

Abstract
Background
We recently discovered that Tubastatin-A, histone deacetylase (HDAC6) inhibitor, can improve survival in a rodent model of hemorrhagic shock (HS), but mechanisms remain poorly defined. In this study we investigated whether Tubastatin-A could protect intestinal tight junction (TJ) in HS.
Methods
In-vivo study: Wistar-Kyoto rats underwent HS (40% blood loss) followed by Tubastatin-A (70 mg/kg) treatment, without fluid resuscitation. The experimental groups were: (1) sham (no hemorrhage, no treatment), (2) control (hemorrhage, without treatment), and (3) treatment (hemorrhage with Tubastatin-A administration). Three hours after hemorrhage, ileum was harvested. Whole cell lysate were analyzed for acetylated α-tubulin (Ac-tubulin), total tubulin, acetylated histone 3 at lysine 9 (Ac-H3K9), β-actin, claudin-3 and zonula occludens 1 (ZO-1) proteins by western blot. Histological effects of Tubastatin-A on small bowel were examined. In-vitro study: human intestinal epithelial cells (Caco-2) were divided into 3 groups: (1) sham (normoxia), (2) control (anoxia, no treatment), (3) treatment (anoxia, treatment with Tubastatin-A). After 12 hours in a anoxia chamber, the cells were examined for Ac-tubulin and Ac-H3K9, cellular viability, cytotoxicity, claudin-3 and ZO-1 protein expression, and transwell permeability study.
Results
Tubastatin-A treatment significantly attenuated HS-induced decreases of Ac-tubulin, Ac-H3K9, ZO-1 and claudin-3 proteins in small bowel in-vivo (P<0.05). In cultured Caco-2 cells, anoxia significantly decreased cellular viability (P<0.001) and increased cytotoxicity (P<0.001) compared to the sham group, while Tubastatin-A treatment offered significant protection (P<0.0001). Moreover, expression of claudin-3 was markedly decreased in-vitro compared to the sham group, whereas this was significantly attenuated by Tubastatin-A (P<0.05). Finally, anoxia markedly increased the permeability of Caco-2 monolayer cells (P<0.05), while Tubastatin-A significantly attenuated the alteration (P<0.05).
Conclusion
Inhibition of HDAC6 can induce Ac-tubulin and Ac-H3K9, promote cellular viability, and prevent the loss of intestinal TJ proteins during HS and anoxia.
Keywords: Intestine, hemorrhagic shock, Tubastatin-A, tight junction, claudin-3
INTRODUCTION
Hemorrhagic shock (HS) is the leading cause of trauma related mortality, accounting for 30–40% of the deaths. 1, 2 Early deaths due to hemorrhage are caused by exsanguinating cardiac arrest, but even if the victim survives the early post-injury period the tissue damage and hypoperfusion triggers a systemic inflammatory response syndrome (SIRS), which can lead to multi-organ dysfunction syndrome (MODS). 3 Laboratory and clinical research strongly suggests that ischemia-reperfusion of the gastrointestinal tract plays a pivotal role in this process, and gut-driven inflammatory response is a major cause of distant organ injury.4–6
The intestinal mucosa is lined with epithelial cells that are connected by tight junctions (TJs) to form an active barrier. TJs are composed of transmembrane proteins including claudins, occludin and junctional adhesion molecules (JAMs) that interact with junctional adaptors such as the zonula occludens (ZO) proteins.4 These proteins form a network linking TJs to the actin cytoskeleton, which is critical for the function of TJs in the regulation of epithelial permeability.4 ZO-1, the first identified TJ protein, establishes a link between the transmembrane protein occludin and the actin cytoskeleton, 7 which is critical to the junction assembly.8, 9 Claudin and occluding proteins regulate the diffusion of ions and solutes across the epithelium. 10 One of the major claudin proteins is claudin-3. Although the exact role of claudin-3 is not completely clear, it appears to be important in the formation and function of TJs.10, 11 Hypoperfusion or ischemia can cause loss of TJs and disruption of the intestinal barrier integrity, which leads to increased permeability 5, 12 enhanced potential for bacterial translocation, systemic inflammation, and distant organ damage.5, 13 Recent studies from our group 14 and others 5 have shown that HS leads to an impairment of the gut barrier due to the loss of TJ proteins. We have also shown that treatment with a pan-histone deacetylase (HDAC) inhibitor, valproic acid (VPA), can stabilize the intestinal claudin-3, maintain the mucosal TJ integrity, and prevent harmful gut-derived substances from getting into the systemic circulation. 14
To date, 18 HDAC isoforms have been identified that are grouped into four classes: the Zn2+ dependent hydrolases class I, II and IV, and NAD+-dependent class III sirtuins.15,16 The class II HDACs have been subdivided into class IIa (HDAC4, 5, 7 and 9) and IIb (HDAC6 and 10) based on domain organization. 16 Class IIb HDACs are distinguished from the class IIa subfamily in possessing tandem deacetylase domains. HDAC6 is unique among the classical HDAC family in being a cytoplasmic enzyme that regulates many important biological processes, including cell migration, immune synapse formation, viral infection, and the degradation of misfolded proteins. 17 HDAC6 also regulates immune synapse formation, promotes HSP90 chaperone function and inhibits T-reg function, 15, 18–20 which makes it a promising target for modulating the inflammatory and immune responses.15
A recent study has identified HDAC6 as a target for protection and regeneration following nervous system injury. 21 We have found that inhibition of HDAC 6 improves survival and attenuates stress responses in a lethal septic model, 22, 23 and promotes survival in a rodent model of HS (accepted for publication in Journal of Trauma and Acute Care Surgery). However, the effects of HDAC6 inhibition on organ injury following HS have not been studied. We hypothesized that Tubastatin-A, an inhibitor of HDAC6, could preserve claudin-3 in TJs of small intestine. Our study aim is to determine whether and how Tubastatin-A protects small bowel TJ function using in-vivo and in-vitro models.
MATERIALS AND METHODS
1. Animals
This study adhered to the principles stated in The Guide for the Care and Use of Laboratory Animals, and was approved by the Institutional Animal Care and Use Committee. Male Wistar Kyoto rats (237–286 grams) were purchased from Charles River Laboratories. Rats were allowed food and water ad libitum.
2. Surgical Procedure
On the day of experimentation, Tubastatin-A (70mg/kg, Calbiochem, San Diego, CA) solution was prepared freshly by dissolving it in dimethyl sulfoxide (DMSO; 1μl/g animal body weight). 24 Anesthesia was induced with 4% isoflurane (Abbott Laboratories, North Chicago, IL) mixed with air in an induction chamber, and maintained by delivering 0.8–1.5% isoflurane via the nose cone using a veterinary multi-channel anesthesia delivery system and vaporizer (Kent Scientific Corporation, Torrington, CT). Body temperature was maintained with an automated heating pad by monitoring anus temperature. After injecting 0.2mL of 0.25% bupivacaine (APP pharmaceuticals, LLC. Schaumburg, IL) for local anesthesia, an incision was made over the left femoral vessels. The femoral artery was dissected and cannulated with polyethylene 50 catheters (Clay Adams, Sparks, MD) for creating hemorrhage, obtaining blood samples, and hemodynamic monitoring (Ponemah Physiology Platform, Gould Instrument Systems, Valley View, OH).
3. Sub-lethal HS protocol and Small Bowel Tissue Harvest
A sub-lethal HS (40% blood loss) protocol was selected to ensure that all rats would survive until the end of the study for measurement of proteins. The volume of hemorrhage was based on each animal’s estimated total blood volume which was calculated as follows: estimated total blood volume (mL) = weight (g) ×0.06 (mL/g) + 0.77. 25 After obtaining baseline arterial blood samples, 40% of the total blood volume was withdrawn over 10 minutes, followed by a 30 minute period of un-resuscitated shock, after which the animals were treated with either Tubastatin-A as described above (treatment group), or DMSO (1μl/g animal body weight, 24 control gorup), without additional resuscitation fluids (n=5/group). The dose of Tubastatin-A was calculated according to the dose of suberoylanilide hydroxamic acid 24 and the molecular weights of these two HDAC inhibitors, to achieve comparable tissue concentrations (per gram of animals’ body weight). This allowed us to cross compare the results, and maintain internal consistency. Following treatment, arterial catheters were removed and the femoral arteries were ligated. Skin incisions were closed with silk sutures, and animals were recovered from anesthesia.
The rats were sacrificed 6 hours after hemorrhage (5.5 hours after completion of treatment), and ileum was harvested. For western blot analysis, the sample was rinsed with cold saline, frozen in liquid nitrogen, and stored at −80°C. For morphologic study, the sample was fixed by immersion in 10% buffered formalin, embedded in paraffin, sliced into 5-μm sections, and stained with hematoxylin and eosin (H&E). Histologic change was examined by a pathologist blinded to the group allocation of the samples. The samples were also obtained from normal rats as a sham group (anesthetized, but no instrumentation, hemorrhage or treatment).
4. Cell Culture
Human intestinal epithelial cells (Caco-2) were grown at 37°C, 5% CO2, 95% air and relative humidity using Dulbecco’s Modified Essential Medium (DMEM) supplemented with 10% fetal bovine serum, 1% non-essential amino acids, and 0.05% penicillin/streptomycin/amphotericin. Cells were grown until 80–90% confluence, and then divided into 3 groups: (1) sham (nomoxia), (2) control (anoxia, no treatment), and (3) treatment (anoxia, and treatment with 5 μM Tubastatin-A). 26 To mimic the HS conditions in-vivo, we used a Modular Incubator Chamber (Billups-Rothenberg, Inc., Del Mar, CA) to create an anoxia environment (95% nitrogen and 5% carbon dioxide).
5. Western blotting
Caco-2 cells were seeded at 0.5 × 104 cells/cm2 on 10cm culture dish (Cell treat Scientific Products, MA) for 36 hours, followed by serum starvation 12 hours and with/without anoxia for 12 hours. Caco-2 cells or rat small bowel tissue (25mg wet weight per sample) was homogenized by using an ultrasonic homogenizer (Branson Ultrasonics Corp, CT), and cell lysates were prepared using the Whole Cell Extraction Kit (Millipore, Temecula, CA) according to the manufacturer’s instructions. Total protein concentration for each sample was determined by the Bradford method (Bio-Rad Laboratories, Hercules, CA). Equal amounts of proteins were separated on 8% or 12% sodium dodecyl sulphate-polyacrylamide gel electrophoresis gels and transferred onto nitrocellulose membranes (Bio-Rad Laboratories, Hercules, CA). The membranes were blocked in Tris-Buffered Saline-Tween 20 (TBS-T) containing 5% milk and then incubated with the primary antibody diluted in TBS-T containing 4% bovine serum albumin (Sigma-Aldrich, St. Louis, MO) at 4°C overnight. Primary antibodies were diluted as follows: Ac-tubulin antibody (Lys 40, 1:1000) and tubulin antibody (1:1000) from Cell Signaling Technology (Danvers, MA), anti-acetyl-Histone 3 (lys 9, 1:5000) from Millipore, anti-β-actin antibody (1:3000) from Sigma-Aldrich, rabbit anti ZO-1 (1ug/ml) from Invitrogen Corporation (Camarillo, CA), and rabbit polyclonal anti-claudin-3 antibody (1:500) from Novus Biologicals (Littleton, CO). The primary antibody was detected by incubating membranes with Licor IRDye 680RD Donkey anti-Mouse or anti-Rabbit secondary antibodies (Li-Cor, Inc, diluted 1:3000 in TBS-T with 5% milk) at room temperature for 1 hour. Signal detection was performed using the Odyssey CLx Infrared Imaging System (Li-Cor, Inc), and detected bands were quantitatively analyzed by using the Image Studio Lite Ver 4.0 (Li-Cor, Inc, Lincoln, NE).
6. Cellular cytotoxicity measured by the lactate dehydrogenase (LDH) leakage assay
Caco-2 cells (passages 26–40) were seeded at 0.5 × 104 cells/cm2 on polycarbonate 12-well (Corning Costar Corporation, Corning, NY) for 24 hours, following serum free starvation for 12 hours. Then cells were treated with glucose free DMEM with or without anoxia for 12 hours. Sham cells were treated in the same way except that they were not subjected to anoxia or treatment. Cytotoxicity was determined by quantifying LDH leakage into the supernatant using LDH assay kit following the manufacturer’s specifications (Roche Diagnostics Corporation, Indianapolis, IN) at 490 nm on a microplate reader (SpectraMax Plus, Molecular Devices, LLC).
7. Measurement of cellular viability/MTT assay
The viability of Caco-2 cells was determined by measuring of mitochondrial respiration, assessed by the reduction of 3-(4, 5-dimethylthiazol-2yl)-2,5-diphenyl-tetrazolium bromide (MTT, Sigma, St. Louis, MO) to formazan. Briefly, Caco-2 cells (passages 26–40) were seeded at 0.5 × 104 cells/cm2 on polycarbonate 96 well. After 24 hours of incubation, cells were starved for 12 hours, treated with or without anoxia for 12 hours. Cells were then treated with 20 μl MTT solution (5 mg/mL) for 4 hours at 37°C, the absorbance was recorded at 590 nm with a micro plate reader (SpectraMax Plus, Molecular Devices, LLC).
8. Fluorescence staining and confocal microscopy
Caco-2 cells were seeded at 0.5 × 104 cells/cm2 on the 12 well culture plate containing gelatin coated coverslips for 24 hours, followed by starvation and anoxia as before. The cells were washed twice with PBS and fixed in 3% formaldehyde fixative solution (Electron Microscopy Sciences, Hatfield, PA) in PBS for 20 minutes at room temperature. After washing with PBS, cells were permeabilized with 0.2% Triton X-100 in wash buffer (0.1% BSA in PBS) for 5 min and washed again. Non-specific background staining was blocked for 45 min in blocking buffer (PBS containing 4% BSA and 0.2% Triton X-100) at room temperature. The primary antibodies were diluted according to the manufacturer’s instructions: Rabbit anti ZO-1 (Mid, 2.5ug/mL) from Invitrogen Corporation, (Camarillo, CA), and Rabbit Polyclonal Claudin-3 Antibody (1:500) from Novus Biologicals (Littleton, CO), then incubated at room temperature for 1 hour. After washing, Rhodamine (TRITC)-conjugated AffiniPure Goat anti-Rabbit IgG (Jackson ImmunoResearch Inc. PA, USA) diluted in dilution buffer were added and incubated for 1 h at room temperature in the dark. Diluted Hoechst (H33342 Sigma Bb-2261) solution was added to each well and incubated 15 minutes at room temperature. Finally, coverslips were rinsed once with PBS and once with water. One drop of ProLong Gold antifade reagent (Molecular Probes Invitrogen, Carlsbad, CA) was added, and the coverslip was mounted with the cells facing towards the microscope slide. The specimens were examined using a confocal microscope (Nikon A-1 Spectral Confocal, Nikon Corporation, Tokyo, Japan) through a high numerical aperture with 60× oil immersion objective.
9. Transwell Study
Caco-2 enterocytes were plated on permeable filters in 12-well Transwell bicameral chambers (COSTAR, Corning, NY). Briefly, 0.5 × 105 cells/well in 0.5 ml 10% Fetal Bovine Serum (FBS) DMEM were added to the upper chamber of each filter membrane in the Transwell. To the lower chamber, 1.5 ml of cell-free DMEM (10% FBS) was added. The transwell was incubated at 37°C in an atmosphere of 5% CO2 in air and 90% humidity, and the medium was changed every other day. The cell monolayers were used for experiments after about 14–21 days in culture to ensure that there is a complete differentiation into enterocyte-like cell. 27 Cells were serum free starved for 12 hours, treated with or without anoxia for 12 hours, and then washed twice with Hank’s Balanced Salt Solution (HBSS) Buffer (pH 7.4). Medium in each insert was replaced with 0.5 ml of HBSS Buffer containing 1mg/ml fluorescein isothiocyanate (FITC) – labeled dextran (4 kDa, Sigma-Aldrich, St Louis, MO). Medium in the bottom well was replaced with serum-free HBSS Buffer alone. After 0.5 hour, 50 μl samples were removed form the bottom compartment and added to 96 well black plates (Falcon) for spectrofluorometric determination of FITC-dextran concentration. The permeability of monolayers was expressed as an intensity of fluorensence (relative fluorescence units, direct concentration) on the reader (GloMax®-Multi+ Microplate Multimode Reader, Promega). All experiments were repeated three times.
10. Statistical Analysis
All continuous variables are expressed as means ± standard deviation (SD). Data were analyzed using GraphPad Prism 6.0 statistical software (GraphPad Software, Inc. CA, USA). Differences among three or more groups were assessed using one way analysis of variance (ANOVA) followed by Tukey post hoc testing for multiple comparisons. Student’s ttest was used to compare the differences between two groups. Welch’s correction was used between populations with unequal variances and unequal sample sizes. In all cases, statistical significance was defined as P<0.05.
RESULTS
1. In-vivo sub-lethal HS model and physiologic parameters
In the sublethal HS model (40% blood loss), 100% of the animals in all groups survived until the end of the experiment, which eliminated any survival bias (i.e., loss of samples due to deaths). There were no significant differences in body weight (sham: 242.6±2.7 g, control: 260.4±7.3 g, Tubastatin-A: 256.7±5.6 g) and cannulation time (control: 37.8±15.7 minutes, Tubastatin-A: 24.8±2.2 minutes) between groups (P>0.05). Forty percent blood loss resulted in significant shock, which was reflected in the laboratory values tested at baseline and end of shock, such as hemoglobin (from 11.98±0.61 g/dL to 8.00±0.75 g/dL and from 13.15±0.41 g/dL to 9.45±0.45 g/dL in control and Tubastatin-A group respectively, P=0.013 and P=0.003) and serum lactate (from 1.58±0.15 mmol/L to 3.60±0.60 mmol/L and from 1.62±0.15 to 2.82±0.34 mmol/L in control and Tubastatin-A group respectively, P=0.026 and P=0.020). There were no significant differences between groups in hemoglobin and serum lactate.
2. Effect of Tubastatin-A on acetylation of α-tubulin and histone 3 at lysine 9 (H3K9) in-vivo
Acetylation of histone 3 lysine 9 (H3K9) and tubulin in rodent HS intestinal tissue was examined by western blots with anti-acetylated H3K9 and anti-acetylated tubulin antibodies, respectively. As shown in Figure 1A, H3K9 was acetylated in the sham group and hemorrhage decreased the level of acetylation. Treatment with Tubastatin-A restored the acetylation of H3K9 back to normal levels (P=0.046). As for α-tubulin protein, no acetylation was detected in both the sham and control groups. However, Tubastatin-A caused a significant increase in α-tubulin acetylation (Figure 1B, P<0.001).
Figure 1.

Effect of Tubastatin-A on acetylation levels of α-tubulin and histone H3 at lysine 9 (H3K9) after HS. (A) Tubastatin-A increases the levels of Ac-H3K9 after HS (*P=0.046). (B) Tubastatin-A increases the levels of Ac-α-tubulin after HS (*P<0.001). Protein bands quantified by densitometry were expressed as mean values ± SD. Ac-H3K9: acetylated histone 3 at lysine 9; Ac-α-Tubulin: acetylated α-Tubulin; Sham: no hemorrhage and no treatment; Control: hemorrhage, no treatment; Tub-A: Tubastatin-A treatment, n=5/group.
3. Tubastatin-A protects small bowel TJ proteins from loss following HS in-vivo
As shown in Figure 2A, levels of claudin-3 protein in the intestine were significantly decreased after HS compared to sham animals (P=0.002). Post-shock administration of Tubastatin-A attenuated the loss of claudin-3 (P=0.002). Likewise, HS significantly decreased ZO-1 protein expression (P=0.004), which was prevented by Tubastatin-A treatment (P=0.017, Figure 2B). Histologic analysis showed that HS caused marked villous bluting, diffuse surface epithelial injury damage, focal epithelial surface necrosis, and moderate to marked intraepithelial and stromal lymphocytic infiltration. Tubastatin-A treatment attenuated these alterations (Figure 2C and 2D).
Figure 2.

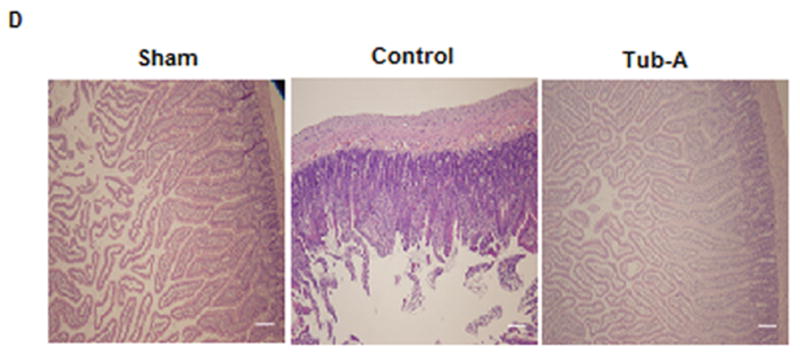
Tubastatin-A protects small bowel tissue TJ proteins from loss. HS induced loss of claudin-3 protein in intestine tissue (*P=0.002, control vs. sham, 2A). Post-shock administration of Tubastatin-A attenuated HS-induced loss of claudin-3 (*P=0.002, Tub-A vs. control, 2A). HS decreased ZO-1 expression (*P=0.004, control vs. sham, 2B), and Tubastatin-A prevented the ZO-1 protein from loss (*P=0.017, Tub-A vs. control, 2B). Histologic analysis shown that HS caused marked villous bluting, diffuse surface epithelial damage, focal epithelial surface necrosis, and moderate to marked intraepithelial and stromal lymphocytic infiltration. Tubastatin-A treatment attenuated these alteration (2C). Sham: no hemorrhage, no treatment; Control: hemorrhage, without treatment; Tub-A: Tubastatin-A treatment; n=5/group, scales in 2C and 2D: 200 μm.
4. Effect of Tubastatin-A on acetylation of α-tubulin and H3K9 in the cultured intestinal Caco-2 cells
In the rest experiments, intestinal Caco-2 cell culture in anoxia condition was employed to mimic HS in-vivo. Similar results were found in the cultured cells as those in small bowel tissue (Figure 3A, 3B). These results indicate that inhibition of HDAC6 could induce acetylation of H3K9 as well as α-tubulin proteins.
Figure 3.

Tubastatin-A induces acetylation of both H3K9 and α-tubulin in Caco-2 cells after anoxia. (A) Tubastatin A induces aceylation of H3K9 after anoxia (*P=0.002 compared to Sham, *P=0.008 compared to control), no differences were found between sham and control group. (B) Tubastatin A induces acetylation of α-tubulin (*P=0.004 compared to sham, *P=0.004 compared to control). No differences were found between sham and control. Protein bands quantified by densitometry were expressed as mean values ± SD. Ac-H3K9: acetylated histone 3 at lysine 9; Ac-α-Tubulin: acetylated α-Tubulin; Sham: no anoxia and no treatment; Control: anoxia, no treatment; Tub-A: Tubastatin-A treatment.
5. Tubastatin-A protects cultured intestinal Caco-2 cells against anoxia
We investigated the cytotoxicity of Tubastatin-A on Caco-2 cells by (i) testing the leakage of LDH enzyme, and (ii) MTT assay for cell viability. The cell culture medium served as a negative control (N.C.). Hypoxic condition caused a significant increase in the leakage of LDH (control vs. sham, P<0.0001), which was significantly attenuated by treatment with Tubastatin-A (Tubastatin-A vs. control, P<0.0001) (Figure 4A). MTT assay further confirmed that anoxia decreased the viability of the Caco-2 cells (control vs. sham, P<0.0001), while Tubastatin-A treatment significantly restored the viability (Tubastatin-A vs. control, P<0.001, Figure 4B and 4C).
Figure 4.

Tubastatin-A protects Caco-2 cells against anoxia (12 hours) injury. Anoxia condition caused significant increased leakage of LDH (*control vs. sham, P<0.0001, 4A), while incubation with Tubastatin-A significantly attenuated the increase (*Tub-A vs. control, P<0.0001, 4A). MTT assay showed that anoxia had a significant decrease of cellular viability compare to sham cells (*control vs. sham, P<0.0001, 3B), while Tubastatin-A treatment significantly restored cellular viability (*Tub-A vs. control, P<0.0001, Figure 4B, 4C). Tub-A: Tubastatin-A treatment; Sham: no anoxia and no treatment; Control: anoxia without treatment; N.C.: negative control.
6. Tubastatin-A protects TJ proteins from loss during anoxia in the cultured intestinal Caco-2 cells
Anoxia caused a significant decrease in cellular claudin-3 protein expression compared to sham cells (P=0.003), while incubation with Tubastatin-A significantly attenuated this loss (P=0.002, Figure 5A). However, no significant changes were noted in the ZO-1 protein expression (Figure 5B).
Figure 5.

Tubastatin-A protects claudin-3 protein from loss in Caco-2 cells in anoxia condition. Representative western blots and densitometry analysis (A, B) as well as immunofluorescence staining (C, D) of claudin-3 and ZO-1 in Caco-2 cells treated with different conditions. Control cells showed significant decrease of claudin-3 compared with sham (*P=0.003), Tubastatin-A treatment protected claudin-3 from loss (*P=0.002 vs. control, A). No significant difference in the expression of ZO-1 protein between the sham, control, and Tubastatin-A treatment groups (B). Normal Caco-2 cells had strong membrane and weak cytoplasmic signals of claudin-3 protein, anoxia caused pronounced loss, while treatment with Tubastatin-A increased the stainings (C). ZO-1 protein strongly expressed in the cellular membrane, while anoxia didn’t result in significant loss and Tubastatin-A treated cells exhibited a moderate change compared to control cells (D). Values are expressed as mean ± SD from three independent experiments. Original magnification 200×; red color: claudin-3 and ZO-1 signal; blue color: nuclear Hoechst staining. Sham: no anoxia injury; Control: anoxia without treatment; Tub-A: Tubastatin-A treatment.
In addition, immunostaining and confocal microscope were performed to examine the TJ protein expression in the intestinal Caco-2 cells in the presence or absence of anoxia, with or without Tubastatin-A. Under normoxic condition, strong membrane and weak cytoplasmic staining of claudin-3 protein were detected. After anoxia insult for 12 hours, Caco-2 cells showed pronounced loss of claudin-3 protein, while Tubastatin-A treatment normalized the staining pattern (Figure 5C). As for ZO-1 protein, immunostaining and confocal study found that ZO-1 protein was strongly expressed in the cellular membrane; however, neither anoxia nor Tubastatin-A treatment caused any significant changes in its expression (Figure 5D).
These results suggested that Tubastatin-A could protect intestinal TJ protein loss, and that claudin 3 is more sensitive than ZO-1 to anoxia insult and Tubastatin-A treatment.
7. Tubastatin-A protects cell permeability during anoxia in the cultured intestinal Caco-2 cells
To further investigate the effect of Tubastatin-A on cell permeability, the leakage of FITC-dextran on the Caco-2 monolayer cells was analyzed using transwell assay after exposed to 12 hours hyoxic condition. As shown in Figure 6, anoxia markedly increased the FITC-dextran permeability (relative fluorescence units: 6421± 279 vs. 2259 ±513, for control and sham groups respectively; P=0.0002). Tubastatin-A incubation significantly decreased the permeability compared to the control group (4763±306 vs. 6421± 279, P=0.016).
Figure 6.
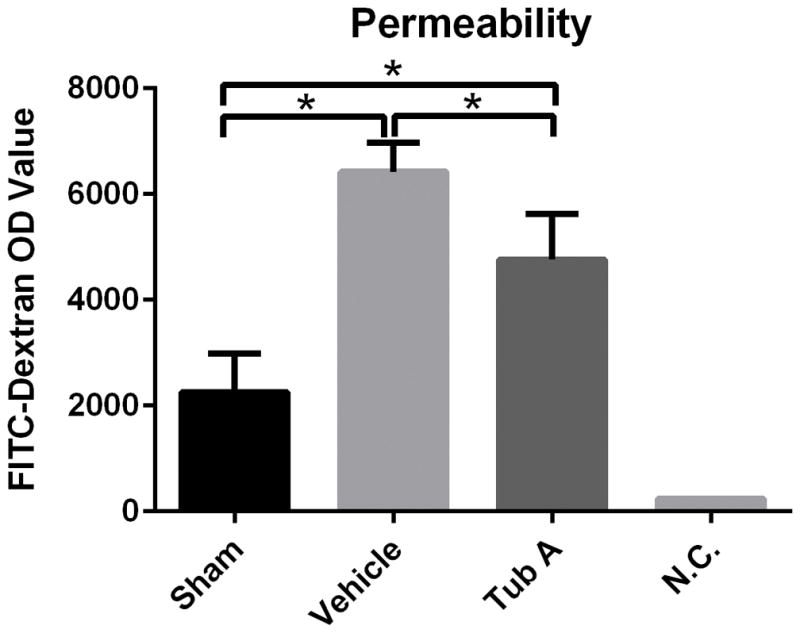
Tubastatin-A decreases Caco-2 cell monolayer permeability during anoxia condition. Anoxia markedly increased the FITC-dextran permeability compare to sham group (*P=0.0002). Tubastatin-A treatment significantly decreased the permeability compared to control group(*P=0.016). Tub-A: Tubastatin-A treatment; Sham: no anoxia and no treatment; Control: anoxia with DMSO treatment; N.C.: negative control.
DISCUSSION
TJs are critical for maintaining cell–cell adhesions in epithelial and endothelial cellular sheets. They act as a primary barrier to the diffusion of solutes through the intercellular space, create a boundary between the apical and the basolateral plasma membrane domains, and recruit various cytoskeletal as well as signaling molecules at their cytoplasmic surface.4 Normally, TJs are anchored to the cell via TJ proteins and filamentous actin (F-actin) cytoskeleton. Hypoperfusion, or ischemia, can cause disruption of F-actin cytoskeleton with subsequent TJ loss and barrier failure.4, 12
In a rodent model of lethal HS (50% blood loss), we recently found that the massive blood loss caused animals died within 3 hours and Tubastatin-A treatment significantly prolonged animal lives (accepted for publication in Journal of Trauma and Acute Care Surgery). In this present project we focused on the small bowel TJ functions by using sub-lethal HS model (40% blood loss). Thuijls et al. reported that in their hemorrhage model (a 30–40% blood loss), loss of the TJ proteins (claudin-3 and ZO-1) was observed as early as 1 hour after induction of hemorrhage, and disruption of this intestinal barrier resulted in translocation of toxins and bacteria, 5 which in turn could cause systemic inflammation. In our study, 40% of the blood volume was withdrawn in 10 minutes, which was followed by 30 min of un-resuscitated shock before administration of Tubastatin-A (70mg/kg) treatment. The degree of ischemia in this model was sufficient to cause a significant disruption in the intestinal TJ, as evidenced by the loss of claudin 3 and ZO-1 proteins. Treatment with Tubastatin-A was associated with normal expression of these proteins, suggesting better preservation of the intestinal barrier integrity during shock.
To study the mechanism in an in-vitro model, we chose the transformed Caco-2 cell line, which is phenotypically similar to small bowel enterocytes and is commonly used for these types of experiments. 28, 29 HS is characterized by tissue hypo-perfusion, which leads to an insufficient delivery of oxygen and nutrient to the cells. To mimic HS in-vivo, Caco-2 cells were placed in an anaerobic environment before we examined the expression and localization of TJ proteins, with or without Tubastatin-A treatment. Anoxia resulted in a significant loss of claudin-3 protein as seen in the Western blot and immunofluorescence examinations, which may be the reason for the leakage of LDH (Figure 3A) and a decrease in cellular viability (MTT assay, Figure 3B). Moreover, the transwell study of the monolayer Caco-2 cells confirmed that anoxia could significantly increase the permeability of the cellular layer, which was attenuated by Tubastatin-A. These findings strongly suggested that inhibition of HDAC6 could protect the intestinal lining during shock, but additional in-vivo assays will have to be performed to ensure that these findings are reproducible in a more comprehensive (intact) biological system.
It is well known that histone acetyltransferase (HAT) and HDAC enzymes control the addition and removal of acetyl groups to maintain a dynamic balance in protein acetylation. 30, 31 Inhibition of HDAC promotes acetylation at specific lysine residues on histones and other nuclear and cytoplasmic proteins, which in turn modulates pivotal cellular functions such as chromosome remodeling, gene transcription, and cell proliferation. 30, 31 It has previously been reported that Tubastatin-A can inhibit HDAC6-mediated α-tubulin deacetylation in cytosol. 32 In addition to the acetylation of α-tubulin protein, our results reveal for the first time that inhibition of HDAC6 with Tubastatin-A can cause hyperacetylation of nuclear histone H3 at K9 in rat small bowel tissues in-vivo (Figure 1A) and Caco-2 cells in-vitro (Figure 3A). These data raise some interesting possibilities such as: (1) it is possible that HDAC6 is not limited to the cytosol as previously reported, but could also somehow translocate into the nucleus; (2) Tubastatin-A in the concentration used in our study may interact with other HDACs in the nucleus in addition to HDAC6 in the cytosol. More experiments are being designed to test these possibilities.
Another potential advantage of using Tubasatin A is that it appears to have minimal adverse effects. Unlike other histone deacetylases, HDAC6 is an excellent drug target. 33 Selective inhibition of HDAC6 does not significantly change gene expression signatures in microarray analysis, alter cell cycle progression, nor lead to an aberrant mitotic spindle formation. 32 Deletion of HDAC6 in mice does not impair normal development or major organ functions, 34 suggesting that reversible HDAC6 inhibition in clinical settings would not cause major side effects in contrast to inhibition of other HDACs, in particular class I HDACs. Using the leakage of LDH enzyme as an indicator of cytotoxic effects and MTT assay for cellular viability, the current study has demonstrated that inhibition of HDAC6 results in better membrane integrity and higher viability of the intestinal epithelial cells (Figure 4). We have previously shown that treatment with VPA (300 mg/kg) can prevent claudin-3 loss in an identical model of HS. 14 The present study demonstrates that treatment with 70 mg/kg of Tubastatin-A achieves the same results. All of these repeatable and positive results indicate that it may be feasible for us to treat post-HS cellular dysfunctions with Tubastatin-A in the future.
However, this study has several limitations that must be acknowledged. The sample size of Caco-2 cell culture was relatively small, which may explain the lack of significant difference in the expression of ZO-1 protein between the different groups (Figure 5B). The animal model had a relatively brief period of observation, and the long-term implications of these findings need to be further validated. It is not clear what off-target effects of Tubstatin A could be in the treatment of HS.
In summary, we have presented in-vivo and in-vitro evidence that treatment with Tubastatin-A can attenuate intestinal TJ loss following HS and cellular anoxia. The protective effects of Tubastatin-A could result from acetylation of the α-tubulin and the histone H3K9. Further studies should be done to understand the precise cyto-protective mechanisms that are activated by HDAC6 inhibition, which can facilitate the development of more effective and specific therapeutic interventions for HS.
Acknowledgments
This research was funded by a grant from NIH 2-R01-GM-084127-06-A1to HBA. Data presented at the Academic Surgical Congress, February 3, 2015, Las Vegas, Nevada.
Footnotes
Author Contribution: YLi and HBA designed this study, for which HBA secured funding. ZC and WH carried out the animal experiments and obtained the data. ZC, WH, BL, IH, YL, and TB performed the tissue analysis. XD performed the pathologic examination of histological effects of Tubastatin-A treatment on small bowel. BP made the figure 2D. VN and PG evaluated the paper and preformed the necessary changes. ZC and YLi wrote the manuscript, and YLi and HBA made critical revisions.
References
- 1.Evans JA, van Wessem KJ, McDougall D, Lee KA, Lyons T, Balogh ZJ. Epidemiology of traumatic deaths: comprehensive population-based assessment. World J Surg. 2010;34(1):158–63. doi: 10.1007/s00268-009-0266-1. [DOI] [PubMed] [Google Scholar]
- 2.Gann DS, Drucker WR. Hemorrhagic shock. J Trauma Acute Care Surg. 2013;75(5):888–95. doi: 10.1097/TA.0b013e3182a686ed. [DOI] [PubMed] [Google Scholar]
- 3.Wilson M, Davis DP, Coimbra R. Diagnosis and monitoring of hemorrhagic shock during the initial resuscitation of multiple trauma patients: a review. J Emerg Med. 2003;24(4):413–22. doi: 10.1016/s0736-4679(03)00042-8. [DOI] [PubMed] [Google Scholar]
- 4.Tsukita S, Furuse M, Itoh M. Multifunctional strands in tight junctions. Nat Rev Mol Cell Bio. 2001;2(4):285–93. doi: 10.1038/35067088. [DOI] [PubMed] [Google Scholar]
- 5.Thuijls G, de Haan JJ, Derikx JP, Daissormont I, Hadfoune M, Heineman E, Buurman WA. Intestinal cytoskeleton degradation precedes tight junction loss following hemorrhagic shock. Shock. 2009;31(2):164–9. doi: 10.1097/SHK.0b013e31817fc310. [DOI] [PubMed] [Google Scholar]
- 6.Hassoun HT, Kone BC, Mercer DW, Moody FG, Weisbrodt NW, Moore FA. Post-injury multiple organ failure: the role of the gut. Shock. 2001;15(1):1–10. doi: 10.1097/00024382-200115010-00001. [DOI] [PubMed] [Google Scholar]
- 7.Fanning AS, Jameson BJ, Jesaitis LA, Anderson JM. The tight junction protein ZO-1 establishes a link between the transmembrane protein occludin and the actin cytoskeleton. J Biol Chem. 1998;273(45):29745–53. doi: 10.1074/jbc.273.45.29745. [DOI] [PubMed] [Google Scholar]
- 8.McNeil E, Capaldo CT, Macara IG. Zonula occludens-1 function in the assembly of tight junctions in Madin-Darby canine kidney epithelial cells. Mol Biol Cell. 2006;17(4):1922–32. doi: 10.1091/mbc.E05-07-0650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Umeda K, Matsui T, Nakayama M, Furuse K, Sasaki H, Furuse M, Tsukita S. Establishment and characterization of cultured epithelial cells lacking expression of ZO-1. J Biol Chem. 2004;279(43):44785–94. doi: 10.1074/jbc.M406563200. [DOI] [PubMed] [Google Scholar]
- 10.Krug SM, Schulzke JD, Fromm M. Tight junction, selective permeability, and related diseases. Semin Cell Dev Biol. 2014 doi: 10.1016/j.semcdb.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 11.Morin PJ. Claudin Proteins in Human Cancer: Promising New Targets for Diagnosis and Therapy. Cancer Res. 2005;65(21):9603–6. doi: 10.1158/0008-5472.CAN-05-2782. [DOI] [PubMed] [Google Scholar]
- 12.Fink MP, Delude RL. Epithelial barrier dysfunction: a unifying theme to explain the pathogenesis of multiple organ dysfunction at the cellular level. Crit Care Clin. 2005;21(2):177–96. doi: 10.1016/j.ccc.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 13.Fink MP. Intestinal epithelial hyperpermeability: update on the pathogenesis of gut mucosal barrier dysfunction in critical illness. Curr Opin Crit Care. 2003;9(2):143–51. doi: 10.1097/00075198-200304000-00011. [DOI] [PubMed] [Google Scholar]
- 14.Li Y, Liu B, Dillon ST, Fukudome EY, Kheirbek T, Sailhamer EA, Velmahos G, deMoya M, Libermann TA, Alam HB. Identification of a novel potential biomarker in a model of hemorrhagic shock and valproic acid treatment. J Surg Res. 2010;159(1):474–81. doi: 10.1016/j.jss.2009.04.011. [DOI] [PubMed] [Google Scholar]
- 15.Shakespear MR, Halili MA, Irvine KM, Fairlie DP, Sweet MJ. Histone deacetylases as regulators of inflammation and immunity. Trends Immunol. 2011;32(7):335–43. doi: 10.1016/j.it.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 16.Li Y, Alam HB. Modulation of acetylation: creating a pro-survival and anti-inflammatory phenotype in lethal hemorrhagic and septic shock. J Biomed Biotechnol. 2011;2011:523481. doi: 10.1155/2011/523481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Valenzuela-Fernandez A, Cabrero JR, Serrador JM, Sanchez-Madrid F. HDAC6: a key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends Cell Biol. 2008;18(6):291–7. doi: 10.1016/j.tcb.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 18.de Zoeten EF, Wang L, Butler K, Beier UH, Akimova T, Sai H, Bradner JE, Mazitschek R, Kozikowski AP, Matthias P, et al. Histone deacetylase 6 and heat shock protein 90 control the functions of Foxp3(+) T-regulatory cells. Mol Cell Biol. 2011;31(10):2066–78. doi: 10.1128/MCB.05155-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kovacs JJ, Murphy PJ, Gaillard S, Zhao X, Wu JT, Nicchitta CV, Yoshida M, Toft DO, Pratt WB, Yao TP. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell. 2005;18(5):601–7. doi: 10.1016/j.molcel.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 20.Serrador JM, Cabrero JR, Sancho D, Mittelbrunn M, Urzainqui A, Sanchez-Madrid F. HDAC6 deacetylase activity links the tubulin cytoskeleton with immune synapse organization. Immunity. 2004;20(4):417–28. doi: 10.1016/s1074-7613(04)00078-0. [DOI] [PubMed] [Google Scholar]
- 21.Rivieccio MA, Brochier C, Willis DE, Walker BA, D’Annibale MA, McLaughlin K, Siddiq A, Kozikowski AP, Jaffrey SR, Twiss JL, et al. HDAC6 is a target for protection and regeneration following injury in the nervous system. Proc Natl Acad Sci U S A. 2009;106(46):19599–604. doi: 10.1073/pnas.0907935106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao T, Li Y, Bronson RT, Liu B, Velmahos GC, Alam HB. Selective histone deacetylase-6 inhibition attenuates stress responses and prevents immune organ atrophy in a lethal septic model. Surgery. 2014;156(2):235–42. doi: 10.1016/j.surg.2014.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao T, Li Y, Liu B, Halaweish I, Mazitschek R, Alam HB. Selective inhibition of histone deacetylase 6 alters the composition of circulating blood cells in a lethal septic model. J Surg Res. 2014;190(2):647–54. doi: 10.1016/j.jss.2014.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Y, Liu B, Zhao H, Sailhamer EA, Fukudome EY, Zhang X, Kheirbek T, Finkelstein RA, Velmahos GC, deMoya M, et al. Protective effect of suberoylanilide hydroxamic acid against LPS-induced septic shock in rodents. Shock. 2009;32(5):517–23. doi: 10.1097/SHK.0b013e3181a44c79. [DOI] [PubMed] [Google Scholar]
- 25.Lee HB, Blaufox MD. Blood volume in the rat. J Nucl Med. 1985;26(1):72–6. [PubMed] [Google Scholar]
- 26.Butler KV, Kalin J, Brochier C, Vistoli G, Langley B, Kozikowski AP. Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J Am Chem Soc. 2010;132(31):10842–6. doi: 10.1021/ja102758v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sappington PL, Yang R, Yang H, Tracey KJ, Delude RL, Fink MP. HMGB1 B box increases the permeability of Caco-2 enterocytic monolayers and impairs intestinal barrier function in mice. Gastroenterology. 2002;123(3):790–802. doi: 10.1053/gast.2002.35391. [DOI] [PubMed] [Google Scholar]
- 28.Cruz N, Qi L, Alvarez X, Berg RD, Deitch EA. The Caco-2 cell monolayer system as an in vitro model for studying bacterial-enterocyte interactions and bacterial translocation. J Burn Care Rehabil. 1994;15(3):207–12. doi: 10.1097/00004630-199405000-00002. [DOI] [PubMed] [Google Scholar]
- 29.Swank GM, Lu Q, Xu DZ, Michalsky M, Deitch EA. Effect of acute-phase and heat-shock stress on apoptosis in intestinal epithelial cells (Caco-2) Crit Care Med. 1998;26(7):1213–7. doi: 10.1097/00003246-199807000-00023. [DOI] [PubMed] [Google Scholar]
- 30.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403(6765):41–5. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 31.Parbin S, Kar S, Shilpi A, Sengupta D, Deb M, Rath SK, Patra SK. Histone deacetylases: a saga of perturbed acetylation homeostasis in cancer. J Histochem Cytochem. 2014;62(1):11–33. doi: 10.1369/0022155413506582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc Natl Acad Sci U S A. 2003;100(8):4389–94. doi: 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Witt O, Deubzer HE, Milde T, Oehme I. HDAC family: What are the cancer relevant targets? Cancer Lett. 2009;277(1):8–21. doi: 10.1016/j.canlet.2008.08.016. [DOI] [PubMed] [Google Scholar]
- 34.Patel J, Pathak RR, Mujtaba S. The biology of lysine acetylation integrates transcriptional programming and metabolism. Nutr Metab (Lond) 2011;8:12. doi: 10.1186/1743-7075-8-12. [DOI] [PMC free article] [PubMed] [Google Scholar]