carr MJ. Effect of nociceptin in acid-evoked cough and airway sensory nerve activation in guinea pigs. Am. J. Respir. Crit. Care Med. 2006;173:271–275. doi: 10.1164/rccm.200507-1043OC. [DOI] [PubMed] [Google Scholar]
- 123.de Guglielmo G, Martin-Fardon R, Teshima K, Ciccocioppo R, Weiss F. MT-7716, a potent NOP receptor agonist, preferentially reduces ethanol seeking and reinforcement in post-dependent rats. Addict. Biol. 2015;20:643–651. doi: 10.1111/adb.12157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Teshima K, Minoguchi M, Tounai S, Ashimori A, Eguchi J, Allen CN, Shibata S. Nonphotic entrainment of the circadian body temperature rhythm by the selective ORL1 receptor agonist W-212393 in rats. Br. J. Pharmacol. 2005;146:33–40. doi: 10.1038/sj.bjp.0706311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zaveri NT, Jiang F, Olsen Cris M, Deschamps Jeffrey R, Parrish D, Polgar W, Toll L. A novel series of piperidin-4-yl-1,3-dihydroindol-2-ones as agonist and antagonist ligands at the nociceptin receptor. J. Med.Chem. 2004;47:2973–2976. doi: 10.1021/jm034249d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zaveri N, Jiang F, Olsen C, Polgar W, Toll L. Small-molecule agonists and antagonists of the opioid receptor-like receptor (ORL1, NOP): ligand-based analysis of structural factors influencing intrinsic activity at NOP. AAPS J. 2005;7:E345–352. doi: 10.1208/aapsj070234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hayashi S, Nakata E, Morita A, Mizuno K, Yamamura K, Kato A, Ohashi K. Discovery of {1-[4-(2-{hexahydropyrrolo[3,4-c]pyrrol-2(1H)-yl}-1H-benzimidazol-1-yl)piperidin-1-yl]cyclooctyl}methanol, systemically potent novel non-peptide agonist of nociceptin/orphanin FQ receptor as analgesic for the treatment of neuropathic pain: design, synthesis, and structure-activity relationships. Bioorg. Med. Chem. 2010;18:7675–7699. doi: 10.1016/j.bmc.2010.07.034. [DOI] [PubMed] [Google Scholar]
- 128.Khroyan TV, Polgar WE, Orduna J, Montenegro J, Jiang F, Zaveri NT, Toll L. Differential effects of nociceptin/orphanin FQ (NOP) receptor agonists in acute versus chronic pain: Studies with bifunctional NOP/mu receptor agonists in the sciatic nerve ligation chronic pain model in mice. J. Pharmacol. Exp. Ther. 2011;339:687–693. doi: 10.1124/jpet.111.184663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Vang D, Paul JA, Nguyen J, Tran H, Vincent L, Yasuda D, Zaveri NT, Gupta K. Small-molecule nociceptin receptor agonist ameliorates mast cell activation and pain in sickle mice. Haematologica. 2015;12:1517–1525. doi: 10.3324/haematol.2015.128736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Murphy NP. The nociceptin/orphanin FQ system as a target for treating alcoholism. CNS Neurol. Disord. Drug Targets. 2010;9:87–93. doi: 10.2174/187152710790966713. [DOI] [PubMed] [Google Scholar]
- 131.Economidou D, Cippitelli A, Stopponi S, Braconi S, Clementi S, Ubaldi M, Martin-Fardon R, Weiss F, Massi M, Ciccocioppo R. Activation of brain NOP receptors attenuates acute and protracted alcohol withdrawal symptoms in the rat. Alcoholism: Clin. Exp. Res. 2011;35:747–755. doi: 10.1111/j.1530-0277.2010.01392.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Martin-Fardon R, Zorrilla EP, Ciccocioppo R, Weiss F. Role of innate and drug-induced dysregulation of brain stress and arousal systems in addiction: Focus on corticotropin-releasing factor, nociceptin/orphanin FQ, and orexin/hypocretin. Brain Res. 2010;1314:145–161. doi: 10.1016/j.brainres.2009.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Rodi D, Zucchini S, Simonato M, Cifani C, Massi M, Polidori C. Functional antagonism between nociceptin/orphanin FQ (N/OFQ) and corticotropin-releasing factor (CRF) in the rat brain: evidence for involvement of the bed nucleus of the stria terminalis. Psychopharmacology. 2008;196:523–531. doi: 10.1007/s00213-007-0985-7. [DOI] [PubMed] [Google Scholar]
- 134.Khroyan TV, Polgar W, Toll L, Jiang F, Zaveri NT. AT-202, a nociceptin receptor agonist, blocks drug- and stress-induced reinstatement of extinguished cocaine conditioned place preference. Society for Neuroscience Annual Meeting; San Diego, CA. 2013.pp. 733–724. [Google Scholar]
- 135.Przydzial MJ, Heisler LK. Nociceptin/orphanin FQ peptide receptor as a therapeutic target for obesity. Mini Rev. Med. Chem. 2008;8:796–811. doi: 10.2174/138955708784912139. [DOI] [PubMed] [Google Scholar]
- 136.Calo G, Guerrini R, Rizzi A, Salvadori S, Burmeister M, Kapusta DR, Lambert DG, Regoli D. UFP-101, a peptide antagonist selective for the nociceptin/orphanin FQ receptor. CNS Drug Rev. 2005;11:97–112. doi: 10.1111/j.1527-3458.2005.tb00264.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kawamoto H, Ozaki S, Itoh Y, Miyaji M, Arai S, Nakashima H, Kato T, Ohta H, Iwasawa Y. Discovery of the first potent and selective small molecule opioid receptor-like (ORL1) antagonist: 1-[(3R,4R)-1-cyclooctylmethyl-3- hydroxymethyl-4-piperidyl]-3-ethyl-1, 3-dihydro-2H-benzimidazol-2-one (J-113397) J. Med. Chem. 1999;42:5061–5063. doi: 10.1021/jm990517p. [DOI] [PubMed] [Google Scholar]
- 138.Ozaki S, Kawamoto H, Itoh Y, Miyaji M, Azuma T, Ichikawa D, Nambu H, Iguchi T, Iwasawa Y, Ohta H. In vitro and in vivo pharmacological characterization of J-113397, a potent and selective non-peptidyl ORL1 receptor antagonist. Eur. J. Pharmacol. 2000;402:45–53. doi: 10.1016/s0014-2999(00)00520-3. [DOI] [PubMed] [Google Scholar]
- 139.Zaratin PF, Petrone G, Sbacchi M, Garnier M, Fossati C, Petrillo P, Ronzoni S, Giardina GA, Scheideler MA. Modification of nociception and morphine tolerance by the selective opiate receptor-like orphan receptor antagonist (−)-cis-1-methyl-7-[[4-(2,6-dichlorophenyl)piperidin-1-yl]methyl]-6,7,8,9- tetrahydro-5H-benzocyclohepten-5-ol (SB-612111) J. Pharmacol. Exp. Ther. 2004;308:454–461. doi: 10.1124/jpet.103.055848. [DOI] [PubMed] [Google Scholar]
- 140.Spagnolo B, Carrà G, Fantin M, Fischetti C, Hebbes C, McDonald J, Barnes TA, Rizzi A, Trapella C, Fanton G, Morari M, Lambert DG, Regoli D, Calò G. Pharmacological characterization of the nociceptin/orphanin FQ Receptor antagonist SB-612111 [(−)-cis-1-methyl-7-[[4-(2,6-dichlorophenyl)piperidin-1-yl]methyl]-6,7,8,9-tetrahydro-5H-benzocyclohepten-5-ol]: In vitro studies. J. Pharmacol. Exp. Ther. 2007;321:961–967. doi: 10.1124/jpet.106.116764. [DOI] [PubMed] [Google Scholar]
- 141.Rizzi A, Gavioli EC, Marzola G, Spagnolo B, Zucchini S, Ciccocioppo R, Trapella C, Regoli D, Calo G. Pharmacological characterization of the nociceptin/orphanin FQ receptor antagonist SB-612111 [(−)-cis-1-methyl-7-[[4-(2,6-dichlorophenyl)piperidin-1-yl]methyl]-6,7,8,9-tetrah ydro-5H-benzocyclohepten-5-ol]: in vivo studies. J. Pharmacol. Exp. Ther. 2007;321:968–974. doi: 10.1124/jpet.106.116780. [DOI] [PubMed] [Google Scholar]
- 142.Gavioli EC, Marzola G, Guerrini R, Bertorelli R, Zucchini S, De Lima TC, Rae GA, Salvadori S, Regoli D, Calo G. Blockade of nociceptin/orphanin FQ-NOP receptor signalling produces antidepressant-like effects: pharmacological and genetic evidences from the mouse forced swimming test. Eur. J. Neurosci. 2003;17:1987–1990. doi: 10.1046/j.1460-9568.2003.02603.x. [DOI] [PubMed] [Google Scholar]
- 143.Gavioli EC, Calo G. Antidepressant- and anxiolytic-like effects of nociceptin/orphanin FQ receptor ligands. Naunyn-Schmiedebergs Arch. Pharmacol. 2006;372:319–330. doi: 10.1007/s00210-006-0035-8. [DOI] [PubMed] [Google Scholar]
- 144.Redrobe J, Calo G, Regoli D, Quirion R. Nociceptin receptor antagonists display antidepressant-like properties in the mouse forced swimming test. Naunyn-Schmiedeberg Arch. Pharmacol. 2002;365:164–167. doi: 10.1007/s00210-001-0511-0. [DOI] [PubMed] [Google Scholar]
- 145.Vitale G, Ruggieri V, Filaferro M, Frigeri C, Alboni S, Tascedda F, Brunello N, Guerrini R, Cifani C, Massi M. Chronic treatment with the selective NOP receptor antagonist [Nphe 1, Arg 14, Lys 15]N/OFQ-NH 2 (UFP-101) reverses the behavioural and biochemical effects of unpredictable chronic mild stress in rats. Psychopharmacology (Berl) 2009;207:173–189. doi: 10.1007/s00213-009-1646-9. [DOI] [PubMed] [Google Scholar]
- 146.Bigoni R, Calo G, Rizzi A, Guerrini R, De Risi C, Hashimoto Y, Hashiba E, Lambert DG, Regoli D. In vitro characterization of J-113397, a non-peptide nociceptin/orphanin FQ receptor antagonist. Naunyn-Schmiedebergs Arch. Pharmacol. 2000;361:565–568. doi: 10.1007/s002100000220. [DOI] [PubMed] [Google Scholar]
- 147.Koizumi M, Sakoori K, Midorikawa N, Murphy NP. The NOP (ORL1) receptor antagonist Compound B stimulates mesolimbic dopamine release and is rewarding in mice by a non-NOP-receptor-mediated mechanism. Br. J. Pharmacol. 2004;143:53–62. doi: 10.1038/sj.bjp.0705906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Marti M, Mela F, Fantin M, Zucchini S, Brown JM, Witta J, Di Benedetto M, Buzas B, Reinscheid RK, Salvadori S, Guerrini R, Romualdi P, Candeletti S, Simonato M, Cox BM, Morari M. Blockade of nociceptin/orphanin FQ transmission attenuates symptoms and neurodegeneration associated with Parkinson's disease. J. Neurosci. 2005;25:9591–9601. doi: 10.1523/JNEUROSCI.2546-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Marti M, Mela F, Veronesi C, Guerrini R, Salvadori S, Federici M, Mercuri NB, Rizzi A, Franchi G, Beani L, Bianchi C, Morari M. Blockade of nociceptin/orphanin FQ receptor signaling in rat substantia nigra pars reticulata stimulates nigrostriatal dopaminergic transmission and motor behavior. J. Neurosci. 2004;24:6659–6666. doi: 10.1523/JNEUROSCI.0987-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Volta M, Marti M, McDonald J, Molinari S, Camarda V, Pela M, Trapella C, Morari M. Pharmacological profile and antiparkinsonian properties of the novel nociceptin/orphanin FQ receptor antagonist 1-[1-cyclooctylmethyl-5-(1-hydroxy-1-methyl-ethyl)-1,2,3,6-tetrahydro-pyridin-4-y l]-3-ethyl-1,3-dihydro-benzoimidazol-2-one (GF-4) Peptides. 2010;31:1194–1204. doi: 10.1016/j.peptides.2010.03.015. [DOI] [PubMed] [Google Scholar]
- 151.Marti M, Trapella C, Morari M. The novel nociceptin/orphanin FQ receptor antagonist Trap-101 alleviates experimental parkinsonism through inhibition of the nigro-thalamic pathway: positive interaction with L-DOPA. J. Neurochem. 2008;107:1683–1696. doi: 10.1111/j.1471-4159.2008.05735.x. [DOI] [PubMed] [Google Scholar]
- 152.Shinkai H, Ito T, Iida T, Kitao Y, Yamada H, Uchida I. 4-Aminoquinolines: novel nociceptin antagonists with analgesic activity. J. Med. Chem. 2000;43:4667–4677. doi: 10.1021/jm0002073. [DOI] [PubMed] [Google Scholar]
- 153.Goto Y, Arai-Otsuki S, Tachibana Y, Ichikawa D, Ozaki S, Takahashi H, Iwasawa Y, Okamoto O, Okuda S, Ohta H, Sagara T. Identification of a novel spiropiperidine opioid receptor-like 1 antagonist class by a focused library approach featuring 3D-pharmacophore similarity. J. Med. Chem. 2006;49:847–849. doi: 10.1021/jm0509851. [DOI] [PubMed] [Google Scholar]
- 154.Fischetti C, Camarda V, Rizzi A, Pel√† M, Trapella C, Guerrini R, McDonald J, Lambert DG, Salvadori S, Regoli D, Calo G. Pharmacological characterization of the nociceptin/orphanin FQ receptor non peptide antagonist Compound 24. Eur. J. Pharmacol. 2009;614:50–57. doi: 10.1016/j.ejphar.2009.04.054. [DOI] [PubMed] [Google Scholar]
- 155.Thompson AA, Liu W, Chun E, Katritch V, Wu H, Vardy E, Huang XP, Trapella C, Guerrini R, Calo G, Roth BL, Cherezov V, Stevens RC. Structure of the nociceptin/orphanin FQ receptor in complex with a peptide mimetic. Nature. 2012;485:395–399. doi: 10.1038/nature11085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ogawa M, Hatano K, Kawasumi Y, Ishiwata K, Kawamura K, Ozaki S, Ito K. Synthesis and evaluation of 1-[(3R,4R)-1-cyclooctylmethyl-3-hydroxymethyl-4-piperidyl]-3-[11C]ethyl-1,3-dihyd ro-2H-benzimidazol-2-one as a brain ORL1 receptor imaging agent for positron emission tomography. Nucl. Med. Biol. 2003;30:51–59. doi: 10.1016/s0969-8051(02)00352-9. [DOI] [PubMed] [Google Scholar]
- 157.Ogawa M, Hatano K, Kawasumi Y, Wichmann J, Ito K. Synthesis and in vivo evaluation of [11C]methyl-Ro 64-6198 as an ORL1 receptor imaging agent. Nucl. Med. Biol. 2001;28:941–947. doi: 10.1016/s0969-8051(01)00260-8. [DOI] [PubMed] [Google Scholar]
- 158.Pike VW, Rash KS, Chen Z, Pedregal C, Statnick MA, Kimura Y, Hong J, Zoghbi SS, Fujita M, Toledo MA, Diaz N, Gackenheimer SL, Tauscher JT, Barth VN, Innis RB. Synthesis and evaluation of radioligands for imaging brain nociceptin/orphanin FQ peptide (NOP) receptors with positron emission tomography. J. Med. Chem. 2011;54:2687–2700. doi: 10.1021/jm101487v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Pedregal C, Joshi EM, Toledo MA, Lafuente C, Diaz N, Martinez-Grau MA, Jimenez A, Benito A, Navarro A, Chen Z, Mudra DR, Kahl SD, Rash KS, Statnick MA, Barth VN. Development of LC-MS/MS-based receptor occupancy tracers and positron emission tomography radioligands for the nociceptin/orphanin FQ (NOP) receptor. J. Med. Chem. 2012;55:4955–4967. doi: 10.1021/jm201629q. [DOI] [PubMed] [Google Scholar]
- 160.Lohith TG, Zoghbi SS, Morse CL, Araneta MD, Barth VN, Goebl NA, Tauscher JT, Pike VW, Innis RB, Fujita M. Retest imaging of [11C]NOP-1A binding to nociceptin/orphanin FQ peptide (NOP) receptors in the brain of healthy humans. Neuroimage. 2014;87:89–95. doi: 10.1016/j.neuroimage.2013.10.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Hostetler ED, Sanabria-Bohórquez S, Eng W, Joshi AD, Patel S, Gibson RE, O'Malley S, Krause SM, Ryan C, Riffel K, Bi S, Okamoto O, Kawamoto H, Ozaki S, Ohta H, de Groot T, Bormans G, Depré M, de Hoon J, De Lepeleire I, Reynders T, Cook JJ, Burns HD, Egan M, Cho W, van Laere K, Hargreaves RJ. Evaluation of [18F]MK-0911, a positron emission tomography (PET) tracer for opioid receptor-like 1 (ORL1), in rhesus monkey and human. Neuroimage. 2013;68:1–10. doi: 10.1016/j.neuroimage.2012.11.053. [DOI] [PubMed] [Google Scholar]
- 162.Zhang L, Drummond E, Brodney MA, Cianfrogna J, Drozda SE, Grimwood S, Vanase-Frawley MA, Villalobos A. Design, synthesis and evaluation of [3H]PF-7191, a highly specific nociceptin opioid peptide (NOP) receptor radiotracer for in vivo receptor occupancy (RO) studies. Bioorg. Med. Chem. Lett. 2014;24:5219–5223. doi: 10.1016/j.bmcl.2014.09.069. [DOI] [PubMed] [Google Scholar]
- 163.Micheli L, Di Cesare Mannelli L, Guerrini R, Trapella C, Zanardelli M, Ciccocioppo R, Rizzi A, Ghelardini C, Calò G. Acute and subchronic antinociceptive effects of nociceptin/orphanin FQ receptor agonists infused by intrathecal route in rats. Eur. J. Pharmacol. 2015;754:73–81. doi: 10.1016/j.ejphar.2015.02.020. [DOI] [PubMed] [Google Scholar]
- 164.Zhang Y, Simpson-Durand CD, Standifer KM. Nociceptin/orphanin FQ peptide receptor antagonist JTC-801 reverses pain and anxiety symptoms in a rat model of post-traumatic stress disorder. Br. J. Pharmacol. 2015;172:571–582. doi: 10.1111/bph.12701. [DOI] [PMC free article] [PubMed] [Google Scholar]