

Abstract
Replica exchange molecular dynamics is a multicanonical simulation technique commonly used to enhance the sampling of solvated biomolecules on rugged free energy landscapes. While replica exchange is relatively easy to implement, there are many unanswered questions about how to use this technique most effciently, especially because it is frequently the case in practice that replica exchange simulations are not fully converged. A replica exchange cycle consists of a series of molecular dynamics steps of a set of replicas moving under different Hamiltonians or at different thermodynamic states followed by one or more replica exchange attempts to swap replicas among the different states. How the replica exchange cycle is constructed affects how rapidly the system equilibrates. We have constructed a Markov state model of replica exchange (MSMRE) using long molecular dynamics simulations of a host–guest binding system as an example, in order to study how different implementations of the replica exchange cycle can affect the sampling effciency. We analyze how the number of replica exchange attempts per cycle, the number of MD steps per cycle, and the interaction between the two parameters affects the largest implied time scale of the MSMRE simulation. The infinite swapping limit is an important concept in replica exchange. We show how to estimate the infinite swapping limit from the diagonal elements of the exchange transition matrix constructed from MSMRE “simulations of simulations” as well as from relatively short runs of the actual replica exchange simulations.
1. INTRODUCTION
Molecular dynamics (MD) is a computer simulation technique that is widely employed to study the behavior of biological systems at atomic resolution, but this method is limited by much shorter time scales of most simulations compared to those of many biochemical processes, such as conformational equilibria of proteins and nucleic acids, or the catalytic cycles of enzymes, which often occur over a wide range of time scales from milliseconds to seconds or longer.1–3 Powerful enhanced conformational sampling techniques have emerged to address this challenge. Umbrella sampling and path sampling methods are two notable examples. In umbrella sampling, one tessellates a region of the conformational space of interest by localizing potentials to explore it thoroughly.4–7 In path sampling, one places interfaces or biasing potentials along chosen reaction coordinates to facilitate barrier crossings, and samples within transition regions that connect stable states.8–18 Typically, these enhanced sampling techniques launch independent swarms of simulation threads each at a region between interfaces or each at a modified Hamiltonian containing a different biasing potential. These techniques often suffer from slow convergence because of the notorious dificulty to achieve equilibration at each state, especially when the biasing potentials strongly localize the system in a limited range of reduced coordinates. Other enhanced conformational sampling techniques are based on generalized ensemble formulations. In addition to the sampling of the conformational space, such techniques also produce a random walk in the thermodynamic and/or Hamiltonian parameter spaces.19,20
Generalized ensemble enhanced sampling can be classified as either serial, such as serial tempering/simulated tempering and Hamiltonian hopping, or parallel sampling. The serial implementations run a single Monte Carlo (MC) or MD thread and update the thermodynamic and Hamiltonian parameters periodically.21,22 However, the single thread infrequently reaches high free energy states because the probability of visiting a particular state is determined by the free energy of that state. To solve this problem, the serial implementations usually add a free energy weight to the Hamiltonian of each state and iteratively adjust those energy weights to equalize the state populations. The determination of optimal free energy weights can be problematic, especially when they slowly converge due to rare conformational transitions.
Parallel replica exchange (RE) algorithms eliminate the need for prior determination of free energy weights.23–26 RE techniques run as many MC/MD threads (replicas) as there are states of the system included in the generalized ensemble, and exchange replicas’ state assignments periodically. The probabilities of exchanges are controlled by microscopic reversibility requirements of not only the configurational space of each replica but the combinatorial set of assignments of states to replicas. Because of the thermodynamic equivalence of replicas and the equality of the numbers of replicas and states, the replica exchange method guarantees that each replica will visit each state with equal probability without the prior knowledge of free energy weights. Replica exchange algorithms provide some of the most powerful conformational sampling tools; under favorable circumstances, they can yield converged results orders of magnitude faster than conventional approaches.
The RE method is one of the most popular enhanced sampling techniques to study the molecular behavior of systems in chemistry, physics, and biology. This technique has been successfully applied to protein folding, protein–ligand binding, conformational free energy estimation, protein structure refinement, etc.25,27–35 Adaptive and enhanced RE approaches have been introduced, including solute tempering RE,36,37 resolution RE,38 asynchronous RE,39 free energy perturbation RE,40 etc.41–47 New exchange algorithms and proposal schemes have also been suggested and examined.48–55 In recent years, researchers have shown an increased interest in analyzing the effciency of RE techniques along with their growing adoption.41,50,56–74 Several groups have developed RE simulation models to study this sampling algorithm, since explicit RE simulations are usually too computationally expensive to study the RE parameter space systematically.60,63,69,74
Following previous work in our group, we have constructed a Markov state model of RE simulations (MSMRE) using the heptanoate β-cyclodextrin binding complex, a host–guest system, as an example. Host–guest systems provide attractive alternatives to protein ligand binding systems for the investigation of molecular recognition. Since these systems are small-sized, convergence of the binding afinity estimate can be achieved with relatively modest computational cost.75 In a previous study, our group used the MSMRE idea to construct a kinetic network model inspired by protein folding to analyze several properties of temperature RE simulations.63 In the present study, we built Markov state models (MSM) based on long MD simulations of heptanoate binding to β-cyclodextrin at multiple Hamiltonian states, then implemented the transition matrices of MSM into MSMRE to generate Markov chains, which imitate the respective MD processes in explicit RE simulations. The MSMRE model described in this work is a more detailed “simulations of simulations” analysis compared with our previous kinetic network model of RE simulations, and it retains many of the important features of real replica exchange simulations, although the non-Markovian effects are lost. The MSMRE simulations are able to mimic weeks-long RE simulations within hours on a desktop by substituting a Markov chain model for the real kinetics of MD simulations that forms the foundation of RE simulations. This powerful tool enables us to analyze how the sampling effciency of replica exchange depends on the construction of the replica exchange cycle, which in this work includes (i) the comparison of two different exchange proposal schemes; (ii) the number of exchange attempts per cycle; (iii) the length of the MD simulation per cycle; (iv) and finally the interaction between the number of exchange attempts per cycle and the length of the MD simulation per cycle.
2. METHODOLOGY AND SIMULATIONS
2.1. Replica Exchange
In replica exchange simulations, sampling is performed by a Markov chain alternating between two components: (i) updates of molecular configurations using MC or MD independently for each replica at a fixed thermodynamic and/or Hamiltonian state, which will be called the “move” process; (ii) updates of thermodynamic and/or Hamiltonian parameters (referred to as “thermodynamic state” below) assignments to replicas, by means of a series of coordinated attempted swaps of thermodynamic states among replicas according to the usual MC algorithm, which will be called the “exchange” process. The move and exchange processes together will be referred to as a RE cycle throughout this paper.
The exchange process in a RE cycle—the sampling of the state permutation space—must satisfy the detailed balance condition
| (1) |
where PRE is the joint probability distribution of a RE configuration, is the conformational coordinates of the mth replica, and {S} represents the Sth combination of replicas and thermodynamic states in the state permutation space. Since each replica associates with one thermodynamic state, there are M! possible permutations of replica configurations with thermodynamic states. One can expand {S} as an array
| (2) |
where s[m] is the index of the thermodynamic state occupied by the mth replica. Both {S} and {S′} are the permutations of the set {1, 2, 3, …, M – 1, M}. TSS′ is the transition probability from state S to S′ when all of the conformational coordinates are fixed.
PRE is simply the product of the probabilities of each replica configuration at the thermodynamic state according to the permutation {S}
| (3) |
In this study, we assume that the distribution of replica configurations is described by the canonical ensemble at each thermodynamic state, and their probabilities are represented by the corresponding Boltzmann distributions. For the mth replica at the s[m]th thermodynamic state
| (4) |
where Zs[m] is the partition function of the s[m]th thermodynamic state, is the energy of the system with configuration at the s[m]th thermodynamic state, and βs[m] = 1/(kBTs[m]) is the inverse temperature. TSS′ in eq 1 is the product of the probability of the trial move αSS′ and the probability of accepting this trial move AccpSS′. For the sake of simplicity, we only discuss the symmetric trial move (αSS′ = αS′S); hence, the ratio of the acceptance probabilities is
| (5) |
The standard realization of eq 5 is to apply the Metropolis MC acceptance criteria
| (6) |
In temperature RE simulations—the basic RE approach—the Hamiltonian function is universal, namely, ; in Hamiltonian RE simulations, all the temperatures are fixed, namely, βs[m] ≡ β. More generally, both temperature and Hamiltonian parameters can be swapped at the same time.
The nearest neighbor exchange scheme is the standard scheme for the sampling of state permutation space in RE simulations. This most commonly employed scheme attempts to exchange pairs at the {(1st, 2nd), (3rd, 4th), …} thermodynamic states in one RE cycle and then attempts to exchange replica pairs at the {(2nd, 3rd), (4th, 5th), …} states in the next RE cycle. Exchanges are accepted with the probability given by eq 6. Its name comes from the fact that the exchanges are only attempted between two replicas associated with adjacent thermodynamic states. Since the replicas with adjacent thermodynamic states usually have large overlap in their conformational distributions, attempting exchanges between them minimizes the rejection probability, which is desirable for better sampling of state permutation space. However, there is a tradeoff between the larger acceptance probability when the replica exchange is limited to attempted exchanges only between nearest neighbors and the large number of replica exchange steps required to reach “distant” thermodynamic states when the exchange attempts are restricted to nearest neighbors. Furthermore, the nearest neighbor exchange proposal scheme requires the prior identification of neighboring states, and might become impractical in the context of new replica exchange techniques being developed for grid computing.39
The independence sampling algorithm, recently proposed by Chodera and Shirts, is the second proposal scheme we consider.50 This algorithm attempts to exchange two replicas that are randomly picked from the set with a uniform probability. The acceptance ratio for exchange follows the same Metropolis criterion as in eq 6. The independence sampling does not require the prior identification of neighboring states but usually has lower acceptance probability compared with the nearest neighbor exchange. To overcome this drawback, Chodera and Shirts suggested that M3 to M5 exchanges be attempted in one RE cycle, where M is the total number of replicas.50
2.2. Heptanoate β-Cyclodextrin Binding System
In this study, the test system used to illustrate the problem is the binding of a guest molecule (heptanoate) to a host molecule β-cyclodextrin) in implicit solvation (OPLS-AA/AGBNP2)— a problem we studied previously.33,75 β-Cyclodextrin (βCD), a well know host molecule, is a frustum-cone-shaped cyclic polymer with a hydrophobic interior core. The narrow opening of βCD is laced with primary hydroxyls, and the wider opening is laced with secondary hydroxyls. The ligand, heptanoate, is a common guest molecule with a hydrophilic carboxylate group and hydrophobic alkyl groups. Both the primary and secondary hydroxyl groups of βCD can coordinate the carboxylate group of heptanoate when the alkyl group of heptanoate stays in the hydrophobic cavity of βCD. The two orientations of heptanoate shown in Figure 1 correspond to two metastable states at each thermodynamic state: labeled “UP” in the left panel and “DOWN” in the right panel. The sampling problems associated with sampling the two binding modes of heptanoate/βCD have features in common with the two-state behavior of other sampling problems in computational biophysics (e.g., the protein folding problem), which makes the heptanoate/βCD complex dynamics a good choice to study the effciency of RE simulations using MSMRE simulations. Our MSMRE models follow the kinetics of the joint distribution value of the binding energy and the metastable conformational state of each replica, i.e., binding “UP” versus binding “DOWN” at each thermodynamic state.
Figure 1.
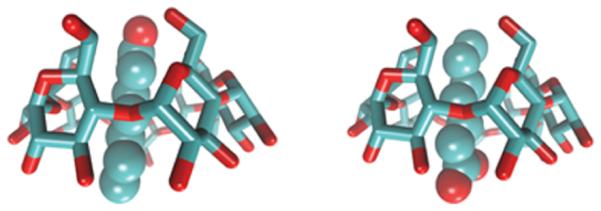
Two orientations of heptanoate in the heptanoate β-cyclodextrin binding complex. Both the primary and the secondary hydroxyl groups of βCD can form a hydrogen bond with the carboxylate group of heptanoate.
The binding energy distribution analysis method (BEDAM) is applied to study the binding of the heptanoate/βCD complex, and provides a benchmark for the MSMRE models.32,33,75,76 BEDAM is a free energy method based on RE simulations in which the interaction between ligand and acceptor is scaled by the factor λ changing gradually from zero to one. Here we chose 12 λ values: 0.0, 0.001, 0.002, 0.004, 0.04, 0.1, 0.25, 0.5, 0.8, 0.9, 0.95, 1.0. The heptanoate molecule is completely free to change its orientation relative to the βCD molecule at the λ = 0 thermodynamic state, which corresponds to no interaction between the host and guest at all, but it has less freedom as λ increases. At the four largest λ thermodynamic states, the heptanoate molecule has a large barrier to flipping its orientation through MD simulations because of the interactions between the heptanoate and βCD molecules which hinders the flip while the guest is bound within the host. We note that no flips between the “UP” and “DOWN” states are observed at λ = 0.8, 0.9, 0.95, 1.0 thermodynamic states and therefore the free energy barriers are effectively infinite at these states.
2.3. Simulations of Replica Exchange Simulations: MSMRE
To analyze the effciency of different RE algorithms, we developed tools to simulate RE simulations in our previous research.63,69 In this work, the basic idea of the model is to replace the explicit MD simulations of biomolecules generated by the integration of Newton’s laws of motion with Markov chains generated by transition matrices. The transition matrices are constructed from the data of reference MD simulations. This approach, which we refer to as the Markov state model of replica exchange (MSMRE), is an extension of the two-state kinetic network model introduced by Zheng et al.63,69 and molecular dynamics meta-simulator introduced by Smith et al.74
2.3.1. Construction of MSMRE Transition Matrices
To prepare the MSMRE models, we ran one set of 72 ns MD simulations independently at 300 K starting from the “UP” metastable state at each thermodynamic state and the other set of 72 ns MD simulations starting from the “DOWN” metastable state. We collected the binding energy and metastable state of each observation during these reference simulations for each thermodynamic state to construct Markov state models. The time scales of the flip of the heptanoate binding mode between “UP” and “DOWN” are much longer than 72 ns at the four largest λ states. No flips of the orientation of heptanoate were observed at those thermodynamic states. In the Supporting Information, we plot the binding energy distribution for each simulation. The pictures show that, at the four largest λ states, the simulations starting from different metastable states have significantly different binding energy distributions and are not converged. In contrast, the binding energy distributions of the eight remaining λ states are converged, as can be seen in the pictures.
We built up the transition matrices mimicking explicit MD simulations in the following way. First, we constructed a 50-bin histogram of binding energies at each thermodynamic state, and labeled each observation generated by the reference simulation with an array {ei, ci}, where i is the time index of the observation, ei is the index of the bin which this observation belongs to in the histogram, and ci = 0 if the observation is in the “UP” metastable state; ci = 1 if the observation is in the “DOWN” metastable state. Then, we assigned each observation to the kth discrete state according to k = ei + 50ci. Lastly, we defined a (100 × 100) state-to-state transition matrix, T(m) (where (m) stands for the “move” matrix), based on the analysis of the time series of discrete states for each thermodynamic state. In this work, we simply used the time interval between two binding energy records in the reference simulations, 0.5 ps, as the lag time for the construction of transition matrices.
The transition matrices need to be calibrated before they are used in MSMRE models. Since the matrices are constructed from finite MD simulations, the estimates of the density of states at different thermodynamic states are not identical, which will cause the outputs of the MSMRE simulation to differ from the equilibrium probabilities solved from the transition matrices. In other words, the inputs and outputs of MSMRE are different statistical ensembles. To make the MSMRE models self-consistent, we adjusted the transition matrices so that their equilibrium distributions agree with the corresponding WHAM estimate determined numerically; this insures that the estimates of the density of states at all the thermodynamic states are the same. Table 1 lists the largest implied time scale of every transition matrix used in our MSMRE models, corresponding to the transition between the “UP” and “DOWN” metastable states at each thermodynamic state.
Table 1.
Largest Implied Time Scales (IT) of the Transition Matrices Used in One-Dimensional MSMRE Models
| λ | 0.000 | 0.001 | 0.002 | 0.004 | 0.040 | 0.100 | 0.250 | 0.500 | 0.800 | 0.900 | 0.950 | 1.000 |
| IT (ps) | 7.1 | 9.1 | 19.2 | 77.1 | 148.8 | 188.3 | 100.3.6 | 2403.0 |
2.3.2. Construction of MSMRE Models
The moves in MSMRE are fulfilled by performing the following steps based on the transition matrices T(m) and the binding energy ensembles collected from the reference simulations:
Decide which discrete state the initial observation belongs to. Suppose it is the ith discrete state.
Choose the next discrete state j according to the probability of moving from the ith state to the jth state, specifically .
Randomly pick one (energy) observation from those (energy values) belonging to the jth discrete state as the next sampling.
The move process chooses the next observation (binding energy and conformational state {ei, ci}) from the database at each thermodynamic state according to the probability where nj is the number of observations in the jth discrete state of that thermodynamic state.
The exchanges in MSMRE use either the nearest neighbor exchange scheme or the independence sampling scheme. This part of MSMRE is the same as explicit RE simulations. Like explicit RE simulations, the move and exchange processes constitute a RE cycle of MSMRE. They were wrapped together by a program written in the C++ language. At the end of each RE cycle, the observation at each thermodynamic state is recorded as the output of the MSMRE simulation at cycle n, when n runs from 1, 2, …, N, where N is the total number of cycles in the MSMRE simulation.
2.3.3. The Effciency of RE Simulations
In this work, we use Markov state modeling (MSM) to describe the RE simulation kinetic network, and then measure the effciency of the RE simulations by examining the properties of the eigenvalues and eigenvectors of the corresponding transition matrix.77 The first step to build up a transition matrix from a RE simulation is clustering the simulation data. Suppose a system with n metastable states in conformational space is running at M thermodynamic states which are coupled by the RE algorithm. There are (M!nM) labeled states in the RE network model of this discretized system if one keeps track of each thermodynamic state and each replica label. The factor M! corresponds to the possible permutations of the way replicas are assigned to thermodynamic states, and the factor nM reflects the fact that the replica at each thermodynamic state occupies one of the n metastable states. However, one can average replica labels, in other words, project the (M!nM) labeled states onto (nM) unlabeled states due to the degeneracy of states and exponentially increasing scale of the labeled state space; this results in a substantial reduction in the number of states. In this study, we drastically simplified the labeling of the conformational state space of the RE simulations by only considering the heptanoate binding mode (“UP” or “DOWN”) at the largest four λ thermodynamic states. Since, at λ = 0.8, 0.9, 0.95, 1.0, each thermodynamic state has two metastable states, the RE system is considered to have 24 = 16 possible conformational states in total. In fact, the equilibration of populations among these 16 states is the rate limiting process for the full system. Since the transition rates via direct MD moves between the “UP” and “DOWN” metastable states are zero at the largest four λ thermodynamic states, in RE or MSMRE simulations, the transitions at these thermodynamic states can only occur by the exchanges with the replicas at the smaller λ states. This mimics the real system for which the barriers to the conformational transition of the ligand from binding in the “UP” mode to the “DOWN” mode are very large when the guest is confined within the host as it is for this alchemical process, and these conformational transitions are forbidden at the four largest λ states because of the strong coupling between the ligand and receptor.
After clustering every snapshot (observation) of a RE simulation to one of 16 unlabeled states (in each thermodynamic state, a conformational state is assigned without regard to the replica label), the RE kinetic network is described via a discretized master equation in the unlabeled state space, for which the solution is
| (7) |
where represents the vector probability of the unlabeled states at time Δt and T(Δt) is the row-normalized transition matrix. Then, one can investigate how effciently a RE simulation performs by determining the largest implied time scale of the transition matrix T(Δt) constructed from the RE trajectory, which represents the relaxation time of the slowest mode of the RE system.
2.3.4. Two-Dimensional MSMRE Model
By following the same procedures, we also constructed a set of databases and transition matrices for 64 thermodynamic states, which include the combinations of 4 temperatures (252, 267, 283, and 300 K) and 16 λ values (0.0, 0.001, 0.002, 0.004, 0.01, 0.04, 0.07, 0.1, 0.2, 0.4, 0.6, 0.7, 0.8, 0.9, 0.95, and 1.0). To measure the effciency of this two-dimensional MSMRE model, we simplified the labeled state space into 212 = 4096 unlabeled states by only considering the orientation of the heptanoate molecule at the largest three λ thermodynamic states at each temperature. (212 corresponds to two metastable states for each of 12 thermodynamic states—λ = 0.9, 0.95, 1.0 at each of four temperatures.) In two-dimensional MSMRE simulations, only the independence sampling scheme was used.
3. RESULTS AND DISCUSSION
As introduced in section 2.1, there are two components in a RE cycle: (i) the MD or MC simulation of each replica at a fixed thermodynamic state (the move process), which includes conformational relaxation at that thermodynamic state; (ii) the attempted swaps of replicas (the exchange process), which is the relaxation in the replica and thermodynamic state permutation space. There are two sets of random variables sampled in RE simulations: one corresponds to the conformations of replicas at each thermodynamic state, and the other corresponds to the permutations of replicas among thermodynamic states. The two components of a RE cycle each correspond to the sampling of one set of variables from the joint distribution, while the other set of variables is frozen. The goal of this section is to examine how the effciency of RE simulations depends on the two sampling components and how they interact using MSMRE models. We refer to the choice of the number of moves (MD steps) and the number of exchanges (replica exchange attempts) per RE cycle together as the “packing” of the moves and exchanges in the cycle.
3.1. Proposal Schemes and the Number of Exchange Attempts per RE Cycle
In this subsection, we examine how the number of exchange attempts per RE cycle ne affects the effciency of RE simulations when the MD period per RE cycle is 0.5 ps long by using the transition matrices built up from the MD simulations with 0.5 ps time resolution. As shown in Figure 2, the largest implied time scale decreases to a plateau value as the number of exchange attempts per RE cycle ne increases. Notice the largest implied time scale is expressed in units of RE cycles. We refer to the plateau value as the infinite swapping limit which corresponds to an infinite number of exchange attempts per RE cycle, as described in ref 51. The nearest neighbor exchange proposal scheme leads to faster equilibration and is therefore more effcient especially when the number of exchange attempts per RE cycle ne is small. With only one exchange attempt per cycle, the nearest neighbor exchange proposal scheme is approximately twice as effcient as the independence sampling scheme. In contrast, in the infinite swapping limit, the largest implied time scale is independent of the proposal scheme. The advantage of the nearest neighbor exchange scheme at small ne for this particular example arises from its higher exchange acceptance ratio (see Table S1 in the Supporting Information for the acceptance ratios) and the specific spacing of λ values in this model. The relative advantage/disadvantage of the nearest neighbor exchange scheme compared with the independence sampling will vary depending on the configuration-space overlap of neighboring thermodynamic states and the total number of thermodynamic states.
Figure 2.
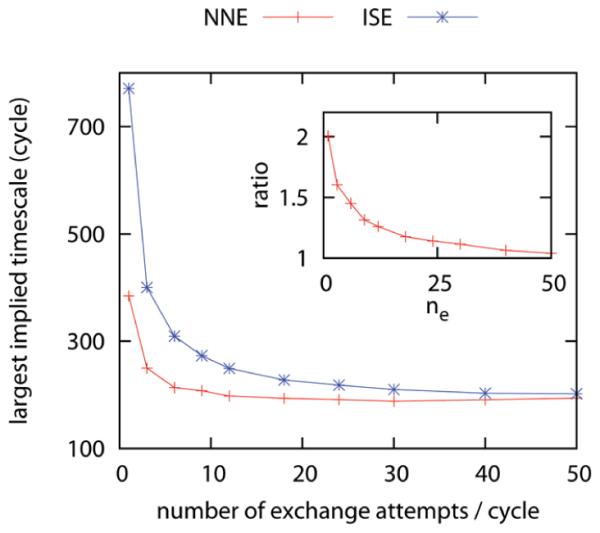
Dependence of the largest implied time scale on the number of exchange attempts per RE cycle ne for the one-dimensional MSMRE simulations using different proposal schemes (NNE, nearest neighbor exchange; ISE, independence sampling). The MD period per RE cycle is fixed at 0.5 ps. Note that the largest implied time scales of the two proposal schemes converge to the same limit. The inset shows the ratio of the largest implied time scales of MSMRE simulations using these two proposal schemes.
The kinetics of the labeled states in RE simulations explains why different proposal schemes have the same effciency in the infinite swapping limit. As discussed in previous work,63,72 the labeled states of RE simulations are connected by both move and exchange processes. One can cluster the labeled states into islands so that the labeled states in one island are connected only by exchange processes; states in different islands are connected through at least one move process. Figure 3 shows an example of clustering a three-replica RE system by their connections. In this example, each replica (represented by the position of the letter or number in the array—see the figure legend) moves via operations of the molecular dynamics transition matrix (arrows drawn in blue) between the “UP” and “DOWN” metastable states at its assigned thermodynamic state, and then can exchange (arrows drawn in red) to another thermodynamic state via an exchange. Since exchange processes cannot change the conformational state of a replica, the number of islands corresponds to the total possible number of ways replicas can be assigned to conformational states, namely, NM when there are M replicas and N metastable states at each thermodynamic state. Thus, there are 23 = 8 islands at the corners of a cube in Figure 3 connected by blue arrows. The number of labeled states in every island is the total number of possible permutations without repetition of replicas and thermodynamic states: M! = 3! = 6. Note that within an island the number of unique metastable state labels determines the degeneracy of the states. For example, the six states in the “DDD” island are all degenerate with respect to the conformational label, while the six states in the “DDU” island are partially degenerate. Figure 3 also shows the exchange connections between labeled states for the nearest neighbor exchange (solid red arrows) and the independence sampling (solid and dashed red arrows) proposal schemes. The proposal scheme does not affect the island clustering but only affects the equilibrium pathways connecting labeled states within each individual island. At the limit of many attempts per RE cycle— the infinite swapping limit—the probability of staying at each labeled state in an island has reached equilibrium in each cycle of replica exchange; therefore, only the MD moves between islands determine the relaxation of the full RE system.
Figure 3.
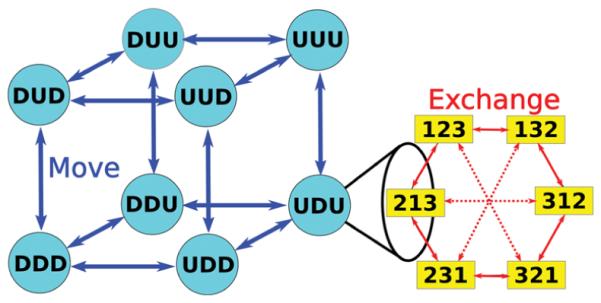
Kinetic network model of RE product state space. In this picture, the nth position of the letter or number represents the metastable state or the thermodynamic state of the nth replica, respectively. The cube on the left shows the possible combinations of replicas and metastable states for a RE system with three replicas, namely, 23 = 8 vertexes. Each vertex of the cube is called an island. For example, UDU represents an island in which the 1st replica is at the “UP” metastable state, the 2nd replica is at the “DOWN” metastable state, and the 3rd replica is at the “UP” metastable state. Since exchanges cannot change the conformational state of a replica, the RE system can only travel to different islands via MD simulations (or moves) represented by blue arrows. On the right side, we show the degeneracy of one island. The states in each island are the possible permutations without repetition of replicas and thermodynamic states; therefore, there are 3! = 6 labeled states in each island. For example, “132” means the 1st replica occupies the 1st thermodynamic state, the 2nd replica occupies the 3rd thermodynamic state, and the 3rd replica occupies the 2nd thermodynamic state. The labeled states (yellow boxes in the figure above) in an island are solely connected by exchanges. Solid red arrows stand for the connections made by the nearest neighbor exchange scheme; i.e., the RE system can switch between the two labeled states connected by solid red arrows through one successful swap of the nearest neighbor exchange scheme. All red arrows, including the solid and dashed arrows, represent the connections made by the independence sampling scheme.
Prior studies have noted the importance of increasing the number of exchange attempts per RE cycle ne to make sure RE systems are “well mixed”.50,51 Chodera and Shirts suggested that M3 to M5 exchanges be attempted in one RE cycle, where M is the total number of replicas.50 A similar discussion of the structure of RE states space was reported recently by Doll et al., and the approach to the plateau value as shown in Figure 2 was termed “infinite swapping”.51–53 If one calculates the equilibrium probabilities of all labeled states in an island and then exchanges to the next labeled state according to those probabilities, the infinite swapping limit is reached in one exchange attempt. However, the number of states in one island increases factorially and becomes overwhelming when the number of replicas increases. For example, for a RE system with 12 replicas, there were (12! = 4.79 × 108) labeled states in every island. Doll et al. proposed the “partial infinite swapping” technique to deal with this factorial explosion.51 For the host– guest binding problem we are considering, the effciency of RE simulations converges rapidly for both proposal schemes even though the number of labeled states in each island is huge. This effect may be explained by the fact that, for each island, only a very small number of states have nontrivial probabilities. Figure 2 suggests that the probabilities of those states have converged to close to their equilibrium values after relaxation corresponding to ~50 exchange attempts per RE cycle on average. The results shown in Figure 2 are for an MSMRE model with fixed MD period per cycle (0.5 ps). As we discuss later, these results are independent of the MD period per cycle.
3.2. The Infinite Swapping Limit
We now propose a way to determine how many exchange attempts per RE cycle are required to reach the infinite swapping limit by observing the trajectories of replicas through state space during simulations of replica exchange. This approach makes it possible to compare proposal schemes and in favorable circumstances estimate the respective number of exchange attempts per RE cycle ne required to reach the infinite swapping limit through short test runs of RE simulations. The basic idea is as follows. In the infinite swapping limit, the probability of a labeled state {S} in one island (see Figure 3) is
| (8) |
where the denominator is the sum of the probabilities of all the labeled states in the same island including {S}. Suppose at the beginning of a set of exchange processes the mth replica is at the αth thermodynamic state and Pm,α(ne) represents the probability for this replica to be at the αth state after ne exchange attempts. After a large number of exchange attempts
| (9) |
where the sum includes every labeled state in that island which has the mth replica at the αth thermodynamic state, namely, s[m] = α. Since the dynamical behaviors of all replicas at a thermodynamic state are the same, we can omit the replica index in Pm,α(ne). If one follows a single replica trajectory within the exchange process per cycle and records the time evolution of the replica through the state space, one can construct an M × M transition matrix T(e) (where (e) stands for the “exchange” matrix) for a set of exchanges of a RE simulation with M thermodynamic states. In that case, Pα(ne) is the diagonal element of the matrix; represents the probability that a labeled replica is at the αth thermodynamic state after one RE cycle consisting of ne exchange attempts given that the replica is at the αth thermodynamic state at the beginning of the RE cycle.78 The precision in the estimate of can be increased by averaging over the trajectories of all the replicas.
According to eq 9, converges to an asymptote when the number of exchange attempts per cycle ne approaches the infinite swapping limit; therefore, the dependence of the diagonal matrix elements on the number of exchange attempts per cycle ne provides a way to determine how many exchange attempts per cycle are required to reach the infinite swapping limit. In practice, an estimate of the limit is required to apply this approach. The value of can be estimated by the unbinned WHAM method (UWHAM).79 Recently, we developed a replica exchange-like algorithm called “RE-SWHAM” which solves the UWHAM equations stochastically.80 The RE-SWHAM algorithm uses a similar replica exchange procedure as MSMRE to resample the raw data generated from parallel simulations. See the Supporting Information for the differences between the RESWHAM and MSMRE methods.
Suppose wαk is the normalized UWHAM weight of the kth data element at the αth thermodynamics state, which satisfies , where M is the total number of thermodynamic states and N is the total number of observations. We showed previously that the probability of the kth data element appearing in the database of the αth thermodynamic state is (wαkN/M) during the RE-SWHAM analysis.80 Therefore, for a RE-SWHAM analysis, the asymptote of is
| (10) |
Because the MSMRE models have been calibrated so that their equilibrium distributions agree with the corresponding UWHAM (or RE-SWHAM) estimate numerically, and because the limit is independent of the MD period per cycle (which we discuss below), eq 10 can be used to estimate the asymptote of and therefore can be used to provide an estimate of the number of exchange attempts per cycle ne required to reach the infinite swapping limit in both MSMRE “simulations of simulations” and the actual MD or MC replica exchange simulations.
Figure 4a shows averaged over all 12 thermodynamic states versus the number of exchange attempts per RE cycle ne in one-dimensional MSMRE simulations. As mentioned previously, for the specific spacing of the thermodynamic states in this model problem, the nearest neighbor exchange reaches the infinite swapping limit first. Also from Figure 4a, it can be seen that the number of exchange attempts per RE cycle ne required to reach the infinite swapping limit is about 40 for the nearest neighbor exchange proposal scheme and 100 for the independence sampling, respectively. Figure 4b shows versus the number of exchange attempts per RE cycle ne in two-dimensional MSMRE simulations (described in section 2.3.4). is the average over the replicas at the largest three λ states of each temperature. In Figure 4b, the function plateaus as ne approaches ~2500. The inset plot of Figure 4b shows the dependence of the largest implied time scale on the number of exchange attempts per RE cycle ne. Note both axes are in logarithmic scale in the inset plot. In each picture of Figure 4, the asymptote of average estimated by eq 10 is shown as a horizontal black line. As demonstrated by these two examples, one can choose the best proposal scheme and estimate the number of exchange attempts per cycle ne required to reach the infinite swapping limit by examining the dependence of on the number of exchange attempts per cycle ne before running RE simulations in production mode.
Figure 4.
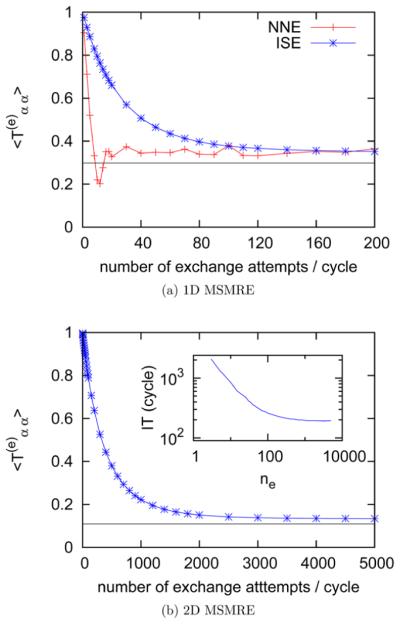
Dependence of the average of diagonal matrix elements on the number of exchange attempts per cycle ne. is the average probability that a labeled replica is at the αth thermodynamic state after the exchange process given that it is at the αth thermodynamic state before the exchange process. Plot a shows the dependence of on the exchange attempts per RE cycle ne for one-dimensional MSMRE simulations using different proposal schemes (NNE, nearest neighbor exchange; ISE, independence sampling). Plot b shows the dependence of on the number of exchange attempts per RE cycle ne for the two-dimensional MSMRE simulations using the independence sampling scheme, and the inset shows the dependence of the largest implied time scale (IT) on the number of exchange attempts per RE cycle ne. The horizontal black lines in plots a and b are the estimates of calculated by eq 10.
3.3. Length of the MD Period per RE Cycle
In this subsection, we first examine how the largest implied time scale depends on the number of exchange attempts per RE cycle ne at the MD f ull relaxation limit. We define the time scale to reach the MD full relaxation limit for the model host–guest problem as follows. At the eight smallest λ thermodynamic states, the MD simulation period is long enough to equilibrate over the whole configurational space via MD moves without exchanges; at the four largest λ thermodynamic states, the MD simulation is only long enough to equilibrate within each of the metastable states (UP and DOWN) but not long enough to equilibrate between the UP and DOWN conformational states. Referring to the time scales in Table 1, the MD full relaxation limit corresponds to an MD period per RE cycle τ longer than the largest implied time scale at the λ = 0.5 state, 2.4 ns, which corresponds to the longest relaxation for the UP to DOWN transition by means of conformational relaxation within a thermodynamic state without coupling to additional relaxation pathways using replica exchange. To model the MD full relaxation limit, we changed the transition matrices in MSMRE to choose the next observation based on the equilibrium probability in the full configurational space (or in the metastable state for the largest four λ states) so that adjacent MD moves are completely uncorrelated.
Figure 5a shows the dependence of the largest implied time scale in units of RE cycles on the number of exchange attempts per RE cycle ne at the MD full relaxation limit. The results are similar to those results shown in Figure 2 obtained from the MSMRE simulations with the MD duration per RE cycle τ equal to 0.5 ps. Both figures show the largest implied time scale reaches a plateau within 50 exchange attempts per cycle, which implies that the number of exchange attempts per RE cycle ne required to reach the infinite swapping limit does not depend on the MD period per RE cycle τ. In the Supporting Information, we plot the dependence of averaged over all 12 thermodynamic states on the number of exchange attempts per RE cycle ne for five one-dimensional MSMRE simulations using the independence sampling scheme. These MSMRE simulations use different lengths of the MD period per RE cycle ranging from 0.5 ps to the MD full relaxation limit. The overlapping of all five curves further demonstrates that the number of exchange attempts per RE cycle ne required to reach the infinite swapping limit does not depend on the MD period per RE cycle τ. This can be understood by the following argument. In each RE cycle, one can construct an exchange transition matrix based on the proposal scheme and the instantaneous energies of replicas. The largest implied time scale of the exchange transition matrix determines the number of exchange attempts required to fully relax the RE system in that RE cycle. Therefore, the number of exchange attempts required to reach the infinite swapping limit is determined by the average of the largest implied time scale of the exchange transition matrix determined by all possible combinations of energy at each thermodynamic state. The probability of an energy combination appearing in a RE cycle is proportional to the product of the Boltzmann factor and the density of states of each energy in that combination, and therefore does not depend on the MD period per RE cycle.
Figure 5.
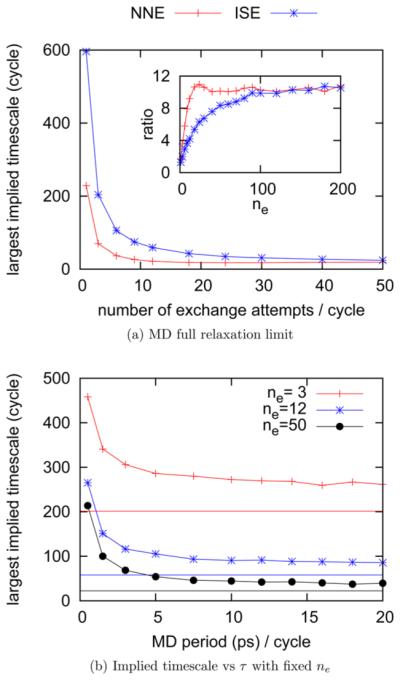
(a) At the MD full relaxation limit, the dependence of the largest implied time scale on the number of exchange attempts per RE cycle ne for the one-dimensional MSMRE simulations using different proposal schemes (NNE, nearest neighbor exchange; ISE, independence sampling). The inset plots the ratio of the largest implied time scales when τ is 0.5 ps to those at the MD full relaxation. (b) The dependence of the largest implied time scale on the MD period per RE cycle τ for the one-dimensional MSMRE simulations using the independence sampling scheme when the number of exchange attempts per RE cycle ne is 3, 12, or 50. The horizontal lines are the respective largest implied time scales at the MD full relaxation limit.
Both the exchange processes and the MD move processes relax the RE system. Compared with Figure 2, the implied time scales in Figure 5a at the MD full relaxation limit are much smaller in units of RE cycles, since there is more conformational relaxation at all thermodynamic states due to the long MD move duration per RE cycle. The inset of Figure 5a shows the ratio of the largest implied time scales when τ is 0.5 ps to those at the MD full relaxation limit. This ratio increases with increasing number of exchange attempts per RE cycle ne, approaching a plateau value of ~10× as ne approaches the infinite swapping limit. Figure 5b shows the dependence of the largest implied time scale in units of RE cycles on the MD period per RE cycle τ for the one-dimensional MSMRE simulations using the independence sampling scheme when the number of exchange attempts per RE cycle ne is 3, 12, or 50. As can be seen from Figure 5b, the largest implied time scale decreases when the MD period per RE cycle τ increases, and every curve converges toward the respective MD full relaxation limit. Although it appears that each curve is already close to converged when τ increases to ~20 ps, the largest implied time scale will continue decreasing until reaching the MD full relaxation limit when τ is longer than 2.4 ns. The reason for this is that conformational relaxation at every λ state makes some contribution to the implied time scale even though the implied time scale is dominated by the state with the fastest conformational relaxation time as ne approaches the infinite swapping limit.
3.4. Packing Moves and Exchanges in RE Simulations
We have used the MSMRE models with independence sampling scheme to investigate how the two components of a RE cycle—the configurational relaxation within each thermodynamic state via moves and the multicanonical relaxation via exchanges—combine to equilibrate RE systems. We showed that either increasing the number of exchange attempts per RE cycle ne or increasing the MD period per RE cycle τ can shorten the largest implied time scale of MSMRE simulations in units of RE cycles. As listed in Table 2, for the host–guest binding affnity model system with one-dimensional replica exchange and 12 thermodynamic states, the largest implied time scale is 771.1 RE cycles when the number of exchange attempts per cycle ne is one and the MD period per RE cycle τ is 0.5 ps, and decreases to 19.2 RE cycles when ne reaches the infinite swapping limit and τ reaches the MD full relaxation limit.
Table 2.
Largest Implied Time Scales of the One-Dimensional MSMRE Simulations Using the Independence Sampling Scheme under Four Conditions
| largest implied time scale (cycle) | ne =1 | infinite swapping limit |
|---|---|---|
| τ = 0.5 ps | 771.1 | 202.1 |
| MD full relaxation limit | 595.8 | 19.2 |
Because performing the MD simulation is the most computationally intensive part of a replica exchange cycle, in the following discussion, we assume the time for the multicanonical exchange in a RE cycle is negligible compared with the time to perform MD simulations and change the unit of the largest implied time scale to MD simulation time in order to provide a more intuitive understanding of the relationship between packing and effciency. Figure 6 shows the largest implied time scale calculations for ~80 one-dimensional MSMRE simulations using the independence sampling scheme, which include different combinations of the MD period per RE cycle τ ranging from 0.5 to 10 ps and the number of exchange attempts per RE cycle ne ranging from one to the “infinite” swapping limit.
Figure 6.
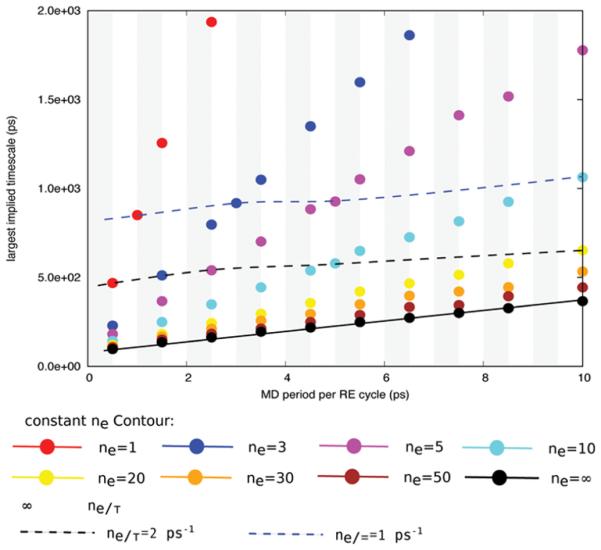
Largest implied time scales of one-dimensional MSMRE simulations using different combinations of the number of exchange attempts per RE cycle ne and the MD period per RE cycle τ. All of these one-dimensional MSMRE simulations use the independence sampling scheme. The vertical borders between white and gray regions are the constant τ contours. The constant τ contours show that increasing the number of exchange attempts per RE cycle ne increases the sampling effciency of RE simulations until ne reaches the infinite swapping limit. Each color marks a constant ne contour. The constant ne contours show that increasing the MD period per RE cycle τ increases the largest implied time scale in units of MD simulation time, namely, decreases the sampling effciency of RE simulations. The dashed lines are the constant ne/τ contours, which show that the largest implied time scale increases monotonously when the number of exchange attempts per RE cycle ne and the MD period per RE cycle τ both increase while the ratio of ne over τ is fixed.
Figure 6 summarizes our results about the impact of the packing (i.e., the concerted change in both the number of exchange attempts per RE cycle ne and the MD period per RE cycle τ) on the sampling effciency of RE simulations. The vertical borders between gray and white regions in Figure 6 are the constant MD period (τ) contours. The data at each constant τ contour (vertical lines) show that increasing the number of exchange attempts per RE cycle ne increases the sampling effciency of RE simulations until ne reaches the infinite swapping limit (the black dots). When the MD period per RE cycle is 0.5 ps, increasing the number of exchange attempts from one to the infinite swapping limit leads to a 4-fold effciency gain. In the Supporting Information Figures S6 and S7, we plot the corresponding results for two-dimensional MSMRE simulations. When the MD period per RE cycle is 0.5 ps, increasing the number of exchange attempts from one to the infinite swapping limit leads to a 25-fold effciency gain for the two-dimensional MSMRE model with 64 replicas (described in section 2.3.4), and a 40-fold effciency gain for another two-dimensional MSMRE model with 128 replicas. This result is consistent with the intuitive idea that the effciency gain obtained by going to the infinite swapping limit becomes larger as the system size (number of states) of the replica exchange simulation increases.
Each broken line of colored dots in Figure 6 marks a contour corresponding to a constant number of exchange attempts per RE cycle ne. If one connects each data point and the origin with a straight line (not shown in Figure 6), the slope of this line is the largest implied time scale in units of RE cycles of that MSMRE simulation, which corresponds to the height of the respective data point in Figure 5b. The constant ne contours show that, although increasing the MD period per RE cycle τ decreases the largest implied time scale in units of RE cycles, it increases the largest implied time scale in units of MD simulation time; in other words, the RE simulation is less effcient when the MD length per cycle is increased at a fixed number of exchange attempts per cycle. Compared with exchange processes, MD move processes are less effcient at relaxing the RE system. This conclusion is in agreement with the results reported previously by Roitberg et al.67,70
Finally, we examine how the sampling effciency of RE simulations changes when both the number of exchange attempts per RE cycle ne and the MD period per RE cycle τ are altered at the same time; we refer to this as packing moves and exchanges into the RE cycle. In Figure 6, the dashed lines are constant ne/τ contours, which correspond to a fixed ratio of exchange attempts/MD period. For the one-dimensional MSMRE model, the largest implied time scale monotonically increases when the number of exchange attempts per RE cycle ne and the MD period per RE cycle τ both increase while the ratio of ne/τ is fixed at 1 or 2 ps−1. As the fixed ratio ne/τ increases, the constant ne/τ contour lines approach the infinite swapping limit curve (i.e., the dashed contour lines approach the solid black infinite swapping limit contour line in Figure 6). In the Supporting Information, Figures S6 and S7, we show the corresponding results for two-dimensional MSMRE simulations. In these examples, the constant ne/τ curves also monotonically increase when ne and τ both increase while the ratio of ne/τ is fixed at 1 or 2 ps−1
To compare the sampling effciency of RE simulations using different combinations of the number of exchange attempts per RE cycle ne and the MD period per RE cycle τ, we introduce the concept of “the average continuous MD period” (or “lifetime”78,81) at the αth thermodynamic state, , which represents the average simulation time that a replica stays at the αth thermodynamic state before it is exchanged to another state. In a RE simulation, the probability density function of a lifetime only has nonzero values at time nτ, where n is a positive integer and τ is the MD period per RE cycle. The lifetime at the αth thermodynamic state is a function of the number of exchange attempts per RE cycle ne and the MD period per RE cycle τ, namely,
| (11) |
where is the αth diagonal element of the exchange transition matrix for ne exchange attempts, which was discussed above in section 3.2. The trajectory of a single replica in the space of thermodynamic state indexes resembles a random walk on a chain or a network.41,62 Equation 11 shows the explicit dependence of the lifetime of a replica on the number of exchange attempts per RE cycle ne and the MD period per RE cycle τ at the αth thermodynamic state.
In Figure 7a, we show the correlations between the largest implied time scales and the average lifetimes for ~600 one-dimensional MSMRE simulations using the independence sampling scheme. For each simulation, the average lifetime is the mean value of the average continuous MD periods over 12 λ thermodynamic states, which ranges from 0.5 to 100 ps. Figure 7b shows the correlation between the largest implied time scales and the average lifetimes for ~200 two-dimensional MSMRE simulations (described in section 2.3.4). For each simulation, the average lifetime is the mean value of the average continuous MD period over the largest three λ thermodynamic states at each temperature. The results in Figure 7a and b show that the average lifetime provides a measure of the sampling effciency comparable to that of thelargest implied time scale as the packing of moves and exchanges into a RE cycle is changed.
Figure 7.
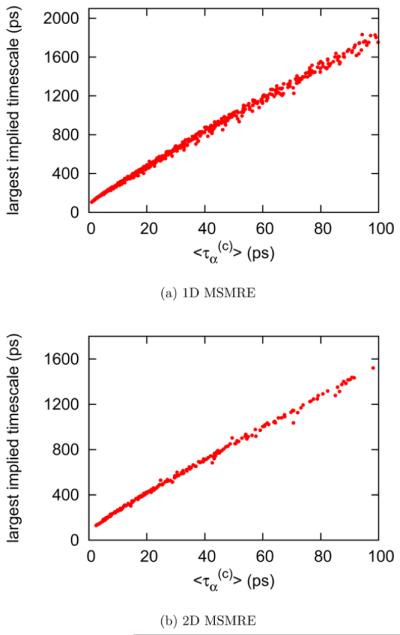
Correlation between the largest implied time scale and the average lifetime. Plot a shows the correlation between the largest implied time scale and the average lifetime for ~600 one-dimensional MSMRE simulations using the independence sampling scheme, where is the average lifetime over 12 λ thermodynamic states. Plot b shows the correlation between the largest implied time scale and the average lifetime for ~200 two-dimensional MSMRE simulations using the independence sampling scheme, where is the average lifetime over the largest three λ thermodynamic states at each temperature. Both results suggest that the average lifetime is a good indicator for the sampling effciency of RE simulations which differ by the way moves and exchange attempts are packed into the replica exchange cycles.
There are a few observations of interest concerning the average lifetime . We note that it corresponds to an explicit expression describing how the effciency of replica exchange varies with both the number of exchange attempts per RE cycle ne and the MD period per RE cycle τ. While the effciency increases as decreases to the infinite swapping limit as expected, the effciency is also predicted to increase as the MD move period decreases without an apparent limit. This is of course unphysical, as the effciency must eventually plateau if the MD move period is short enough. Formulas for the effciency of replica exchange in the fast exchange limit (both ne going to the infinite swapping limit and τ going to zero simultaneously) have been derived.63,72 We have tried to investigate the behavior of the MSMRE model in this limit but were unable to converge the MSMRE simulations for very short MD periods below 50 fs. We also note that the average lifetime can be related to the mean first passage times (MFPTs) for the replicas to cross the thermodynamic state space,81 and these MFPTs have been used previously as an effciency diagnostic to analyze ways to optimize replica exchange parameters.41 An advantage of using as a measure of the effciency of the RE packing scheme is that it can be calculated directly from RE simulations without having to construct an MSM.
4. CONCLUSION
We have built Markov state models of replica exchange (MSMRE) to study the sampling effciency of RE simulations as the number of exchange attempts and the MD period are varied in a replica exchange cycle. The MSMRE model replaces the explicit MD simulations generated by the integration of Newton’s equations of motion with Markov chains generated by transition matrices parametrized from independent simulations at each thermodynamic state. This “simulations of simulations” tool can model explicit RE simulations which require months’ of computational time on computer clusters with ones which require hours on a desktop, therefore significantly reducing the time required to develop new adaptive RE algorithms and proposal schemes.
We applied the MSMRE models to study different possible implementations of the replica exchange cycle. Our results show that different exchange proposal schemes converge to the same infinite swapping limit. It is possible to reach the infinite swapping limit with tens of exchange attempts per RE cycle in the one-dimensional system we studied and a few thousand exchange attempts per RE cycle in the two-dimensional system. If the computational cost for exchange is negligible compared with the computational cost to perform MD simulations, which is true for most RE simulations, one should always perform enough exchange attempts per RE cycle to reach the infinite swapping limit. We propose a practical approach to estimate the number of exchange attempts per RE cycle ne required to reach the infinite swapping limit which involves estimating in preproduction runs of replica exchange. We also applied the MSMRE models to study the impact of the MD period per RE cycle τ on the sampling effciency. Our results confirm that MD move processes are less effcient at relaxing RE systems compared with exchange processes. As shown in Figure 6, the effciency of MSMRE simulations decreases when the MD length per cycle is increased at a fixed number of exchange attempts per cycle. Therefore, the MD period per RE cycle in RE simulations should be set as short as possible until the overhead time associated with restarting MD simulations becomes significant. Finally, we applied the MSMRE models to study the packing of moves and exchanges in RE simulations. We introduced the concept of the lifetime of replicas at a thermodynamic state in RE simulations, and found that the average lifetime of replicas can be used as an indicator of the sampling effciency of RE simulations which depends on the way moves and exchange attempts are packed into the replica exchange cycles.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the National Science Foundation (CDI type II 1125332) and the National Institutes of Health (GM30580). The simulations were carried out on the Owlsnest computer cluster of Temple University, which is supported by grants from the National Science Foundation (CNS-09-58854), and Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation (ACI-1053575).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the
ACS Publications website at DOI: 10.1021/acs.jpcb.6b02015.
The binding energy distributions of the heptanoate β-cyclodextrin binding complex at different λ states obtained from the independent MD simulations; the binding energy distributions obtained from the RE-SWHAM method, the MSMRE model, and the explicit RE simulation; the percentage of the “DOWN” metastable state calculated by the RE-SWHAM method, the MSMRE model, and the explicit RE simulation; the estimate of the effciency of RE simulations; the acceptance ratios in MSMRE simulations; the differences between the RE-SWHAM and MSMRE methods; the number of exchange attempts required to reach the infinite swapping limit for the MSMRE simulations using different lengths of the MD period per RE cycle; packing moves and exchange in two-dimensional MSMRE simulations (PDF)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Zwier MC, Chong LT. Reaching biological timescales with all-atom molecular dynamics simulations. Curr. Opin. Pharmacol. 2010;10:745–752. doi: 10.1016/j.coph.2010.09.008. [DOI] [PubMed] [Google Scholar]
- (2).Zuckerman DM. Equilibrium sampling in biomolecular simulations. Annu. Rev. Biophys. 2011;40:41–62. doi: 10.1146/annurev-biophys-042910-155255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Makarov DE. Single Molecule Science: Physical Principles and Models. CRC Press; 2015. p. 59. Chapter 5. [Google Scholar]
- (4).Torrie GM, Valleau JP. Nonphysical sampling distributions in Monte Carlo free-energy estimation: umbrella sampling. J. Comput. Phys. 1977;23:187–199. [Google Scholar]
- (5).Northrup SH, Pear MR, Lee CY, McCammon JA, Karplus M. Dynamical theory of activated processes in globular proteins. Proc. Natl. Acad. Sci. U. S. A. 1982;79:4035–4039. doi: 10.1073/pnas.79.13.4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Law SM, Feig M. Base-flipping mechanism in postmismatch recognition by MutS. Biophys. J. 2011;101:2223–2231. doi: 10.1016/j.bpj.2011.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Dickson A, Warmflash A, Dinner AR. Nonequilibrium umbrella sampling in spaces of many order parameters. J. Chem. Phys. 2009;130:074104. doi: 10.1063/1.3070677. [DOI] [PubMed] [Google Scholar]
- (8).Dellago C, Bolhuis PG, Chandler D. Efficient transition path sampling: application to Lennard-Jones cluster rearrangements. J. Chem. Phys. 1998;108:9236–9245. [Google Scholar]
- (9).Zuckerman DM, Woolf TB. Dynamic reaction paths and rates through importance-sampled stochastic dynamics. J. Chem. Phys. 1999;111:9475–9484. [Google Scholar]
- (10).van Erp TS, Moroni D, Bolhuis PG. A novel path sampling method for the calculation of rate constants. J. Chem. Phys. 2003;118:7762–7774. [Google Scholar]
- (11).Faradjian AK, Elber R. Computing time scales from reaction coordinates by milestoning. J. Chem. Phys. 2004;120:10880–10889. doi: 10.1063/1.1738640. [DOI] [PubMed] [Google Scholar]
- (12).Allen RJ, Warren PB, ten Wolde PR. Sampling rare switching events in biochemical networks. Phys. Rev. Lett. 2005;94:018104. doi: 10.1103/PhysRevLett.94.018104. [DOI] [PubMed] [Google Scholar]
- (13).Zhang BW, Jasnow D, Zuckerman DM. Efficient and verified simulation of a path ensemble for conformational change in a united-residue model of calmodulin. Proc. Natl. Acad. Sci. U. S. A. 2007;104:18043–18048. doi: 10.1073/pnas.0706349104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Zwier MC, Adelman JL, Kaus JW, Pratt AJ, Wong KF, Rego NB, Suarez E, Lettieri S, Wang DW, Grabe M, et al. WESTPA: an interoperable, highly scalable software package for weighted ensemble simulation and analysis. J. Chem. Theory Comput. 2015;11:800–809. doi: 10.1021/ct5010615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Zheng L, Chen M, Yang W. Random walk in orthogonal space to achieve efficient free-energy simulation of complex systems. Proc. Natl. Acad. Sci. U. S. A. 2008;105:20227–20232. doi: 10.1073/pnas.0810631106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Guttenberg N, Dinner AR, Weare J. Steered transition path sampling. J. Chem. Phys. 2012;136:234103. doi: 10.1063/1.4724301. [DOI] [PubMed] [Google Scholar]
- (17).Molloy K, Shehu A. Elucidating the ensemble of functionally-relevant transitions in protein systems with a robotics-inspired method. BMC Struct. Biol. 2013;13:S8. doi: 10.1186/1472-6807-13-S1-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Hawk AT, Konda SSM, Makarov DE. Computation of transit times using the milestoning method with applications to polymer translocation. J. Chem. Phys. 2013;139:064101. doi: 10.1063/1.4817200. [DOI] [PubMed] [Google Scholar]
- (19).Mitsutake A, Sugita Y, Okamoto Y. Generalized-ensemble algorithms for molecular simulations of biopolymers. Biopolymers. 2001;60:96–123. doi: 10.1002/1097-0282(2001)60:2<96::AID-BIP1007>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- (20).Okamoto Y. Generalized-ensemble algorithms: enhanced sampling techniques for Monte Carlo and molecular dynamics simulations. J. Mol. Graphics Modell. 2004;22:425–439. doi: 10.1016/j.jmgm.2003.12.009. [DOI] [PubMed] [Google Scholar]
- (21).Marinari E, Parisi G. Simulated tempering: a new Monte Carlo scheme. Europhys. Lett. 1992;19:451–458. [Google Scholar]
- (22).Lyubartsev AP, Martsinovski AA, Shevkunov SV, Vorontsov-Velyaminov PN. New approach to Monte Carlo calculation of the free energy: method of expanded ensembles. J. Chem. Phys. 1992;96:1776–1783. [Google Scholar]
- (23).Swendsen RH, Wang JS. Replica Monte Carlo simulation of spin-glasses. Phys. Rev. Lett. 1986;57:2607–2609. doi: 10.1103/PhysRevLett.57.2607. [DOI] [PubMed] [Google Scholar]
- (24).Hansmann UHE. Parallel tempering algorithm for conformational studies of biological molecules. Chem. Phys. Lett. 1997;281:140–150. [Google Scholar]
- (25).Sugita Y, Okamoto Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999;314:141–151. [Google Scholar]
- (26).Sugita Y, Kitao A, Okamoto Y. Multidimensional replica-exchange method for free-energy calculations. J. Chem. Phys. 2000;113:6042–6051. [Google Scholar]
- (27).Khandogin J, Brooks CL., III Linking folding with aggregation in Alzheimer’s β-amyloid peptides. Proc. Natl. Acad. Sci. U. S. A. 2007;104:16880–16885. doi: 10.1073/pnas.0703832104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Faraldo-Gomez JD, Roux B. Characterization of conformaional equilibria through Hamiltonian and temperature replica-exchange simulations: assessing entropic and environmental effects. J. Comput. Chem. 2007;28:1634–1647. doi: 10.1002/jcc.20652. [DOI] [PubMed] [Google Scholar]
- (29).Zheng W, Andrec M, Gallicchio E, Levy RM. Recovering kinetics from a simplified protein folding model using replica exchange simulations: a kinetic network and effective stochastic dynamics. J. Phys. Chem. B. 2009;113:11702–11709. doi: 10.1021/jp900445t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Itoh SG, Damjanović A, Brooks BR. pH replica-exchange method based on discrete protonation states. Proteins: Struct., Funct., Genet. 2011;79:3420–3436. doi: 10.1002/prot.23176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Zheng W, Gallicchio E, Deng N, Andrec M, Levy RM. Kinetic network study of the diversity and temperature dependence of Trp-Cage folding pathways: combining transition path theory with stochastic simulations. J. Phys. Chem. B. 2011;115:1512–1523. doi: 10.1021/jp1089596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Lapelosa M, Gallicchio E, Levy RM. Conformational transitions and convergence of absolute binding free energy calculations. J. Chem. Theory Comput. 2012;8:47–60. doi: 10.1021/ct200684b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Gallicchio E, Levy RM. Prediction of SAMPL3 host-guest affinities with the Binding Energy Distribution Analysis Method (BEDAM) J. Comput.-Aided Mol. Des. 2012;26:505–516. doi: 10.1007/s10822-012-9552-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Morrow BH, Koenig PH, Shen JK. Self-assembly and bilayer-micelle transition of fatty acids studied by replica-exchange constant pH molecular dynamics. Langmuir. 2013;29:14823–14830. doi: 10.1021/la403398n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Gallicchio E, Deng N, He P, Wickstrom L, Perryman AL, Santiago DN, Forli S, Olson AJ, Levy RM. Virtual screening of integrase inhibitors by large scale binding free energy calculations: the SAMPL4 challenge. J. Comput.-Aided Mol. Des. 2014;28:475–490. doi: 10.1007/s10822-014-9711-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Liu P, Kim B, Friesner RA, Berne BJ. Replica exchange with solute tempering: a method for sampling biological systems in explicit water. Proc. Natl. Acad. Sci. U. S. A. 2005;102:13749–13754. doi: 10.1073/pnas.0506346102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Wang L, Friesner RA, Berne BJ. Replica exchange with solute scaling: a more efficient version of Replica Exchange with Solute Tempering (REST2) J. Phys. Chem. B. 2011;115:9431–9438. doi: 10.1021/jp204407d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Lyman E, Zuckerman DM. Resolution exchange simulation with incremental coarsening. J. Chem. Theory Comput. 2006;2:656–666. doi: 10.1021/ct050337x. [DOI] [PubMed] [Google Scholar]
- (39).Gallicchio E, Levy RM, Parashar M. Asynchronous replica exchange for molecular simulations. J. Comput. Chem. 2008;29:788–794. doi: 10.1002/jcc.20839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Jiang W, Roux B. Free Energy Perturbation Hamiltonian Replica-Exchange Molecular Dynamics (FEP/H-REMD) for absolute ligand binding free energy calculations. J. Chem. Theory Comput. 2010;6:2559–2565. doi: 10.1021/ct1001768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Nadler W, Meinke JH, Hansmann UHE. Folding proteins by first-passage-times-optimized replica exchange. Phys. Rev. E: Stat. Nonlin. Soft Matter Phys. 2008;78:061905. doi: 10.1103/PhysRevE.78.061905. [DOI] [PubMed] [Google Scholar]
- (42).Arrar M, de Oliveira CAF, Fajer M, Sinko W, McCammon JA. w-REXAMD: a Hamiltonian replica exchange approach to improve free energy calculations for systems with kinetically trapped conformations. J. Chem. Theory Comput. 2013;9:18–23. doi: 10.1021/ct300896h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Vogel T, Li YW, Wüst T, Landau DP. Generic, hierarchical framework for massively parallel Wang-Landau sampling. Phys. Rev. Lett. 2013;110:210603. doi: 10.1103/PhysRevLett.110.210603. [DOI] [PubMed] [Google Scholar]
- (44).Cole DJ, Tirado-Rives J, Jorgensen WL. Enhanced Monte Carlo sampling through replica exchange with solute tempering. J. Chem. Theory Comput. 2014;10:565–571. doi: 10.1021/ct400989x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Zang T, Yu L, Zhang C, Ma J. Parallel continuous simulated tempering and its applications in large-scale molecular simulations. J. Chem. Phys. 2014;141:044113. doi: 10.1063/1.4890038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Lee J, Miller BT, Damjanovií A, Brooks BR. Enhancing constant-pH simulation in explicit solvent with a two-dimensional replica exchange method. J. Chem. Theory Comput. 2015;11:2560–2574. doi: 10.1021/ct501101f. [DOI] [PubMed] [Google Scholar]
- (47).Vogel T, Perez D. Towards an optimal flow: density-of-states-informed replica-exchange simulations. Phys. Rev. Lett. 2015;115:190602. doi: 10.1103/PhysRevLett.115.190602. [DOI] [PubMed] [Google Scholar]
- (48).Brenner P, Sweet CR, VonHandorf D, Izaguirre JA. Accelerating the replica exchange method through an efficient all-pairs exchange. J. Chem. Phys. 2007;126:074103. doi: 10.1063/1.2436872. [DOI] [PubMed] [Google Scholar]
- (49).Ballard AJ, Jarzynski C. Replica exchange with non-equilibrium switches. Proc. Natl. Acad. Sci. U. S. A. 2009;106:12224–12229. doi: 10.1073/pnas.0900406106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Chodera JD, Shirts MR. Replica exchange and expanded ensemble simulations as Gibbs sampling: simple improvements for enhanced mixing. J. Chem. Phys. 2011;135:194110. doi: 10.1063/1.3660669. [DOI] [PubMed] [Google Scholar]
- (51).Plattner N, Doll JD, Dupuis P, Wang H, Liu Y, Gubernatis JE. An infinite swapping approach to the rare-event sampling problem. J. Chem. Phys. 2011;135:134111. doi: 10.1063/1.3643325. [DOI] [PubMed] [Google Scholar]
- (52).Dupuis P, Liu Y, Plattner N, Doll JD. On the infinite swapping limit for parallel tempering. Multiscale Model. Simul. 2012;10:986–1022. [Google Scholar]
- (53).Plattner N, Doll JD, Meuwly M. Overcoming the rare-event sampling problem in biological systems with infinite swapping. J. Chem. Theory Comput. 2013;9:4215–4224. doi: 10.1021/ct400355g. [DOI] [PubMed] [Google Scholar]
- (54).Kondo HX, Taiji M. Enhanced exchange algorithm without detailed balance condition for replica exchange method. J. Chem. Phys. 2013;138:244113. doi: 10.1063/1.4811711. [DOI] [PubMed] [Google Scholar]
- (55).Lu J, Vanden-Eijnden E. Infinite swapping replica exchange molecular dynamics leads to a simple simulation patch using mixture potentials. J. Chem. Phys. 2013;138:084105. doi: 10.1063/1.4790706. [DOI] [PubMed] [Google Scholar]
- (56).Kofke DA. On the acceptance probability of replica-exchange Monte Carlo trials. J. Chem. Phys. 2002;117:6911. [Google Scholar]
- (57).Predescu C, Predescu M, Ciobanu CV. On the efficiency of exchange in parallel tempering monte carlo simulations. J. Phys. Chem. B. 2005;109:4189–4196. doi: 10.1021/jp045073+. [DOI] [PubMed] [Google Scholar]
- (58).Zuckerman DM, Lyman E. A second look at canonical sampling of biomolecules using replica exchange simulation. J. Chem. Theory Comput. 2006;2:1200–1202. doi: 10.1021/ct600297q. [DOI] [PubMed] [Google Scholar]
- (59).Trebst S, Troyer M, Hansmann UHE. Optimized parallel tempering simulations of proteins. J. Chem. Phys. 2006;124:174903. doi: 10.1063/1.2186639. [DOI] [PubMed] [Google Scholar]
- (60).Hritz J, Oostenbrink C. Optimization of replica exchange molecular dynamics by fast mimicking. J. Chem. Phys. 2007;127:204104. doi: 10.1063/1.2790427. [DOI] [PubMed] [Google Scholar]
- (61).Nadler W, Hansmann UHE. Generalized ensemble and tempering simulations: a unified view. Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys. 2007;75:026109. doi: 10.1103/PhysRevE.75.026109. [DOI] [PubMed] [Google Scholar]
- (62).Nadler W, Hansmann UHE. Dynamics and optimal number of replicas in parallel tempering simulations. Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys. 2007;76:065701. doi: 10.1103/PhysRevE.76.065701. [DOI] [PubMed] [Google Scholar]
- (63).Zheng W, Andrec M, Gallicchio E, Levy RM. Simulating replica exchange simulations of protein folding with a kinetic network model. Proc. Natl. Acad. Sci. U. S. A. 2007;104:15340–15345. doi: 10.1073/pnas.0704418104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Abraham MJ, Gready JE. Ensuring mixing efficiency of replica-exchange molecular dynamics simulations. J. Chem. Theory Comput. 2008;4:1119–1128. doi: 10.1021/ct800016r. [DOI] [PubMed] [Google Scholar]
- (65).Denschlag R, Lingenheil M, Tavan P. Efficiency reduction and pseudo-convergence in replica exchange sampling of peptide folding-unfolding equilibria. Chem. Phys. Lett. 2008;458:244–248. [Google Scholar]
- (66).Nymeyer H. How efficient is replica exchange molecular dynamics? An analytic approach. J. Chem. Theory Comput. 2008;4:626–636. doi: 10.1021/ct7003337. [DOI] [PubMed] [Google Scholar]
- (67).Sindhikara D, Meng Y, Roitberg AE. Exchange frequency in replica exchange molecular dynamics. J. Chem. Phys. 2008;128:024103. doi: 10.1063/1.2816560. [DOI] [PubMed] [Google Scholar]
- (68).Zhang C, Ma J. Comparison of sampling efficiency between simulated tempering and replica exchange. J. Chem. Phys. 2008;129:134112. doi: 10.1063/1.2988339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Zheng W, Andrec M, Gallicchio E, Levy RM. Simple continuous and discrete models for simulating replica exchange simulations of protein folding. J. Phys. Chem. B. 2008;112:6083–6093. doi: 10.1021/jp076377+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Sindhikara DJ, Emerson DJ, Roitberg AE. Exchange often and properly in replica exchange molecular dynamics. J. Chem. Theory Comput. 2010;6:2804–2808. doi: 10.1021/ct100281c. [DOI] [PubMed] [Google Scholar]
- (71).Machta J. Strengths and weaknesses of parallel tempering. Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys. 2009;80:056706. doi: 10.1103/PhysRevE.80.056706. [DOI] [PubMed] [Google Scholar]
- (72).Rosta E, Hummer G. Error and efficiency of replica exchange molecular dynamics simulations. J. Chem. Phys. 2009;131:165102. doi: 10.1063/1.3249608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Grossfield A, Zuckerman DM. Quantifying uncertainty and sampling quality in biomolecular simulations. Annu. Rep. Comput. Chem. 2009;5:23–48. doi: 10.1016/S1574-1400(09)00502-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Smith DB, Okur A, Brooks BR. MDMS: Molecular Dynamics Meta-Simulator for evaluating exchange type sampling methods. Chem. Phys. Lett. 2012;545:118–124. doi: 10.1016/j.cplett.2012.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Wickstrom L, He P, Gallicchio E, Levy RM. Large scale affinity calculations of cyclodextrin host-guest complexes: under-standing the role of reorganization in the molecular recognition process. J. Chem. Theory Comput. 2013;9:3136–3150. doi: 10.1021/ct400003r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Gallicchio E, Lapelosa M, Levy RM. The Binding Energy Distribution Analysis Method (BEDAM) for the estimation of protein-ligand binding affinities. J. Chem. Theory Comput. 2010;6:2961–2977. doi: 10.1021/ct1002913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Bowman GR, Noé F, Pande VS. An Introduction to Markov State Models and Their Application to Long Timescale Molecular Simulation (Advances in Experimental Medicine and Biology) Springer; 2013. [Google Scholar]
- (78).Levy RM, Dai W, Deng N, Makarov DE. How long does it take to equilibrate the unfolded state of a protein? Protein Sci. 2013;22:1459–1465. doi: 10.1002/pro.2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Tan Z, Gallicchio E, Lapelosa M, Levy RM. Theory of binless multi-state free energy estimation with applications to protein-ligand binding. J. Chem. Phys. 2012;136:144102. doi: 10.1063/1.3701175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Zhang BW, Xia J, Tan Z, Levy RM. A stochastic solution to the unbinned WHAM equations. J. Phys. Chem. Lett. 2015;6:3834–3840. doi: 10.1021/acs.jpclett.5b01771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Dai W, Sengupta AM, Levy RM. First passage times, lifetimes, and relaxation times of unfolded proteins. Phys. Rev. Lett. 2015;115:048101. doi: 10.1103/PhysRevLett.115.048101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.