

Abstract
Background
Hepatocellular carcinoma (HCC) is the third leading cause of cancer-related deaths. The average survival and 5-year survival rates of HCC patients still remains poor. Thus, there is an urgent need to better understand the mechanisms of cancer progression in HCC and to identify useful biomarkers to predict prognosis.
Methods
Public data portals including Oncomine, The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) profiles were used to retrieve the HCC-related microarrays and to identify potential genes contributed to cancer progression. Bioinformatics analyses including pathway enrichment, protein/gene interaction and text mining were used to explain the potential roles of the identified genes in HCC. Quantitative real-time polymerase chain reaction analysis and Western blotting were used to measure the expression of the targets. The data were analysed by SPSS 20.0 software.
Results
We identified 80 genes that were significantly dysregulated in HCC according to four independent microarrays covering 386 cases of HCC and 327 normal liver tissues. Twenty genes were consistently and stably dysregulated in the four microarrays by at least 2-fold and detection of gene expression by RT-qPCR and western blotting showed consistent expression profiles in 11 HCC tissues compared with corresponding paracancerous tissues. Eleven of these 20 genes were associated with disease-free survival (DFS) or overall survival (OS) in a cohort of 157 HCC patients, and eight genes were associated with tumour pathologic PT, tumour stage or vital status. Potential roles of those 20 genes in regulation of HCC progression were predicted, primarily in association with metastasis. INTS8 was specifically correlated with most clinical characteristics including DFS, OS, stage, metastasis, invasiveness, diagnosis, and age.
Conclusion
The significantly dysregulated genes identified in this study were associated with cancer progression and prognosis in HCC, and might be potential therapeutic targets for HCC treatment or potential biomarkers for diagnosis and prognosis.
Electronic supplementary material
The online version of this article (doi:10.1186/s13046-016-0403-2) contains supplementary material, which is available to authorized users.
Keywords: Hepatocellular carcinoma, Microarray, Progression, Prognosis
Background
Hepatocellular carcinoma (HCC) is the third leading cause of cancer-related deaths [1]. There are 750,000 new cases of HCC and nearly 700,000 deaths each year, making this a particularly lethal form of cancer [2]. Over the past decade major progress has been made in our understanding of the risk factors and molecular pathways driving liver carcinogenesis, and these advances have led to substantial opportunities for HCC prevention, surveillance, early diagnosis, prediction of prognosis, and therapy [1]. However, the average survival of HCC patients is normally between 6 and 20 months [3], and long-term prognosis is poor with reported 5-year survival rates ranging from 17 to 53 % [4]. Thus, there is an urgent need to better understand the mechanism of cancer progression and development in HCC and to identify useful biomarkers for diagnosis and prognosis.
High-throughput profiling technologies such as microarrays and, more recently, next-generation sequencing have become invaluable tools for biomedical research, and large amounts of data generated by those tools, including mRNA expression, DNA methylation, and microRNA expression, are collected in public archives such as the major public projects The Cancer Genome Atlas (TCGA) [5] and the International Cancer Genome Consortium [6], and the most prominent primary data archives, ArrayExpress [7], Gene Expression Omnibus (GEO) [8], Oncomine [9] and the databases of the International Nucleotide Sequence Database Collaboration [10]. The wide range of those databases, the various ways in which publicly archived gene expression data are being used in support of new studies, and reuse of these public data can be very powerful [11]. In particular, reusing of the data has the potential to predict treatment response and disease progression and was advantageous to develop precision therapies [12]. For example, based on data retrieved from Oncomine, TCGA, and GEO, Liu et al. identified several genes associated with ovarian cancer progression [13] and drug resistance [14]. In a similar manner, we identified that upregulation of E2F transcription factor 3 is associated with poor prognosis in HCC [15]. In the present study, using data of mRNA expression, DNA methylation, and clinical data retrieved from Oncomine, GEO, and the TCGA cohort, we identified a group of genes associated with cancer progression and prognosis in HCC.
Methods
Samples
All patients who underwent curative hepatectomy for primary HCC at the First Affiliated Hospital of Guangxi Medical University between March 2015 and September 2015 were eligible for inclusion in this study. Total of 11 HCCs and the matched paracancerous tissues were collected during surgery and stored in a liquid nitrogen tank until use for mRNA isolation and protein extraction. The study was endorsed by the Ethics Committee of Guangxi Medical University and was performed according to the Declaration of Helsinki, 2013 edition. All patients received an explanation of the aims of the study and signed informed consent.
mRNA isolation and quantitative real-time polymerase chain reaction (RT-qPCR) analysis
Total RNA from 11 HCC and their matched paracancerous tissues was isolated using a miRNeasy Mini Kit (Qiagen, Hilden, Germany). RNA was quantified by spectrophotometry on a NanoDrop 2000 (Thermo Scientific, DE, USA). A total of 2 μg RNA was subjected to cDNA synthesis using the miScript II RT Kit (Qiagen, Hilden, Germany). RT-qPCR was performed with the QuantiFast SYBR Green PCR Kit (Qiagen, Hilden, Germany). Data were collected with the StepOnePlus Real-Time PCR System (ABI, CA, USA) according to the manufacturer’s instructions. The gene expression was compared in each HCC sample and the matched paracancerous tissue, and then the homogeneity of variance in all samples was analysed using the t-test. The RT-qPCR gene-specific primers were as follows: TBCE: forward primer, 5′-AGGCCAACAGATGTTCTCCAG-3′, reverse primer, 5′-CAGGGGGTTTCTTAGGCAGG-3′; INTS8: forward primer, 5′-AACTGAGAGTTCTACTGCTGGA-3′, reverse primer, 5′-GCTGCGCCCAAATCATAGC-3′; VIPR1: forward primer, 5′-TGCTGGGACACCATCAACTC-3′, reverse primer, 5′-TTGTCCGGAAAGAAGGCGAA-3′; CLEC4M: forward primer, 5′-TACTTCATGTCTAACTCCCAGCG-3′, reverse primer, 5′-GCTCCTCAGCAGTTTTGATTACG-3′; MARCO: forward primer, 5′-GGGGACACAGGACTTCAAGG-3′, reverse primer, 5′-CCCTGTTCTCCCTTCACACC-3′; DNASE1L3: forward primer, 5′-AGCCCTTTGTGGTCTGGTTC-3′, reverse primer, 5′-CGTCCGTGTAGACCTCAACC-3′; CRHBP: forward primer, 5′-AAATCCTCAGCAGGTTGCGA-3′, reverse primer, 5′-AAGGCGTCATCTTGGAAGGG-3′; FCN2: forward primer, 5′-CTGCAAGGACCTGCTAGACC-3′, reverse primer, 5′-TGTCATTCCCCAGCCAGAAC-3′; GAPDH (used as the control): forward primer, 5′-GAAGGTGAAGGTCGGAGT-3′, reverse primer, 5′-GAAGATGGTGATGGGATTT-3′.
Protein extraction and western blotting
Total protein was extracted from HCC and paracancerous tissues with RIPA lysis buffer (Solarbio, Beijing, China) and proteinconcentration was determined using an Enhanced BCA Protein Quantification Kit (KeyGEN BioTECH, Jiangsu, China). Then the samples were separated by Novex NuPAGE SDS-PAGE Gel System (Thermo Fisher Scientific, MA, USA) and were transferred to the PVDF membrane using the Bio-Rad Criterion System (Bio-Rad, CA, USA). Membranes were blocked with 8 % non-fat dry milk in PBS containing 0.1 % Tween-20 (0.1 % TBST, pH7.4) for 1 h. Membranes were incubated with antibodies specific for human INTS8 (rabbit polyclonal antibody, 1:750 dilutions; Proteintech, Hubei, China) and GAPDH (rabbit polyclonal antibody, 1:1,000 dilution; Boster, Hubei, China) overnight at 4 °C. After 3 washings with 0.1 % TBST for 5 min, horseradish peroxidase-conjugated goat anti-rabbit secondary antibodies (1:5,000 dilution; Bioss, Beijing, China) were applied, followed by washings with 0.1 % TBST for 5 min each at room temperature (RT). The bound immunocomplexes were detected using ECL+ reagent (GE Healthcare Bio-Sciences, NJ, USA) with a FluorChem M system (Proteinsimple, CA, USA).
Gene expression profiles
The genes significantly dysregulated in HCC were identified based on the 4 microarrays, Chen Liver microarray (104 HCCs vs. 76 liver tissues), Roessler Liver microarray (22 HCCs vs. 21 liver tissues), Roessler Liver 2 microarray (225 HCCs vs. 220 liver tissues) and Wurmbach Liver microarray (35 HCCs vs. 10 liver tissues), which are all deposited in Oncomine database (https://www.oncomine.org/resource/login.html) [9]. The 4 microarrays together covering total of 386 cases of HCCs and 327 cases of normal liver tissues. The rank for a gene is the median rank for that gene across each of the analyses. DNA methylation, mRNA expression, and clinical data of 379 HCC patients in a TCGA cohort were retrieved from cBioPortal for Cancer Genomics (http://cbioportal.org) [16, 17], but only 157 samples with matched gene expression data, prognosis data and most of the other clinical data were used to analyze the clinical importance of the target genes. mRNA expression data associated with HCC metastasis were retrieved from microarray GDS3091 [18] and GDS274 [19], which were deposited in the GEO profiles databases (http://www.ncbi.nlm.nih.gov/geoprofiles/) [8].
Bioinformatics analyses
Enrichment of the biological process and cellular component of a group of genes was determined using the DAVID online tool (http://david.abcc.ncifcrf.gov/) [20, 21]. Protein/gene-protein/gene interaction analysis was performed using the GeneMANIA online tool (http://www.genemania.org/) [22, 23]. Function prediction based on text mining was performed using the Coremine Medical online database (http://www.coremine.com/medical/) [24].
Data analysis
The data were analysed by SPSS 20.0 software. The mRNA expression of a gene is presented as the mean ± SD. Homogeneity of variance was analysed using the t-test. Expression values of a gene were dichotomised into high and low expression using the median as a cutoff for analysis of clinical importance in a TCGA cohort, as described in a previous study [25]. The probability of survival and its significance was calculated using the Kaplan-Meier method and log-rank test, respectively. A Cox proportional hazard model was performed for multivariate analysis of prognosis. The correlation between gene expression and clinicopathologic characteristics was evaluated by Pearson’s χ2 test (two-sided). The correlation between DNA methylation and gene expression was analysed using bivariate correlations. P values < 0.05 were considered to indicate statistically significant differences.
Results
Retrieval of significantly dysregulated genes in HCC
Four independent microarrays deposited in the Oncomine database were selected to identify genes associated with cancer development and progression in HCC. These microarrays were Chen Liver Statistics covering 104 cases of HCC and 76 cases of liver tissue, Roessler Liver Statistics covering 22 cases of HCC and 21 cases of liver tissue, Roessler Liver 2 Statistics covering 225 cases of HCC and 220 cases of liver tissue, and Wurmbach Liver Statistics covering 35 cases of HCC and 10 cases of liver tissues. Based on analysis of these four independent microarrays, 40 genes that were significantly upregulated (P < 1.36E-10) and 40 genes that were significantly downregulated (P < 1.31E-10) in HCC were retrieved (Fig. 1). Analysis of the 80 genes by the DAVID online tool indicated that cell cycle was the top biological process, covering 17 genes, and microtubule cytoskeleton was the top cellular component, covering 14 genes (Additional file 1: Table S1).
Fig. 1.
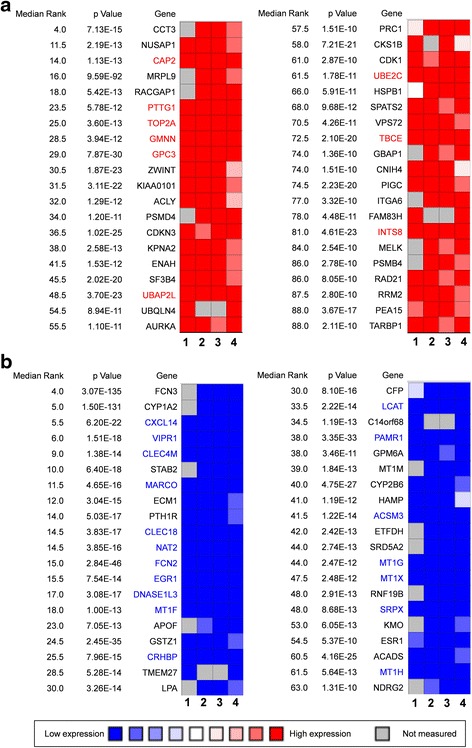
The 80 genes that were significantly dysregulated in hepatocellular carcinomas according to four independent microarrays retrieved from the Oncomine database. a The top 40 genes that were significantly upregulated in four microarrays. b The top 40 genes that were significantly downregulated in four microarrays. The four microarrays cover a total of 386 cases of hepatocellular carcinomas and 327 cases of normal liver tissue: (1) Chen Liver Statistics, 104 cases of hepatocellular carcinoma and 76 cases of liver tissue; (2) Roessler Liver Statistics, 22 cases of hepatocellular carcinoma and 21 cases of liver tissue; (3) Roessler Liver 2 Statistics, 225 cases of hepatocellular carcinoma and 220 cases of liver tissue; (4) Wurmbach Liver Statistics, 35 cases of hepatocellular carcinoma and 10 cases of liver tissue. The rank for a gene is the median rank for that gene across each of the analyses. The P value given for a gene is for the median-ranked analysis. The genes labelled in red and in blue were significantly and consistently up- and downregulated in the four microarrays, respectively
Among the 80 genes that were dysregulated in HCCs according to four independent microarrays covering a total of 386 cases of HCC and 327 cases of normal liver tissues, nine genes (CAP2, PTTG1, TOP2A, GMNN, GPC3, UBE2C, UBAP2L, TBCE, and INTS8) were consistently and stably upregulated and 18 genes (CXCL14, VIPR1, CLEC4M, MARCO, CLEC1B, NAT2, FCN2, EGR1, DNASE1L3, MT1F, CRHBP, LCAT, PAMR1, ACSM3, MT1G, MT1X, SRPX, and MT1H) were consistently and stably downregulated in HCC, by least 2-fold (Fig. 1; Table 1). Among the above 27 genes, seven genes—CAP2, GMNN, PTTG1, TBCE, TOP2A, UBE2C, and FCN2—encode proteins associated with cell cycle and microtubule cytoskeleton (Additional file 1: Table S1). Protein/gene-protein/gene interaction analysis was performed to further explain the interrelationships of these genes in HCC. As shown in Additional file 2: Figure S2, the 27 proteins/genes directly/indirectly interacted with each other via co-localisation, genetic interactions, shared common pathways, and protein domains, and, in particular, co-expression, and 10 of them—VIPR1, DNASE1L3, SRPX, MT1H, CXCL14, CLEC4M, CRHBP, GPC3, NAT2, and MARCO—interacted with at least 14 other genes, more than half of all the genes in the interaction network (Additional file 2: Figure S2). Moreover, these genes were also those that were dysregulated at least 4-fold in HCC (Table 1).
Table 1.
Genes that were stably and consistently dysregulated in 386 cases of hepatocellular carcinoma compared with 327 cases of normal liver tissues according to four independent microarrays retrieved from the Oncomine database, and their associations with hepatocellular carcinoma
| Gene | Independent microarray data (Fold change) | No. of articlesa | Associations with hepatocellular carcinoma | ||||
|---|---|---|---|---|---|---|---|
| Direction of regulation | Chen Liver | Roessler Liver | Roessler Liver 2 | Wurmbach Liver | |||
| TBCE b | Up | 2.125 | 2.403 | 2.822 | 2.419 | - | - |
| INTS8 b | Up | 2.393 | 3.102 | 2.340 | 2.115 | - | - |
| UBAP2L b | Up | 2.108 | 2.959 | 2.819 | 2.742 | - | - |
| GMNN b | Up | 3.362 | 7.340 | 4.696 | 3.394 | 1 | Potential oncogene [38] |
| UBE2C | Up | 4.733 | 3.661 | 3.422 | 5.113 | 4 | Cancer progression and poor prognosis [39] |
| PTTG1 | Up | 4.688 | 4.741 | 5.773 | 10.622 | 9 | Angiogenesis, progression, and poor prognosis [40, 41], therapeutic target [42] |
| CAP2 | Up | 3.526 | 4.254 | 5.790 | 8.569 | 10 | Multistage hepatocarcinogenesis [43], early detection [44] |
| TOP2A | Up | 2.663 | 11.236 | 8.292 | 13.321 | 11 | Early age onset, shorter patient survival and chemoresistance [45] |
| GPC3 | Up | 16.826 | 26.693 | 28.236 | 76.162 | 199 | Diagnosis [29], cell proliferation and invasion [28]; prediction of recurrence [30] |
| VIPR1 b | Down | 9.979 | 5.310 | 7.202 | 4.855 | - | - |
| CLEC4M b | Down | 28.107 | 9.276 | 4.361 | 36.431 | - | - |
| MARCO b | Down | 11.333 | 6.107 | 3.984 | 20.154 | - | - |
| DNASE1L3 b | Down | 8.386 | 12.378 | 7.653 | 10.303 | - | - |
| PAMR1 b | Down | 2.726 | 2.381 | 2.473 | 2.917 | - | - |
| ACSM3 b | Down | 2.902 | 6.135 | 4.836 | 11.262 | - | - |
| CLEC1B b | Down | 6.600 | 6.605 | 4.748 | 36.770 | 1 | Downregulated in a cohort of 65 pairs of human HCCs [46] |
| MT1F b | Down | 14.107 | 18.140 | 15.749 | 9.680 | 1 | Inhibition of cancer growth [47] |
| CRHBP b | Down | 16.565 | 7.020 | 4.822 | 46.837 | 1 | Downregulated in a cohort of 65 pairs of human HCCs [46] |
| LCAT b | Down | 4.917 | 8.507 | 8.064 | 7.435 | 1 | LCAT activity correlated with serum albumin and serum bilirubin level [48] |
| MT1X b | Down | 10.812 | 11.558 | 8.227 | 6.903 | 1 | HCC-related [49] |
| SRPX b | Down | 4.929 | 5.104 | 5.879 | 7.202 | 1 | Proliferation, migration and invasiveness [50] |
| MT1H b | Down | 13.846 | 9.037 | 8.473 | 7.723 | 1 | Potential tumour suppressor [51] |
| FCN2 b | Down | 10.881 | 9.089 | 6.299 | 44.688 | 2 | HBV- and HCV-related HCC [52], FCN2 haplotypes associate with HCC [53] |
| CXCL14 b | Down | 12.903 | 9.667 | 10.940 | 13.977 | 4 | Potential diagnostic marker [54]; rs2237062 polymorphism influences HBV-related HCC progression [52, 55]; potential tumour suppressor [56] |
| MT1G b | Down | 13.065 | 11.134 | 11.160 | 11.187 | 4 | Tumour suppressor gene [51, 57], biomarker [58] |
| EGR1 | Down | 3.541 | 10.547 | 6.769 | 9.241 | 12 | Critical for hepatocarcinogenesis [59] |
| NAT2 | Down | 8.024 | 16.088 | 13.999 | 36.890 | 14 | NAT2 polymorphism is risk factor for developing HCC [60], NAT2 activity is critical in smoking-related hepatocarcinogenesis [61] |
aNo. of articles was based on a search in the PubMed database
bpoorly studied genes in HCC
Measurement of gene expression at mRNA and protein level
Among the 27 genes, the associations of seven with HCC are relatively well studied and described in published papers. However, the relationship of the remaining 20 genes with HCC was poorly understood, and these genes were selected for further analyses (Table 1). The expression of eight genes that were randomly selected from the 20 genes was measured by RT-qPCR in 11 tissues of HCC patients compared with matched paracancerous tissues. As shown in Fig. 2a, the expression of TBCE and INTS8 was increased, whereas that of VIPR1, CLEC4M, MARCO, DNASE1L3, CRHBP, and FCN2 was decreased in HCC tissues, although the changes in TBCE and VIPR1 expression were not statistically significant. Compared with the average expression in paracancerous tissues, the expression of INTS8 in HCC was upregulated with 2.06-fold and the expression of CLEC4M, MARCO, DNASE1L3, CRHBP, and FCN2 was downregulated with 3.83-, 5.70-, 5.63-, 3.87-, and 8.94-fold, respectively. All results of gene expression determined by RT-qPCR were completely consistent with their expression identified by the four independent microarrays (Fig. 1; Table 1). Furthermore, a significant increase at the protein level of INTS8 was observed in HCC tissues compared with corresponding paracancerous tissues (Fig. 2b), which was consistent with its expression at the mRNA level.
Fig. 2.
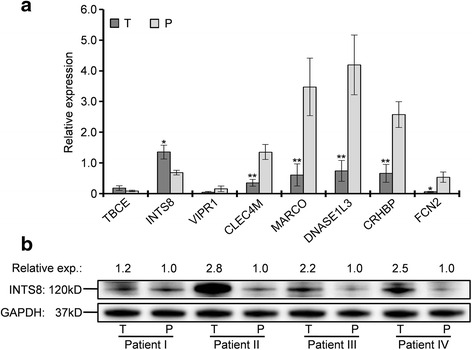
Measurement of gene expression at mRNA and protein level. a mRNA expression of genes in 11 tissues of HCC patients compared with matched paracancerous tissue. * P < 0.05; ** P < 0.01. b Protein expression of INTS8 in four tissues of HCC patients compared with expression in corresponding paracancerous tissues. The intensity of protein bands was measured by Image J software.. T, HCC tissue; P, paracancerous tissue
Analysis of clinical importance
The clinical importance in HCC of the 20 selected genes (Table 1) was evaluated on the basis of TCGA clinical data. A total of 379 HCC patient samples with clinical data in a cohort of TCGA were retrieved. Among these, 157 samples with mRNA expression values were selected for analysis of the relationship between genes and clinical characteristics. The expression values of a gene were categorised as high or low according to the median value in accordance with a previous study [25].
A total of 11 genes were associated with DFS and/or OS (Table 2); among those, low expression of ACSM3 and CXCL14 was associated with poor DFS, and low expression of CRHBP, DNASE1L3, FCN2, MT1X, and VIPR1 was associated with poor OS (Fig. 3, Table 2). Four genes were associated with both DFS and OS: high expression of INTS8 in HCC patients, and low expression of LCAT, MARCO, and PAMR1, was associated with poor DFS and OS (Fig. 4, Table 2). To elucidate whether any of the above genes was an independent factor for predicting patient survival, we performed multivariate analyses of tumour stage, tumour pathologic PT, tumour residual, tumour status, vital status, age, gender, and the 11 genes by a Cox proportional hazards model (Table 3). We found that stage (P = 0.050), tumour status (P = 0.001), DNASE1L3 expression (P = 0.042), and INTS8 expression (P = 0.023) were independent risk prognostic factors for OS in HCC patients, although no gene was found to be an independent prognostic factor for DFS (data not shown).
Table 2.
The associations of 11 genes with disease-free survival (DFS) and/or overall survival (OS) of patients with hepatocellular carcinoma in a TCGA cohort, analysed using Kaplan-Meier survival plots
| DFS (Median) | OS (Median) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 95 % Confidence Interval | 95 % Confidence Interval | ||||||||
| Estimate | Std. Error | Lower Boundary | Upper Boundary | Estimate | Std. Error | Lower Boundary | Upper Boundary | ||
| ACSM3 | H | 24.800 | 7.587 | 9.930 | 39.670 | ||||
| L | 14.400 | 2.391 | 9.714 | 19.086 | |||||
| Overall | 19.300 | 3.428 | 12.581 | 26.019 | |||||
| CXCL14 | H | 29.300 | 10.355 | 9.004 | 49.596 | ||||
| L | 16.400 | 2.222 | 12.045 | 20.755 | |||||
| Overall | 19.300 | 3.428 | 12.581 | 26.019 | |||||
| INTS8 | H | 14.400 | 2.408 | 9.681 | 19.119 | 21.700 | 5.048 | 11.805 | 31.595 |
| L | 27.200 | 4.266 | 18.839 | 35.561 | 53.300 | 10.531 | 32.659 | 73.941 | |
| Overall | 19.300 | 3.428 | 12.581 | 26.019 | 37.800 | 8.792 | 20.568 | 55.032 | |
| LCAT | H | 29.300 | 5.287 | 18.937 | 39.663 | 55.600 | 13.029 | 30.063 | 81.137 |
| L | 14.800 | 2.129 | 10.628 | 18.972 | 21.700 | 5.133 | 11.640 | 31.760 | |
| Overall | 19.300 | 3.428 | 12.581 | 26.019 | 37.800 | 8.792 | 20.568 | 55.032 | |
| MARCO | H | 24.800 | 6.094 | 12.856 | 36.744 | 53.300 | 16.525 | 20.911 | 85.689 |
| L | 15.600 | 1.710 | 12.248 | 18.952 | 23.300 | 5.664 | 12.199 | 34.401 | |
| Overall | 19.300 | 3.428 | 12.581 | 26.019 | 37.800 | 8.792 | 20.568 | 55.032 | |
| PAMR1 | H | 29.300 | 7.881 | 13.853 | 44.747 | 69.500 | 7.445 | 54.908 | 84.092 |
| L | 16.400 | 3.616 | 9.312 | 23.488 | 21.100 | 1.762 | 17.647 | 24.553 | |
| Overall | 19.300 | 3.428 | 12.581 | 26.019 | 37.800 | 8.792 | 20.568 | 55.032 | |
| CRHBP | H | 55.600 | 13.080 | 29.964 | 81.236 | ||||
| L | 27.500 | 7.523 | 12.754 | 42.246 | |||||
| Overall | 37.800 | 8.792 | 20.568 | 55.032 | |||||
| DNASE1L3 | H | 55.600 | 6.310 | 43.232 | 67.968 | ||||
| L | 23.300 | 5.103 | 13.298 | 33.302 | |||||
| Overall | 37.800 | 8.792 | 20.568 | 55.032 | |||||
| FCN2 | H | 53.300 | 12.677 | 28.453 | 78.147 | ||||
| L | 30.600 | 10.341 | 10.331 | 50.869 | |||||
| Overall | 37.800 | 8.792 | 20.568 | 55.032 | |||||
| MT1X | H | 58.800 | 12.301 | 34.690 | 82.910 | ||||
| L | 23.300 | 5.997 | 11.546 | 35.054 | |||||
| Overall | 37.800 | 8.792 | 20.568 | 55.032 | |||||
| VIPR1 | H | 51.300 | 7.615 | 36.374 | 66.226 | ||||
| L | 20.600 | 5.643 | 9.540 | 31.660 | |||||
| Overall | 37.800 | 8.792 | 20.568 | 55.032 | |||||
The gene expression and survival data of 157 HCC patients in a TCGA cohort were used for the analysis. Expression values of a gene were dichotomised into high and low expression using the median as a cutoff
H high expression, L low expression
Fig. 3.
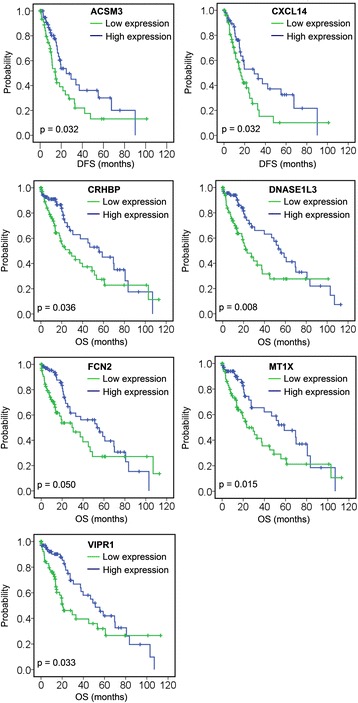
Association of seven genes (ACSM3, CXCL14, CRHBP, DNASE1L3, FCN2, MT1X, and VIPR1) with DFS or OS, analysed using Kaplan-Meier survival plots. The survival data of 157 HCC patients in a TCGA cohort were used for the analysis. Expression values of a gene were dichotomised into high expression (blue line) and low expression (green line) using the median as a cutoff
Fig. 4.
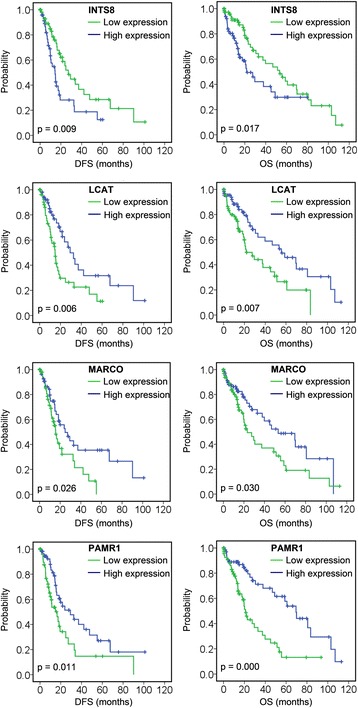
Association of INTS8, LCAT, MARCO, and PAMR1 with DFS and OS, analysed using Kaplan-Meier survival plots. The survival data of 157 HCC patients in a TCGA cohort were used for the analysis. Expression values of a gene were dichotomised into high expression (blue line) and low expression (green line) using the median as a cutoff
Table 3.
Multivariate analysis of prognosis of 157 HCC patients in a TCGA cohort using Cox proportional hazard model
| Factors | B | SE | Wald | df | Sig. | Exp(B) | 95.0 % CI | |
|---|---|---|---|---|---|---|---|---|
| Lower | Upper | |||||||
| CRHBP | -.267 | 1.272 | .044 | 1 | .834 | .766 | .063 | 9.268 |
| DNASE1L3 | -.969 | .476 | 4.140 | 1 | .042 | .379 | .149 | .965 |
| FCN2 | .517 | .896 | .333 | 1 | .564 | 1.676 | .290 | 9.704 |
| INTS8 | .204 | .090 | 5.175 | 1 | .023 | 1.227 | 1.029 | 1.463 |
| LCAT | .030 | .194 | .024 | 1 | .877 | 1.031 | .704 | 1.509 |
| MARCO | -.070 | .859 | .007 | 1 | .935 | .932 | .173 | 5.020 |
| MT1X | .788 | 1.051 | .561 | 1 | .454 | 2.198 | .280 | 17.256 |
| PAMR1 | -.158 | .236 | .448 | 1 | .503 | .854 | .538 | 1.355 |
| VIPR1 | .194 | .287 | .459 | 1 | .498 | 1.215 | .692 | 2.131 |
| Stage (I/II–III) | .901 | .463 | 3.784 | 1 | .050 | 2.462 | .993 | 6.101 |
| PT (1–2/3–4) | -.223 | .426 | .273 | 1 | .601 | .800 | .347 | 1.844 |
| Residual (R0/R1–2) | -.175 | .586 | .089 | 1 | .765 | .839 | .266 | 2.649 |
| Tumour status (free/with) | 1.300 | .404 | 10.359 | 1 | .001 | 3.669 | 1.662 | 8.097 |
| Vital status (dead/alive) | −13.599 | 55.193 | .061 | 1 | .805 | .000 | .000 | 1.2e + 41 |
| Age | .000 | .014 | .001 | 1 | .981 | 1.000 | .974 | 1.028 |
| Gender | -.276 | .312 | .781 | 1 | .377 | .759 | .412 | 1.399 |
PT AJCC Tumour Pathologic PT
Six genes were associated with tumour pathologic PT and tumour stage (Table 4); among these, high expression of INTS8 and UBAP2L, and low expression of ACSM3, FCN2, LCAT, and MT1G, was significantly associated with metastatic tumour and late stage (P ≤ 0.05). In particular, UBAP2L was markedly and highly expressed in T2 tumours (72.5 % vs. 27.5 %) and LCAT was lowly expressed in T2 tumours (30.0 % vs. 70.0 %) and highly expressed in T1 tumours (72.6 % vs. 27.4 %). In addition, LCAT was highly expressed in stage I tumours (71.2 % vs. 28.8 %).
Table 4.
Associations of genes expression with AJCC tumour pathologic PT, tumour stage, age and gender in 157 patients with hepatocellular carcinoma
| Factors | No. of patients | ACSM3 | FCN2 | INTS8 | LCAT | MT1G | UBAP2L | ||||||
| High | Low | High | Low | High | Low | High | Low | High | Low | High | Low | ||
| PT | 157 | P = 0.037 | P = 0.026 | P = 0.046 | P = 0.000 | P = 0.016 | P = 0.004 | ||||||
| T1 | 62 (39.5 %) | 39 (62.9 %) | 23 (37.1 %) | 39 (62.9 %) | 23 (37.1 %) | 23 (37.1 %) | 39 (62.9 %) | 45 (72.6 %) | 17 (27.4 %) | 39 (62.9 %) | 23 (37.1 %) | 22 (35.5 %) | 40 (64.5 %) |
| T2 | 40 (25.5 %) | 20 (50.0 %) | 20 (50.0 %) | 13 (32.5 %) | 27 (67.5 %) | 24 (60.0 %) | 16 (40.0 %) | 12 (30.0 %) | 28 (70.0 %) | 13 (32.5 %) | 27 (67.5 %) | 29 (72.5 %) | 11 (27.5 %) |
| T3 | 46 (29.3 %) | 16 (34.8 %) | 30 (65.2 %) | 22 (47.8 %) | 24 52.2 %) | 28 (60.9 %) | 18 (39.1 %) | 18 (39.1 %) | 28 (60.9 %) | 21 (45.7 %) | 25 54.3 %) | 23 (50.0 %) | 23 (50.0 %) |
| T4 | 9 (5.7 %) | 4 (44.4 %) | 5 (55.6 %) | 4 (44.4 %) | 5 (55.6 %) | 4 (44.4 %) | 5 (55.6 %) | 4 (44.4 %) | 5 (55.6 %) | 6 (66.7 %) | 3 (33.3 %) | 5 (55.6 %) | 4 (44.4 %) |
| Stage | 143 | P = 0.016 | P = 0.032 | P = 0.026 | P = 0.000 | P = 0.037 | P = 0.009 | ||||||
| I | 59 (41.3 %) | 36 (61.0 %) | 23 (39.0 %) | 36 (61.0 %) | 23 (39.0 %) | 23 (39.0 %) | 36 (61.0 %) | 42 (71.2 %) | 17 (28.8 %) | 37 (62.7 %) | 22 (37.3 %) | 22 (37.3 %) | 37 (62.7 %) |
| II | 36 (25.2 %) | 19 (52.8 %) | 17 (47.2 %) | 12 (33.3 %) | 24 (66.7 %) | 22 (61.1 %) | 14 (38.9 %) | 12 (33.3 %) | 24 (66.7 %) | 13 (36.1 %) | 23 (63.9 %) | 25 (69.4 %) | 11 (30.6 %) |
| III | 48 (33.6 %) | 16 (33.3 %) | 32 66.7 %) | 25 (52.1 %) | 23 (47.9 %) | 30 (62.5 %) | 18 (37.5 %) | 18 (37.5 %) | 30 (62.5 %) | 23 (47.9 %) | 25 (52.1 %) | 25 (52.1 %) | 23 (47.9 %) |
| CXCL14 | GMNN | INTS8 | MT1F | MT1G | SPRX | ||||||||
| High | Low | High | Low | High | Low | High | Low | High | Low | High | Low | ||
| Agea | 157 | P = 0.031 | P = 0.031 | P = 0.005 | P = 0.013 | P = 0.031 | P = 0.031 | ||||||
| < 65 | 80 (51.0 %) | 47 (58.8 %) | 33 (41.3 %) | 47 (58.8 %) | 33 (41.3 %) | 49 (61.2 %) | 31 (38.8 %) | 47 (58.8 %) | 32 (40.0 %) | 47 (58.8 %) | 33 (41.3 %) | 47 (58.8 %) | 33 (41.3 %) |
| ≥ 65 | 77 (49.0 %) | 32 (41.6 %) | 45 (58.4 %) | 32 (41.6 %) | 45 (58.4 %) | 30 (39.0 %) | 47 (61.0 %) | 32 (41.6 %) | 46 (59.7 %) | 32 (41.6 %) | 45 (58.4 %) | 32 (41.6 %) | 45 (58.4 %) |
| CLEC1B | CRHBP | FCN2 | MT1G | TBCE | |||||||||
| High | Low | High | Low | High | Low | High | Low | High | Low | ||||
| Gender | 157 | P = 0.003 | P = 0.019 | P = 0.043 | P = 0.003 | P = 0.019 | |||||||
| Female | 62 (39.5 %) | 22 (35.5 %) | 40 (64.5 %) | 24 (38.7 %) | 38 (61.3 %) | 25 (61.0 %) | 37 (39.0 %) | 22 (35.5 %) | 40 (64.5 %) | 24 (38.7 %) | 38 (61.3 %) | ||
| Male | 95 (60.5 %) | 57 (60.0 %) | 38 (40.0 %) | 55 (57.9 %) | 40 (42.1 %) | 54 (56.8 %) | 41 (43.2 %) | 57 (60.0 %) | 38 (40.0 %) | 55 (57.9 %) | 40 (42.1 %) | ||
aAge was dichotomised into < 65 and ≥ 65 using the median as a cutoff. PT, AJCC Tumour Pathologic PT. Expression values of a gene were dichotomised into high and low expression using the median as a cutoff. P value determined using Pearson’s χ2 test (2-sided)
Ten genes were associated with age and gender. As shown in Table 4, we found that six genes—CXCL14, GMNN, INTS8, MT1F, MT1G, and SPRX—were expressed at low levels in HCC patients aged ≥ 65 years. Expression of five genes was related to the gender of HCC patients. Except for FCN2, which is lowly expressed in male HCC patients, the other four genes, CLEC1B, CRHBP, MT1G, and TBCE, were all lowly expressed in female HCC patients. In addition, PAMR1 and MT1X were closely related to the vital status; both showed low expression in 60.3 % (38/63) of HCC patients with dead status, compared with high expression in 57.4 % (54/94) of patients with alive status (P = 0.022).
Potential roles of the genes in HCC progression
The potential roles of the 20 genes in HCC were predicted on the basis of Coremine Medical mining. As shown in Fig. 5, the associations of the genes with diagnosis, prognosis, drug resistance, recurrence, metastasis, and invasiveness of HCC was comprehensively analysed. The results indicated that, with the exception of PAMR1, the other 19 genes were all associated with at least one factor contributing to cancer progression, and many of the genes, for example GMNN, CXCL14, MT1G, MT1X, SPRX, and VIPR1, were closely associated with almost all of the factors included in this analysis. Most of the genes were extensively associated with several factors. For example, 15 genes (including INTS8, LCAT, MARCO, and DANSE1L3) were associated with diagnosis, 14 genes (including INTS8, MARCO, CRHBP, and VIPR1) were associated with metastasis, and 13 genes (including LCAT, MARCO, FCN2, and CXCL14) were associated with prognosis.
Fig. 5.
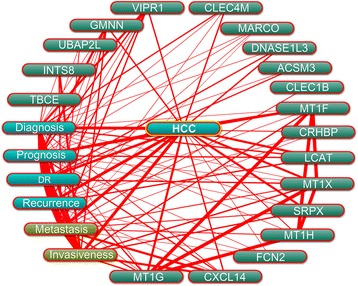
Association of the genes with HCC characteristics was determined by text mining using Coremine Medical and probabilistic scoring (P < 0.05). HCC: hepatocellular carcinoma, DR: drug resistance
Based on the gene expression in two independent GEO microarrays corresponding to HCC metastasis, the association of the genes CLEC4M, CRHBP, MARCO, MT1X, SRPX, UBAP2L, and VIPR1 with metastasis was further analysed; unfortunately, data for the other genes were unavailable. The expression of CRHBP, LCAT, and SPRX was significantly dysregulated in nine HCCs with venous metastasis compared with 11 HCC without (Fig. 6a). Genes VIPR1, LCAT, BAP2L, CLEC4M, CRHBP, and SRPX were significantly dysregulated in 32 HCCs with portal vein tumour thrombus metastasis and 33 HCCs with intrahepatic spread metastasis compared with 22 HCCs with no metastasis (Fig. 6b&c). In particular, LCAT was highly expressed in HCC patients with venous metastasis and patients with portal vein tumour thrombus metastasis, and SRPX was lowly expressed in HCC patients with venous metastasis and patients with intrahepatic spread metastasis (Fig. 6).
Fig. 6.
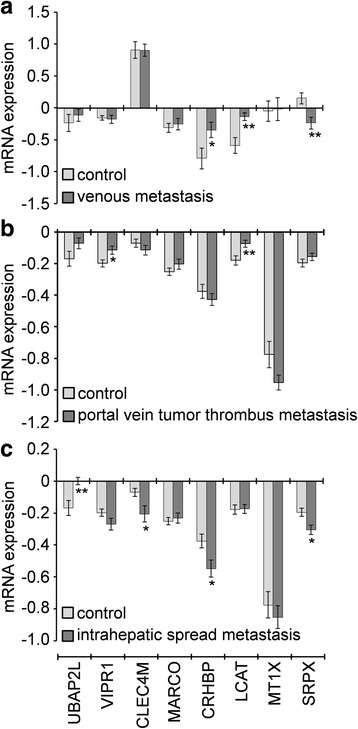
mRNA expression of the genes in HCC patients with and without metastasis according to microarray data retrieved from the GEO online database. a Microarray data GDS3091 [18] cover nine HCCs with venous metastasis and 11 without as controls. b, c Microarray data GDS274 [19] cover 32 HCCs with portal vein tumour thrombus metastasis, 33 with intrahepatic spread metastasis, and 22 HCCs with no metastasis as controls. *, P < 0.05; **, P < 0.01
Correlation of DNA methylation with mRNA expression of the target genes
DNA methylation and mRNA expression data from 379 HCC patients in a TCGA cohort were retrieved and the correlations between them were analysed using bivariate correlations. Among the 20 genes that are poorly studied in HCC (Table 1), DNA methylation data of CLEC1B and SRPX were not available. DNA methylation was negatively correlated with the mRNA expression for eight genes, ACSM3, INTS8, LCAT, MT1X, CRHBP, MARCO, PAMR1, and VIPR1. In particular, high methylation of the first four genes was significantly correlated with lower mRNA expression (Fig. 7), indicating that the expression of these genes in HCC might be regulated by DNA methylation.
Fig. 7.
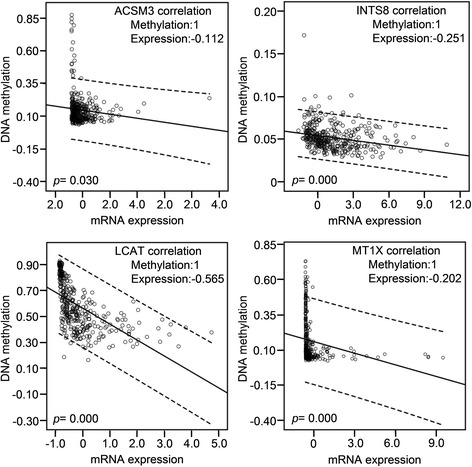
DNA methylation of four genes was significantly and negatively correlated with their mRNA expression. Data for gene expression and DNA methylation in 379 HCCs were retrieved from a TCGA cohort. The correlation between DNA methylation and gene expression was analysed using bivariate correlations
Discussion
Cancer is frequently considered to be a disease of the cell cycle because alterations in different families of cell cycle regulators cooperate in tumour development. Molecular analysis of human tumours has shown that cell cycle regulators are frequently mutated in human neoplasms, underscoring the importance of maintaining cell cycle commitment in the prevention of human cancer [26]. Abnormal expression of cell cycle controllers, particularly G1/S-phase transition, is often implicated in the pathogenesis of most human cancers, including HCC. For example, vaccinia-related kinase 1 promotes HCC by controlling the levels of cell cycle regulators associated with G1/S transition [27]. In this study, 80 genes that were significantly dysregulated in HCC were identified based on four independent microarrays covering a total of 386 cases of hepatocellular carcinoma and 327 cases of normal liver tissues (Fig. 1), and biological process annotation of these genes revealed that 17 of these genes were implicated in cell cycle functions (Additional file 1: Table S1). These results suggested that these genes might contribute to cancer progression and development in HCC at least in part through regulation of the cell cycle.
Twenty-seven genes were further identified to be consistently dysregulated in all four microarrays by at least 2-fold (Table 1). The expression of eight of these genes (TBCE, INTS8, VIPR1, CLEC4M, MARCO, DNASE1L3, CRHBP, and FCN2) was confirmed in 11 tissues of HCC patients compared with matched paracancerous tissues by RT-qPCR (Fig. 2a). Seven of the 27 genes (UBE2C, PTTG1, CAP2, TOP2A, GPC3, EGR1, and NAT2) have been well studied in HCC (Table 1). For example, GPC3 plays critical roles in cell proliferation and invasion through the induction of apoptosis [28] and is a biomarker for diagnosis [29] and recurrence [30]. Protein/gene-protein/gene interaction analyses indicated that these 27 proteins/genes strongly interacted with each other, and 10 of them interacted with at least half of all the genes (Additional file 2: Figure S2). Moreover, six of these genes were related to the cell cycle in HCC (Additional file 1: Table S1). Together, these results indicate that the genes identified in this study might play crucial roles in HCC progression, probably functioning as a group.
Biomarkers not only have prognostic implications, but are also helpful for measurement of treatment responses and surveillance for tumour recurrence and for guiding clinical decisions [31]. Thus, prognostic biomarkers for HCC patients are necessary and crucial, and there is an ongoing search for predictive biomarkers. In this study, a group of genes associated with DFS and OS (Table 2) were identified in 157 HCC patients. Among these genes, low expression of ACSM3 and CXCL14 was associated with poor DFS, low expression of CRHBP, DNASE1L3, FCN2, MT1X, and VIPR1 was associated with poor OS (Fig. 3, Table 2), high expression of INTS8 was associated with poor DFS and OS, and low expression of LCAT, MARCO, and PAMR1 was associated with poor DFS and OS (Fig. 4, Table 2). Furthermore, DNASE1L3 and INTS8 were identified as independent risk prognostic factors for OS (Table 3). There are few reports of the association of these genes with prognosis in HCC or in other cancers. Previous studies indicate that downregulation of CXCL14 is associated with prognosis in gastric cancer patients [32], MT1X may aid in the prognostic discrimination of oral squamous cell carcinoma cases [33], and MARCO expression is associated with breast cancer survival and risk of recurrence [34].
Twenty genes that have been less studied in HCC (Table 1) were further evaluated to predict their potential roles in HCC progression. Coremine medical mining suggested that most of those genes were associated with diagnosis, prognosis, drug resistance, recurrence, metastasis, and invasiveness. In particular, 13, 14, and 15 genes were potentially associated with prognosis, metastasis, and diagnosis in HCC, respectively (Fig. 5). The association of these genes with prognosis appears to have clinical importance, as 11 genes were shown to be associated with DFS or/and OS (Table 2, Fig. 3 & 4). The role of these genes in metastasis was further confirmed by gene expression analysis, which showed that five genes were significantly dysregulated in HCC with venous metastasis, portal vein tumour thrombus metastasis, or intrahepatic spread metastasis, compared with the appropriate controls. Specifically, LCAT was highly expressed in HCC patients with venous metastasis and patients with portal vein tumour thrombus metastasis, and SRPX was lowly expressed in HCC patients with venous metastasis and patients with intrahepatic spread metastasis (Fig. 6), suggesting that these two genes might be closely related to HCC metastasis. There are few studies on LCAT and SRPX in cancer metastasis, with only one reported that SRPX is upregulated in gastric cancer cells after depletion of TWIST, which promoted the epithelial-mesenchymal transition that occurs during the initial steps of tumour metastasis [35].
INTS8 encodes a subunit of the integrator complex that is involved in the cleavage of small nuclear RNAs, and its association with cancer is poorly understood. Limited studies indicate that INTS8 contains mutations in peripheral T cell lymphoma compared with non-malignant samples from 12 patients [36], and a combination of INTS8 with SULF1, ATP6V1C1, and GPR172A can be used to discriminate gastric carcinomas from adjacent noncancerous tissues [37]. In this study, we found that, potentially regulated by demethylation (Fig. 7), INTS8 was significantly and consistently upregulated at least 2.115-fold in HCC according to four independent microarrays (Fig. 1; Table 1) and that INTS8 mRNA was upregulated 2.06-fold on average in 11 tissues of HCC patients compared with corresponding paracancerous tissues, with a similar expression profile at the protein level (Fig. 2). Based on the clinical importance analysis of 157 HCC patients in a TCGA cohort, we found that high expression of INTS8 was associated with poor DFS and OS (Fig. 4, Table 2), and was an independent risk prognostic factor for OS (Table 3). Moreover, high expression of INTS8 was associated with metastatic tumours and late stage (Table 4), and with younger HCC patients (<65 years old) (Table 4). In addition, text mining indicated that INTS8 was closely related with metastasis, invasiveness, and diagnosis (Fig. 5). The above results strongly indicate that this gene is indeed upregulated in HCC, where it might play crucial roles in HCC cancer progression and development, and is a potential biomarker for diagnosis and, in particular, prognosis.
Conclusion
In summary, by means of data retrieved from six independent microarrays, RT-qPCR and western blotting detection in 11 pairs of tissues, clinical importance analyses in a cohort of 157 patients, and bioinformatics analyses including biological process annotation, protein interaction and text mining, we have identified a group of genes that are significantly dysregulated in HCC and might be associated with cancer progression, development, and, in particular, prognosis. These genes could be potential therapeutic targets for HCC treatment, and might be useful biomarkers for diagnosis and prognosis.
Acknowledgements
None.
Funding
The present study was supported by the National Natural Science Foundation of China (grant nos. 81360448), the Natural Science Foundation of Guangxi (nos. 2014GXNSFAA118139), Key Laboratory of High-Incidence-Tumor Prevention and Treatment (Guangxi Medical University), Ministry of Education (nos. GK2015-ZZ03 and GK2014-ZZ03) and Guangxi Outstanding Teachers Training Project for Colleges.
Availability of data and materials
None.
Authors’ contributions
FY and XL performed most analysis. FY wrote the manuscript. LS provided the clinical samples. TL performed the RT-qPCR and western blotting. TP helped collect samples and revise manuscript. YN and SL performed mRNA and protein isolation. XZ and XQ designed the study and helped draft manuscript. All authors reviewed the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
None.
Ethics approval and consent to participate
The study was endorsed by the Ethics Committee of Guangxi Medical University and was performed according to the Declaration of Helsinki, 2013 edition. All patients received an explanation of the aims of the study and signed informed consent. We are free to use ovarian cancer data in TCGA by meeting its freedom-to-publish criteria: A marker paper has been published on that tumour type.
Additional files
Biological process and cellular component annotation of the 80 genes associated with HCC development and progression by DAVID online tool. (PDF 167 kb)
Protein/gene-protein/gene interaction network of the 27 genes that were stably and consistently dysregulated in 386 cases of hepatocellular carcinoma compared with 327 cases of normal liver tissue according to the four independent microarrays retrieved from the Oncomine database. (PDF 306 kb)
Footnotes
Fuqiang Yin, Lipei Shu and Xia Liu are co-first authors.
Contributor Information
Fuqiang Yin, Email: yinfq@mail2.sysu.edu.cn.
Lipei Shu, Email: shulipei888@163.com.
Xia Liu, Email: realliuxia@sina.com.
Ting Li, Email: 516225191@qq.com.
Tao Peng, Email: pengtaocn@hotmail.com.
Yueli Nan, Email: 331578797@qq.com.
Shu Li, Email: 369363170@qq.com.
Xiaoyun Zeng, Email: zxyxjw@21cn.com.
Xiaoqiang Qiu, Email: xqqiu9999@sina.com.
References
- 1.Yang JD, Roberts LR. Hepatocellular carcinoma: A global view. Nat Rev Gastroenterol Hepatol. 2010;7:448–58. doi: 10.1038/nrgastro.2010.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miki D, Ochi H, Hayes CN, Aikata H, Chayama K. Hepatocellular carcinoma: towards personalized medicine. Cancer Sci. 2012;103:846–50. doi: 10.1111/j.1349-7006.2012.02242.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Byam J, Renz J, Millis JM. Liver transplantation for hepatocellular carcinoma. Hepatobiliary Surg Nutri. 2013;2:22–30. doi: 10.3978/j.issn.2304-3881.2012.11.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singhal A, Jayaraman M, Dhanasekaran DN, Kohli V. Molecular and serum markers in hepatocellular carcinoma: predictive tools for prognosis and recurrence. Crit Rev Oncol Hematol. 2012;82:116–40. doi: 10.1016/j.critrevonc.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 5.Cancer Genome Atlas Research N. Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013;45:1113–20. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.International Cancer Genome C. Hudson TJ, Anderson W, Artez A, Barker AD, Bell C, et al. International network of cancer genome projects. Nature. 2010;464:993–8. doi: 10.1038/nature08987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rustici G, Kolesnikov N, Brandizi M, Burdett T, Dylag M, Emam I, et al. ArrayExpress update--trends in database growth and links to data analysis tools. Nucleic Acids Res. 2013;41:D987–90. doi: 10.1093/nar/gks1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, et al. NCBI GEO: archive for functional genomics data sets--update. Nucleic Acids Res. 2013;41:D991–5. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, et al. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6:1–6. doi: 10.1016/S1476-5586(04)80047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coordinators NR. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2015;43:D6–17. doi: 10.1093/nar/gku1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rung J, Brazma A. Reuse of public genome-wide gene expression data. Nat Rev Genet. 2013;14:89–99. doi: 10.1038/nrg3394. [DOI] [PubMed] [Google Scholar]
- 12.Kannan L, Ramos M, Re A, El-Hachem N, Safikhani Z, Gendoo DM, et al. Public data and open source tools for multi-assay genomic investigation of disease. Briefings in bioinformatics. 2016;17:603–15. [DOI] [PMC free article] [PubMed]
- 13.Liu X, Gao Y, Zhao B, Li X, Lu Y, Zhang J, et al. Discovery of microarray-identified genes associated with ovarian cancer progression. Int J Oncol. 2015;46:2467–78. doi: 10.3892/ijo.2015.2971. [DOI] [PubMed] [Google Scholar]
- 14.Liu X, Gao Y, Lu Y, Zhang J, Li L, Yin F. Downregulation of NEK11 is associated with drug resistance in ovarian cancer. Int J Oncol. 2014;45:1266–74. doi: 10.3892/ijo.2014.2503. [DOI] [PubMed] [Google Scholar]
- 15.Zeng X, Yin F, Liu X, Xu J, Xu Y, Huang J, et al. Upregulation of E2F transcription factor 3 is associated with poor prognosis in hepatocellular carcinoma. Oncol Rep. 2014;31:1139–46. doi: 10.3892/or.2014.2968. [DOI] [PubMed] [Google Scholar]
- 16.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:11. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Can Dis. 2012;2:401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Budhu A, Forgues M, Ye QH, Jia HL, He P, Zanetti KA, et al. Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell. 2006;10:99–111. doi: 10.1016/j.ccr.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 19.Ye QH, Qin LX, Forgues M, He P, Kim JW, Peng AC, et al. Predicting hepatitis B virus-positive metastatic hepatocellular carcinomas using gene expression profiling and supervised machine learning. Nat Med. 2003;9:416–23. doi: 10.1038/nm843. [DOI] [PubMed] [Google Scholar]
- 20.da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 21.da Huang W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mostafavi S, Ray D, Warde-Farley D, Grouios C, Morris Q. GeneMANIA: a real-time multiple association network integration algorithm for predicting gene function. Genome Biol. 2008;9(Suppl 1):S4. doi: 10.1186/gb-2008-9-s1-s4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zuberi K, Franz M, Rodriguez H, Montojo J, Lopes CT, Bader GD, et al. GeneMANIA prediction server 2013 update. Nucleic Acids Res. 2013;41:W115–22. doi: 10.1093/nar/gkt533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Leeuw N, Dijkhuizen T, Hehir-Kwa JY, Carter NP, Feuk L, Firth HV, et al. Diagnostic interpretation of array data using public databases and internet sources. Hum Mutat. 2012;33:930–40. doi: 10.1002/humu.22049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hedditch EL, Gao B, Russell AJ, Lu Y, Emmanuel C, Beesley J, et al. ABCA transporter gene expression and poor outcome in epithelial ovarian cancer. J Natl’ Cancer Institute. 2014; 106 [DOI] [PMC free article] [PubMed]
- 26.D’Andrilli G, Kumar C, Scambia G, Giordano A. Cell cycle genes in ovarian cancer: steps toward earlier diagnosis and novel therapies. Clin Cancer Res. 2004;10:8132–41. doi: 10.1158/1078-0432.CCR-04-0886. [DOI] [PubMed] [Google Scholar]
- 27.Lee N, Kwon JH, Kim YB, Kim SH, Park SJ, Xu W, et al. Vaccinia-related kinase 1 promotes hepatocellular carcinoma by controlling the levels of cell cycle regulators associated with G1/S transition. Oncotarget. 2015;6:30130–48. doi: 10.18632/oncotarget.4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pan Z, Chen C, Long H, Lei C, Tang G, Li L, et al. Overexpression of GPC3 inhibits hepatocellular carcinoma cell proliferation and invasion through induction of apoptosis. Mol Med Rep. 2013;7:969–74. doi: 10.3892/mmr.2013.1279. [DOI] [PubMed] [Google Scholar]
- 29.Yu JP, Xu XG, Ma RJ, Qin SN, Wang CR, Wang XB, et al. Development of a Clinical Chemiluminescent Immunoassay for Serum GPC3 and Simultaneous Measurements Alone With AFP and CK19 in Diagnosis of Hepatocellular Carcinoma. J Clin Lab Anal. 2015;29:85–93. [DOI] [PMC free article] [PubMed]
- 30.Wang Y, Shen Z, Zhu Z, Han R, Huai M. Clinical values of AFP, GPC3 mRNA in peripheral blood for prediction of hepatocellular carcinoma recurrence following OLT: AFP, GPC3 mRNA for prediction of HCC. Hepat Mon. 2011;11:195–9. [PMC free article] [PubMed] [Google Scholar]
- 31.Wong KF, Xu Z, Chen J, Lee NP, Luk JM. Circulating markers for prognosis of hepatocellular carcinoma. Expert Opinion Med Diagnos. 2013;7:319–29. doi: 10.1517/17530059.2013.795146. [DOI] [PubMed] [Google Scholar]
- 32.Hu C, Lin F, Zhu G, Xue X, Ding Y, Zhao Z, et al. Abnormal hypermethylation of promoter region downregulates chemokine CXC ligand 14 expression in gastric cancer. Int J Oncol. 2013;43:1487–94. doi: 10.3892/ijo.2013.2078. [DOI] [PubMed] [Google Scholar]
- 33.Brazao-Silva MT, Rodrigues MF, Eisenberg AL, Dias FL, de Castro LM, Nunes FD, Faria PR, Cardoso SV, Loyola AM, de Sousa SCOM. Metallothionein gene expression is altered in oral cancer and may predict metastasis and patient outcomes. Histopathology. 2015;67:358–67. doi: 10.1111/his.12660. [DOI] [PubMed] [Google Scholar]
- 34.Bergamaschi A, Tagliabue E, Sorlie T, Naume B, Triulzi T, Orlandi R, Russnes HG, Nesland JM, Tammi R, Auvinen P, Kosma V-M, Ménard S, Børresen-Dale A-L. Extracellular matrix signature identifies breast cancer subgroups with different clinical outcome. J Pathol. 2008;214:357–67. doi: 10.1002/path.2278. [DOI] [PubMed] [Google Scholar]
- 35.Feng MY, Wang K, Shi QT, Yu XW, Geng JS. Gene expression profiling in TWIST-depleted gastric cancer cells. Anat Rec. 2009;292:262–70. doi: 10.1002/ar.20802. [DOI] [PubMed] [Google Scholar]
- 36.Simpson HM, Khan RZ, Song C, Sharma D, Sadashivaiah K, Furusawa A, Liu X, Nagaraj S, Sengamalay N, Sadzewicz L, Luke J. Concurrent Mutations in ATM and Genes Associated with Common gamma Chain Signaling in Peripheral T Cell Lymphoma. PLoS One. 2015;10:e0141906. doi: 10.1371/journal.pone.0141906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheng L, Zhang Q, Yang S, Yang Y, Zhang W, Gao H, Deng X, Zhang Q. A 4-gene panel as a marker at chromosome 8q in Asian gastric cancer patients. Genomics. 2013;102:323–30. doi: 10.1016/j.ygeno.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 38.Kim HE, Kim DG, Lee KJ, Son JG, Song MY, Park YM, et al. Frequent amplification of CENPF, GMNN and CDK13 genes in hepatocellular carcinomas. PLoS One. 2012;7:e43223. doi: 10.1371/journal.pone.0043223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ieta K, Ojima E, Tanaka F, Nakamura Y, Haraguchi N, Mimori K, et al. Identification of overexpressed genes in hepatocellular carcinoma, with special reference to ubiquitin-conjugating enzyme E2C gene expression. Int J Cancer. 2007;121:33–8. doi: 10.1002/ijc.22605. [DOI] [PubMed] [Google Scholar]
- 40.Fujii T, Nomoto S, Koshikawa K, Yatabe Y, Teshigawara O, Mori T, et al. Overexpression of pituitary tumor transforming gene 1 in HCC is associated with angiogenesis and poor prognosis. Hepatology. 2006;43:1267–75. doi: 10.1002/hep.21181. [DOI] [PubMed] [Google Scholar]
- 41.Su MC, Hsu HC, Liu YJ, Jeng YM. Overexpression of pituitary tumor-transforming gene-1 in hepatocellular carcinoma. Hepatogastroenterology. 2006;53:262–5. [PubMed] [Google Scholar]
- 42.Liang M, Chen X, Liu W, Li S, Li C, Jiang L, et al. Role of the pituitary tumor transforming gene 1 in the progression of hepatocellular carcinoma. Cancer Biol Ther. 2011;11:337–45. doi: 10.4161/cbt.11.3.14102. [DOI] [PubMed] [Google Scholar]
- 43.Shibata R, Mori T, Du W, Chuma M, Gotoh M, Shimazu M, et al. Overexpression of cyclase-associated protein 2 in multistage hepatocarcinogenesis. Clin Cancer Res. 2006;12:5363–8. doi: 10.1158/1078-0432.CCR-05-2245. [DOI] [PubMed] [Google Scholar]
- 44.Sakamoto M, Mori T, Masugi Y, Effendi K, Rie I, Du W. Candidate molecular markers for histological diagnosis of early hepatocellular carcinoma. Intervirology. 2008;51(Suppl 1):42–5. doi: 10.1159/000122603. [DOI] [PubMed] [Google Scholar]
- 45.Wong N, Yeo W, Wong WL, Wong NL, Chan KY, Mo FK, et al. TOP2A overexpression in hepatocellular carcinoma correlates with early age onset, shorter patients survival and chemoresistance. Int J Cancer. 2009;124:644–52. doi: 10.1002/ijc.23968. [DOI] [PubMed] [Google Scholar]
- 46.Ho DW, Kai AK, Ng IO. TCGA whole-transcriptome sequencing data reveals significantly dysregulated genes and signaling pathways in hepatocellular carcinoma. Frontiers Med. 2015;9:322–30. doi: 10.1007/s11684-015-0408-9. [DOI] [PubMed] [Google Scholar]
- 47.Lu DD, Chen YC, Zhang XR, Cao XR, Jiang HY, Yao L. The relationship between metallothionein-1F (MT1F) gene and hepatocellular carcinoma. Yale J Biol Med. 2003;76:55–62. [PMC free article] [PubMed] [Google Scholar]
- 48.Tahara D, Nakanishi T, Akazawa S, Yamaguchi Y, Yamamoto H, Akashi M, et al. Lecithin-cholesterol acyltransferase and lipid transfer protein activities in liver disease. Metabolism. 1993;42:19–23. doi: 10.1016/0026-0495(93)90166-L. [DOI] [PubMed] [Google Scholar]
- 49.Gui T, Dong X, Li R, Li Y, Wang Z. Identification of hepatocellular carcinoma-related genes with a machine learning and network analysis. J Computational Biol. 2015;22:63–71. doi: 10.1089/cmb.2014.0122. [DOI] [PubMed] [Google Scholar]
- 50.Lin ZY, Chuang WL. Genes responsible for the characteristics of primary cultured invasive phenotype hepatocellular carcinoma cells. Biomed Pharmacother. 2012;66:454–8. doi: 10.1016/j.biopha.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 51.Udali S, Guarini P, Ruzzenente A, Ferrarini A, Guglielmi A, Lotto V, et al. DNA methylation and gene expression profiles show novel regulatory pathways in hepatocellular carcinoma. Clin Epigen. 2015;7:43. doi: 10.1186/s13148-015-0077-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou X, Zhu HQ, Lu J. Regulation of gene expression in HBV- and HCV-related hepatocellular carcinoma: integrated GWRS and GWGS analyses. Int J Clin Experimental Med. 2014;7:4038–50. [PMC free article] [PubMed] [Google Scholar]
- 53.Hoang TV, Toan NL, le Song H, Ouf EA, Bock CT, Kremsner PG, et al. Ficolin-2 levels and FCN2 haplotypes influence hepatitis B infection outcome in Vietnamese patients. PLoS One. 2011;6:e28113. doi: 10.1371/journal.pone.0028113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sun H, Chua MS, Yang D, Tsalenko A, Peter BJ, So S. Antibody Arrays Identify Potential Diagnostic Markers of Hepatocellular Carcinoma. Biomark Insights. 2008;3:1–18. doi: 10.4137/bmi.s595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gu X, Wang H, Wang A, Dou T, Qi P, Ji Q, et al. An intronic polymorphism rs2237062 in the CXCL14 gene influences HBV-related HCC progression in Chinese population. Mol Biol Rep. 2012;39:797–803. doi: 10.1007/s11033-011-0801-7. [DOI] [PubMed] [Google Scholar]
- 56.Wang W, Huang P, Zhang L, Wei J, Xie Q, Sun Q, et al. Antitumor efficacy of C-X-C motif chemokine ligand 14 in hepatocellular carcinoma in vitro and in vivo. Cancer Sci. 2013;104:1523–31. doi: 10.1111/cas.12279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kanda M, Nomoto S, Okamura Y, Nishikawa Y, Sugimoto H, Kanazumi N, et al. Detection of metallothionein 1G as a methylated tumor suppressor gene in human hepatocellular carcinoma using a novel method of double combination array analysis. Int J Oncol. 2009;35:477–83. doi: 10.3892/ijo_00000448. [DOI] [PubMed] [Google Scholar]
- 58.Ji XF, Fan YC, Gao S, Yang Y, Zhang JJ, Wang K. MT1M and MT1G promoter methylation as biomarkers for hepatocellular carcinoma. World JGastroenterol. 2014;20:4723–9. doi: 10.3748/wjg.v20.i16.4723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lu D, Han C, Wu T. Microsomal prostaglandin E synthase-1 promotes hepatocarcinogenesis through activation of a novel EGR1/beta-catenin signaling axis. Oncogene. 2012;31:842–57. doi: 10.1038/onc.2011.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Agundez JA, Olivera M, Ladero JM, Rodriguez-Lescure A, Ledesma MC, Diaz-Rubio M, et al. Increased risk for hepatocellular carcinoma in NAT2-slow acetylators and CYP2D6-rapid metabolizers. Pharmacogenetics. 1996;6:501–12. doi: 10.1097/00008571-199612000-00003. [DOI] [PubMed] [Google Scholar]
- 61.Farker K, Schotte U, Scheele J, Hoffmann A. Impact of N-acetyltransferase polymorphism (NAT2) in hepatocellular carcinoma (HCC)--an investigation in a department of surgical medicine. Exp Toxicol Pathol. 2003;54:387–91. doi: 10.1078/0940-2993-00275. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
None.