

Abstract
In Drosophila, many studies have examined the short or long term evolution occurring across synonymous sites. Few, however, have examined both the recent and long-term evolution to gain a complete view of this selection. Here we have analyzed Drosophila ananassae DNA polymorphism and divergence data using several different methods, and have identified evidence of positive selection favoring preferred codons in both recent and long-term evolutionary time scale. Further in D. ananassae, the strength of selection for preferred codons was stronger on the X chromosome compared to the autosomes. We show that this stronger selection is not due to higher gene expression of X linked genes. Analysis of the selectively neutral introns indicated the X chromosome also had a preference for GC over AT nucleotides, potentially from GC biased gene conversions (gcBGC) that can also affect the base composition of synonymous sites. Thus selection for preferred codons and gcBGC both seem to be partially responsible for shaping the D. ananassae synonymous site evolution.
Introduction
Despite the redundancy of the genetic code many organisms do not use an equal frequency of each codon. This phenomenon is termed codon usage bias (CUB) and has been observed in both single cellular and multicellular organisms (Hershberg and Petrov 2008). CUB can be explained by selective forces that favor specific codons to maximize the translational accuracy (Akashi 1994; Drummond and Wilke 2008) and translational efficiency (Gouy and Gautier 1982; Ikemura 1985) of each gene. These codons that are selectively advantageous are termed “preferred” codons due to their preference in usage across the genome-wide coding sequences. Selection for CUB could also be driven by a coevolutionary mechanism, evidenced by a recent study showing the frequent modifications of the tRNA anticodons that could have driven the unexpected shifts in genomic codon usage along several Drosophila lineages (Zaborske et al. 2014). On the other hand, non-selective forces such as the genome-wide bias in mutation can also cause patterns of codon usage (Chen et al. 2004). For example in Drosophila, excluding the willistoni lineage (Singh et al. 2006; Heger and Ponting 2007), most favored codons end with a G or C nucleotide (Vicario et al. 2007) suggesting that non-selective forces such as GC biased gene conversion (gcBGC) could generate the same signals as CUB (Marais 2003; Galtier et al. 2006) by causing higher GC content. So, these most frequently observed codons could result from selective and/or non-selective forces.
Theoretical models have been developed to examine the population genetics of CUB. Using the standard mutation-selection-drift models, these population genetic models assume selection favoring preferred codons while stochastic factors such as mutation and demography allow the random accumulation of preferred or unpreferred codons across the synonymous sites (Li 1987; Bulmer 1991; McVean and Charlesworth 1999; Zeng and Charlesworth 2009). Positive selection favoring the preferred codons over unpreferred codons causes the preferred codons to reach high frequency and ultimately fix in a population (Akashi 1997). On the other hand since the strength of selection (Nes; Ne, effective population size and s, selection coefficient) is thought to be weak (Nes ≈ 1) for synonymous sites (Li 1987; Bulmer 1991; Akashi 1995; Chamary et al. 2006; Yang and Nielsen 2008), mutation can result in accumulation of weakly deleterious unpreferred codons that segregate at a low frequency in the population. In addition, due to the weak selective force on synonymous sites (Nes ≈ 1) genetic drift is predicted to be a non-trivial force shaping the CUB in populations with small effective population sizes (Ohta 1973). Evidence shows that demographic events alone are able to skew the frequency distribution of nearly neutral synonymous variants and cause spurious inferences of CUB when they are not considered (Zeng and Charlesworth 2009). Thus an appropriate population genetic model that parameterizes the strength of selection, mutational bias, and demography across synonymous sites would be necessary to study the evolution of CUB (Zeng and Charlesworth 2009; Zeng 2010; Zeng and Charlesworth 2010).
Temporal variation in the pattern of selection for CUB can be examined using different population genetic data. For example, polymorphism data obtained from a single species examines the short-term evolutionary history and limits the temporal window of evolution to recent events. On the other hand orthologous genes from two closely related species or comparative genomic data from more divergent taxa can be analyzed to infer the long-term evolution. Different methods have been developed to test for selection shaping either the recent or long-term evolution of CUB at synonymous sites. Specific population genetic models have been developed to analyze synonymous polymorphisms and infer evidence of recent selection for CUB (Hartl et al. 1994; McVean and Charlesworth 1999; Comeron 2006; Cutter and Charlesworth 2006; Zeng and Charlesworth 2010; Haddrill et al. 2011; Qiu et al. 2011; de Procé et al. 2012; Campos et al. 2013). Long-term selection on synonymous sites can be examined using methods such as counting and comparing the number of fixed preferred and unpreferred mutations (Akashi 1995; Akashi 1996; DuMont et al. 2004), or using phylogenetic data and codon model based likelihood to estimate the strength of selection for CUB (McVean and Vieira 2001; Nielsen et al. 2007).
In order to gain a complete understanding of the CUB of an organism, it is important to examine both the recent and long term evolution occurring across the synonymous sites. For example in D. melanogaster, analysis of polymorphism data showed evidence of selection for preferred codons (Carlini and Stephan 2005; Zeng and Charlesworth 2009; Zeng and Charlesworth 2010; Andolfatto et al. 2011; Campos et al. 2013) while analyzing between-species data indicated a relaxation of selection for preferred codons (Akashi 1995; Akashi 1996; McVean and Vieira 2001; DuMont et al. 2004; Nielsen et al. 2007; Singh et al. 2007; Bauer DuMont et al. 2009). This suggests that the D. melanogaster lineage had reduced selection across the synonymous sites or even positive selection for unpreferred codons at some genes, and only recently has there been an increased selection for preferred codons.
CUB has been extensively studied in various species of Drosophila (Akashi 1994; Akashi 1995; Akashi 1996; Akashi and Schaeffer 1997; Powell et al. 2003; Maside et al. 2004; Haddrill et al. 2011; de Procé et al. 2012; Campos et al. 2013). However, less is known about CUB within the lineage leading to the ananassae subgroup. In D. ananassae genome-wide studies have shown evidence of CUB, specifically for the significant preference of preferred codons (Vicario et al. 2007), but analyses were limited to simple codon usage statistics such as frequency of optimal codons (Ikemura 1981), effective number of codons (Wright 1990), and codon adaptation index (Sharp and Li 1987) values (Heger and Ponting 2007; Singh et al. 2008). Choi and Aquadro (2014) conducted a population genetic analysis of synonymous site polymorphisms in D. ananassae, but this was based on a small number of genes involved in the regulation of the germline stem cell. Further, some of those genes had evidence of adaptive protein evolution, which is known to have a negative correlation with CUB (Akashi 1994; Akashi 1996; Betancourt and Presgraves 2002; Kim 2004; Andolfatto 2007; Drummond and Wilke 2008; Marion de Procé et al. 2009; Sella et al. 2009; Haddrill et al. 2011). Thus, in order to gain a better understanding of synonymous site evolution within the ananassae subgroup, we analyzed D. ananassae polymorphism and divergence data for 33 coding DNA sequences that show no prior evidence of departure from selective neutrality. Intron sequences were also analyzed to infer the gcBGC and demographic forces that may have influenced CUB by causing apparent changes in codon preference. For D. ananassae, results showed significant evidence of selection for preferred codons in both recent and long-term evolutionary time scales. Using an explicit population genetic model we estimated the strength of selection for preferred codon usage, mutation bias, and demography using both synonymous and intron polymorphisms. We show that the analytically estimated strength of selection (Nes) for preferred codons is higher on the X chromosome versus the autosomes, however, gcBGC could partially explain this apparent pattern of higher selection for preferred codons.
Material and Methods
Forty-three D. ananassae genes with population data were obtained from the study of sex-biased genes by Grath et al. (2009). The population dataset consists of the ancestral Bangkok strain for D. ananassae (Vogl et al. 2003; Das et al. 2004; Schug et al. 2007) with D. atripex or D. phaeopleura used as the close outgroup. For each gene the population genetic data had been generated by sequencing on average 11 isofemale lines (minimum of 8 to maximum of 12 lines; Grath et al. 2009). To polarize the fixed differences occurring only in D. ananassae, sequences from D. bipectinata a third distant outgroup species were obtained from the genome sequence of the modENCODE project (Chen et al. 2014).
Selection on synonymous sites is thought to be weaker than selection on nonsynonymous sites. Consequently due to the tight linkage between the two sites, the Hill-Robertson effect (Hill and Robertson 1966) could lead to decreased efficiency of selection on synonymous sites (McVean and Charlesworth 2000; Betancourt and Presgraves 2002; Andolfatto 2007). Using the MK-test (McDonald and Kreitman 1991) and the multi-locus HKA test (Hudson et al. 1987; J. Hey [https://bio.cst.temple.edu/~hey/software/software.htm]) we identified loci that have potentially experienced positive selection at the amino acid level. This left us with a set of 33 loci that are close to neutrality and were subsequently used for our CUB analysis (Online Resource 1 for complete list of genes identified as neutral). We compared the Fop and ENC statistics between genes with and without evidence of positive selection and found no significant difference between the two groups (Mann-Whitney U test FDR corrected p-value = 1.0 for both statistics). This suggested that these genes with positive selection might not be strongly affected by the Hill-Robertson effect. Nonetheless, we chose conservatively to exclude genes with evidence of positive selection to make our study comparable to other Drosophila synonymous site evolution studies, which have used the same method of excluding loci under positive selection.
In Drosophila gcBGC will lead to higher GC content and can lead to apparent increases in preference for preferred codons that end in G or C nucleotides. This makes it difficult to determine whether selective or non-selective mutational forces shaped the increased preference in specific codons. However, selection for preferred codons is expected to act only on the synonymous sites while gcBGC will increase GC content in both synonymous and intron sequences. Thus intron sequences were also analyzed for evidence of gcBGC. Further, we distinguished genes originating from the autosomes and sex chromosome for potential sex differences in the mutations rate (Bachtrog 2008). For the intron analysis, out of the total 33 genes analyzed for CUB, 16 genes out of the total 23 autosomal genes had introns while 6 genes out of the total 10 X chromosomal genes had introns. Due to the lower number of X-linked introns an additional 10 X-linked intron sequences from the same ancestral Bangkok strain were obtained from the study by Das et al. (2004). These 10 X-linked intron sequences lacked any outgroup sequences and only the polymorphism was analyzed for them.
Within the melanogaster group, codon usage is quite conserved among the different species (Vicario et al. 2007). Thus the codon usage table of D. melanogaster (Shields et al. 1988; Akashi 1995) was used for all codon usage analyses. Codons that were most frequently used were designated as preferred and the rest as unpreferred codons. Codon usage statistics such as the frequency of optimal codon (Fop) and effective number of codons (ENC) were measured using the program codonW (J. Peden, http://codonw.sourceforge.net/).
Following Akashi (1995; 1997) the Frequency Distribution and Divergence (FDD) test (Akashi 1999) was conducted to determine if the frequency distributions of the preferred and unpreferred mutations were significantly different. The unfolded frequency distribution of D. ananassae-specific preferred and unpreferred polymorphism and fixed mutations were polarized using D. atripex and D. bipectinata as outgroup sequences. We focused on those changes specifically along the D. ananassae lineage from an ancestral unpreferred to derived preferred mutations (U>P) or from an ancestral preferred to derived unpreferred (P>U) mutations. Using the same method gcBGC was inferred using the intron sequences that had an outgroup sequence. The frequency distributions of the ancestral GC to derived AT (GC>AT) changes or the ancestral AT to derived GC (AT>GC) changes were analyzed.
Per-site fixation of preferred and unpreferred variants were estimated using the method of DuMont et al. (2004). This method is analogous to the estimation of dN and dS (Nei and Gojobori 1996): using reconstructed parsimonious ancestral sequence we estimated the effective number of preferred and unpreferred sites and the number of D. ananassae lineage-specific fixed preferred and unpreferred mutations. The numbers of lineage-specific preferred and unpreferred fixations per preferred and unpreferred sites were then compared using a 2×2 Fisher’s exact test.
The autosomal and X chromosomal synonymous polymorphisms were analyzed together using the maximum-likelihood method of Zeng and Charlesworth (2009) as modified by Haddrill et al. (2011), to jointly estimate these parameters: (1) strength of selection Nes, where s corresponds to selection coefficient of favoring the preferred codons over unpreferred codons; (2) mutation bias κ as the ratio of preferred to unpreferred codon mutation rate over unpreferred to preferred codon mutation rate; and (3) instantaneous demographic change Na/Nb, where Nb and Na corresponds to population size before and after the instantaneous change respectively. To estimate selection for preferred codons and gcBGC jointly, synonymous and intron polymorphisms were analyzed together using the method of Zeng and Charlesworth (2009) as extended by Zeng and Charlesworth (2010). Synonymous sites were used to model the preference for preferred versus unpreferred variants while introns were used to model the preference for GC versus AT variants. Specifically, intron sites were used to estimate: (1) Nes, where s corresponds to the selection coefficient favoring the GC over AT nucleotide; (2) κ, ratio of GC to AT nucleotide mutation rate over AT to GC nucleotide mutation rate; and (3) the demography parameter Na/Nb. The extended Zeng and Charlesworth (2010) method was used to analyze the autosomal and X chromosomal data separately. We note at the time of our analysis there was no method to analyze the autosomal and X chromosomal synonymous and intron data all together. The Zeng and Charlesworth (2009) method does not require any outgroup sequences and fits the observed frequency distribution of synonymous or intron data to a population genetic model (reviewed in Zeng 2012). Approximating a chi-square distribution, significance of each model was determined by calculating the twice the difference in log-likelihood of the complex versus the simple model. Fit of each model was also assessed by calculating the Akaike Information Criterion (AIC) statistic.
All statistical tests resulting in a p-value were corrected for multiple hypotheses testing using the false discovery rate (FDR) controlling method of Benjamini and Hochberg (1995) as implemented in the program R (https://www.r-project.org).
Results
Due to the nature of our dataset we initially examined if there were any gross differences in the codon usage statistics for the 21 female-biased genes, 40 male-biased genes and 59 non-sex biased genes identified in Grath et al. (2009) (Online Resource 2). In D. melanogaster, based on codon usage statistics, male-biased genes were previously shown to have lower codon usage bias (CUB) compared to both female and non-sex biased genes (Hambuch and Parsch 2005). However for our D. ananassae dataset, the nonparametric Kruskal-Wallis test showed no significant difference in Fop (H = 0.66, df =2, FDR corrected p-value = 0.927) and ENC (H = 0.23, df = 2, FDR corrected p-value = 1.0) values among the three sex-biased gene categories. We then examined a reduced set of 43 genes that had both polymorphism and divergence data that we analyzed in this study. The nonparametric Kruskal-Wallis test showed no significant difference in Fop (H = 1.38, df =2, FDR corrected p-value = 0.501) and ENC (H = 1.29, df = 2, FDR corrected p-value = 0.524) values. That Hambuch and Parsch (2005) detected a significant difference in CUB could be due to the much larger number of genes they analyzed.
For the following population genetic analysis, of the 43 genes with polymorphism and divergence data, we analyzed 33 genes that were close to neutrality. We note that for gene CG18266 an appropriate ortholog could not be found in the distant outgroup D. bipectinata genome, thus this gene was excluded from the analyses that required polarizing fixed differences.
Population genetic models for CUB predict different shapes of the frequency distribution for preferred and unpreferred mutations across synonymous sites (Akashi 1999; McVean and Charlesworth 2000). Strong CUB selection for preferred codons will shift the preferred mutation frequency distribution towards high frequency derived variants, whereas the frequency of unpreferred mutations will be shifted towards rare variants. We have plotted the Frequency Distribution and Divergence (FDD) of U>P and P>U changes in our D. ananassae population genetic data in Figure 1. Consistent with expectations of selection for preferred codons, there were more P>U mutations (169 polymorphisms and 149 fixed variants for a total of 318 variants) compared to U>P mutations (27 polymorphisms and 92 fixed variants for a total of 119 variants). Compared to the U>P mutations, more P>U mutations were segregating as singletons (8.4% of the total U>P mutations segregating as singletons versus 19.8% of the total P>U mutations segregating as singletons) while there were fewer P>U mutations fixed (77.3% of the total U>P mutations are fixed versus 46.8% of the total P>U mutations are fixed) (Figure 1). Further, the U>P mutations were segregating at a significantly higher frequency than the P>U mutations (Wilcoxon’s W = 13224.5, FDR corrected p-value = 8.05×10−7).
Figure 1.
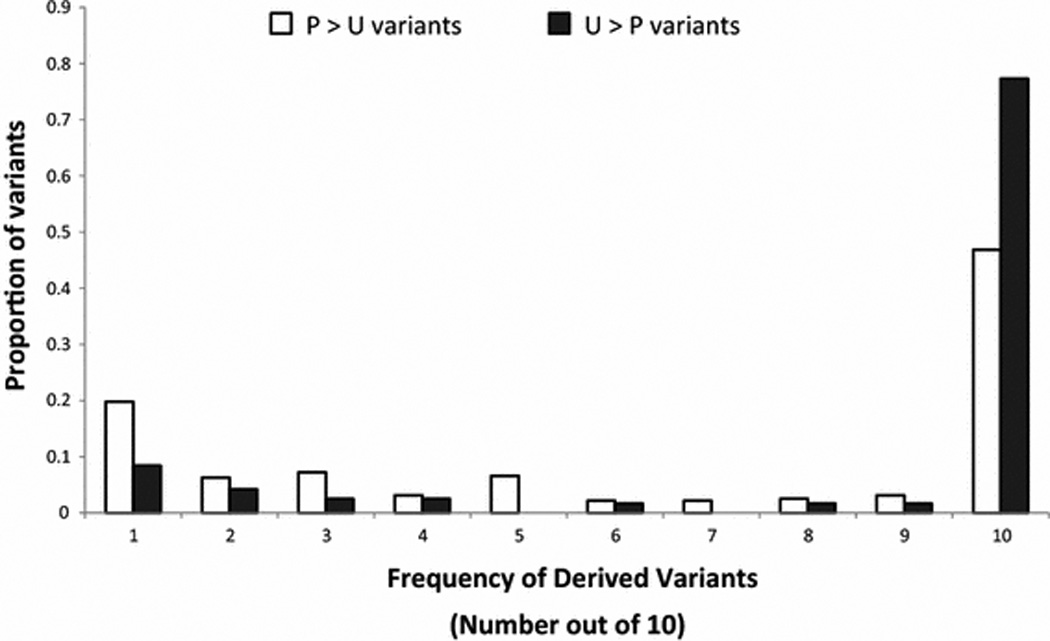
The proportion of segregating polymorphic and fixed preferred and unpreferred variants. Within and between species DNA variation are shown for 10 alleles where 1 equals to singletons and 10 equals to variants fixed specifically for D. ananassae.
With the majority of Drosophila preferred codons ending with a G or C nucleotides, inferred selection for preferred codons could in fact represent artifacts from GC biased gene conversion (gcBGC) skewing the base composition towards higher GC content. Unlike CUB, which only affects synonymous sites, gcBGC would affect both coding and intron regions. Thus intron sequences can be analyzed to distinguish the contributions of gcBGC. We compared the FDD of GC>AT and AT>GC changes at 22 introns that had outgroup sequences. There was no significant difference (Wilcoxon’s W = 2693.5, FDR corrected p-value = 0.184) between the frequency distribution of the 84 GC>AT and 85 AT>GC changes (Online Resource 3A). FDD of GC>AT and AT>GC changes at synonymous sites were also examined and they were not different from the FDD of U>P and P>U changes, since preferred codons end with G or C nucleotides in D. ananassae (Online Resource 3B).
Analysis of the D. ananassae FDD of preferred and unpreferred mutations indicated the preferred codons were segregating at a significantly higher frequency than unpreferred codons. Although selection for preferred codon usage predicts this pattern across synonymous sites (Akashi 1997), a considerable proportion of the P>U variants (46.8%) were fixed (Figure 1) suggesting the possibility of selection favoring the fixation of unpreferred codons in some of the analyzed genes. To examine the possibility of some genes having selection favoring the fixation of unpreferred codons we used the method of DuMont et al. (2004) to estimate the number of preferred and unpreferred fixations per preferred and unpreferred sites for each gene. The number of fixed preferred variants per preferred sites and fixed unpreferred variants per unpreferred sites were then compared to detect the long-term evolution occurring on synonymous sites. Results showed that most genes had the ratio of fixed preferred variants per preferred sites over fixed unpreferred variants per unpreferred sites (RP/U) greater than one. Fourteen genes had RP/U significantly greater than one (p < 0.05) before multiple hypothesis correction, and 12 genes remained significant (p < 0.05) after FDR correction (Table 1).
Table 1.
Numbers of fixed preferred, unpreferred substitutions and number of preferred, unpreferred sites.
| Gene | P sites | P fixations | U sites | U fixations | aRP/U |
bFET p-value |
|---|---|---|---|---|---|---|
| CG9723 | 52 | 11 | 245 | 6 | 8.638 | 0.000044* |
| CG10920 | 38 | 9 | 261 | 10 | 6.182 | 0.000472* |
| CG7387 | 48 | 7 | 237 | 4 | 8.641 | 0.000971* |
| CG13189 | 29 | 6 | 201 | 5 | 8.317 | 0.00168* |
| CG15717 | 24 | 5 | 148 | 3 | 10.278 | 0.0031* |
| CG7840 | 37 | 6 | 177 | 4 | 7.176 | 0.00416* |
| CG10035 | 39 | 6 | 150 | 3 | 7.692 | 0.00497* |
| CG3024 | 36 | 7 | 180 | 7 | 5.000 | 0.00616* |
| CG1749 | 63 | 8 | 142 | 3 | 6.011 | 0.00665* |
| CG6459 | 30 | 6 | 138 | 5 | 5.520 | 0.00957* |
| CG12276 | 51 | 6 | 144 | 3 | 5.647 | 0.0155* |
| CG3085 | 44 | 4 | 218 | 3 | 6.606 | 0.0209* |
| CG9135 | 65 | 7.5 | 230 | 7.5 | 3.538 | 0.0283 |
| CG6981 | 13 | 3 | 101 | 3 | 7.769 | 0.0307 |
| CG6971 | 36 | 5 | 110 | 4 | 3.819 | 0.0555 |
| CG2577 | 22 | 3 | 229 | 12 | 2.602 | 0.156 |
| CG5499 | 15 | 1 | 56 | 0 | N.A | 0.222 |
| CG2222 | 19 | 2 | 125 | 5 | 2.632 | 0.251 |
| CG9383 | 25 | 5 | 100 | 11 | 1.818 | 0.333 |
| CG10252 | 31 | 3 | 99 | 5 | 1.916 | 0.407 |
| CG6036 | 57 | 7 | 136 | 10 | 1.670 | 0.409 |
| CG5272 | 37 | 1 | 108 | 8 | 0.365 | 0.454 |
| CG8277 | 37 | 0 | 112 | 4 | 0 | 0.573 |
| CG15336 | 17 | 2 | 56 | 4 | 1.647 | 0.627 |
| CG1239 | 60 | 4 | 104 | 10 | 0.693 | 0.773 |
| CG3509 | 51 | 4 | 120 | 12 | 0.784 | 0.782 |
| CG10853 | 29 | 1 | 69 | 1 | 2.379 | 1 |
| CG11981 | 23 | 1 | 112 | 6 | 0.812 | 1 |
| CG18418 | 30 | 1 | 179 | 7 | 0.852 | 1 |
| CG5915 | 27 | 0 | 102 | 2 | 0 | 1 |
| CG4593 | 19 | 1 | 117 | 6 | 1.026 | 1 |
| CG11379 | 16 | 1 | 145 | 8.5 | 1.066 | 1 |
| Total | 1120 | 134 | 4651 | 182 | 3.057 | 7.07E-18* |
Ratio of fixed preferred variants per preferred sites over fixed unpreferred variants per unpreferred sites.
Significance was determined after the two-tailed Fisher’s Exact Test (FET) and shown are the resulting p-values.
Significant p-value < 0.05 after correcting for multiple hypothesis testing using the method of Benjamini-Hochberg (1995).
N.A, not appilicable.
The presented analyses thus far have examined sequence divergence to determine selection occurring across long evolutionary time scale. To infer recent selection occurring across synonymous sites we used the method of Zeng and Charlesworth (2009) which does not require an outgroup to polarize ancestral or derived variants to estimate the strength of selection for CUB. The dataset was divided into autosomes and X chromosome using both coding and intron sequences to estimate the parameters: strength of selection for preferred codons across coding sequences, strength of selection favoring the GC versus AT nucleotides across intron sequences (Zeng and Charlesworth 2010), mutation bias, and population size variation (i.e. demography). A summary of the data used in this analysis is given in Online Resource 4 and results using the model are shown in Table 2 listed from the model with lowest to highest AIC value.
Table 2.
Estimates of selection, mutation, and demography for synonymous sites and introns in autosomes and X chromosome.
| a) Autosomes | ||||||||
|---|---|---|---|---|---|---|---|---|
| Model | γcod | γint | κcod | κint | g | τ | lnL | ΔAIC |
| L0 (γint = 0) | 1.425 | - | 3.306 | 1.672 | - | - | −8444.861 | 0 |
| L0 | 1.425 | 0.447 | 3.307 | 2.569 | - | - | −8443.994 | 0.266 |
| L1 | 1.412 | 0.443 | 3.267 | 2.559 | 1.046 | 0.15 | −8443.984 | 4.246 |
| L0 (γcod = 0) | - | 0.447 | 0.833 | 2.569 | - | - | −8462.24 | 34.758 |
| b) X chromosome | ||||||||
|---|---|---|---|---|---|---|---|---|
| Model | γcod | γint | κcod | κint | g | τ | lnL | ΔAIC |
| L1 | 1.287 | 0.76 | 2.155 | 3.033 | 2.07 | 0.31 | −6021.439 | 0 |
| L0 | 1.509 | 0.868 | 2.656 | 3.353 | - | - | −6023.904 | 0.93 |
| L0 (γint = 0) | 1.521 | - | 2.698 | 1.443 | - | - | −6027.873 | 6.868 |
| L0 (γcod = 0) | - | 0.876 | 0.616 | 3.383 | - | - | −6032.664 | 16.45 |
N1 and N2 are the effective population sizes before and after a demographic event. L0 is a model without a demographic event and L1 is a model with demographic event. γcod = 2N1scod and γint = 2N1sint, where scod and sint are the selection coefficients against heterozygous carriers of the unpreferred variants for coding region and AT variants for intron regions respectively. κcod and κint, is the mutation bias towards unpreferred variants and AT variants respectively. From the L1 model, g = N1/N2 and τ is the time since the demographic event in units of 2N2 generations. lnL, log-likelihood estimate of the model. ΔAIC, difference in the Akaike Information Criterion of the model in question with the model with the smallest AIC value
No significant demographic changes were detected in the autosomal data consisting of both synonymous and intron polymorphisms (Table 2a row 2 versus row 3, FDR corrected p-value = 1.0). While for the X chromosome data the model forcing a change in population size had the lowest AIC scores, this was still not a significantly better fit than the less parameterized model assuming no change in demography (Table 2b row 2 versus row 1, FDR corrected p-value = 0.167). When the autosomes and X chromosome were analyzed separately there was significant evidence of selection for preferred codons (Autosome: Table 2a row 2 versus row 4, FDR corrected p-value = 1.5e-8; X chromosome: Table 2b row 2 versus row 4, FDR corrected p-value = 2.0e-4).
In the autosomal intron dataset the original model was not a significantly better fit than the less parameterized model forcing no preference for GC variants (γint = 0) (Table 2a row 1 versus row 2, FDR corrected p-value = 0.317). On the other hand in the X chromosomal introns, the original model was a significantly better fit than the less parameterized model forcing γint=0 (Table 2b row 3 versus row 2, FDR corrected p-value = 0.016). This result contrasts with our FDD analysis of intron sites that had not found any evidence of gcBGC. We note, however, that the FDD test was based on 6 intron sequences while the maximum-likelihood model was based on 16 intron sequences. Thus, a lack of sequences and loss of power in the former analysis is a likely reason for this difference.
As our previous intron analysis suggested potential effects of biased mutation rate or gcBGC on the X chromosome, we examined if this would have led to different evolutionary patterns between the autosomes and X chromosome synonymous sites. Synonymous polymorphism data was analyzed using a modified version of the Zeng and Charlesworth (2009) method to handle the X chromosome and autosome data together (Haddrill et al. 2011). This method estimated the same parameters from Table 2 for coding sequences and results are shown in Table 3 listed from the model with lowest to highest AIC values.
Table 3.
Estimates of selection, mutation, and demography for synonymous sites on the autosomes and X chromosome.
| Model | γA | γX | κ | λ | g | τ | lnL | ΔAIC |
|---|---|---|---|---|---|---|---|---|
| L0 (λ = 0.75) | 1.38 | 2.24 | 3.29 | - | - | - | −8227.88 | 0 |
| L0 | 1.37 | 1.86 | 1.48 | 0.90 | - | - | −8226.88 | 0.01 |
| L1 | 1.26 | 1.68 | 0.80 | 0.93 | 1.71 | 4.64 | −8226.74 | 3.73 |
| L0 (γX = γA) | 1.45 | - | 3.36 | 1.20 | - | - | −8229.96 | 4.17 |
| L0 (γX = λγA) | - | 1.78 | 1.67 | 0.84 | - | - | −8248.41 | 41.07 |
| L0 (γA = 0) | - | 0.35 | 1.70 | 0.94 | - | - | −8250.16 | 44.56 |
| L0 (γX = 0) | −0.25 | - | 2.38 | 0.94 | - | - | −8261.42 | 67.08 |
N1 and N2 are the effective population sizes of autosomes before and after a demographic event, respectively. L0 is a model without a demographic event and L1 is a model with demographic event. γA = 2N1sA and γX = 2N1sX, where sA and sX are the selection coefficients against heterozygous carriers of the unpreferred variants; κ, mutation bias towards unpreferred variants; λ, ratio of effective population size of X chromosome to autosome. From the L1 model, g = N1/N2 and τ, time since the demographic event in units of 2N2 generations. lnL, log-likelihood estimate of the model. ΔAIC, difference in the Akaike Information Criterion of the model in question with the model with the smallest AIC value.
Concordant with the intron data, jointly analyzing the autosomal and X chromosome data with a model assuming an instantaneous change in the population size was not a significantly better fit than the less parameterized model assuming no demographic variation (Table 3 row 2 versus row 3, FDR corrected p-value = 1.0). We thus carried out the rest of our analysis without demography as an additional parameter.
Estimates of selection for preferred codons were significantly different from zero for both autosomal (Table 3 row 2 versus row 6, FDR corrected p-value = 1.45e-10) and X chromosomal (Table 3 row 2 versus row 7, FDR corrected p-value = 2.72e-15) polymorphisms. We next examined if there were any differences in the strength of selection for preferred codons between the autosomes and X chromosome. A model assuming equal coefficients of selection (s) for preferred codons between the autosomes and X chromosome (sA = sX) was not significantly better fit than the original model that parameterized sA and sX separately (Table 3 row 2 versus row 5, FDR corrected p-value = 6.5e-10). Further, a model forcing equal intensity of selection (γ = Nes) between the autosome and X chromosome (γA = γX) was significantly worse fit than the original model that parameterized γA and γX separately (Table 3 row 2 versus row 4, FDR corrected p-value = 0.034).
We then estimated if there were differences in the effective population sizes (Ne) for the X chromosome and autosomes, as difference in Ne can affect the selection intensity (Nes). With no sexual selection males and females have equal reproductive successes and the ratio of X chromosome to autosome effective population size (λ) is expected to be 3/4 (Wright 1931). Our original model incorporated λ as a free parameter and estimated it as 0.9, however, this was not significantly better fit than a model forcing λ = 0.75 and has one less parameter to be estimated (Table 3 row 1 versus row 2, FDR corrected p-value = 0.275).
Discussion
We find that selection on codon usage in D. ananassae favors the use of preferred codons, corroborating previous studies (Heger and Ponting 2007; Singh et al. 2008; Choi and Aquadro 2014). Further, in this study we have used specific population genetic models and tests to examine synonymous site evolution across both short- and long-term evolutionary time scales, and found significant evidence of selection for preferred codons on both temporal scales.
We analyzed selection across synonymous sites while factoring in demography as an additional possible parameter that could influence the selection on synonymous polymorphisms (Zeng and Charlesworth 2009). Analyzing both the autosome and X chromosome coding and intron sequences, there was no significant evidence for a change in population size in the ancestral Bangkok population of D. ananassae. Compared to previous studies, however, the same X chromosome intron dataset used in this study (Das et al. 2004) had site frequency based tests (Das et al. 2004) and coalescent-based methods (Heled and Drummond 2008) suggesting a population expansion in the Bangkok population of D. ananassae. The fact that the method we used in this study jointly estimated the parameters of selection, mutation, and demography may have allowed selection and mutation to be distinguished from demography, and account for our lack of evidence for a population expansion.
Using the polymorphism data we then estimated the strength of selection (γ = Nes) for preferred codons on the X chromosome (γX) and found it to be significantly higher than on the autosomes (γA) (Table 3: γA=1.38 vs γX=2.24). Further, the underlying selective coefficients of γX and γA were significantly different from one another (sA ≠ sX) suggesting that selection for CUB is significantly higher on the X chromosome compared to the autosomes. Compared to other Drosophila studies that have used the same method, our estimates of γA and γX in D. ananassae (γA=1.38; γX=2.24) were lower than D. miranda (γA=1.56; γX=2.64) and D. pseudoobscura (γA=1.77; γX=2.92) (Haddrill et al. 2011); comparable only to the autosomes in D. melanogaster (γA=1.36; γX=1.53) (Campos et al. 2013); and higher than the γX in D. americana (γX =1.55) (de Procé et al. 2012). If the selection coefficient (s) for preferred codons is the same across all Drosophila species, this observed difference in γA and γX could be due to differences in the effective population sizes (Ne) among species. On the other hand, non-selective forces such as biased gene conversion may confound these estimates. Notably in Drosophila rates of substitution are heterogeneous and base composition has not reached equilibrium among different lineages (Singh et al. 2009). Thus our interpretation of the observed patterns of γ among the different Drosophila species is preliminary and would benefit from additional data and study.
We found that the X chromosomal introns from Das et al. (2004) had evidence of increased preference for GC variants over AT variants (Table 2) likely from GC biased gene conversion (gcBGC). Population genetic theory predicts nucleotides under gcBGC to appear falsely as alleles under positive selection (Duret and Galtier 2009; Nagylaki 1983), thus biasing the background allele frequency spectrum. In fact, when we had forced our model to include demographic changes, the X chromosome data indicated a very recent increase in population size (Table 2b). However, this was not a significantly better fit then our less parameterized model that assumed no demographic change.
Our polymorphism analysis also revealed that the ratio of X chromosome to autosome effective population size (λ) was not significantly different from the expected ratio of 3/4 (Table 3) (Wright 1931). Our estimate of λ was consistent with results of Grath et al. (2009). Additionally, because theory predicts λ to exceed the expected value of 3/4 with population expansions (Pool and Nielsen 2007), our estimate of λ = 0.75 in D. ananassae is consistent with the view that the Bangkok population of D. ananassae did not have a recent change in population size.
Our estimates of the intensity of selection, γ (=Nes), for preferred codons were higher for the X chromosome versus the autosomes (Table 3), which is in line with previous studies (Singh et al. 2005a; Singh et al. 2008; Haddrill et al. 2011; Campos et al. 2013). Further, theory predicts the ratio of the selective coefficients on the X chromosome to the autosome (sX/sA) would equal 4/3 (≈ 1.33), when males and females have an equal chance of mating and selection for CUB is semidominant (Vicoso and Charlesworth 2009). As γX and γA both share the term Ne their ratios would reduce to sX/sA and since we had evidence that λ = 3/4 in D. ananassae, males and females appear to have an equal mating chance resulting in sX/sA to equal 4/3 (≈ 1.33). However, with our estimated γA = 1.38 and γX = 2.24 in D. ananassae, the observed ratio sX/sA was 1.62 which is greater than the predicted ratio of 1.33. Our result is consistent with Haddrill et al. (2011) who found sX/sA = 1.65 in D. pseudoobscura and sX/sA = 1.69 in D. miranda. Thus in D. ananassae the ratio of selection coefficient (s) for preferred codons on the X chromosome compared to the autosome (sX/sA) is also higher than theoretical predictions of sX/sA.
Why the X chromosome would have a higher selection coefficient than the autosomes is unknown. As codon bias is strongest among genes with higher gene expression (Duret and Mouchiroud 1999) X chromosomal genes could be expressed at higher expression than autosomal genes. We investigated this possibility by reanalyzing the microarray data of Grath et al. (2009) focusing on genes with sex unbiased gene expression. Examination of all six biological and two technical replicates for the 63 unbiased genes showed no significant gene expression differences (Mann-Whitney U test FDR corrected p-value > 0.05) between the X and autosomal genes. Thus the higher selection on the X chromosome was not likely to be due to overall differences in gene expression.
On the other hand, Haddrill et al. (2011) have proposed differences in the effective rate of recombination between the X chromosome and autosomes as a potential cause for the observed difference. Because of an absence of male recombination in most species of Drosophila (Orr-Weaver 1995) autosomes are expected to have a reduced rate of recombination compared to X chromosomes (Langley et al. 1988), ultimately leading to a difference in the “effective” recombination rate. gcBGC is thought to occur during recombination when repair of double strand breaks favors G or C basepairs over A or T basepairs (Marais 2003). As most preferred codons end with G or C nucleotides in Drosophila, the higher recombination rate on the X chromosome compared to the autosome could lead to higher levels of gcBGC on the X chromosome. This would then subsequently lead to apparent increased selection for preferred codons on the X chromosome.
Studies in D. ananassae, however, have shown evidence of recombination occurring regularly in males (Tobari 1993). Here, the male recombination is expected to increase the effective recombination rate on the autosomes in D. ananassae and will result in higher levels of gcBGC across the autosomes of this species. Our analysis of D. ananassae autosomal and X chromosomal intron data revealed that only the X-linked introns had significant evidence of preference for GC variants over AT variants consistent with gcBGC (Table 2). This suggested that the X chromosome has elevated gcBGC events and the joint effects from gcBGC and selection for preferred codons have both led to patterns of variation that might inflate estimates of selection for preferred codons on the X. As male recombination would elevate effective recombination rates on the autosomes for D. ananassae an elevation of gcBGC should also occur in the autosomes as well. Thus the potentially higher gcBGC effects on the X chromosome alone cannot fully explain the higher selection for CUB in the X chromosome versus autosome. We note, however, recent evidence from D. melanogaster has shown that gene conversions can occur at sites that were not associated with crossing-overs during recombination (Comeron et al. 2012). Thus, rates of recombination maybe a poor predictor of gcBGC and it is possible that in D. ananassae the X chromosome has higher levels of gcBGC despite the lower effective recombination rate than the autosomes.
Finally, our evidence of gcBGC in D. ananassae is interesting as support for gcBGC from previous Drosophila studies has been inconclusive. Although several studies have supported significant gcBGC effects in Drosophila (Marais et al. 2001; Singh et al. 2005b; Haddrill and Charlesworth 2008) recent studies based on whole genome level recombination rates and polymorphisms from D. melanogaster have suggested otherwise (Comeron et al. 2012; Robinson et al. 2014). Robinson et al. (2014) has suggested the different amount of gcBGC between the X chromosome and autosomes could be due to a shift in the mutational processes. Here, analysis of short introns from the X chromosome and autosomes of D. ananassae, which were previously shown to be close to neutrality in Drosophila (Haddrill et al. 2005; Halligan and Keightley 2006; Singh et al. 2009; Robinson et al. 2014), would indicate any potential difference in the mutational process.
In conclusion we find evidence of both recent and long-term selection for preferred codons in D. ananassae, and that the strength of this selection is higher on the X chromosome versus the autosomes. Differences in recombination rate associated gcBGC do not appear to be sufficient to explain this difference in selection between the autosomes and X chromosomes in D. ananassae. Future studies examining the D. ananassae genome-wide recombination rate and generation of whole genome polymorphism data would help elucidate the cause of this difference in strength of selection between the autosomes and X chromosome.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of Health grant number R01GM095793 to C.F.A. and Daniel A. Barbash, and a Cornell Center for Comparative and Population Genomics (3CPG) Scholar Award to J.Y.C. We thank Kai Zeng for reprogramming his software for our polymorphism analysis. We thank Sonja Grath for assisting our analysis of the D. ananassae microarray data. We also thank Vanessa Bauer DuMont and the two reviewers for helpful discussions and comments.
References
- Akashi H. Synonymous codon usage in Drosophila melanogaster: natural selection and translational accuracy. Genetics. 1994;136:927–935. doi: 10.1093/genetics/136.3.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi H. Inferring weak selection from patterns of polymorphism and divergence at “silent” sites in Drosophila DNA. Genetics. 1995;139:1067–1076. doi: 10.1093/genetics/139.2.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi H. Molecular evolution between Drosophila melanogaster and D. simulans: reduced codon bias, faster rates of amino acid substitution, and larger proteins in D. melanogaster . Genetics. 1996;144:1297–1307. doi: 10.1093/genetics/144.3.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi H. Codon bias evolution in Drosophila. Population genetics of mutation-selection drift. Gene. 1997;205:269–278. doi: 10.1016/s0378-1119(97)00400-9. [DOI] [PubMed] [Google Scholar]
- Akashi H. Inferring the fitness effects of DNA mutations from polymorphism and divergence data: statistical power to detect directional selection under stationarity and free recombination. Genetics. 1999;151:221–238. doi: 10.1093/genetics/151.1.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi H, Schaeffer SW. Natural selection and the frequency distributions of “silent” DNA polymorphism in Drosophila . Genetics. 1997;146:295–307. doi: 10.1093/genetics/146.1.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andolfatto P. Hitchhiking effects of recurrent beneficial amino acid substitutions in the Drosophila melanogaster genome. Genome Res. 2007;17:1755–1762. doi: 10.1101/gr.6691007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andolfatto P, Wong KM, Bachtrog D. Effective population size and the efficacy of selection on the X chromosomes of two closely related Drosophila species. Genome Biol Evol. 2011;3:114–128. doi: 10.1093/gbe/evq086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachtrog D. Evidence for male-driven evolution in Drosophila . Mol. Biol. Evol. 2008;25:617–619. doi: 10.1093/molbev/msn020. [DOI] [PubMed] [Google Scholar]
- Bauer DuMont VL, Singh ND, Wright MH, Aquadro CF. Locus-specific decoupling of base composition evolution at synonymous sites and introns along the Drosophila melanogaster and Drosophila sechellia lineages. Genome Biol Evol. 2009;1:67–74. doi: 10.1093/gbe/evp008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J R Stat Soc Series B Stat Methodol. 1995;57:289–300. [Google Scholar]
- Betancourt AJ, Presgraves DC. Linkage limits the power of natural selection in Drosophila . Proc. Natl. Acad. Sci. U.S.A. 2002;99:13616–13620. doi: 10.1073/pnas.212277199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulmer M. The selection-mutation-drift theory of synonymous codon usage. Genetics. 1991;129:897–907. doi: 10.1093/genetics/129.3.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos JL, Zeng K, Parker DJ, Charlesworth B, Haddrill PR. Codon usage bias and effective population sizes on the X chromosome versus the autosomes in Drosophila melanogaster . Mol. Biol. Evol. 2013;30:811–823. doi: 10.1093/molbev/mss222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlini DB, Stephan W. In vivo introduction of unpreferred synonymous codons into the Drosophila Adh gene results in reduced levels of ADH protein. Genetics. 2003;163:239–243. doi: 10.1093/genetics/163.1.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamary JV, Parmley JL, Hurst LD. Hearing silence: non-neutral evolution at synonymous sites in mammals. Nat. Rev. Genet. 2006;7:98–108. doi: 10.1038/nrg1770. [DOI] [PubMed] [Google Scholar]
- Chen SL, Lee W, Hottes AK, Shapiro L, McAdams HH. Codon usage between genomes is constrained by genome-wide mutational processes. Proc. Natl. Acad. Sci. U.S.A. 2004;101:3480–3485. doi: 10.1073/pnas.0307827100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z-X, Sturgill D, Qu J, Jiang H, Park S, Boley N, Suzuki AM, Fletcher AR, Plachetzki DC, FitzGerald PC, et al. Comparative validation of the D. melanogaster modENCODE transcriptome annotation. Genome Res. 2014;24:1209–1223. doi: 10.1101/gr.159384.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JY, Aquadro CF. The coevolutionary period of Wolbachia pipientisinfecting Drosophila ananassae and its impact on the evolution of the host germline stem cell regulating genes. Mol Biol Evol. 2014;31:2457–2471. doi: 10.1093/molbev/msu204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comeron JM. Weak selection and recent mutational changes influence polymorphic synonymous mutations in humans. Proc. Natl. Acad. Sci. U.S.A. 2006;103:6940–6945. doi: 10.1073/pnas.0510638103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comeron JM, Ratnappan R, Bailin S. The many landscapes of recombination in Drosophila melanogaster . PLoS Genet. 2012;8:e1002905. doi: 10.1371/journal.pgen.1002905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutter AD, Charlesworth B. Selection intensity on preferred codons correlates with overall codon usage bias in Caenorhabditis remanei . Curr. Biol. 2006;16:2053–2057. doi: 10.1016/j.cub.2006.08.067. [DOI] [PubMed] [Google Scholar]
- Das A, Mohanty S, Stephan W. Inferring the Population Structure and Demography of Drosophila ananassae From Multilocus Data. Genetics. 2004;168:1975–1985. doi: 10.1534/genetics.104.031567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond DA, Wilke CO. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell. 2008;134:341–352. doi: 10.1016/j.cell.2008.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuMont VB, Fay JC, Calabrese PP, Aquadro CF. DNA variability and divergence at the notch locus in Drosophila melanogaster and D. simulans: a case of accelerated synonymous site divergence. Genetics. 2004;167:171–185. doi: 10.1534/genetics.167.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L, Mouchiroud D. Expression pattern and, surprisingly, gene length shape codon usage in Caenorhabditis, Drosophila, and Arabidopsis . Proc. Natl. Acad. Sci. U.S.A. 1999;96:4482–4487. doi: 10.1073/pnas.96.8.4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L, Galtier N. Biased gene conversion and the evolution of mammalian genomic landscapes. Annu Rev Genomics Hum Genet. 2009;10:285–311. doi: 10.1146/annurev-genom-082908-150001. [DOI] [PubMed] [Google Scholar]
- Galtier N, Bazin E, Bierne N. GC-biased segregation of noncoding polymorphisms in Drosophila . Genetics. 2006;172:221–228. doi: 10.1534/genetics.105.046524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouy M, Gautier C. Codon usage in bacteria: correlation with gene expressivity. Nucleic Acids Res. 1982;10:7055–7074. doi: 10.1093/nar/10.22.7055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grath S, Baines JF, Parsch J. Molecular evolution of sex-biased genes in the Drosophila ananassae subgroup. BMC Evol. Biol. 2009;9:291. doi: 10.1186/1471-2148-9-291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddrill PR, Charlesworth B, Halligan DL, Andolfatto P. Patterns of intron sequence evolution in Drosophila are dependent upon length and GC content. Genome Biol. 2005;6:R67. doi: 10.1186/gb-2005-6-8-r67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddrill PR, Charlesworth B. Non-neutral processes drive the nucleotide composition of non-coding sequences in Drosophila. Biol. Lett. 2008;4:438–441. doi: 10.1098/rsbl.2008.0174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddrill PR, Zeng K, Charlesworth B. Determinants of synonymous and nonsynonymous variability in three species of Drosophila . Mol. Biol. Evol. 2011;28:1731–1743. doi: 10.1093/molbev/msq354. [DOI] [PubMed] [Google Scholar]
- Halligan DL, Keightley PD. Ubiquitous selective constraints in the Drosophila genome revealed by a genome-wide interspecies comparison. Genome Res. 2006;16:875–884. doi: 10.1101/gr.5022906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambuch TM, Parsch J. Patterns of synonymous codon usage in Drosophila melanogaster genes with sex-biased expression. Genetics. 2005;170:1691–1700. doi: 10.1534/genetics.104.038109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl DL, Moriyama EN, Sawyer SA. Selection intensity for codon bias. Genetics. 1994;138:227–234. doi: 10.1093/genetics/138.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heger A, Ponting CP. Variable Strength of Translational Selection Among 12 Drosophila Species. Genetics. 2007;177:1337–1348. doi: 10.1534/genetics.107.070466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heled J, Drummond AJ. Bayesian inference of population size history from multiple loci. BMC Evol. Biol. 2008;8:289. doi: 10.1186/1471-2148-8-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberg R, Petrov DA. Selection on codon bias. Annu. Rev. Genet. 2008;42:287–299. doi: 10.1146/annurev.genet.42.110807.091442. [DOI] [PubMed] [Google Scholar]
- Hill WG, Robertson A. The effect of linkage on limits to artificial selection. Genet. Res. 1966;8:269–294. [PubMed] [Google Scholar]
- Hudson RR, Kreitman M, Aguadé M. A test of neutral molecular evolution based on nucleotide data. Genetics. 1987;116:153–159. doi: 10.1093/genetics/116.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikemura T. Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes: a proposal for a synonymous codon choice that is optimal for the E. coli translational system. J. Mol. Biol. 1981;151:389–409. doi: 10.1016/0022-2836(81)90003-6. [DOI] [PubMed] [Google Scholar]
- Ikemura T. Codon usage and tRNA content in unicellular and multicellular organisms. Mol. Biol. Evol. 1985;2:13–34. doi: 10.1093/oxfordjournals.molbev.a040335. [DOI] [PubMed] [Google Scholar]
- Kim Y. Effect of strong directional selection on weakly selected mutations at linked sites: implication for synonymous codon usage. Mol. Biol. Evol. 2004;21:286–294. doi: 10.1093/molbev/msh020. [DOI] [PubMed] [Google Scholar]
- Langley CH, Montgomery E, Hudson R, Kaplan N, Charlesworth B. On the role of unequal exchange in the containment of transposable element copy number. Genet. Res. 1988;52:223–235. doi: 10.1017/s0016672300027695. [DOI] [PubMed] [Google Scholar]
- Li WH. Models of nearly neutral mutations with particular implications for nonrandom usage of synonymous codons. J. Mol. Evol. 1987;24:337–345. doi: 10.1007/BF02134132. [DOI] [PubMed] [Google Scholar]
- Marais G, Mouchiroud D, Duret L. Does recombination improve selection on codon usage? Lessons from nematode and fly complete genomes. Proc. Natl. Acad. Sci. U.S.A. 2001;98:5688–5692. doi: 10.1073/pnas.091427698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marais G. Biased gene conversion: implications for genome and sex evolution. Trends Genet. 2003;19:330–338. doi: 10.1016/S0168-9525(03)00116-1. [DOI] [PubMed] [Google Scholar]
- Marion de Procé S, Halligan DL, Keightley PD, Charlesworth B. Patterns of DNA-sequence divergence between Drosophila miranda and D. pseudoobscura. J. Mol. Evol. 2009;69:601–611. doi: 10.1007/s00239-009-9298-2. [DOI] [PubMed] [Google Scholar]
- Maside X, Lee AW, Charlesworth B. Selection on codon usage in Drosophila americana . Curr. Biol. 2004;14:150–154. doi: 10.1016/j.cub.2003.12.055. [DOI] [PubMed] [Google Scholar]
- McDonald JH, Kreitman M. Adaptive protein evolution at the Adh locus in Drosophila . Nature. 1991;351:652–654. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- McVean GA, Charlesworth B. The effects of Hill-Robertson interference between weakly selected mutations on patterns of molecular evolution and variation. Genetics. 2000;155:929–944. doi: 10.1093/genetics/155.2.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVean GAT, Charlesworth B. A population genetic model for the evolution of synonymous codon usage: patterns and predictions. Genet Res. 1999;74:145–158. [Google Scholar]
- McVean GA, Vieira J. Inferring parameters of mutation, selection and demography from patterns of synonymous site evolution in Drosophila . Genetics. 2001;157:245–257. doi: 10.1093/genetics/157.1.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagylaki T. Evolution of a finite population under gene conversion. Proc. Natl. Acad. Sci. U.S.A. 1983;80:6278–6281. doi: 10.1073/pnas.80.20.6278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- Nielsen R, Bauer DuMont VL, Hubisz MJ, Aquadro CF. Maximum likelihood estimation of ancestral codon usage bias parameters in Drosophila . Mol. Biol. Evol. 2007;24:228–235. doi: 10.1093/molbev/msl146. [DOI] [PubMed] [Google Scholar]
- Ohta T. Slightly deleterious mutant substitutions in evolution. Nature. 1973;246:96–98. doi: 10.1038/246096a0. [DOI] [PubMed] [Google Scholar]
- Orr-Weaver TL. Meiosis in Drosophila: seeing is believing. Proc Natl Acad Sci U S A. 1995;92:10443–10449. doi: 10.1073/pnas.92.23.10443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pool JE, Nielsen R. Population size changes reshape genomic patterns of diversity. Evolution. 2007;61:3001–3006. doi: 10.1111/j.1558-5646.2007.00238.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell JR, Sezzi E, Moriyama EN, Gleason JM, Caccone A. Analysis of a shift in codon usage in Drosophila . J. Mol. Evol. 2003;(57 Suppl 1):S214–S225. doi: 10.1007/s00239-003-0030-3. [DOI] [PubMed] [Google Scholar]
- de Procé SM, Zeng K, Betancourt AJ, Charlesworth B. Selection on codon usage and base composition in Drosophila americana . Biol. Lett. 2012;8:82–85. doi: 10.1098/rsbl.2011.0601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu S, Zeng K, Slotte T, Wright S, Charlesworth D. Reduced efficacy of natural selection on codon usage bias in selfing Arabidopsis and Capsella species. Genome Biol Evol. 2011;3:868–880. doi: 10.1093/gbe/evr085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MC, Stone EA, Singh ND. Population genomic analysis reveals no evidence for GC-biased gene conversion in Drosophila melanogaster. Mol. Biol. Evol. 2014;31:425–433. doi: 10.1093/molbev/mst220. [DOI] [PubMed] [Google Scholar]
- Schug MD, Smith SG, Tozier-Pearce A, McEvey SF. The genetic structure of Drosophila ananassae populations from Asia, Australia and Samoa. Genetics. 2007;175:1429–1440. doi: 10.1534/genetics.106.066613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sella G, Petrov DA, Przeworski M, Andolfatto P. Pervasive natural selection in the Drosophila genome? PLoS Genet. 2009;5:e1000495. doi: 10.1371/journal.pgen.1000495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PM, Li WH. The codon Adaptation Index--a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987;15:1281–1295. doi: 10.1093/nar/15.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonsen KL, Churchill GA, Aquadro CF. Properties of statistical tests of neutrality for DNA polymorphism data. Genetics. 1995;141:413–429. doi: 10.1093/genetics/141.1.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh ND, Arndt PF, Clark AG, Aquadro CF. Strong evidence for lineage and sequence specificity of substitution rates and patterns in Drosophila. Mol. Biol. Evol. 2009;26:1591–1605. doi: 10.1093/molbev/msp071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh ND, Arndt PF, Petrov DA. Minor shift in background substitutional patterns in the Drosophila saltans and willistoni lineages is insufficient to explain GC content of coding sequences. BMC Biol. 2006;4:37. doi: 10.1186/1741-7007-4-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh ND, Bauer DuMont VL, Hubisz MJ, Nielsen R, Aquadro CF. Patterns of mutation and selection at synonymous sites in Drosophila . Mol. Biol. Evol. 2007;24:2687–2697. doi: 10.1093/molbev/msm196. [DOI] [PubMed] [Google Scholar]
- Singh ND, Davis JC, Petrov DA. X-linked genes evolve higher codon bias in Drosophila and Caenorhabditis . Genetics. 2005a;171:145–155. doi: 10.1534/genetics.105.043497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh ND, Arndt PF, Petrov DA. Genomic heterogeneity of background substitutional patterns in Drosophila melanogaster. Genetics. 2005b;169:709–722. doi: 10.1534/genetics.104.032250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh ND, Larracuente AM, Clark AG. Contrasting the efficacy of selection on the X and autosomes in Drosophila . Mol. Biol. Evol. 2008;25:454–467. doi: 10.1093/molbev/msm275. [DOI] [PubMed] [Google Scholar]
- Tobari YN. Drosophila ananassae: genetical and biological aspects. Japan Scientific Societies Press. 1993 [Google Scholar]
- Vicario S, Moriyama EN, Powell JR. Codon usage in twelve species of Drosophila . BMC Evol. Biol. 2007;7:226. doi: 10.1186/1471-2148-7-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicoso B, Charlesworth B. Effective population size and the faster-X effect: an extended model. Evolution. 2009;63:2413–2426. doi: 10.1111/j.1558-5646.2009.00719.x. [DOI] [PubMed] [Google Scholar]
- Vogl C, Das A, Beaumont M, Mohanty S, Stephan W. Population subdivision and molecular sequence variation: theory and analysis of Drosophila ananassae data. Genetics. 2003;165:1385–1395. doi: 10.1093/genetics/165.3.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright F. The “effective number of codons” used in a gene. Gene. 1990;87:23–29. doi: 10.1016/0378-1119(90)90491-9. [DOI] [PubMed] [Google Scholar]
- Wright S. Evolution in Mendelian Populations. Genetics. 1931;16:97–159. doi: 10.1093/genetics/16.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Nielsen R. Mutation-selection models of codon substitution and their use to estimate selective strengths on codon usage. Mol. Biol. Evol. 2008;25:568–579. doi: 10.1093/molbev/msm284. [DOI] [PubMed] [Google Scholar]
- Zaborske JM, DuMont VLB, Wallace EWJ, Pan T, Aquadro CF, Drummond DA. A nutrient-driven tRNA modification alters translational fidelity and genome-wide protein coding across an animal genus. PLoS Biol. 2014;12:e1002015. doi: 10.1371/journal.pbio.1002015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng K. A simple multiallele model and its application to identifying preferred-unpreferred codons using polymorphism data. Mol. Biol. Evol. 2010;27:1327–1337. doi: 10.1093/molbev/msq023. [DOI] [PubMed] [Google Scholar]
- Zeng K. The application of population genetics in the study of codon usage bias. In: Cannarozzi GM, Schneider A, editors. Codon Evolution: Mechanisms and Models. OUP Oxford: 2012. [Google Scholar]
- Zeng K, Charlesworth B. Estimating selection intensity on synonymous codon usage in a nonequilibrium population. Genetics. 2009;183:651–662. doi: 10.1534/genetics.109.101782. 1SI-23SI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng K, Charlesworth B. Studying patterns of recent evolution at synonymous sites and intronic sites in Drosophila melanogaster . J. Mol. Evol. 2010;70:116–128. doi: 10.1007/s00239-009-9314-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.