

Abstract
Specificity of protein degradation by cellular proteasomes comes from tetra-ubiquitin recognition. We carry out molecular dynamics simulations to characterize how the ubiquitin receptor Rpn10 recognizes in the 26S proteasome K48-linked tetra-ubiquitin. In the binding pose, ubiquitin and Rpn10 interact primarily through hydrophobic patches. However, K48-linked tetra-ubiquitin mostly assumes a closed form in solution prior to binding, in which its hydrophobic patches are not exposed to solvent. Likewise, the hydrophobic ubiquitin interacting motifs (UIMs) of Rpn10 are mostly protected prior to binding. As a result, ubiquitin recognition in the proteasome requires refolding of both K48-linked tetra-ubiquitin and Rpn10. Simulations suggest that conserved complementary electrostatic patterns of Rpn10 and ubiquitins guide protein association (stage 1 in the recognition process), which induces refolding (stage 2), and then facilitates formation of hydrophobic contacts (stage 3). The simulations also explain why Rpn10 has higher affinity for K48-linked tetra-ubiquitin than for mono-ubiquitin and K48-linked di- and tri-ubiquitins. Simulation results expand on the current view that the flexible arm of Rpn10 acts as an extended fragment of α-helices and flexible coils in the recognition process.
Graphical abstract
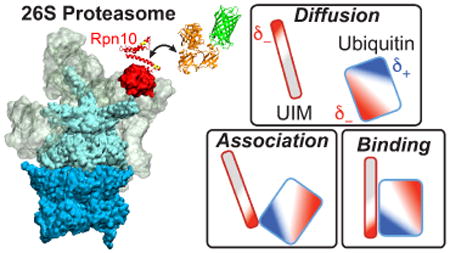
Introduction
How proteins recognize and bind to each other in the crowded cell is an intriguing fundamental question. To address this question from first-principles, it is necessary to have knowledge of structure, energetics, and dynamics of the system during diffusion, association and dissociation characterizing recognition processes. For globular protein partners, typical models of recognition include lock-and-key, induced fit, and conformational selection, where recognition leads systems to stable states characterized by distinct minima in free energy.1 However, these models are poorly suited to systems where recognition partners are disordered proteins or the recognition processes involve protein refolding. In the latter case, ordered and disordered proteins can bind into complexes of well determined structures or into “fuzzy” complexes, with many possible system states, each characterized by shallow minima in free energy.2
In the present study, we examine the recognition of ubiquitin-based proteins by the partially disordered flexible arm of the ubiquitin receptor Rpn10 of the 26S proteasome (Fig. 1A). Ubiquitin recognition is a key step within the ubiquitin 26S proteasome proteolytic pathway.3 Protein degradation by the proteasome is substrate unspecific, so that any protein marked by K48-linked tetra-ubiquitin tag within the ubiquitin 26S proteasome proteolytic pathway is degraded; substrate degradation can occur after the ubiquitin tag of the substrate is bound to the ubiquitin recognition subunits of the 26S proteasome. There are two proteasome subunits currently known to be associated with ubiquitin recognition: Rpn10 and Rpn13.4 It was shown recently that knockout of Rpn13 has almost no effect on the 26S proteasome function,5 indicating the importance of the Rpn10 receptor for ubiquitin recognition. Therefore, we focus on Rpn10 in our study.
Figure 1.
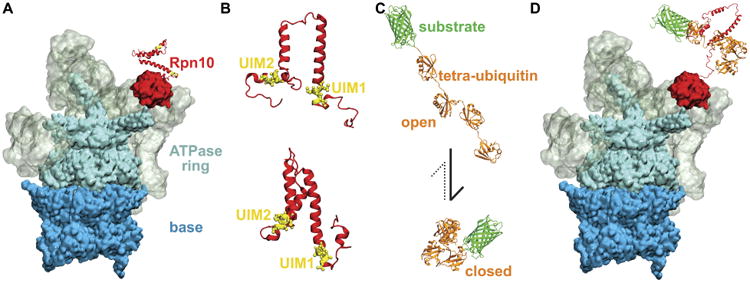
Recognition of K48-linked tetra-ubiquitin by the Rpn10 receptor of 26S proteasome. A. 26S proteasome prior to recognition of the tetra-ubiquitin tag. Different colors mark the main functional components of the proteasome, namely base (blue), ATPase ring (cyan), regulatory particle (grey), and Rpn10 (red). B. Representative structures of Rpn10 flexible arms from NMR experiment (top) and from MD simulations performed in the present study (bottom). UIMs of Rpn10 become protected in MD simulations, while the whole flexible arm becomes more compact than observed in NMR-derived structures. C. K48-linked tetraubiquitin tags (orange) in open and closed forms, covalently bound to an arbitrary substrate (green). The dominant form of K48-linked tetra-ubiquitin in solution is the closed form. D. 26S proteasome after the recognition process, during which the Rpn10 flexible arm and K48-linked tetra-ubiquitin refolded and bound to each other through their hydrophobic interfaces.
Rpn10 consists of a globular part that binds to the 26S proteasome and a flexible arm part that contains either one ubiquitin-interacting motif (UIM) (as observed in yeast or S. pombe Rpn10) or two UIMs (as observed in human Rpn10, which is often called S5a), as shown in Fig. 1B. Previous studies identified several relevant structures of Rpn10 and ubiquitin species: an extended arm of Rpn10,6 K48-linked tetra-ubiquitin in a closed state,7 and complexes of Rpn10 arm and mono-ubiquitin6 or K48-linked di-ubiquitin.8 Binding of Rpn10 to mono- and di-ubiquitin is seemingly simple and mediated through hydrophobic interactions. Ubiquitin binds the UIM through the hydrophobic patch composed of Leu8, Ile44 and Val70 residues; these residues are also implicated in ubiquitin binding to many other proteins.9
Even though ubiquitin binds many of its partners through hydrophobic patches, K48-linked tetra-ubiquitin and di-ubiquitin mostly assume closed forms in solution10 (Fig. 1C-bottom), in which the hydrophobic patches are protected (unexposed to solvent). Our results reveal that hydrophobic UIMs of Rpn10, which bind to hydrophobic patches of ubiquitin, are likewise mostly protected prior to binding. As all the key hydrophobic interfaces are protected before binding, refolding of both K48-linked tetra-ubiquitin and Rpn10 to reveal hydrophobic patches emerges as a necessary step of the binding process, as summarized in Fig. 1. Furthermore, as it is unlikely that protected hydrophobic patches can initiate refolding and efficiently search for each other, other types of interactions between ubiquitins and Rpn10 may be responsible for the initial stage of their mutual recognition. These functionally important interactions could involve other conserved residues, such as those identified in the immediate proximity of UIMs in Ref.11
It is usually hypothesized that long-range and short-range interactions contribute to protein recognition. First, long-range electrostatics, which can act at distances up to 50 Å, are considered to contribute to kinetics of protein recognition by accelerating protein association by several orders of magnitude.12,13 Once initiated, the long-range electrostatics can hold potential proteins within a certain range to allow sufficient rotations into a proper orientation for binding.14 Second, shorter-range hydrophobic interactions, salt bridges and van der Waals interactions, are considered to determine the specificity and affinity of protein-protein interactions.15
In the present study, we carry out atomistic molecular dynamics (MD) simulations of Rpn10 and ubiquitin/K48-linked tetra-ubiquitin to determine how they recognize and eventually bind to each other with high affinity. We identify that electrostatic interactions and refolding processes are essential for recognition of ubiquitin and K48-linked tetra-ubiquitin by the flexible Rpn10 proteins. Furthermore, we establish why K48-linked tetra-ubiquitin is the preferred tag protein of Rpn10,16 even though Rpn10 can simultaneously bind only to two ubiquitin units at most, through its hydrophobic UIMs. In the absence of experimental data, our computational results provide a valuable means to achieve a comprehensive description of Rpn10-K48-linked tetra-ubiquitin recognition and binding.
Methods
In order to characterize mutual recognition of ubiquitin and Rpn10 receptor of the 26S proteasome, we carried out atomistic MD simulations of ubiquitin species and Rpn10 fragments in several relevant states. The present section first describes molecular modeling of Rpn10 fragments and ubiquitin species before and after the recognition process, which aims to characterize solvent exposure of charged and hydrophobic groups, as well as the charge-charge and hydrophobic interactions at initial and final stages of binding. Then, steered MD simulations that investigate the role of charge-charge interactions during Rpn10-ubiquitin unbinding are described. Finally, data analysis steps are outlined that quantify the importance of charge-charge and hydrophobic interactions for binding and validate simulation results with respect to experimental data.
Atomic Models
To investigate the Rpn10-ubiquitin recognition process, structures that represent initial and final stages of this process were prepared on the basis of NMR solution structures of the Rpn10 flexible arm (pdbID: 1YX46), K48-linked tetra-ubiquitin in the closed state (pdbID: 2O6V7), and the Rpn10 flexible arm bound to K48-linked di-ubiquitin (pdbID: 2KDE8) from H. sapiens (human). The Rpn10 flexible arm, also called only the flexible arm, contains residues 196-306. Homology models of Rpn10 flexible arms of S. cerevisiae (yeast) and S. pombe were prepared with Modeller17 and SCWRL 4.0,18 based on the structure of the homologous fragment of human Rpn10. The prepared protein structures were solvated in TIP3P water19 and 0.15 M NaCl with solvate and ionize VMD plugins.20
In order to examine interactions of Rpn10 and K48-linked tetra-ubiquitin, three different complexes of these proteins were prepared. In all of the prepared complexes, K48-linked tetraubiquitin is in the open form, prepared by bringing together two K48-linked di-ubiquitins in the open form (pdbID: 2KDE). Rpn10 was prepared in the complex such that UIM1 binds to the distal ubiquitin U1, as observed in the structure pdbID:2KDE. The three complexes differ by the position of UIM2, which binds to either ubiquitin U2 (case 1), ubiquitin U3 (case 2), or the proximal ubiquitin U4 (case 3). The structure in case 1 is directly based on pdbID:2KDE. In order to obtain structures for cases 2 and 3, the structure of case 1 was first modified by aligning the UIM2-containing helix into binding poses with ubiquitins U3 or U4. This alignment results in structures of Rpn10:K48-linked tetra-ubiquitin complexes in which Rpn10 helices containing UIM1 and UIM2 are in proper positions with respect to their ubiquitin binding partners, but the remaining helices and unstructured coils are in unphysical conformations (one stretched-out bond). These remaining helices and unstructured coils of Rpn10 were then relaxed in short simulations performed in vacuum, in which K48-linked tetra-ubiquitin and UIM1- and UIM2- containing helices of Rpn10 are kept restrained and the remaining α-helices of Rpn10 have secondary structure restraints applied.
Molecular Dynamics Simulations
MD simulations of the above described structures were carried out in order to characterize initial and final states of the recognition process and to define the processes that are necessary to occur as the system transitions between the two states. The MD simulations were performed with NAMD2,21 using the CHARMM22 force field with CMAP corrections,22–24 CHARMM27 force field,22 or the CHARMM36 force field,23,25,26 as summarized in Table S1. Four simulations in CHARMM22/CMAP and CHARMM27 force fields are equivalent to each other (as these protein force fields are differing only in lipid parameters not relevant here), whereas the two simulations in the CHARMM36 force field are not equivalent (as CHARMM36 is a distinct force field with parameters that correct the α-helical bias of CHARMM22/CMAP26). The simulations were performed in the NPT ensemble, at a pressure of 1 atm and a temperature of 310 K. The constant temperature was controlled by Langevin dynamics and the constant pressure was controlled by the Nose-Hoover Langevin piston method.27,28 The long-range electrostatic forces were calculated by means of the particle-mesh-Ewald (PME) method.29 The time step was set to 2 fs; long-range interactions were evaluated every 2 fs (van der Waals) and 4 fs (Coulombic). Each prepared system was minimized for 5,000 steps, followed by 5 ns of equilibration with harmonic constraints on heavy atoms [k = 1 kcal/(mol Å2)], and 200 ns production run.
Steered MD Simulations
To induce unbinding of mono-ubiquitin from a typical UIM (UIM2 of human Rpn10), we performed two 40 ns steered MD (SMD) simulations along distance-based collective variables in NAMD. The force was applied to the center of mass (COM) distances between hydrophobic contacts of UIM2 (IAYAM residues) and ubiquitin (residues 8, 44, and 70) with a pulling spring force constant of 1 kcal/mol/Å2. The two selected hydrophobic groups were separated by 20 Å during the course of the simulations.
Data Analysis
In order to quantify the relative importance of hydrophobic and electrostatic interactions for the overall binding affinity, all the inter-residue interactions in MD simulations of Rpn10-ubiquitin complexes were examined employing a contact map analysis. Here, our contact map analysis procedure is described in detail. First, a rough screening of interacting residues was performed with the g_mdmat tool in GROMACS,30 which provided a matrix of average values of minimal distances between all the pairs of protein residues. Then, our tcl scripts processed the obtained matrix and the associated MD trajectory to provide a list of pairs of residues that remain within 7 Å of each other for more than 40% of the total simulation time. The selected threshold numbers are chosen with a considerably wide range, in order to identify residue pairs exhibiting the most significant constant and transient interactions. In comparison to our selected threshold numbers, most of the NMR-observed interactions occur in our simulations for almost 100% of time and at close distances. Finally, the identified pairs of interacting residues were examined visually to determine types of interaction (hydrophobic, electrostatic).
In order to validate MD simulations with respect to existing experimental data, nuclear Overhauser enhancement (NOE) distances were calculated from simulations and compared to their experimental counterparts, reported earlier.6,8 NOE distances between selected atoms were calculated as 〈1/r6〉−1/6, where r-values are instantaneous distances between two atoms.31 All the performed analyses (interacting residues, NOE distances) were performed for the last 150 ns of each production run.
Images of protein structures were prepared in VMD.20 Most of the representative simulation structures were obtained through cluster analysis of MD trajectories with the g_cluster tool in GROMACS.32 Electrostatic potentials of proteins were evaluated with the APBS Electrostatics plugin in VMD.33 The reported interaction energies were computed with a modified generalized Born (GB) model.34
Results
MD simulations were carried out to characterize the recognition of mono-ubiquitin and K48-linked tetra-ubiquitin by Rpn10. It has been suggested that the interaction between the two proteins is primarily due to hydrophobic interactions.6 However, as pointed out above, prior to binding to Rpn10, K48-linked tetra-ubiquitin exists mostly in the compact form, shown in Fig. 1C, in which the protein's hydrophobic patches are protected (unexposed to solvent).7 According to our simulations, the hydrophobic UIMs of Rpn10 are likewise mostly protected. Therefore, association and recognition of tetra-ubiquitin and Rpn10 must involve refolding and opening of both proteins, where in the case of Rpn10 the refolding involves mainly the flexible arm (Fig. 2). Our simulations suggest that initial association of Rpn10 and ubiquitins is guided by their conserved complementary electrostatic patterns (stage 1 of the recognition process), which induces their refolding (stage 2) and establishment of hydrophobic interactions between the two proteins (stage 3). The simulations carried out explain also why Rpn10 has higher affinity for K48-linked tetra-ubiquitin than for K48-linked di- and tri-ubiquitins.
Figure 2.
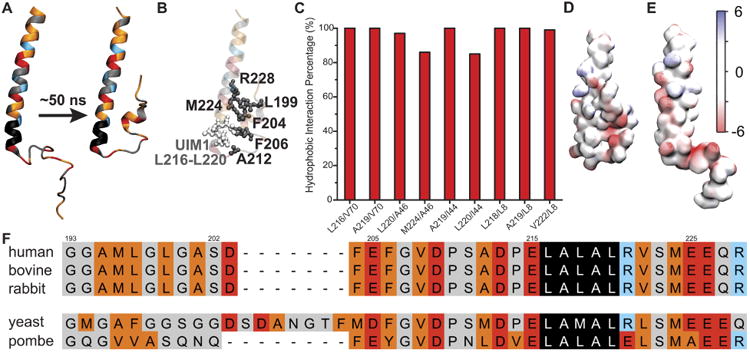
Properties of the flexible arm of human Rpn10. A. The starting structure (left) and a representative structure (right) of an Rpn10 fragment from MD simulations. Amino acids marked in red are negatively charged, blue are positively charged, orange are hydrophobic, black comprise UIM1, and grey cover the rest. B. Interacting nonpolar residues/groups of Rpn10 fragment; the structure is the same as in panel A (right). C. Percentage of time residue pairs (respective residues represented by their center-of-mass) shown in panel B remained within 7 Å of each other. D. Electrostatic properties of the Rpn10 fragment with protected UIM1. E. Electrostatic properties of Rpn10 fragment without protected UIM1 (reported earlier6). The surface of the fragment is colored according to the color bar with electrostatic energy given in units of kBT/e, where kB is Boltzmann's constant, T is the temperature, and e is the charge of an electron. F. Aligned sequences of Rpn10 flexible arms. The color code is the same as in panel A. Note: in panel A, B, D, and E, residues 196-244 of Rpn10 are shown. The alignment of the full sequences is provided in Fig. S2.
Properties of Rpn10 Flexible Arm
To investigate how hydrophobic UIMs of Rpn10 recognize their binding partners, we characterize first the properties of the Rpn10 flexible arm by itself. Previous NMR experiments determined that the Rpn10 flexible arm is composed of well-defined non-interacting α-helices connected by flexible disordered linkers;6 flexibility of the Rpn10 fragment was proposed to facilitate its binding to poly-ubiquitins.
Flexibility of a UIM-containing Rpn10 fragment, i.e., of the flexible arm, was examined in MD simulations. Figure 2A shows a typical conformational change of this fragment: UIM1 of human Rpn10 is seen to engage during the simulations immediately in interactions with nearby nonpolar residues and becomes protected, i.e., less exposed to solvent and shielded from interactions with other molecules. The nonpolar residues in contact with the UIM are highlighted in Fig. 2B. The identified residue contacts persist over more than 80% of the simulation time, as shown in Fig. 2C. A similar UIM protection is also observed in simulations of yeast and S. pombe fragments of Rpn10, homologous to the simulated fragment of human Rpn10 (Fig. S1).
Figure 2D-E clearly shows that the Rpn10 fragment (flexible arm) in the NMR-resolved structure (E) assumes a more extended form than seen in MD simulations (D). Despite their structural differences, experimental and simulated NOE distances for selected atom pairs are in a good agreement (only atom pairs of structured α-helices have reported NOE distances). In fact, 93.9% of all NOEs are satisfied in the MD simulation ensemble, when allowing for 0.25 Å beyond the upper or lower bounds in NMR distance ranges, as is commonly done35 (Supplementary Table S2). We conclude from the comparison that the ensemble of structures arising from simulations is actually consistent with the NMR measurements.
Atoms of the flexible coil segments of the Rpn10 fragment (flexible arm) did not display NOE interactions.6 This observation may seem to contradict the simulation results presented in Fig. 2C, which show persistent hydrophobic contacts between hydrophobic residues of UIM1 and the flexible coil. However, contacts between specific atom pairs of UIM1 and the flexible coil interchange over the course of the simulation, as there are no strong directional interactions that stabilize them. As NOE interactions should not be observed for such dynamically interchanging interactions, our simulated ensembles actually do not contradict the NOE data and likely exhibit realistic conformations of the Rpn10 flexible arm.
Two consequences can be inferred from the observed protection of UIMs. First, the Rpn10 fragment is not as extended or as flexible (on a timescale of several nanoseconds) as assumed.6 Second, UIMs are partially occluded from exposure to ubiquitin. Since both ubiquitin species and UIMs have hydrophobic patches that are protected, their mutual recognition has to involve refolding that is guided by other surface amino acids. Functionally important amino acids are often conserved. Figs. 2F, S2 show that Rpn10 has multiple conserved residues besides UIMs, many of which are negatively charged. Furthermore, many other conserved acidic residues are distributed along the flexible arm of Rpn10, resulting in its high net negative charge (-25 to -26 for species with two UIMs, or -7 to -10 for species with a single UIM, as shown in Table 1 and Fig. S2).
Table 1.
Number of basic and acidic residues and total charge, qtot, of Rpn10 flexible arms at neutral pH.
| Rpn10 arm | Nbasic | Nacidic | qtot |
|---|---|---|---|
| human | 16 | 41 | -25 |
| bovine | 18 | 44 | -26 |
| rabbit | 16 | 42 | -26 |
| S. cerevisiae | 5 | 15 | -10 |
| S. pombe | 5 | 12 | -7 |
The conserved negatively charged residues in the proximity of UIM should impact ubiquitin recognition. As UIMs are protected by nearby hydrophobic residues, the negatively charged residues simultaneously aggregate around UIM1 (Fig. 2B), thus creating a negatively charged surface mask around the UIM. Furthermore, the arm with aggregated acidic residues (Fig. 2D) is found to have a more intense negative potential than the Rpn10 flexible arm resolved in NMR experiments6 (Fig. 2E).
Electrostatic properties of ubiquitin and their effect on Rpn10 binding
To explore further the role of electrostatics in ubiquitin-Rpn10 refolding and binding, we examine the electrostatic properties of mono-ubiquitin (Fig. 3) and tetra-ubiquitin (Fig. 4). Ubiquitin contains a hydrophobic patch, made of residues Leu8, Ile44 and Val70. Fig. 3 shows that the side of ubiquitin interacting with the UIM of Rpn10 contains the hydrophobic patch surrounded by a positive or neutral potential, while the opposite side of the protein has predominantly negative potential, making ubiquitin dipolar in nature.
Figure 3.
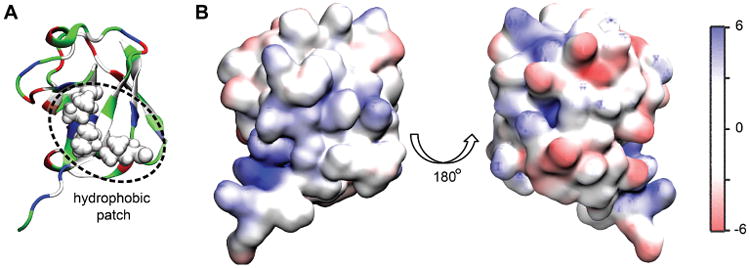
Electrostatic properties of mono-ubiquitin. A. Face of mono-ubiquitin containing the hydrophobic patch interacting with the UIM of Rpn10. B. The electrostatic properties of two sides of mono-ubiquitin, the left one corresponding to the orientation displayed in panel A. The units of the potential are the same as in Fig. 2 (kBT/e).
Figure 4.
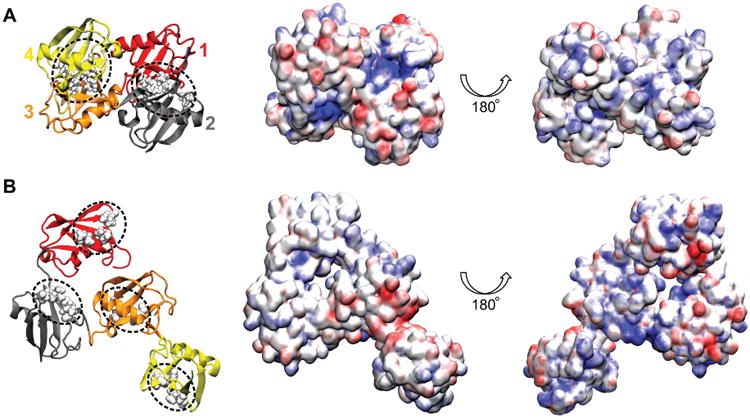
Electrostatic properties of K48-linked tetra-ubiquitin in closed and open conformations. A. Conformation and properties of the closed form of tetra-ubiquitin rendered in surface representation. Hydrophobic patches of ubiquitin units are in contact with each other, and are not exposed to solvent. Shown in the middle and on the right are the electrostatic properties of two sides of the closed form. B. Example of an open conformation of K48-linked tetra-ubiquitin. The structure shown is representative of the largest cluster of configurations obtained in a 200 ns simulation. The images in the middle column correspond to the conformations shown in the left column. The color scale of electrostatic potential is the same as in Fig. 3 (kBT/e).
In the ubiquitin 26S proteasome proteolytic pathway, the 26S proteasome recognizes K48-linked tetra-ubiquitin,16 rather than mono-ubiquitin. Fig. 4 shows two conformations of K48- linked tetra-ubiquitin (closed and open), colored according to their electrostatic properties. In the closed form, which arises predominantly in solution at neutral pH,7 hydrophobic patches are protected. The electrostatic potential of one side of the closed form is primarily negative, except for two very strongly positive concave patches, formed by the C-terminal tails of two nearby ubiquitin units; the opposite side is weakly positive, and has several hydrophobic surfaces exposed.
If UIMs are to bind K48-linked tetra-ubiquitin molecules, they need to either associate with the closed or semi-closed molecules and induce their opening, or associate with the miniscule fraction of open molecules that exist in solution. Fig. 4B shows an exemplary open conformation of K48-linked tetra-ubiquitin, colored according to its electrostatic properties, obtained in a 200 ns equilibration simulation of the fully extended form of K48-linked tetra-ubiquitin. However, as the extended form undergoes a quick hydrophobic collapse, followed by very few conformational changes, a flexible open form of K48-linked tetra-ubiquitin chain likely does not exist in solution by itself.
Binding of mono-ubiquitin to UIM1 of Rpn10. Binding of mono-ubiquitin to UIMs is mediated by hydrophobic interactions.8 However, hydrophobic interactions are usually not long-range or discriminatory enough for molecular recognition. Our MD simulations identified several hydrophobic and polar interactions that stabilize binding between Rpn10 and ubiquitin. Fig. 5B highlights the amino acids of Rpn10 and ubiquitin that are in contact for > 40% of time in MD trajectories. In addition to multiple hydrophobic interactions, several salt bridges are seen to be formed. The most representative, yet transient, salt bridges are characterized in Fig. 5B. These salt bridges likely stabilize the binding pose of Rpn10 and mono-ubiquitin.
Figure 5.
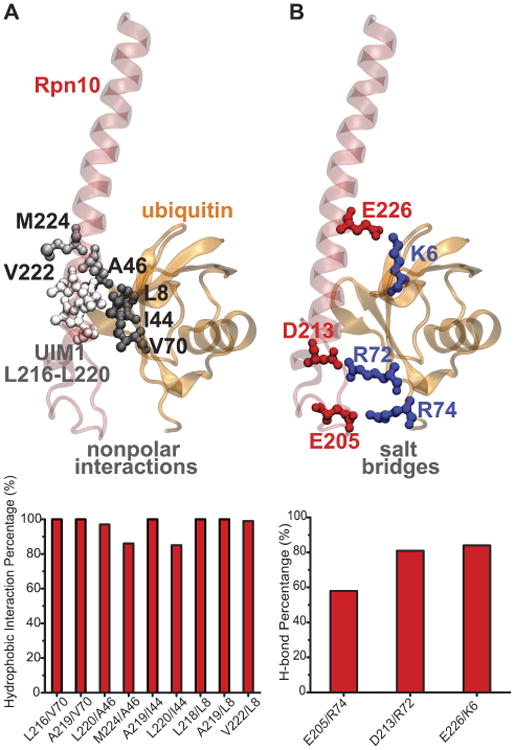
Binding of mono-ubiquitin to Rpn10 UIM1. A. Secondary structure of human Rpn10 UIM1 with mono-ubiquitin bound. UIM1 and nonpolar interactions are highlighted in licorice. The time percentage of significant nonpolar interactions is plotted below. B. Same structure as in panel A with salt bridges highlighted in licorice. The time percentage of salt bridges formed is plotted below. Note: only residues 196-244 of Rpn10 are shown in this figure (same as in Fig. 2).
The structure of mono-ubiquitin bound to UIM1 of human Rpn10 was previously resolved through NMR experiments.6 For the Rpn10 fragment bound to ubiquitin, shown in Fig. 5, average distances of contacts observed in simulations are in good agreement with experimental data; 93.5% of all NOEs are within experimental bounds, when taking into account the 0.25 Å allowance beyond the upper bound or below the lower bound in the NMR distance range.35 The NOE signals were observed for only 12 intermolecular hydrophobic interactions, which are also seen in our MD simulations as shown in Fig. 6. However, NOE signals corresponding to the salt bridges observed in MD simulations are likely difficult to detect through the NOE signals because of their transient nature.
Figure 6.
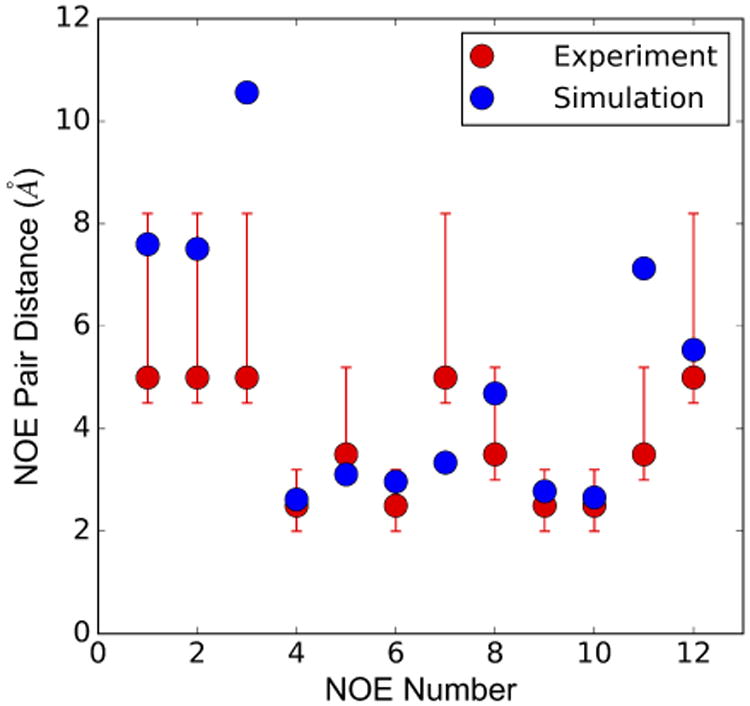
Comparison of simulated NOE pair distances with the NMR data for intermolecular signals between Rpn10 UIM1 and ubiquitin.
Steered MD simulations of ubiquitin-UIM2 unbinding
SMD simulations were performed to investigate the role of charge-charge interactions in unbinding/binding events of ubiquitin and its binding partners. Two independent 40 ns SMD simulations were performed for unbinding of mono-ubiquitin from the UIM2 of human Rpn10, in which hydrophobic groups of the two binding partners were pulled apart by SMD forces. Based on observations of electrostatic patterns on UIMs and mono-ubiquitin, we hypothesized that as hydrophobic groups unbind, charge-charge interactions of UIMs and mono-ubiquitin should persist, if relevant for guiding the process of ubiquitin binding to its partners. Fig. 7 confirms this hypothesis; as hydrophobic groups of ubiquitin and UIM separate, the positively charged groups of ubiquitin and the negatively charged groups of UIM preserve the electrostatic interactions. The role of the electrostatic interactions was observed in two independent simulations, in which the hydrophobic groups separated into different directions.
Figure 7.
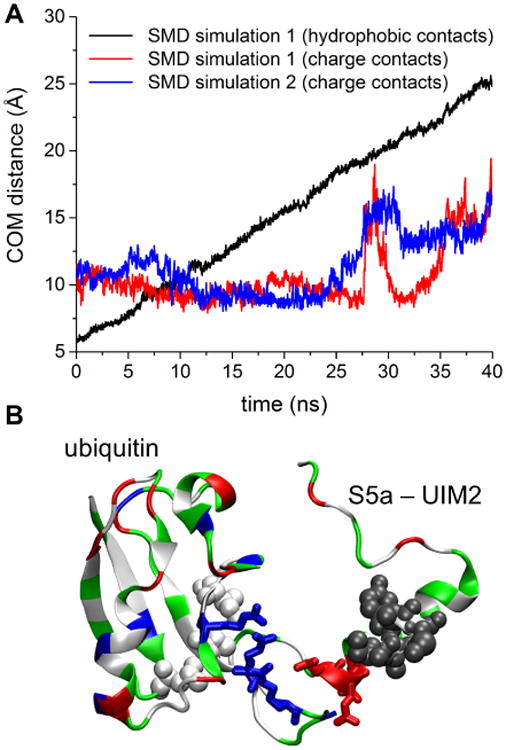
Steered MD simulations of mono-ubiquitin and human Rpn10 UIM2 unbinding. A. Center of mass (COM) distances between hydrophobic contacts of UIM2 (IAYAM residues) and ubiquitin (residues 8, 44, and 70), and charge contacts of UIM2 (acidic residues 283, 284 and 285) and ubiquitin (basic residues 283, 284 and 285) in performed SMD simulations. B. Representative snapshot of the system at the end of one of the SMD simulations. While hydrophobic contacts, shown in white and grey spheres, are broken through the SMD force, the charge contacts, shown in blue and red licorice representation, persist. The pulling is performed along the distance vector between two center of mass groups, where the first group includes residues 8, 44, and 70 of mono-ubiquitin (white vdW spheres) and the second group includes IAYAM residues of Rpn10 UIM2 (grey vdW spheres).
Binding of K48-linked tetra-ubiquitin to Rpn10
While UIMs only bind to single ubiquitin units, the human 26S proteasome binds to K48-linked tetra-ubiquitin with high selective affinity.16 Rpn10 binds K48-linked tetra-ubiquitin with 100-fold higher affinity than K48-linked di-ubiquitin, as summarized in Table S3. The stronger binding to K48-linked tetra-ubiquitin is observed even though the human 26S proteasome has only two UIMs and can only bind to a maximum of two ubiquitin units at the same time. The question arises why Rpn10 has stronger binding to K48-linked tetra-ubiquitin than to di-ubiquitin and tri-ubiquitin.
We suggest that strong binding of human 26S proteasome to K48-linked tetra-ubiquitin is due to additional non-specific interactions between the two UIMs of human Rpn10 and ubiquitin in the case of K48-linked tetra-ubiquitin. To test how the flexible arm of Rpn10 binds to K48-linked tetra-ubiquitin, we examined three separate complexes in MD simulations as shown in Fig. 8, in which UIM1 always binds to the distal ubiquitin (U1), and UIM2 binds to either U2 (case 1), U3 (case 2), or the proximal ubiquitin U4 (case 3). The system in case 1 is similar to the Rpn10 fragment binding to K48-linked di-ubiquitin, which was also examined (case 4). The simulations revealed that in cases 1 and 4, Rpn10 has weaker interactions with ubiquitin chains than in cases 2 and 3, as Rpn10 remains in more compact conformations. In cases 2 and 3, the Rpn10 arm is more stretched out, with Rpn10 binding to all ubiqutin units simultaneously, resulting in more favorable net interaction energies. The additional (non-UIM) interactions are indeed transient, but they still contribute to a tighter overall binding of Rpn10 to K48-linked tetra-ubiquitin, in agreement with experimental results.
Figure 8.
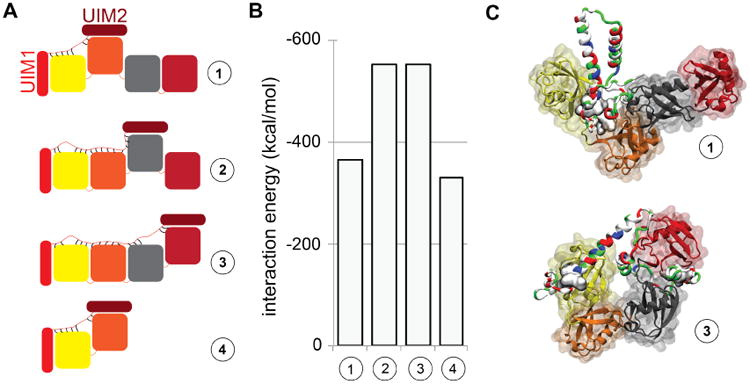
Rpn10 flexible arm binding to K48-linked tetra-ubiquitin. A. Schematic views of the systems examined in MD simulations. Additional interactions (see text) between the Rpn10 flexible arm and ubiquitin subunits are represented through short black lines. B. Interaction energies between the Rpn10 fragment and K48-linked tetra-ubiquitin or di-ubiquitin for binding combinations shown in panel A, computed with a modified generalized Born (GB) model.34 C. Representative snapshot of Rpn10 binding to neighboring ubiquitins (top) and non-neighboring ubiquitins (bottom).
Discussion
In the present study, two questions were addressed by means of MD simulations. First, we asked how ubiquitin and Rpn10, its receptor within the ubiquitin 26S proteasome proteolytic pathway, recognize each other. Second, we asked how the presence of multiple ubiquitin units and the chain architecture in K48-linked tetra-ubiquitin leads to its enhanced binding to Rpn10. Our simulations suggest that the binding process requires refolding (opening) of both K48-linked tetra-ubiquitin and the Rpn10 flexible arm. Furthermore, charge interactions likely guide binding processes and stabilize bound states that involve ubiquitin. The simulations also characterized several conformations of Rpn10 bound to K48-linked tetra-ubiquitin.
Prior to the present simulation study, the C-terminal tail of Rpn10 was thought to be a flexible arm made of several non-interacting α-helices connected by disordered coils. Instead, our simulations show that the Rpn10 flexible arm is not as flexible or as extended as previously thought. Furthermore, helical UIMs interact with the nearby disordered coils, which reduces their exposure to solution, and also, in some cases, surrounds them with a negatively charged surface layer (Fig. 2). Even though the structural picture that emerges from simulations is different from previous conclusions based on NMR data, our results are in agreement with those data (NOEs).
Based on the distinct charge patterns observed on ubiquitins and Rpn10, a hypothesis summarized in Fig. 9 can be built on how mono- and poly-ubiquitins get recognized by Rpn10 and other proteins. Mono- and poly-ubiquitins have distinct regions of positive potential surrounding the hydrophobic patch involved in binding. The rest of the ubiquitin surface contains primarily regions of negative charge. Given that the flexible arm of Rpn10 is strongly negatively charged (especially the N-terminal sides of the two UIMs), the following three stage scenario for the recognition of Rpn10 and ubiquitins suggests itself. In stage 1, the negatively charged arm of Rpn10 preferentially seeks the positively charged surfaces of ubiquitins, and avoids the negatively charged surfaces of ubiquitins during diffusion and association stages of the binding process, as shown schematically in Fig. 9. In stage 2, the negative charges near the UIMs are recognized by the positively charged C-terminal tail of ubiquitin units, which induces opening/refolding of Rpn10 and K48-linked tetraubiquitin. Once nearby and partially exposed, in stage 3 the hydrophobic regions of UIM and ubiquitin finally lock into the most favorable binding pose.
Figure 9.
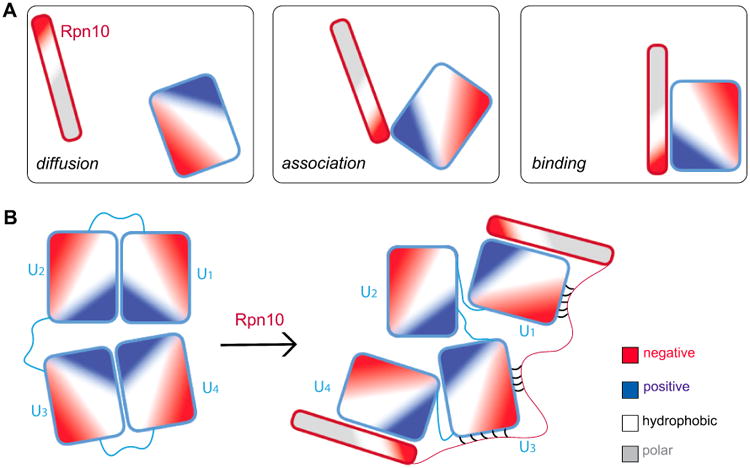
Schematic binding process of Rpn10 (red-framed rod) to ubiquitins (blue-framed rectangles). A. Three stages of the binding process of Rpn10 fragment and mono-ubiquitin. B. Binding of Rpn10 fragment to K48-linked tetra-ubiquitin.
The above scenario is supported by present and previous studies of ubiquitin receptors.6,8,11,36 We note that each of the UIMs of human, rabbit, mouse, yeast and S. pombe has conserved negative charges nearby. SMD simulations of mono-ubiquitin-UIM unbinding showed that these conserved charges guide Rpn10 and ubiquitin towards hydrophobic binding (Fig. 7). Furthermore, even though the ubiquitin-interacting surface of the second proteasomal ubiquitin receptor Rpn13 differs greatly from the one of Rpn10, it also binds ubiquitin with a surface that contains multiple negatively charged residues37 and negative electrostatic potential, as shown in Fig. S3, which can coordinate the positively charged C-terminus of ubiquitin. Multiple other partners of ubiquitin also have distinct patches of negative charge on their surfaces that bind to the hydrophobic patch of ubiquitin, for example protein Vps27 has a UIM that is preceded by multiple acidic residues.11
Our simulations also clarified why 26S proteasome binds to K48-linked tetra-ubiquitin with higher affinity than to K48-linked di-ubiquitin (Table S3), despite the fact that Rpn10 UIMs can only bind directly to two ubiquitin units. The simulated Rpn10 exhibits multiple transient contacts to the K48-linked tetra-ubiquitin chain, which provides additional stabilization to the K48-linked tetra-ubiquitin bound state.
In summary, the simulations carried out indicate that recognition of ubiquitin by Rpn10 receptors is facilitated by the partners flexible nature, involving even refolding to a significant degree, and a combination of hydrophobic and negatively charged residues on the Rpn10 flexible arm. The simulation results provide a comprehensive description of Rpn10-K48-linked tetra-ubiquitin recognition and binding, and indicate that negatively charged residues of ubiquitin receptors are, in general, crucial for their binding to mono-ubiquitin and polyubiquitin chains.
Conclusion
Our results provide insight into structural dynamics of Rpn10 and K48-linked tetra-ubiquitin. The simulations carried out reveal that in the unbound form, namely before the recognition, UIMs of Rpn10 are protected through hydrophobic interactions between the UIMs and their flexible linkers, and that K48-linked tetra-ubiquitin by itself remains in the closed state.
Since hydrophobic interfaces of separated Rpn10 and K48-linked tetra-ubiquitin are protected, recognition and binding processes need to involve significant refolding of the two proteins. A full understanding of the selective affinity of Rpn10 and K48-linked tetra-ubiquitins will have to await explicit simulations of partner refolding preceding recognition and binding. However, the present simulations already reveal the principal recognition steps. Our simulations suggest that ubiquitin recognition is a three stage process: in stage 1, the conserved electrostatic pattern of Rpn10 UIM brings Rpn10 and K48-linked tetra-ubiquitins together. In stage 2, the negative charges near the UIMs are recognized by the positively charged C-terminal tails of the ubiquitin subunits, which induces refolding of the Rpn10 and K48-linked tetra-ubiquitin partners. In stage 3, the formed electrostatic interactions complete recognition and binding through hydrophobic interactions.
The combination of flexibility with electrostatic and hydrophobic interaction for ubiquitin recognition is not unique for the proteasome and, instead, is a general motif in ubiquitin recognition.11 Finally, the simulations explain why Rpn10 has higher affinity for K48-linked tetra-ubiquitin than for K48-linked di- and tri-ubiquitins.
Supplementary Material
Acknowledgments
We gratefully acknowledge Prof. Kylie Walters for data from NMR experiments conducted in her laboratory, and Dr. Eri Sakata for useful discussions. This work was supported by National Science Foundation (NSF) grants PHY0822613 and PHY1430124, as well as by National Institutes of Health (NIH) grant 9P41GM104601. The authors gratefully acknowledge computer time provided by the NSF-funded Extreme Science and Engineering Discovery Environment (XSEDE) MCA93S028, Max Planck Institute Rechenzentrum Garching, and the Computational Science and Engineering Program at the University of Illinois (Taub computing cluster), and a Blue Waters Illinois allocation, which is part of the Blue Waters sustained-petascale computing project supported by the National Science Foundation (awards OCI-0725070 and ACI-1238993) and the state of Illinois. Blue Waters is a joint effort of the University of Illinois at Urbana-Champaign and its National Center for Super-computing Applications. L.V. acknowledges support as a Center for the Physics of Living Cells (CPLC) Postdoctoral Fellow, and T.R. support as a Feodor Lynen von Humboldt Postdoctoral Fellow.
Footnotes
Supporting Information Available: Three tables list simulations performed, compare simulated NOE pair distances with NMR data for S5a/Rpn10 arm structures with and without ubiquitin bound, and list experimentally determined Kd values for binding of 26S proteasome to different ubiquitin species and for binding of human S5a to mono- and K48-linked diubiquitin. Three figures show properties of the flexible arm of the Rpn10 fragment in S. cerevisiae and S. pombe as seen from MD simulations, aligned sequences of Rpn10 flexible arms of human, bovine, rabbit, S. cerevisiae and S. pombe species, and NMR structure of mouse Rpn13 bound to mono-ubiquitin.
This material is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.Csermely P, Palotai R, Nussinov R. Induced Fit, Conformational Selection and Independent Dynamic Segments: An Extended View of Binding Events. Trends Biochem Sci. 2010;35:539–546. doi: 10.1016/j.tibs.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mésáaros B, Simon I, Dosztányi Z. The Expanding View of Protein-Protein Interactions: Complexes Involving Intrinsically Disordered Proteins. Phys Biol. 2011;8:035003. doi: 10.1088/1478-3975/8/3/035003. [DOI] [PubMed] [Google Scholar]
- 3.Finley D. Recognition and Processing of Ubiquitin-Protein Conjugates by the Proteasome. Annu Rev Biochem. 2009;78:477–513. doi: 10.1146/annurev.biochem.78.081507.101607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Förster F, Unverdorben P, Śledź P, Baumeister W. Unveiling the Long-Held Secrets of the 26S Proteasome. Structure. 2013;21:1551–1562. doi: 10.1016/j.str.2013.08.010. [DOI] [PubMed] [Google Scholar]
- 5.Matyskiela ME, Lander GC, Martin A. Conformational Switching of the 26S Proteasome Enables Substrate Degradation. Nat Struc Mol Biol. 2013;20:781–788. doi: 10.1038/nsmb.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Q, Young P, Walters KJ. Structure of S5a Bound to Monoubiquitin Provides a Model for Polyubiquitin Recognition. J Mol Biol. 2005;348:727–739. doi: 10.1016/j.jmb.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 7.Eddins MJ, Varadan R, Fushman D, Pickart CM, Wolberger C. Crystal Structure and Solution NMR Studies of Lys48-linked Tetraubiquitin at Neutral pH. J Mol Biol. 2007;367:204–211. doi: 10.1016/j.jmb.2006.12.065. [DOI] [PubMed] [Google Scholar]
- 8.Zhang N, Wang Q, Ehlinger A, Randles L, Lary JW, Kang Y, Haririnia A, Storaska AJ, Cole JL, Fushman D, et al. Structure of the S5a: K48-Linked Diubiquitin Complex and Its Interactions with Rpn13. Mol Cell. 2009;35:280–290. doi: 10.1016/j.molcel.2009.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hicke L, Schubert HL, Hill CP. Ubiquitin-Binding Domains. Nat Rev Mol Cell Biol. 2005;6:610–621. doi: 10.1038/nrm1701. [DOI] [PubMed] [Google Scholar]
- 10.Ye Y, Blaser G, Horrocks MH, Ruedas-Rama MJ, Ibrahim S, Zhukov AA, Orte A, Klenerman D, Jackson SE, Komander D. Ubiquitin Chain Conformation Regulates Recognition and Activity of Interacting Proteins. Nature. 2012;492:266–270. doi: 10.1038/nature11722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sgourakis NG, Patel MM, Garcia AE, Makhatadze GI, McCallum SA. Conformational Dynamics and Structural Plasticity Play Critical Roles in the Ubiquitin Recognition of a UIM Domain. J Mol Biol. 2010;396:1128–1144. doi: 10.1016/j.jmb.2009.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.von Hippel PH, Berg OG. Facilitated Target Location in Biological Systems. J Biol Chem. 1989;264:675–678. [PubMed] [Google Scholar]
- 13.Zhou HX. Brownian Dynamics Study of the Influences of Electrostatic Interaction and Diffusion on Protein-Protein Association Kinetics. Biophys J. 1993;64:1711–1726. doi: 10.1016/S0006-3495(93)81543-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sheinerman FB, Norel R, Honig B. Electrostatic Aspects of Protein-Protein Interactions. Curr Opin Struc Biol. 2000;10:153–159. doi: 10.1016/s0959-440x(00)00065-8. [DOI] [PubMed] [Google Scholar]
- 15.Janin J. The Kinetics of Protein-Protein Recognition. Proteins. 1997;28:153–161. doi: 10.1002/(sici)1097-0134(199706)28:2<153::aid-prot4>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 16.Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. Recognition of the Polyubiquitin Proteolytic Signal. EMBO J. 2000;19:94–102. doi: 10.1093/emboj/19.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sali A, Blundell TL. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 18.Krivov GG, Shapovalov MV, Dunbrack RL. Improved Prediction of Protein Side-Chain Conformations with SCWRL4. Proteins. 2009;77:778–795. doi: 10.1002/prot.22488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of Simple Potential Functions for Simulating Liquid Water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 20.Humphrey W, Dalke A, Schulten K. VMD: Visual Molecular Dynamics. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 21.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K. Scalable Molecular Dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mackerell AD, Feig M, Brooks CL. Extending the Treatment of Backbone Energetics in Protein Force Fields: Limitations of Gas-Phase Quantum Mechanics in Reproducing Protein Conformational Distributions in Molecular Dynamics Simulation. J Comput Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 23.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, et al. All-atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J Phys Chem B. 1998;102:3586–616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 24.Mackerell AD. Empirical Force Fields for Biological Macromolecules: Overview and Issues. J Comput Chem. 2004;25:1584–1604. doi: 10.1002/jcc.20082. [DOI] [PubMed] [Google Scholar]
- 25.Mackerell AD, Feig M, Brooks CL. Improved Treatment of the Protein Backbone in Empirical Force Fields. J Am Chem Soc. 2004;126:698–699. doi: 10.1021/ja036959e. [DOI] [PubMed] [Google Scholar]
- 26.Best RB, Zhu X, Shim J, Lopes PEM, Mittal J, Feig M, MacKerell AD. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone, Φ, Ψ, and Side-Chain χ1 and χ2 Dihedral Angles. J Chem Theo Comp. 2012;8:3257–3273. doi: 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feller SE, Zhang Y, Pastor RW, Brooks BR. Constant Pressure Molecular Dynamics Simulation: The Langevin Piston Method. J Chem Phys. 1995;103:4613–4621. [Google Scholar]
- 28.Martyna GJ, Tobias DJ, Klein ML. Constant Pressure Molecular Dynamics Algorithms. J Chem Phys. 1994;101:4177–4189. [Google Scholar]
- 29.Darden T, York D, Pedersen L. Particle Mesh Ewald: An Nlog(N) Method for Ewald Sums in Large Systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 30.Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. Journal of Chemical Theory and Computation. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 31.Han W, Wu YD. A Strand-Loop-Strand Structure Is a Possible Intermediate in Fibril Elongation: Long Time Simulations of Amyloid-β Peptide (10-35) J Am Chem Soc. 2005;127:15408–15416. doi: 10.1021/ja051699h. [DOI] [PubMed] [Google Scholar]
- 32.Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJC. GROMACS: Fast Flexible and Free. J Comput Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 33.Hardy DJ, Stone JE, Vandivort KL, Gohara D, Rodrigues C, Schulten K. Fast Molecular Electrostatics Algorithms on GPUs. GPU Computing Gems Emerald Edition. 2011:43–58. [Google Scholar]
- 34.Tanner DE, Chan KY, Phillips JC, Schulten K. Parallel Generalized Born Implicit Solvent Calculations with NAMD. J Chem Theory Comput. 2011;7:3635–3642. doi: 10.1021/ct200563j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou R. Trp-Cage: Folding Free Energy Landscape in Explicit Water. Proc Natl Acad Sci USA. 2003;100:13280–13285. doi: 10.1073/pnas.2233312100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fujiwara K, Tenno T, Sugasawa K, Jee JG, Ohki I, Kojima C, Tochio H, Hiroaki H, Hanaoka F, Shirakawa M. Structure of the Ubiquitin-Interacting Motif of S5a Bound to the Ubiquitin-Like Domain of HR23B. J Biol Chem. 2004;279:4760–4767. doi: 10.1074/jbc.M309448200. [DOI] [PubMed] [Google Scholar]
- 37.Husnjak K, Elsasser S, Zhang N, Chen X, Randles L, Shi Y, Hofmann K, Walters KJ, Finley D, Dikic I. Proteasome Subunit Rpn13 Is a Novel Ubiquitin Receptor. Nature. 2008;453:481–488. doi: 10.1038/nature06926. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.