

Abstract
Objective
Circulating human T effector memory cell (TEM) recognition of non-self MHC molecules on allograft endothelial cells (EC) can initiate graft rejection despite elimination of professional antigen presenting cells necessary for naïve T cell activation. Our prior studies of CD4 TEM have established that engagement of the T cell receptor (TCR) not only activates T cells but also triggers transendothelial migration (TEM) by a process that is distinct from that induced by activating chemokine receptors (CR) on T cells, being slower, requiring microtubule organizing center (MTOC)-directed cytolytic granule polarization to and release from the leading edge of the T cell, and requiring engagement of proteins of the EC lateral border recycling compartment (LBRC). While CD4 TEM may contribute to acute allograft rejection, the primary effectors are alloreactive CD8 TEM. Whether and how TCR engagement affects TEM of human CD8 TEM is unknown.
Approach and Results
We modeled TEM of CD8 TEM across cultured human microvascular EC engineered to present superantigen under conditions of venular shear stress in vitro in a flow chamber. Here we report that TCR engagement can also induce TEM of this population that similarly differs from CR-driven TEM with regard to kinetics, morphological manifestations, and MTOC dynamics as with CD4 TEM. However, CD8 TEM do not require either cytolytic granule release or interactions with proteins of the LBRC.
Conclusions
These results imply that therapeutic strategies designed to inhibit TCR-driven recruitment based on targeting granule release or components of the LBRC will not affect CD8 TEM and are unlikely to block acute rejection in the clinic.
Introduction
Allogeneic transplantation is the most effective treatment for many end-stage organ diseases. Facilitated by modern immunosuppressive regimens, acute allograft rejection rates have fallen dramatically, but have not been completely eliminated. Unlike typical laboratory rodents, adult humans have a high frequency of alloreactive T effector memory cells (TEM) in their circulation and the pre-transplant frequency of donor-specific memory T cells correlates with risk of acute rejection episodes 1,2. Allograft rejection by memory T cells can occur despite depletion of professional antigen presenting cells (APC) from the graft 3 or a need to prime the host immune response in secondary lymphoid organs 4,5. Moreover, TEM are more difficult to suppress than naïve T cells 6,7. Thus, it is important to understand how human TEM sense and are recruited to an allograft to further reduce rejection rates.
We have previously shown that human CD4 and CD8 TEM can be activated by direct recognition of allogeneic class II and class I MHC molecule presentation, respectively, by cultured human endothelial cells (EC) 8 and that human EC may be rejected by adoptively transferred allogeneic T cells in vivo in immunodeficient mouse hosts 9,10. In vitro, EC presentation of antigen to CD4 TEM under conditions of flow not only causes T cell activation, but also induces transendothelial migration (TEM), a model of T cell recruitment 11. Remarkably, this process shares many more features of the interactions of a T cell with an APC than it does with conventional chemotaxis or haptotaxis. Specifically, in response to TCR engagement, CD4 TEM round up instead of flattening out and move their microtubule organizing center (MTOC) and cytosolic granules to the region of contact with the EC rather than into a trailing uropod. Unexpectedly, degranulation proved to be a necessary step in the TEM process, apparently requiring extracellular granzyme A activity to successfully cross the EC monolayer 12. TEM itself begins by pushing a thick cytoplasmic foot-like process, that we have called a transendothelial protrusion (TEP), between adjacent EC. The nucleus then follows the MTOC into the TEP as TEM proceeds 13. The transmigrating T cell engages the EC via LFA-1 binding to endothelial ICAM-1 as well as interactions with EC proteins associated with the lateral border recycling compartment (LBRC) such as PECAM-1 (CD31), CD99, CD112 and CD155 11,14,15. In contrast, chemokine-stimulated TEM of CD4 TEM may use either endothelial ICAM-1 or VCAM-1 and does not require interactions with proteins of the LBRC, degranulation or extracellular granzyme A activity. While CD4 TEM may contribute to rejection, the rejection process appears to correlate with the presence of CD8 cytotoxic T cells 10 which may arise from CD8 TEM 16. Recently, it has been demonstrated in mice that CD8 T cells can be recruited by antigen recognition on EC, triggering rejection independent of professional APC 17. Little is known about TEM by human CD8 TEM.
In the present study, we applied our in vitro flow chamber model that we previously used to analyze TEM by CD4 TEM to study the cell biology of human CD8 TEM recruitment in response to antigen and compare this both to chemokine responses of CD8 TEM and to our prior findings with CD4 TEM. Unexpectedly, we find significant differences between the TCR-mediated responses of these two populations.
Materials and Methods
Materials and Methods are available in the online-only Data Supplement.
Results
We modeled TEM of CD8 TEM across microvascular EC under conditions of venular shear stress in vitro in a flow chamber using TNF-activated, CIITA-transduced monolayers of untransformed HDMEC to present toxic shock syndrome toxin-1 (TSST-1), a superantigen, to allogeneic TEM isolated from PBMC. TSST-1 activates about 5-10% of the T cells, namely those clones which formed their TCR using a Vβ2 gene segment. This increases the frequency of responsive T cells to a level well above that activated by alloantigen (closer to 0.1%), significantly increasing the number of TCR-activated T cells available for analysis and allowing simultaneous analysis of both TCR-triggered Vβ2+ TEM and Vβ2− TEM responding to TNF-induced chemokines via chemokine receptors (CR) in the same field 11. CIITA transduction is used to restore MHC class II expression lost by EC when placed in culture. While MHC class II molecules are not required for allogeneic responses of CD8 T cells, they are necessary for the presentation of superantigen to both CD4 and CD8 T cells. Shear stress is required to stimulate the rapid TEM of T cells, whether the signal is antigen or chemokine 11,18,19.
Using this assay for CD8 TEM, we found this population to share many of the characteristics of CD4 TEM undergoing TEM. Shortly after encounter with superantigen, ZAP70 is phosphorylated at levels well above that induced by chemokines, indicative of TCR signaling. The extent of the phosphorylation varies from cell to cell, but the ranges completely overlap between the two populations, suggesting that signal strengths within these T cell subsets are comparable (Figure 1A). In both cases, only the antigen-activated TEM become circular and rounded-up rather than spread and crawling. In Vβ2+ T cells, the MTOC then locates to a position between the T cell nucleus and EC apical surface rather than trailing in the uropod, the latter being a shared feature of both CD4 and CD8 TEM in response to chemokine (Figure 1B, Supplemental Figure I, and ref. 12). By 15 minutes, TCR-activated cells start inserting transendothelial protrusions (TEPs) between and under the EC. This is followed by transit of the nucleus through the monolayer, thereby completing TCR-driven TEM. As in CD4 TEM, TCR-driven TEM of CD8 TEM is delayed compared to CR-driven TEM (Figure 1C and ref. 11). Treatment of T cells with blebbistatin, an inhibitor of myosin IIA, allows TEP formation but prevents cell body transit across the monolayer during TCR-driven TEM; blebbistatin has no effect on CR-driven TEM (Figure 1D and ref. 13). As previously observed for CD4 TEM12, the MTOC, identified by staining for γ-tubulin, precedes the nucleus across the EC monolayer during CD8 TCR-driven TEM in stark contrast to CR-driven TEM, where the MTOC trails the nucleus (Supplemental Figure 1A-C). Using NFAT translocation to the nucleus as a marker for TEM activated by alloantigen, we find that the MTOC similarly precedes the nucleus in alloantigen-driven TEM of both CD4 and CD8 TEM (Figure 1E).
Figure 1.

Similarities between CD4 and CD8 TEM TEM. A. ZAP70 activation during CR-driven and TCR-driven TEM of CD4 and CD8 TEM. Graph shows quantification of staining for P-ZAP70(Y319) of individual CD4 TEM and CD8 TEM after the indicated durations of flow. CR and TCR denote chemokine- and TCR-driven TEM, respectively. P<0.0001 for all CR and TCR comparisons, and ns for all CR vs CR and TCR vs TCR comparisons. B. MTOC localization during CR- and TCR-driven TEM of CD8 TEM. Graph shows % of MTOC localized between the T cell nucleus (sub-nuclear) and EC apical surface for CR-driven (CR) and TCR-driven (TCR) cells after 5 min flow. N=77 and 75 for CR and TCR, respectively, from 3 separate experiments with different donors. C. Kinetics of CR-driven and TCR-driven TEM. Left graph shows % CR-driven (circles connected by dotted lines) and TCR-driven (squares connected by solid lines) TEM of CD8 TEM at 5, 15, and 30 min. Right graph shows % transendothelial protrusions (TEP) at 5, 15, and 30 min during TCR-driven TEM. Data combined from three experiments with different donors. D. Myosin IIA is required for TCR-driven TEM of CD8 TEM at a step after TEP formation. TEM assays of CD8 TEM treated with blebbistatin (bleb) or vehicle (veh). Left graph shows % TEM of CR-driven TEM, middle graph shows % TEM of TCR-driven TEM, and right graph shows % TEP of TCR-driven TEM. Data combined from three experiments with different donors. E. MTOC precedes the nucleus in alloantigen-driven TEM. Flow assay samples of CD4 and CD8 TEM on HDMEC treated with interferon-γ 72h and TNF 20h stained for CD45 (green), γ-tubulin (MTOC), NFAT, and nuclei (DAPI). The bottom panels represent confocal slices beneath the EC monolayer, and the top panels are slices taken 1.44 μm (CD4) and 1.84 μm (CD8) above the lower panels. Note the nuclear localization of NFAT, indicative of TCR activation. The arrows indicate the MTOC positioned near the front of the nucleus; the nucleus is in the process of traversing the monolayer.
Despite these similarities between the T cell subsets, we also observed important differences between CD4 and CD8 TEM. Unlike CD4 TEM, the cytosolic granules of many (but not all) CD8 TEM, contain granzyme B as well as granzyme A and that when granzyme B is present, it co-localizes to the same granules as granzyme A (20-22 and Supplemental Figure 1D). CD8 TEM granules also contain perforin, a necessary component for cytolysis, which is lacking in CD4 TEM (23 and data not shown). As we had seen with CD4 TEM, TCR signaling causes both the MTOC and cytosolic granules to be relocated to a region in proximity to the T cell-EC apical surface (Supplemental Figure 1). Unlike CD4 TEM, however, treatment with concanamycin A, an inhibitor of vesicular H+-ATPase on granules that thereby prevents acidification and renders them nonfunctional, failed to inhibit TCR-driven TEM of CD8 TEM Figure 2A). In the presence of the serine protease inhibitor AEBSF, neither CR- nor TCR-driven TEM of CD8 TEM was inhibited, in contrast to TCR-driven TEM of CD4 TEM (Figure 2B). Furthermore, again in contrast to our prior studies of CD4 TEM 12 and experiments repeated here in parallel, quantification of granzyme A+ granules in T cells that receive TCR signals indicates that degranulation does not occur during TCR-driven TEM of CD8 TEM (Figure 2C). Interestingly, concanamycin A did inhibit CR-driven TEM of CD8, but not CD4 TEM, to a limited extent (Figure 2A).
Figure 2.
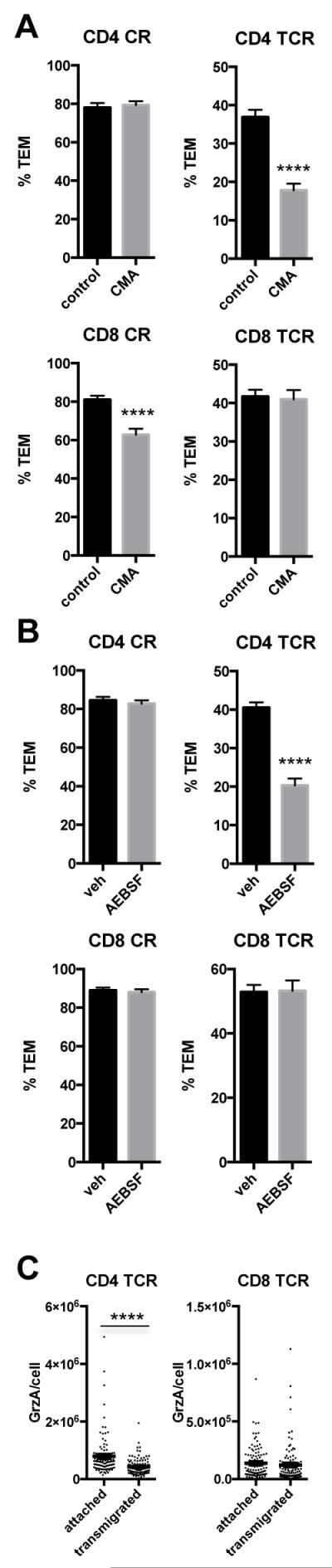
Exocytosis of lytic granules is not necessary for TCR-driven TEM of CD8 TEM. A. TEM assay of CD4 and CD8 TEM treated with concanamycin A (CMA) or vehicle (veh). Upper graphs show CD4, and lower CD8. Left graphs show % TEM of CR-driven TEM, and right graphs show % TEM of TCR-driven TEM. Data combined from three experiments with different donors. B. Graphs show %TEM of assays performed in the presence of serine protease inhibitor AEBSF. C. Quantification of granzyme A in cells responding to antigen after 2 min flow (attached) and after transmigration. Graphs show granzyme A content/cell measured from pictures taken of stained cells; exposure times were 800 and 50 milliseconds for CD4 and CD8, respectively, due to stronger staining of CD8. Data combined from three experiments with different donors. Horizontal and vertical bars within the dots represent mean +/− SEM, respectively. P=0.44 for CD8.
We next interrogated whether components of the LBRC were involved in TEM of CD8 TEM. Such interactions have been shown to be essential for human neutrophil and monocyte TEM 24. While our prior studies showed that CD4 TEM did not utilize LBRC proteins in response to chemokines, they did require their use when TEM was induced by TCR signals 14,15. Using approaches documented previously and repeated in parallel here, knockdown of PECAM-1 (CD31, knockdown confirmed by immunofluorescence staining) or blocking antibodies to CD112 and CD155 (on EC) or CD96 (on T cells) affected CD4 TEM TCR-driven TEM, but had no effect on CD8 TEM TEM in response to chemokine or antigen (Figure 3A-C). Furthermore, treatment of EC with an inhibitor of soluble adenylyl cyclase, an intracellular enzyme that facilitates leukocyte TEM by mobilizing vesicles of the LBRC to the plasma membrane 25, selectively inhibited TCR-driven TEM of CD4 TEM (Figure 3D).
Figure 3.

Components of the lateral border recycling compartment are not necessary for TCR-driven TEM of CD8 TEM. A. TEM assays on EC treated with control and PECAM-1 siRNA. Graphs show, from left to right, % TEM of CD4 TCR-driven TEM, CD8 CR-driven TEM and CD8 TCR-driven TEM. Data combined from three experiments with different donors. B. TEM assays on EC treated with blocking antibodies to CD112 and CD155. Left graph shows % TEM of CD4 TCR-driven TEM, middle graph shows % TEM of CD8 CR-driven TEM, and right graph shows % TEM of CD8 TCR-driven TEM. One representative experiment of three with different donors. C. TEM assays of TEM treated with blocking antibody to CD96. Left graph shows % TEM of CD4 TCR-driven TEM, middle graph shows % TEM of CD8 CR-driven TEM, and right graph shows % TEM of CD8 TCR-driven TEM. Data combined from three experiments with different donors. D. TEM assays of TEM on EC treated with soluble adenylyl cyclase inhibitor KH7. Graphs show, from left to right, % TEM of CD4 CR-driven, CD4 TCR-driven, CD8 CR-driven, and CD8 TCR driven TEM. Data combined from four experiments with different donors.
We next examined the roles of other EC adhesion molecules in TEM, namely ICAM-1, VCAM-1, JAM-A, and JAM-B. TCR-driven TEM of both CD4 and CD8 TEM, as well as CR-driven TEM of CD8 TEM, showed a dependence on EC ICAM-1, as determined in experiments using ICAM-1 blocking mAb on the EC (Figure 4A) as well as knockdown of EC ICAM-1 by siRNA (Figure 4B, Supplemental Figure II). Blocking mAbs to or siRNA knockdown of VCAM-1 reduced CD4 TEM TCR-driven TEM, and knockdown of JAM-A had a small but significant effect on CD4 TEM CR-driven TEM, as reported previously 14, but none of these treatments affected CD8 TEM TEM (Figure 4C-E, Supplemental Figure II). JAM-B knockdown had no effects on TEM (Figure 4F, Supplemental Figure II).
Figure 4.

ICAM-1, but not VCAM-1, is necessary for CR- and TCR-driven TEM of CD8 TEM. A. TEM assays of TEM on EC treated with control IgG (control) and blocking antibodies to ICAM-1 (ICAM-1). From left to right, graphs show % TEM of CD4 CR-driven TEM, CD4 TCR-driven TEM, CD8 CR-driven TEM and CD8 TCR-driven TEM. Data combined from two (CD4) and three (CD8) experiments with different donors. B. TEM assays of TEM on EC transfected with control and ICAM-1 siRNA. Graphs are in the same order as in panel A. Data combined from 3 experiments with different donors. C. TEM assays of TEM on EC treated with control IgG (control) and blocking antibodies to VCAM-1. From left to right, graphs show % TEM of CD4 CR-driven TEM, CD4 TCR-driven TEM, CD8 CR-driven TEM and CD8 TCR-driven TEM. Data combined from three experiments with different donors. D. TEM assays of TEM on EC transfected with control and VCAM-1 siRNA. Graphs are in the same order as in panel C. Data combined from two experiments with different donors. E. TEM assays of TEM on EC transfected with control and JAM-A siRNA. Data combined from two experiments with different donors. F. TEM assays of TEM on EC transfected with control and JAM-B siRNAs. Data combined from two experiments with different donors.
Treatment with a blocking mAb to integrin VLA-4 (α4 subunit, CD49d), the known receptor for VCAM-1, selectively reduced CD4 TEM TCR-driven TEM, while blocking mAb to integrin LFA-1 (αL subunit, CD11a) and mAb to the common LFA-1 and Mac-1 beta subunit (β2, CD18) effectively diminished both CD4 and CD8 TEM TCR-driven TEM (Figure 5A-C). Since the mAb to LFA-1/Mac-1 appeared to be more potent than the mAb to LFA-1 in affecting CD8 TEM TCR-driven TEM, we also tested a blocking mAb to CD11b, the Mac-1 alpha subunit (αM), but found no significant effect (Figure 5D). The blocking mAbs to LFA-1 and VLA-4 had similar effects on total TEM adhesion (Figure 5E). However, in contrast to CD4, CD8 TEM did not show an antigen-induced increase in binding, unless treated with an integrin blocking mAb (Figure 5F).
Figure 5.

Both CD4 and CD8 TEM TCR-driven TEM are inhibited by LFA-1 blocking mAbs. A. TEM assays in the presence of VLA-4 blocking mAb clone PS/2. Graphs show % TEM of data combined from 3 experiments. B. TEM assays in the presence of LFA-1 blocking mAb clone TS1/22. Graphs show % TEM of data combined from 5 experiments with different donors. C. TEM assays in the presence of LFA-1/Mac-1 blocking mAb clone TS1/18. Graphs show % TEM of data combined from at least 3 experiments with different donors. D. TEM assays in the presence of Mac-1 (CD11b) blocking mAb. Graphs show % TEM of data combined from 3 experiments with different donors. E. Adhesion of CD4 and CD8 TEM. Graphs show the combined raw data of 3 (CD4) and 2 (CD8) experiments with different donors. F. Antigen-induced binding of CD4 and CD8 TEM. Graphs show fold enrichment of antigen-specific T cells attached compared to the input (reference) population, mean and sem from 3 experiments with different donors.
Discussion
The fundamental points made by this study is that human CD8 TEM, like CD4 TEM, can be triggered to undergo a kinetically and morphologically distinct TEM process characterized by dramatic rearrangement of organelles to the leading edge of the cell, rather than to a trailing uropod, and an invasion between adjacent ECs led by a blunt TEP. Despite these similarities, CD4 and CD8 TEM TCR-driven TEM do differ in several important ways. Specifically, TCR-driven TEM of CD4 TEM requires granzyme A stored in cytolytic granules to be exocytosed to traverse the endothelium in a process that requires interactions with proteins of the lateral border recycling compartment (LBRC), including PECAM-1, CD99, CD112, and CD155, with their receptors on the T cells 12,14,15. Extracellular enzymatic activity of granzyme A is then required to permit TEM. The target of this serine protease is unknown, but it is possible that it may be needed to mobilize the LBRC to the plasma membrane. As shown here, CD8 TEM do not require either degranulation or engagement of endothelial proteins of the LBRC in order to transmigrate in response to TCR signals, although there is a small but significant effect of concanamycin A on CR-driven TEM of CD8 TEM. The limited effect of concanamycin on human CD8 TEM appears to contradict a prior report showing that granzyme B contributed to antigen-independent transmigration of differentiated CD8 CTL 26. These differences likely reflect a difference between CD8 TEM, which are poised to become CTL, and functionally mature CTL. Only the latter degranulate in response to TCR signals. The modest effect of concanamycin A on CR-driven TEM of CD8 T that we observed could be explained by the presence of a relatively small subset of mature CTL in our freshly isolated peripheral blood human TEM subsets. Moreover, analysis of freshly isolated human CD8 T is likely to be complicated by the heterogeneity of subsets within this population; recent multiparameter phenotyping by CyTOF indicates that there are at least 4 subtypes of human Tc cells, and all lack expression of CCR7, like the TEM used here 27. Nevertheless, the key point is that TCR-driven TEM of CD8 TEM does not appear to be affected by reagents that affect degranulation or TEM via the LBRC, as the same reagents effectively inhibit CD4 TEM in experiments performed in parallel.
The exocytosis of granules appears to be a key difference between TCR-driven TEM of CD4 and CD8 TEM. It was recently shown that human memory CD4 T activated by antibodies for 24 h will actually secrete more granzyme B than CD8 T cells, even though the percentage of granzyme B-containing CD4 T cells is much lower and CD8 T cells contain more intracellular granzyme B per cell 28. While this study compared freshly synthesized granzyme B in CD4 T cells to predominantly pre-made granzyme B in CD8 T cells, it nevertheless indicates that exocytosis of granules is under stricter control in memory CD8 T cells. With regard to transmigrating T cells, strict control of exocytosis in CD8 TEM makes sense teleologically, since a relatively large number of CD8 TEM contain both granzymes A and B as well as perforin and would likely kill the EC if they were to degranulate during TEM. This would be a potentially disastrous consequence if CD8 TEM were being recruited to defend against a reinfection by an intracellular pathogen, the physiological role of this cell population. Interestingly, mature CTL will degranulate when encountering allogeneic EC, causing cell lysis 16,29 highlighting an important difference between CD8 CTL and CD8 TEM. The molecular explanation(s) for the differences in degranulation by CD4 and CD8 TEM are unknown but experiments to determine this difference are an area of active investigation. One potential explanation could be a difference in the strength and/or duration of TCR signaling; it was recently shown that TCR-induced P-ZAP70 in mouse CD8 T cells is completely dephosphorylated within 30 minutes 30. However, our assessment of the extent of ZAP70 phosphorylation suggests that the difference in degranulation between human CD4 and CD8 TEM interacting with antigen presented by EC cannot be explained by differences in the quantitative strength or duration of signaling.
It is interesting to speculate that the lack of degranulation of CD8 TEM during TCR-driven TEM and the lack of the need for interactions with proteins of the LBRC may be related. Other leukocyte cell types that utilize the LBRC for TEM are also dependent on serine proteases similar to granzyme A, e.g., elastase 31 or proteinase 3 32. Perhaps cleavage of an endothelial receptor by a serine protease is required to activate mobilization of the LBRC. With the exception of TCR-driven TEM by CD4 TEM, T cells appear to be independent of this mechanism. Perhaps LFA-1 engagement of EC ICAM-1 provides an alternative signal.
In contrast to our earlier results 11, we now saw that VCAM-1 blocking Abs can selectively inhibit TCR-driven TEM of CD4 TEM. We attribute this discrepancy to the differences among anti-VCAM-1 antibodies. The mAb used in our prior report had a lower affinity than the two mAbs used here. We believe the new finding to be correct both because we had similar effects by siRNA knock down of VCAM-1 in EC and with use of a VLA-4 blocking mAb.
Although not the main focus of this study, we also found that, unlike CD8 TEM, CR-driven TEM of CD4 TEM cells is not inhibited by ICAM-1 blocking antibodies. The lack of effect of ICAM-1 blockade could imply either that ICAM-1 plays no role or that it is redundant in TEM of CR-driven CD4 TEM. Curiously, LFA-1 blocking Abs did have a small inhibitory effect on CR-driven TEM by CD4 TEM cells. This effect of LFA-1 blocking Abs is consistent with the inhibitory effects of JAM-A knockdown, since JAM-A is an alternative ligand for LFA-1 33. Others have reported strong to modest effects of LFA-1 blocking Abs on TEM of freshly isolated human peripheral blood lymphocytes across cytokine-activated human umbilical vein endothelial cells in static assays, with or without a chemotactic gradient 34-36 and approximately 33% inhibition in flow assays with apically presented SDF-1α 18; in all cases, VLA-4 blocking Abs had no effect on their own, but the combination of VLA-4 and LFA-1 blocking Abs was consistently potent. However, SDF-1α recruitment is not restricted to the TEM subset and other T cell subsets may be more dependent upon LFA-1 interactions 19. Interestingly, effector T cells generated by TCR activation and prolonged culture in IL-2 are especially dependent upon LFA-1 for TEM 37.
Not surprisingly, both LFA-1 and VLA-4 blocking Abs inhibited adhesion of CD4 and CD8 TEM cells; a limited number of experiments indicated that using both LFA-1 and VLA-4 antibodies have an additive effect. A curious observation is that the contribution of TCR signals to adhesion is different between CD4 and CD8 TEM. As noted previously 38 and replicated here, TCR signaling enhances adhesion of CD4 TEM, i.e., the proportion of adherent T cells that are antigen specific is higher than the starting population. Such seems not to be the case for adhesion of CD8 TEM to EC, although an effect could be observed in the presence of integrin blocking mAbs.
The differences between CD4 and CD8 TEM detailed here has significant clinical implications, particularly with regard to T cell-mediated graft rejection, a process dependent on the response of host T cells to the vascular endothelium of the allograft 39. Blocking interactions of circulating TEM with proteins of the LBRC that might be effective in blocking recruitment of CD4 TEM may fail due to the independence of CD8 TEM recruitment of these target proteins. In contrast, agents that target common features, such as antibodies to LFA-1 or ICAM-1 can reduce TCR-driven TEM of both CD4 and CD8 TEM and, hence, inhibit graft rejection, but redundancy with VLA-4 (CD 49d/CD29) with VCAM-1 may still permit CR-induced recruitment. Experiments using humanized mice may help to address these questions as a complement to clinical trials.
Supplementary Material
Highlights.
TCR engagement of alloantigen presented by human EC triggers transendothelial migration (TEM) of effector memory CD8 T cells by a process that differs from TEM triggered by chemokine receptors.
While TCR-triggered TEM of human effector memory CD8 T cells resembles that occurring in human effector memory CD4 T cells in several respects, it differs in that CD8 T cells do not require granule exocytosis nor engagement of components of the lateral border recycling compartment to undergo TEM.
Differences in effector memory T cell between TEM triggered by antigen recognition or by chemokines and between CD8 and CD4 T cells suggest that multiple therapeutic approaches will be required to effectively inhibit T cell recruitment to human allografts.
Acknowledgments
We thank Louise Camera-Benson for generating HDMEC and Rita Palmarozza for supplying PBMC. We also thank Dr. Gillian Griffiths, University of Cambridge, for helpful discussions.
Funding
This work was supported by National Institutes of Health Grant R01-HL051014.
Abbreviations List
- CMA
concanamycin A
- CR
chemokine receptor
- EC
endothelial cell
- LBRC
lateral border recycling compartment
- MTOC
microtubule organizing center
- TCR
T cell receptor
- TEM
effector memory T cells
- TEM
transendothelial migration
- TEP
transendothelial protrusion
- TSST-1
toxic shock syndrome toxin 1
Footnotes
Competing Financial Interests
The authors have no conflicting financial interests.
References
- 1.Heeger PS, Greenspan NS, Kuhlenschmidt S, et al. Pretransplant frequency of donor-specific, IFN-gamma-producing lymphocytes is a manifestation of immunologic memory and correlates with the risk of posttransplant rejection episodes. Journal of immunology. 1999;163:2267–2275. [PubMed] [Google Scholar]
- 2.Lakkis FG, Lechler RI. Origin and biology of the allogeneic response. Cold Spring Harbor perspectives in medicine. 2013;3:1–11. doi: 10.1101/cshperspect.a014993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brewer Y, Palmer A, Taube D, et al. Effect of graft perfusion with two CD45 monoclonal antibodies on incidence of kidney allograft rejection. Lancet. 1989;2:935–937. doi: 10.1016/s0140-6736(89)90951-3. [DOI] [PubMed] [Google Scholar]
- 4.Shiao SL, Kirkiles-Smith NC, Shepherd BR, McNiff JM, Carr EJ, Pober JS. Human effector memory CD4+ T cells directly recognize allogeneic endothelial cells in vitro and in vivo. Journal of immunology. 2007;179:4397–4404. doi: 10.4049/jimmunol.179.7.4397. [DOI] [PubMed] [Google Scholar]
- 5.Chalasani G, Dai Z, Konieczny BT, Baddoura FK, Lakkis FG. Recall and propagation of allospecific memory T cells independent of secondary lymphoid organs. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:6175–6180. doi: 10.1073/pnas.092596999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Page AJ, Ford ML, Kirk AD. Memory T-cell-specific therapeutics in organ transplantation. Current opinion in organ transplantation. 2009;14:643–649. doi: 10.1097/MOT.0b013e328332bd4a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pearl JP, Parris J, Hale DA, et al. Immunocompetent T-cells with a memory-like phenotype are the dominant cell type following antibody-mediated T-cell depletion. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2005;5:465–474. doi: 10.1111/j.1600-6143.2005.00759.x. [DOI] [PubMed] [Google Scholar]
- 8.Shiao SL, McNiff JM, Pober JS. Memory T cells and their costimulators in human allograft injury. Journal of immunology. 2005;175:4886–4896. doi: 10.4049/jimmunol.175.8.4886. [DOI] [PubMed] [Google Scholar]
- 9.Schechner JS, Nath AK, Zheng L, et al. In vivo formation of complex microvessels lined by human endothelial cells in an immunodeficient mouse. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:9191–9196. doi: 10.1073/pnas.150242297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abrahimi P, Qin L, Chang WG, et al. Blocking MHC class II on human endothelium mitigates acute rejection. JCI Insight. 2016;1:1–16. doi: 10.1172/jci.insight.85293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manes TD, Pober JS. Antigen presentation by human microvascular endothelial cells triggers ICAM-1-dependent transendothelial protrusion by, and fractalkine-dependent transendothelial migration of, effector memory CD4+ T cells. Journal of immunology. 2008;180:8386–8392. doi: 10.4049/jimmunol.180.12.8386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Manes TD, Pober JS. Polarized granzyme release is required for antigen-driven transendothelial migration of human effector memory CD4 T cells. Journal of immunology. 2014;193:5809–5815. doi: 10.4049/jimmunol.1401665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manes TD, Pober JS. TCR-driven transendothelial migration of human effector memory CD4 T cells involves Vav, Rac, and myosin IIA. Journal of immunology. 2013;190:3079–3088. doi: 10.4049/jimmunol.1201817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Manes TD, Pober JS. Identification of endothelial cell junctional proteins and lymphocyte receptors involved in transendothelial migration of human effector memory CD4+ T cells. Journal of immunology. 2011;186:1763–1768. doi: 10.4049/jimmunol.1002835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Manes TD, Hoer S, Muller WA, Lehner PJ, Pober JS. Kaposi's sarcoma-associated herpesvirus K3 and K5 proteins block distinct steps in transendothelial migration of effector memory CD4+ T cells by targeting different endothelial proteins. Journal of immunology. 2010;184:5186–5192. doi: 10.4049/jimmunol.0902938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dengler TJ, Pober JS. Human vascular endothelial cells stimulate memory but not naive CD8+ T cells to differentiate into CTL retaining an early activation phenotype. Journal of immunology. 2000;164:5146–5155. doi: 10.4049/jimmunol.164.10.5146. [DOI] [PubMed] [Google Scholar]
- 17.Walch JM, Zeng Q, Li Q, et al. Cognate antigen directs CD8+ T cell migration to vascularized transplants. The Journal of clinical investigation. 2013;123:2663–2671. doi: 10.1172/JCI66722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cinamon G, Shinder V, Alon R. Shear forces promote lymphocyte migration across vascular endothelium bearing apical chemokines. Nature immunology. 2001;2:515–522. doi: 10.1038/88710. [DOI] [PubMed] [Google Scholar]
- 19.Manes TD, Pober JS, Kluger MS. Endothelial cell-T lymphocyte interactions: IP[corrected]-10 stimulates rapid transendothelial migration of human effector but not central memory CD4+ T cells. Requirements for shear stress and adhesion molecules. Transplantation. 2006;82:S9–14. doi: 10.1097/01.tp.0000231356.57576.82. [DOI] [PubMed] [Google Scholar]
- 20.Peters PJ, Borst J, Oorschot V, et al. Cytotoxic T lymphocyte granules are secretory lysosomes, containing both perforin and granzymes. The Journal of experimental medicine. 1991;173:1099–1109. doi: 10.1084/jem.173.5.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grossman WJ, Verbsky JW, Tollefsen BL, Kemper C, Atkinson JP, Ley TJ. Differential expression of granzymes A and B in human cytotoxic lymphocyte subsets and T regulatory cells. Blood. 2004;104:2840–2848. doi: 10.1182/blood-2004-03-0859. [DOI] [PubMed] [Google Scholar]
- 22.Bratke K, Kuepper M, Bade B, Virchow JC, Jr., Luttmann W. Differential expression of human granzymes A, B, and K in natural killer cells and during CD8+ T cell differentiation in peripheral blood. Eur J Immunol. 2005;35:2608–2616. doi: 10.1002/eji.200526122. [DOI] [PubMed] [Google Scholar]
- 23.Bade B, Boettcher HE, Lohrmann J, et al. Differential expression of the granzymes A, K and M and perforin in human peripheral blood lymphocytes. Int Immunol. 2005;17:1419–1428. doi: 10.1093/intimm/dxh320. [DOI] [PubMed] [Google Scholar]
- 24.Sullivan DP, Muller WA. Neutrophil and monocyte recruitment by PECAM, CD99, and other molecules via the LBRC. Semin Immunopathol. 2014;36:193–209. doi: 10.1007/s00281-013-0412-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watson RL, Buck J, Levin LR, et al. Endothelial CD99 signals through soluble adenylyl cyclase and PKA to regulate leukocyte transendothelial migration. The Journal of experimental medicine. 2015;212:1021–1041. doi: 10.1084/jem.20150354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prakash MD, Munoz MA, Jain R, et al. Granzyme B promotes cytotoxic lymphocyte transmigration via basement membrane remodeling. Immunity. 2014;41:960–972. doi: 10.1016/j.immuni.2014.11.012. [DOI] [PubMed] [Google Scholar]
- 27.Cheng Y, Wong MT, van der Maaten L, Newell EW. Categorical Analysis of Human T Cell Heterogeneity with One-Dimensional Soli-Expression by Nonlinear Stochastic Embedding. Journal of immunology. 2016;196:924–932. doi: 10.4049/jimmunol.1501928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin L, Couturier J, Yu X, Medina MA, Kozinetz CA, Lewis DE. Granzyme B secretion by human memory CD4 T cells is less strictly regulated compared to memory CD8 T cells. BMC Immunol. 2014;15:36. doi: 10.1186/s12865-014-0036-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Biedermann BC, Pober JS. Human endothelial cells induce and regulate cytolytic T cell differentiation. Journal of immunology. 1998;161:4679–4687. [PubMed] [Google Scholar]
- 30.Yang M, Chen T, Li X, et al. K33-linked polyubiquitination of Zap70 by Nrdp1 controls CD8(+) T cell activation. Nature immunology. 2015;16:1253–1262. doi: 10.1038/ni.3258. [DOI] [PubMed] [Google Scholar]
- 31.Cepinskas G, Sandig M, Kvietys PR. PAF-induced elastase-dependent neutrophil transendothelial migration is associated with the mobilization of elastase to the neutrophil surface and localization to the migrating front. J Cell Sci. 1999;112:1937–1945. doi: 10.1242/jcs.112.12.1937. [DOI] [PubMed] [Google Scholar]
- 32.Kuckleburg CJ, Tilkens SB, Santoso S, Newman PJ. Proteinase 3 contributes to transendothelial migration of NB1-positive neutrophils. Journal of immunology. 2012;188:2419–2426. doi: 10.4049/jimmunol.1102540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ostermann G, Weber KS, Zernecke A, Schroder A, Weber C. JAM-1 is a ligand of the beta(2) integrin LFA-1 involved in transendothelial migration of leukocytes. Nature immunology. 2002;3:151–158. doi: 10.1038/ni755. [DOI] [PubMed] [Google Scholar]
- 34.Van Epps DE, Potter J, Vachula M, Smith CW, Anderson DC. Suppression of human lymphocyte chemotaxis and transendothelial migration by anti-LFA-1 antibody. Journal of immunology. 1989;143:3207–3210. [PubMed] [Google Scholar]
- 35.Oppenheimer-Marks N, Davis LS, Bogue DT, Ramberg J, Lipsky PE. Differential utilization of ICAM-1 and VCAM-1 during the adhesion and transendothelial migration of human T lymphocytes. Journal of immunology. 1991;147:2913–2921. [PubMed] [Google Scholar]
- 36.Ding Z, Xiong K, Issekutz TB. Chemokines stimulate human T lymphocyte transendothelial migration to utilize VLA-4 in addition to LFA-1. Journal of leukocyte biology. 2001;69:458–466. [PubMed] [Google Scholar]
- 37.Shulman Z, Cohen SJ, Roediger B, et al. Transendothelial migration of lymphocytes mediated by intraendothelial vesicle stores rather than by extracellular chemokine depots. Nature immunology. 2012;13:67–76. doi: 10.1038/ni.2173. [DOI] [PubMed] [Google Scholar]
- 38.Manes TD, Shiao SL, Dengler TJ, Pober JS. TCR signaling antagonizes rapid IP-10-mediated transendothelial migration of effector memory CD4+ T cells. Journal of immunology. 2007;178:3237–3243. doi: 10.4049/jimmunol.178.5.3237. [DOI] [PubMed] [Google Scholar]
- 39.Abrahimi P, Liu R, Pober JS. Blood Vessels in Allotransplantation. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2015;15:1748–1754. doi: 10.1111/ajt.13242. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.