

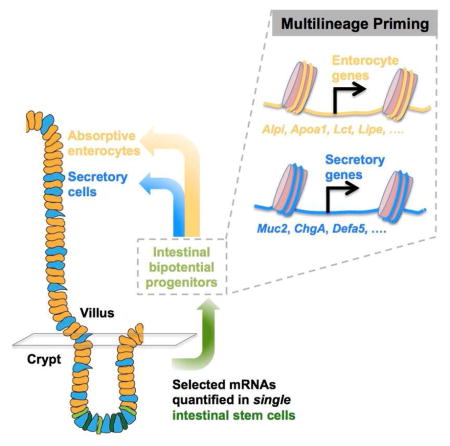
SUMMARY
Lgr5+ intestinal stem cells (ISC) drive epithelial self-renewal, and their immediate progeny – intestinal bipotential progenitors – produce absorptive and secretory lineages via lateral inhibition. To define features of early transit from the ISC compartment, we used a microfluidics approach to measure selected stem- and lineage-specific transcripts in single Lgr5+ cells. We identified two distinct cell populations, one that expresses known ISC markers and a second, abundant population that simultaneously expresses markers of stem and mature absorptive and secretory cells. Single-molecule mRNA in situ hybridization and immunofluorescence verified expression of lineage-restricted genes in a subset of Lgr5+ cells in vivo. Transcriptional network analysis revealed that one group of Lgr5+ cells arises from the other and displays characteristics expected of bipotential progenitors, including activation of Notch ligand and cell-cycle inhibitor genes. These findings define the earliest steps in ISC differentiation and reveal multilineage gene priming as a fundamental property of the process.
Graphical Abstract
INTRODUCTION
Cell turnover in the small bowel relies on pools of 12–15 Wnt-responsive Lgr5+ ISC that lie at the base of each intestinal crypt and replicate daily to produce new ISC and transit-amplifying (TA) progenitors (Barker et al., 2007). Other cells present near crypt tier 4 express a combination of Bmi1, mTert and Hopx1 (Barker et al., 2012) and may represent Paneth cell precursors that are recruited into the stem-cell pool upon epithelial injury (Buczacki et al., 2013). Both Lgr5+ ISC and TA cells replicate briskly, albeit at different rates, and TA cells quickly adopt a single fate – absorptive or secretory – whereas ISC stay multipotent; the basis for these cardinal differences is unknown. In another self-renewing tissue, blood cell progenitors simultaneously activate genes specific to each daughter lineage before distinct cell types are specified, a phenomenon known as multilineage priming (Hu et al., 1997; Miyamoto et al., 2002). Because absorptive and secretory fates are determined by lateral inhibition, a means for reciprocal cell specification (Pellegrinet et al., 2011; Stamataki et al., 2011), it is unclear if the progeny of Lgr5+ ISC traverse a similar phase. Lateral inhibition likely occurs in intestinal bipotential progenitors (IBP), which have never been captured and may represent the earliest, albeit transient, progeny of Lgr5+ ISC.
Lgr5+ cells show a range of GFP signal in Lgr5Gfp mice (Barker et al., 2007) and cells at the center of the crypt base produce larger clones than cells located at the periphery (Ritsma et al., 2014). Not all Lgr5+ cells spawn functional clones in vivo (Kozar et al., 2013) and some of them correspond to non-cycling Paneth-cell precursors (Buczacki et al., 2013). Although these observations suggest that early progenitors might arise among Lgr5+ cells, a recent single-cell mRNA study (Grun et al., 2015) reported that Lgr5hi cells are homogeneous, possibly because the method has low sensitivity for transcripts expressed at low abundance. To overcome this limitation, we measured 185 transcripts for selected stem and lineage-specific markers in single GFP+ (Lgr5+) intestinal crypt cells isolated from the same Lgr5GFP mice (Barker et al., 2007). We identify a distinct population that expresses slightly reduced levels of known ISC transcripts and co-expresses markers of mature secretory cells and enterocytes. Immunofluorescence and single-molecule mRNA in situ hybridization confirmed presence of these cells in vivo, and analysis of transcript networks indicates that they represent early ISC-derived bipotential progenitors.
RESULTS AND DISCUSSION
We used microfluidic quantitative (q) RT-PCR following targeted pre-amplification of 185 genes from defined categories (Suppl. Table S1), including genes previously identified as Lgr5+ cell-specific (Kim et al., 2014; Munoz et al., 2012); targets of various signaling pathways; markers specific to mature enterocytes or secretory cells (Kim et al., 2014); and tissue-restricted transcription factors. To ensure reproducibility and RNA quality, we assessed 3 housekeeping genes (Actb, Gapdh, Hprt) and used two separate primer pairs to measure 5 genes. From Lgr5Gfp mice (Barker et al., 2007), we captured crypt epithelial cells that showed strong GFP fluorescence in flow cytometry (Fig. 1A), but might nevertheless include LGR5+ cells on the verge of ISC exit. Fluorescence microscopy and direct visualization verified recovery of dilute, viable GFP+ singlets (Suppl. Fig. S1A). Following reverse transcription with primers specific to the selected genes and PCR amplification of cDNA, we excluded wells that gave Ct values <13 in qRT-PCR for Actb, further eliminating possible rare doublets. Different primers for each of 5 selected genes gave concordant results (Table S1), indicating a robust protocol.
Figure 1. Targeted mRNA profiles identify two populations of Lgr5+ intestinal crypt cells.

(A) Flow cytometry plot, showing the gates applied to isolate Lgr5hi (green) cells. (B) Heatmap display of k-means (k=2) clustering of linear-transformed Ct values from 183 mRNAs (x-axis, 5 genes are represented by 2 primer sets each) in 192 single Lgr5+ intestinal crypt cells (y-axis). Blue represents absent to low, and yellow to amber colors represent increasing, transcript levels. Genes are ordered by hierarchical clustering with the average linkage method and Euclidean distance. A block of genes that best distinguishes the two cell populations, including most mature villus markers, is boxed. (C) Violin plots showing differential expression of representative stem (Lgr5, Olfm4) and differentiated (Apoa1, Muc2) cell markers in all cells in the two populations – P1 (blue) and P2 (green). (D) T-SNE analysis of the qRT-PCR data, demonstrating discrete Lgr5+ cell populations (blue, green); overlaid colors are from the adjoining k-means clusters. See also Figure S1 and Tables S1 and S2.
We measured levels of all 185 genes in 192 cells captured on 2 separate days and pooled the data for subsequent analyses (Table S2); two genes, Zg16 and Ido1, gave no signal in any cell and were excluded from the analysis. K-means clustering of the RNA data, using the Silhouette measure (Clifford et al., 2011) to identify the best k (Fig. S1B), revealed two distinct cell populations that were roughly equal in size (Fig. 1B) and expressed similar levels of markers historically assigned to quiescent ISC (Fig. S1C). The salient differences between these two populations were modestly higher (2- to 8-fold) expression of ISC markers such as Lgr5 and Olfm4 in one pool and 8- to >100-fold higher expression of many genes in the other (Figs. 1B–C, Padj <10−7 to <10−5). After confirming efficient qPCR by selected primer pairs, we estimated copy numbers of some of the latter mRNAs at 3% to 8% of Hprt copies (Fig. S1D). Cells isolated on different days were similarly distributed in the two pools, and to verify the results from k-means clustering, we used t-distributed Stochastic Neighbor Embedding (t-SNE) (van der Maaten and Hinton, 2008). The two cell populations identified by k-means clustering remained distinct on a t-SNE map (blue and green dots in Fig. 1D) and the high concordance of RNA profiles in each group (Fig. 1B), together with the absence of outliers in t-SNE, strongly supports the absence of cell doublets.
Among the 185 genes we interrogated, 35 genes discriminated the 2 cell populations without ambiguity (ΔCt >3, Padj <10−6, Fig. 1B, shaded in Table S1) and 31 of these transcripts were higher in Population 2. Weighted gene co-expression network analysis (WGCNA) (Zhang and Horvath, 2005) revealed two highly coordinated gene modules in this population (Fig. 2A), compared to the modest connectivity of expressed genes in Population 1 (Fig. S2A), and the transcripts elevated in Population 2 overlapped significantly with these modules (Fig. 2B). Eighteen of the 27 common genes represented secretory or enterocyte-specific markers (Fig. 2C) that were not mutually exclusive, but appeared at similar levels in nearly every cell in Population 2 and were virtually absent in the other cells (Figs. 1B and 2C–D). The simultaneous expression of different lineage programs is reminiscent of multilineage priming in blood progenitors (Hu et al., 1997; Miyamoto et al., 2002) and the lack of any instance of unilineage expression suggests that Population 2 may represent IBP. Single-cell latent variable modeling (scLVM) (Buettner et al., 2015) attributed only 12.2% of the variation to cell replication, and transcript profiles were very similar before and after correcting for cell cycle effects (Fig. S2B). Cell cycle-related transcripts that were increased in IBP included both positive and negative regulators of the cell cycle, and Pcna, Mki67, and targets of Wnt signaling were expressed at comparable levels (Fig. S2C). Thus, the distinct mRNA profiles do not trivially reflect differential mitotic activity and both populations seem to include cycling cells.
Figure 2. Candidate IBP show multilineage priming.

(A) Results of WGCNA, showing network modules of genes that are strongly co-expressed across the cells in Population 2. In contrast, Population 1 showed limited connectivity (Fig. S2A). (B) Overlap of 31 genes showing differentially high expression in IBP with 76 genes showing high network connectivity (see Supplemental Experimental Procedures). (C) ΔCt and connectivity values for the 31 genes that best distinguish Population 2; lineage-specific markers are labeled green. (D) Violin plots showing highly differential expression of markers of each major terminal intestinal cell type in all cells in populations P1 (blue) and P2 (green): Lct and Treh (enterocytes), Cck (endocrine), Spdef (goblet and other secretory cells), and Defa5 (Paneth cells). See also Figure S2.
Superficially, the presence of numerous candidate IBP among Lgr5+ cells contrasts with recent evidence of population homogeneity by single-cell mRNA-seq (Grun et al., 2015). One explanation is that Grun et al examined cells with higher GFP levels than we did. Thus, our Population 1 might represent homogeneous GFPhi ISC, whereas Population 2 may contain cells with modestly lower Lgr5 mRNA (Fig. 1C) and protein levels, i.e., cells leaving the ISC compartment. Another explanation is the low sensitivity of single-cell RNA-seq for low-abundance transcripts, and indeed the method did not reliably capture genes that distinguish ISC from IBP in our qRT-PCR study. Although a few lineage markers – such as Defa5, Muc2, and Ang4 – were detected in some cells, most markers were not (Fig. S2D). Nevertheless, to exclude the possibility that our qRT-PCR signals are spurious, we performed bulk (ensemble) RNA-seq analysis on triplicate samples of Lgr5hi cells sorted using the same parameters as in our single-cell analysis and also queried bulk RNA data from Lgr5hi cells profiled on microarrays (Munoz et al., 2012). Every lineage marker we detected in single cells was represented among the >11,000 genes identified in these ensemble studies (Fig. S3A), compared to <4,000 genes in the single-cell mRNA-seq study (Grun et al., 2015).
In light of the multilineage profiles of putative IBP, transcripts specific to enterocytes or secretory cells might persist in specified progenitors of the other type. This was indeed evident in ensemble analysis of the respective purified progenitors (Fig. S3A), e.g., whereas high Alpi levels are restricted to enterocytes in vivo (Tetteh et al., 2016), levels ~10-fold lower than those found in bulk villus cells are equally abundant in both enterocyte and secretory progenitors. Conversely, we detected many secretory genes in enterocyte progenitors. Because this Atoh1-null population categorically lacks secretory cells (Kim et al., 2014; Yang et al., 2001), genes from this lineage were likely activated in a preceding cell generation, IBP. Together, these observations imply that the earliest cells to leave the ISC compartment activate genes of both intestinal lineages, at levels that elude detection at the current resolution of single-cell RNA-seq.
To confirm our findings by independent methods, first we used single-molecule mRNA in situ hybridization (ISH) with branched DNA (bDNA) signal amplification (Player et al., 2001). Probes for the villus cell markers Alpi, Chga, Neurog3 and Cck gave the expected signals in most (enterocyte) or few (enteroendocrine) wild-type mouse villus cells, respectively, with weaker signals in crypt epithelium and virtually none in the lamina propria; conversely, Lgr5 probes carrying a different chromophore stained only crypt base columnar cells (Fig. S3B). We detected low levels of mature villus-cell marker mRNAs in up to 24.7% of Lgr5-expressing cells (Figs. 3A–B and S3C), greatly exceeding the background of red signals and compatible with the different sensitivities of single-cell qRT-PCR and single-mRNA ISH to detect transcripts of low abundance. Second, we used Atoh1Gfp knock-in mice (Rose et al., 2009) to examine protein levels of ATOH1, a transcription factor whose RNA is restricted to the pool of putative IBP (Fig. S3D). After verifying ATOH1/GFP expression in Lysozyme+ Paneth cells and occasional secretory progenitors positioned higher than crypt tier 5 (red arrow in Fig. 3C), we restricted attention to Lgr5+ cells in the crypt base (open arrows, Fig. 3C, N = 454), which showed distinct populations of ATOH1+ and ATOH1− nuclei (filled or open arrows, respectively, in Figs. 3D–E and S3E). As protein expression must trail new transcripts, the fraction of ATOH1+ cells (23.7%, Fig. 3F) is compatible with that detected by qRT-PCR (47.9%). mRNA ISH and ATOH1/GFP stains did not localize lineage marker-expressing Lgr5+ cells to high crypt tiers, which suggests that cell heterogeneity may originate – perhaps stochastically – among ISC at the crypt bottom and that cells with this feature preferentially exit the ISC compartment. The cells we regard as IBP may, however, correspond to a GFPlow population in vivo (Basak et al., 2014) and their property of multilineage priming is significant irrespective of the precise crypt location.
Figure 3. Expression of lineage markers in Lgr5+ crypt base cells in vivo.

(A) Representative images of single-molecule mRNA ISH for Alpi, ChgA, Cck, Neurog3 (red) and Lgr5 (blue), showing red and blue signals in the same crypt base cells. Cells with arrows pointing to co-expressed red and blue dots are magnified in the respective insets. Scale bars, 15 μm. (B) Fraction of double-positive (DBL+, red and blue) cells and background of extra-epithelial cells with red dots in intestines from 4 mice in 2 experiments. (C) Immunostaining of Atoh1Gfp/Gfp crypts with Lysozyme (red) and GFP (green) Ab and DAPI nuclear stain (blue). GFP (ATOH1) was present in Lysozyme+ Paneth cells (P) at the crypt base and in occasional TA cells (red arrow); only slim columnar cells wedged between Paneth cells (white arrows) were assessed further. (D) Absence (open arrows) or presence (filled arrows) of ATOH1 in a representative z-section of 3 consecutive crypts, with fluorescence channels separated for clarity. (E) Magnified view of a single crypt, showing that ATOH1 signals in some putative IBP are similar to those in neighboring Paneth (P) cells. (F) Fraction of ATOH1/GFP+ cells among 454 columnar DAPI+ nuclei in tiers 0–3 of Atoh1Gfp/Gfp mouse crypts. See also Figure S3.
To examine further the relationship between Populations 1 and 2, we considered that any transition among them is likely not abrupt; rather, transcripts from one cell state might decline while those from the other begin to accumulate. The foregoing cluster analysis (Fig. 1B), which is discrete, would fail to detect such a transition, but the non-branching structure of the t-SNE map (Fig. 1D) permits the use of Principal Curves to infer cell trajectories (Hastie and Stuetzle, 1989). We derived such a Principal Curve, then divided all the cells into 10 groups according to the inferred pseudo-time (Marco et al., 2014), and identified 28 cells at the boundary between the two major populations (Fig. 4A). Average expression of each of the 183 genes in the 10 groups of cells revealed 66 genes that discriminate between ISC and IBP (denoted by a box on the cluster dendrogram and heatmap in Fig. 4B) and, as expected, include nearly every gene that had shown high ΔCt values (Fig. 4C). Whereas ISC and IBP expressed uniformly higher levels of different subsets in this gene group, the 28 boundary cells varied in expression (Fig. 4C), with declining average levels of stem-cell markers, such as Lgr5, and concomitant increase of mature markers (Fig. 4D). Average expression values were similar for different numbers of bins. For example, using 8 bins instead of 10, the histogram of cell numbers identified 12 boundary cells, and mean expression over these 12 cells was highly correlated (R2=0.95) with that in the 28 cells identified using 10 bins. Gradual accumulation of terminal cell markers, as revealed in boundary cells, strongly suggests a cell transition from ISC to putative IBP.
Figure 4. Evidence that Lgr5+ ISC transition into the IBP population.

(A) Principal Curve Analysis (black curve) projected on the t-SNE map from Fig. 1D reveals the relationship of the two populations – based on proximity of gene expression – as a non-branching curve. The 28 boundary cells – determined by unbiased partitioning of the principal curve into 10 bins of equal distance – are now represented in pink. The graph indicates cell numbers in each bin; blue and green denote ISC and IBP, respectively. (B) Heatmap of the global analysis (183 genes X 192 single cells; red=high, green=low expression) partitioned in 10 bins according to the above Principal Curve Analysis. 66 transcripts denoted by a dotted box provide discrimination. (C) The latter transcripts include nearly every gene that distinguished Populations 1 and 2 by ΔCt (Fig. 1B) and the dotted box in B is here expanded and rotated 90° to show the trajectory of expression in ISC (blue), boundary cells (pink), and IBP (green). (D) Average levels of representative IBP-enriched (Lifr, Muc2, Dct, Kit), ISC-enriched (Lgr5, Agr3, Sema4d), and Actb mRNAs in cell groups defined by distance along the Principal Curve. (E) Violin plots for expression of Notch ligand genes Dll1 and Dll4 in all ISC and IBP.
The high census of IBP suggests that they are distinct from the small, label-retaining fraction of Lgr5+ cells. Transcripts recently assigned to the latter – Nfatc3, Nfat5, Cd82 (Buczacki et al., 2013) – were essentially similar in ISC and IBP (Fig. S3F) and may increase only in Paneth-cell precursors. Notably, and in line with recent evidence for extreme plasticity in crypts (Kim et al., 2014; Tian et al., 2011; van Es et al., 2012), IBP may be unstable cells that revert to ISC as readily as they differentiate into absorptive or secretory cells. The latter event occurs as some cells use DLL1 or DLL4 to signal to Notch receptors on their neighbors (Pellegrinet et al., 2011; Stamataki et al., 2011). Because lateral inhibition requires equipotent cells to deliver or respond to Notch signals, increased expression of these ligands is one feature expected in IBP. Indeed, average Dll1 mRNA is higher in IBP and Dll4 increases substantially in most of these cells (Fig. 4E).
In summary, microfluidic qRT-PCR reveals a distinct cell population that seems to represent the earliest progeny of Lgr5+ ISC: putative IBP with multilineage priming and modestly reduced Lgr5/GFP expression. Although multilineage priming was originally inferred from bulk cell populations (Hu et al., 1997; Miyamoto et al., 2002), recent studies suggest that single blood progenitors express genes exclusive to one lineage or another (Paul et al., 2015; Perie et al., 2015). In contrast, our analysis revealed no cell expressing genes specific to just one intestinal lineage (Fig. 1) and enterocyte progenitors continue to express secretory genes (Fig. S3A); these findings likely reflect features particular to lineage specification by lateral inhibition. Levels of certain TF mRNAs – Atoh1, Spdef, Pax4 and Tbx3 – first rise in IBP, where they may initiate the lineage-affiliated programs. Although equal expression of Mki67 and Pcna in ISC and IBP supports the idea that all crypt cells other than Paneth cells and their precursors replicate, high mRNA levels of cell cycle inhibitors Cdkn1a, 1b, 2a and 2b in IBP (Fig. S2C) suggest that they or their immediate progeny may replicate slower than ISC or TA cells.
Despite clear differences in gene activity, IBP are unlikely to show different behaviors than ISC by lineage tracing or in organoids, where even ISC and specified progenitors are difficult to distinguish (Buczacki et al., 2013; Tetteh et al., 2016; van Es et al., 2012). Moreover, no Cre driver or surface marker is likely expressed exclusively in IBP, i.e., not also in ISC or specified progenitors. Thus, our targeted single-cell analysis, reinforced by localization of transcripts in vivo, reveals features of a crucial and transient cell population that is likely difficult to isolate or to characterize by other means.
EXPERIMENTAL PROCEDURES
Isolation of single Lgr5+ ISC
Intestines harvested from Lgr5GFP mice (Barker et al., 2007) were washed with phosphate-buffered saline (PBS). Villi were scraped away using cover slips and the crypt epithelium collected by shaking in 5 mM EDTA for 1 h at 4°C (Kim et al., 2014). Single cells were obtained on 2 separate days by digestion in 5X TrypLE (Invitrogen) for 1 h at 37°C and verified by fluorescence microscopy. GFPhi cells were sorted into individual wells in 96-well plates using a FACSAria II sorter (Becton-Dickinson). Cells from one of the two isolations were also examined visually in microfluidic channels.
Single-cell gene expression analysis by Microfluidic qRT-PCR
The pre-amplification solution in 96 wells included 5 μL of a master mix containing 2.5 μL CellsDirect reaction mix (Invitrogen), 0.5 μL of the primer pool (0.1 μM, Table S1, synthesized at Bioneer), 0.1 μL reverse transcriptase (RT)/Taq polymerase (Invitrogen), and 1.9 μL nuclease-free water. Lysed cells were treated with this mix at 50°C for 1 h, followed by inactivation of RT, activation of Taq at 95°C for 3 min, and 20 cycles of sequence-specific cDNA amplification (15 sec denaturation at 95°C, 15 min annealing and elongation at 60°C). Amplified single-cell cDNAs were first tested in control qRT-PCR reactions for Actb and samples giving Ct values between 13 and 17 were selected for subsequent analysis with the full primer pools, Universal PCR Master Mix (Applied Biosystems) and EvaGreen Binding Dye (Biotium), using 96 X 96 Dynamic Arrays on the BioMark System (Fluidigm). Table S2 lists the Ct values for each gene in each cell, calculated using BioMark Real-Time PCR Analysis software (Fluidigm).
Computational analyses
mRNA levels were estimated by subtracting the Ct values from the background level of 28 (start of the tail of the distribution in the histogram of Ct values), which approximates log2 gene expression levels. K-means clustering was done in MATLAB using the squared Euclidean distance of normalized data (z-scores). To determine the optimal k, we applied every value from 2 to 20, assessed the average Silhouette value (Clifford et al., 2011) for each clustering result (Figure S1B), and selected k=2, which gave the largest mean Silhouette value. Differentially expressed genes were identified using a two-sided Wilcoxon-Mann-Whitney rank sum test implemented in the “coin” package in R. Differences between populations were determined by subtracting median Ct values (equivalent to log2 expression levels). P-values were adjusted for multiple testing (Benjamini and Hochberg, 1995). Violin plots were generated in R using the package “vioplot”. For t-SNE analysis (van der Maaten and Hinton, 2008), we used the MATLAB toolbox for dimensionality reduction (http://homepage.tudelft.nl/19j49/t-SNE.html). The pseudotime of individual cells was estimated as previously described (Marco et al., 2014), fitting a principal curve (Hastie and Stuetzle, 1989) to the single-cell expression data. We used the R package “princurve” with the options “smoother = lowess” and “maxit = 200”. Heatmaps (Fig. 2) were prepared with MultiExperiment Viewer (http://www.tm4.org/mev.html) using the Euclidean distance and average linkage as parameters for unsupervised hierarchical clustering of genes. Latent variable modeling and analysis of co-expression gene networks are described in Supplemental Experimental Procedures.
Analysis of public single-cell mRNA-seq data
Processed mRNA-seq data on 192 isolated Lgr5+ mouse intestinal cells (Grun et al., 2015) were obtained from the Gene Expression Omnibus (GEO accession files: GSE62270_data_counts_Lgr5SC.txt.gz). Violin plots for genes relevant to our study were generated using the Vioplot2 function in R.
Single-mRNA in situ hybridization (ISH) with branched DNA (bDNA) amplification
Intestines from C57BL/6J mice were fixed overnight in 4% paraformaldehyde, embedded in paraffin, and cut in 5 μm sections. ISH was performed twice on 2 intestines each, using Quantigene ViewRNA probes (Affymetrix) for two-color ISH, as described in Supplemental Experimental Procedures. Between 320 and 460 Lgr5+ crypt base cells were counted in at least 50 crypts from each mouse (N=4). Cells were scored as DBL+ when at least one dot for a mature-cell marker mRNA (red) was present in a cell expressing Lgr5 mRNA (blue dots). Background signals were estimated from counts of red dots in 370 to 440 nucleated sub-epithelial cells for each mature-cell marker in each sample.
Supplementary Material
Acknowledgments
Supported by grants R01DK081113, including a supplement from the Office of the Director (NIH Common Fund), the Intestinal Stem Cell Consortium (U01DK103152) of the NIDDK and NIAID, K99DK095983 (T-H.K.), F32DK103453 (U.J.) and P50CA127003; an American-Italian Cancer Foundation fellowship (A.C.); and funds from the Harvard Stem Cell Institute (G-C.Y and S.H.O.), Affymetrix (N.D. and M.N.R.) and the Lind family (R.A.S.). We thank L. Deary and D. Breault for help with microscopy.
Footnotes
AUTHOR CONTRIBUTIONS
T-H.K. and R.A.S. conceived the study; T-H.K., G.G., M.S., N.D. and U.J. performed experiments; A.S. performed computational analyses; T-H.K., M.S., A.C., L.J. and R.A.S. analyzed data; S.H.O. and M.N.R. supervised portions of the study; G-C.Y. supervised computational analyses and R.A.S. provided overall supervision. R.A.S. and G-C.Y. drafted the manuscript, with input from all authors.
The authors declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003–1007. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- Barker N, van Oudenaarden A, Clevers H. Identifying the stem cell of the intestinal crypt: strategies and pitfalls. Cell Stem Cell. 2012;11:452–460. doi: 10.1016/j.stem.2012.09.009. [DOI] [PubMed] [Google Scholar]
- Basak O, van de Born M, Korving J, Beumer J, van der Elst S, van Es JH, Clevers H. Mapping early fate determination in Lgr5+ crypt stem cells using a novel Ki67-RFP allele. EMBO J. 2014;33:2057–2068. doi: 10.15252/embj.201488017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multipe testing. J Royal Stat Soc Series B. 1995;57:289–300. [Google Scholar]
- Buczacki SJ, Zecchini HI, Nicholson AM, Russell R, Vermeulen L, Kemp R, Winton DJ. Intestinal label-retaining cells are secretory precursors expressing Lgr5. Nature. 2013;495:65–69. doi: 10.1038/nature11965. [DOI] [PubMed] [Google Scholar]
- Buettner F, Natarajan KN, Casale FP, Proserpio V, Scialdone A, Theis FJ, Teichmann SA, Marioni JC, Stegle O. Computational analysis of cell-to-cell heterogeneity in single-cell RNA-sequencing data reveals hidden subpopulations of cells. Nat Biotechnol. 2015;33:155–160. doi: 10.1038/nbt.3102. [DOI] [PubMed] [Google Scholar]
- Clifford H, Wessely F, Pendurthi S, Emes RD. Comparison of clustering methods for investigation of genome-wide methylation array data. Frontiers Genet. 2011;2:88. doi: 10.3389/fgene.2011.00088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grun D, Lyubimova A, Kester L, Wiebrands K, Basak O, Sasaki N, Clevers H, van Oudenaarden A. Single-cell messenger RNA sequencing reveals rare intestinal cell types. Nature. 2015;525:251–255. doi: 10.1038/nature14966. [DOI] [PubMed] [Google Scholar]
- Hastie T, Stuetzle W. Principal Curves. J Am Stat Assoc. 1989;84:502–516. [Google Scholar]
- Hu M, Krause D, Greaves M, Sharkis S, Dexter M, Heyworth C, Enver T. Multilineage gene expression precedes commitment in the hemopoietic system. Genes Dev. 1997;11:774–785. doi: 10.1101/gad.11.6.774. [DOI] [PubMed] [Google Scholar]
- Kim TH, Li F, Ferreiro-Neira I, Ho LL, Luyten A, Nalapareddy K, Long H, Verzi M, Shivdasani RA. Broadly permissive intestinal chromatin underlies lateral inhibition and cell plasticity. Nature. 2014;506:511–515. doi: 10.1038/nature12903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozar S, Morrissey E, Nicholson AM, van der Heijden M, Zecchini HI, Kemp R, Tavare S, Vermeulen L, Winton DJ. Continuous clonal labeling reveals small numbers of functional stem cells in intestinal crypts and adenomas. Cell Stem Cell. 2013;13:626–633. doi: 10.1016/j.stem.2013.08.001. [DOI] [PubMed] [Google Scholar]
- Marco E, Karp RL, Guo G, Robson P, Hart AH, Trippa L, Yuan GC. Bifurcation analysis of single-cell gene expression data reveals epigenetic landscape. Proc Natl Acad Sci USA. 2014;111:5643–5650. doi: 10.1073/pnas.1408993111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto T, Iwasaki H, Reizis B, Ye M, Graf T, Weissman IL, Akashi K. Myeloid or lymphoid promiscuity as a critical step in hematopoietic lineage commitment. Dev Cell. 2002;3:137–147. doi: 10.1016/s1534-5807(02)00201-0. [DOI] [PubMed] [Google Scholar]
- Munoz J, Stange DE, Schepers AG, van de Wetering M, Koo BK, Itzkovitz S, Volckmann R, Kung KS, Koster J, Radulescu S, et al. The Lgr5 intestinal stem cell signature: robust expression of proposed quiescent ‘+4’ cell markers. EMBO J. 2012;31:3079–3091. doi: 10.1038/emboj.2012.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul F, Arkin Y, Giladi A, Jaitin DA, Kenigsberg E, Keren-Shaul H, Winter D, Lara-Astiaso D, Gury M, Weiner A, et al. Transcriptional Heterogeneity and Lineage Commitment in Myeloid Progenitors. Cell. 2015;163:1663–1677. doi: 10.1016/j.cell.2015.11.013. [DOI] [PubMed] [Google Scholar]
- Pellegrinet L, Rodilla V, Liu Z, Chen S, Koch U, Espinosa L, Kaestner KH, Kopan R, Lewis J, Radtke F. Dll1- and dll4-mediated notch signaling are required for homeostasis of intestinal stem cells. Gastroenterology. 2011;140:1230–1240. doi: 10.1053/j.gastro.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perie L, Duffy KR, Kok L, de Boer RJ, Schumacher TN. The Branching Point in Erythro-Myeloid Differentiation. Cell. 2015;163:1655–1662. doi: 10.1016/j.cell.2015.11.059. [DOI] [PubMed] [Google Scholar]
- Player AN, Shen LP, Kenny D, Antao VP, Kolberg JA. Single-copy gene detection using branched DNA (bDNA) in situ hybridization. J Histochem Cytochem. 2001;49:603–612. doi: 10.1177/002215540104900507. [DOI] [PubMed] [Google Scholar]
- Ritsma L, Ellenbroek SI, Zomer A, Snippert HJ, de Sauvage FJ, Simons BD, Clevers H, van Rheenen J. Intestinal crypt homeostasis revealed at single-stem-cell level by in vivo live imaging. Nature. 2014;507:362–365. doi: 10.1038/nature12972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose MF, Ren J, Ahmad KA, Chao HT, Klisch TJ, Flora A, Greer JJ, Zoghbi HY. Math1 is essential for the development of hindbrain neurons critical for perinatal breathing. Neuron. 2009;64:341–354. doi: 10.1016/j.neuron.2009.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamataki D, Holder M, Hodgetts C, Jeffery R, Nye E, Spencer-Dene B, Winton DJ, Lewis J. Delta1 expression, cell cycle exit, and commitment to a specific secretory fate coincide within a few hours in the mouse intestinal stem cell system. PLoS One. 2011;6:e24484. doi: 10.1371/journal.pone.0024484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetteh PW, Basak O, Farin HF, Wiebrands K, Kretzschmar K, Begthel H, van den Born M, Korving J, de Sauvage F, van Es JH, et al. Replacement of lost Lgr5-positive stem cells through plasticity of their enterocyte-lineage daughters. Cell Stem Cell. 2016;18:203–213. doi: 10.1016/j.stem.2016.01.001. [DOI] [PubMed] [Google Scholar]
- Tian H, Biehs B, Warming S, Leong KG, Rangell L, Klein OD, de Sauvage FJ. A reserve stem cell population in small intestine renders Lgr5-positive cells dispensable. Nature. 2011;478:255–259. doi: 10.1038/nature10408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Maaten LJP, Hinton GE. Visualizing high-dimensional data using t-SNE. J Mach Learning Res. 2008;9:2579–2605. [Google Scholar]
- van Es JH, Sato T, van de Wetering M, Lyubimova A, Nee AN, Gregorieff A, Sasaki N, Zeinstra L, van den Born M, Korving J, et al. Dll1+ secretory progenitor cells revert to stem cells upon crypt damage. Nat Cell Biol. 2012;14:1099–1104. doi: 10.1038/ncb2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Bermingham NA, Finegold MJ, Zoghbi HY. Requirement of Math1 for secretory cell lineage commitment in the mouse intestine. Science. 2001;294:2155–2158. doi: 10.1126/science.1065718. [DOI] [PubMed] [Google Scholar]
- Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4 doi: 10.2202/1544-6115.1128. Article 17. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.