

Abstract
Background
Immunoactivation is less evident in secondary progressive MS (SPMS) compared to relapsing-remitting disease. MicroRNA (miRNA) expression is integral to the regulation of gene expression; determining their impact on immune-related cell functions, especially CD4+ T cells, during disease progression will advance our understanding of MS pathophysiology. This study aimed to compare miRNA profiles of CD4+ T cells from SPMS patients to healthy controls (HC) using whole miRNA transcriptome next-generation sequencing (NGS). Total RNA was extracted from CD4+ T cells and miRNA expression patterns analyzed using Illumina-based small-RNA NGS in 12 SPMS and 12 HC samples. Results were validated in a further cohort of 12 SPMS and 10 HC by reverse transcription quantitative polymerase chain reaction (RT-qPCR).
Results
The ten most dysregulated miRNAs identified by NGS were selected for qPCR confirmation; five (miR-21-5p, miR-26b-5p, miR-29b-3p, miR-142-3p, and miR-155-5p) were confirmed to be down-regulated in SPMS (p < 0.05). SOCS6 is targeted by eight of these ten miRNAs. Consistent with this, SOCS6 expression is up-regulated in SPMS CD4+ T cells (p < 0.05). This is of particular interest as SOCS6 has previously been shown to act as a negative regulator of T cell activation.
Conclusions
Ninety-seven percent of miRNA candidates identified by NGS were down-regulated in SPMS. The down-regulation of miRNAs and increased expression of SOCS6 in SPMS CD4+ T cells may contribute to reduced immune system activity in progressive MS.
Electronic supplementary material
The online version of this article (doi:10.1186/s13148-016-0253-y) contains supplementary material, which is available to authorized users.
Keywords: Multiple sclerosis, Secondary progressive, MicroRNAs, Immunology, CD4+ T cells, Next-generation sequencing
Background
Multiple sclerosis (MS) is an autoimmune disease characterized by multifocal inflammatory attacks in the CNS [1]. In the relapsing-remitting (RRMS) stage of the disease, CD4+ T cells are among the primary infiltrators moving from the periphery, through the blood-brain barrier, and into the CNS [2]. These cells then initiate an immune response that results in localized demyelination and corresponding symptoms. The later stage of MS, secondary progressive (SPMS), is characterized by compounding neurodegeneration and increasing disability; however, the relevance of inflammation is unclear [3]. As key regulators of gene expression, microRNAs (miRNAs) may be affecting the immune-related functions of CD4+ T cells in SPMS and may help to elucidate the actions of these cells in SPMS.
MiRNAs are short, non-coding RNA molecules (~22 bp) that regulate gene expression at the posttranscriptional stage by targeting the 3′ untranslated region of target genes. Their small size and stable structure make them ideal biomarkers. In recent years, miRNA expression patterns in MS have been the focus of numerous studies, many of which have concentrated on using miRNAs as biomarkers for diagnosis and prognosis [4]. These studies predominantly use easily acquired (and often highly heterogeneous) samples such as whole blood, peripheral blood mononuclear cells (PBMCs), serum, and plasma. Numerous dysregulated miRNAs have been identified, however which cell types are actually responsible for differing miRNA profiles, and the consequences of altered miRNA expression is not clear in many studies. Furthermore, it is likely that these heterogeneous samples are masking the signal of differentially expressed miRNA in specific cell subtypes. To overcome this, we have focused on CD4+ T cells in this study on SPMS.
Next-generation sequencing (NGS) allows for stringent examination of cell-specific miRNA expression profiles as well as discovery of previously uncharacterized miRNAs. Here, we have used small-RNA NGS analysis of CD4+ T cells from SPMS patients and healthy controls (HC). The total coverage approach of NGS generates expression information on all small RNA species including all known and novel miRNAs, as well as other small RNA species (isomiRs and snoRNAs)—a clear advantage over microarray and candidate approach assays. Three previous studies in MS have used NGS to effectively identify miRNA expression profiles in the whole blood [5, 6], serum [6], and PBMCs [7] from RRMS patients. However, NGS techniques have not been used for specific cell types or in SPMS samples.
The miRNA expression profile of CD4+ T cells, either as instigating molecules or by-products of erroneous molecular mechanisms, will provide insight into the function of these cells in SPMS. Here, we used NGS to provide a comprehensive analysis of the miRNA expression profiles of CD4+ T cells from SPMS patients and healthy controls (HC) and confirmed these results using targeted expression assays.
Methods
Sample collection
Whole blood was collected at a single study center from an initial cohort of 12 SPMS patients and 12 HC and a replication cohort of 12 SPMS and 10 HC. All patients were diagnosed with SPMS according to the McDonald criteria [8] and demonstrated EDSS progression without evidence of relapse in the 24 months prior to collection. Controls were age (±5 years) and gender matched (Table 1). The SPMS patient group was free of MS-specific treatments for a minimum period of 6 months prior to collection. Samples were collected at the John Hunter Hospital, and laboratory work was conducted at the Hunter Medical Research Institute, Newcastle.
Table 1.
Details of SPMS and healthy control individuals
| Next generation sequencing | Replication cohort | |||
|---|---|---|---|---|
| SPMS | HC | SPMS | HC | |
| Number | 12 | 12 | 12 | 10 |
| Female | 9 | 9 | 8 | 5 |
| Age in years (mean ± SD) | 60.2 ± 8.3 | 61.3 ± 9.5 | 61.4.0 ± 6.5 | 60.1 ± 5.9 |
| EDSS (mean ± SD) | 6.9 ± 0.9 | NA | 5.9 ± 1.0 | NA |
| Active SPMS | 3 | NA | 4 | NA |
| Disease duration in years (mean ± SD) | 25.6 ± 11.1 | NA | 18.3 ± 6.5 | NA |
| Progression duration (mean ± SD) | 10.8 ± 8.1 | NA | 8.9 ± 6.2 | NA |
EDSS expanded disability status scale, SD standard deviation, NA not applicable
Blood sample processing
PBMCs were isolated from 45 mL of heparinized whole blood by density gradient centrifugation on lymphoprep (Axis-Shield PoC AS, Norway). CD4+ T cells were enriched from the PBMCs using EasySep magnetic negative selection according to the manufacturer’s protocol (StemCell Technologies, Canada). The purity of the CD4+ selection was assessed by flow cytometry using a FITC-conjugated anti-CD4 antibody (anti-human CD4 antibody, clone OTK4, FITC, catalog# 60016FI, StemCell Technologies, Canada) on a BD FACSCanto II flow cytometer and then analyzed using FACSDiva software (BD Biosciences, USA) at the Analytical Biomolecular Research Facility of the University of Newcastle. All samples met a minimum purity threshold of >90 %.
RNA isolation
Total RNA was isolated from the CD4+ T cells using the miRNeasy Mini kit (Qiagen, USA) following the manufacturer’s instructions. The quality of the RNA was assessed using the RNA 6000 Nano kit on a 2100 Bioanalyzer (Agilent Technologies, USA); a RNA integrity number greater than 8 was deemed suitable for sequencing and reverse transcription quantitative polymerase chain reaction (RT-qPCR). Purity was measured on an Epoch spectrophotometer (BioTek, USA), and concentration was measured using the high-sensitivity RNA kit on Qubit 2.0 Fluorometer (Life Technologies, Thermo Fisher Scientific, USA).
miRNA sequencing and analysis
A cohort of 12 SPMS and 12 HC samples was run through NGS at the Diamantina Institute, University of Queensland, Brisbane, Australia. Samples were individually barcoded and then sequenced in two multiplexed pools each containing 12 samples. The sequencing libraries were prepared from 1-μg total RNA, using the TruSeq small RNA preparation kit (Illumina, USA) and sequenced using the 50-bp fragment protocol on the HiSeq 2500 platform. The sequencing generated four to nine million reads per sample, more than sufficient for expression and discovery applications. The sample sequencing reads were demultiplexed using the CASAVA 1.8 software package (Illumina, USA). The Illumina adapter sequences were trimmed from the fastq files using Trimmomatic [9]. All reads were aligned and counted against miRBase 21 [10].
RT-qPCR
Mature miRNA TaqMan assays (Applied Biosystems, Thermo Fisher Scientific, USA) were used to determine expression of the ten most differentially expressed miRNAs in the initial NGS cohort as well as a replication cohort of 12 SPMS and 10 HC (assay IDs in miRNA numerical order: 000397, 000399, 000407, 000408, 000409, 000413, 002223, 000464, 002623, 000524). The small RNA RNU44 (ref: 001094) was used as an endogenous control. RNU44 has previously been demonstrated to be a stable control in CD4+ T cells [11], and its stability has been shown in our 47 samples (mean ± standard deviation Ct value of 23.58 ± 0.63). RNU44 was used for normalization using the ΔCt method. The relative expression (2−ΔCt) of all samples (24 SPMS and 22 HC) was calculated.
Statistical analysis
The two-sample Kolmogorov-Smirnov test (K-S test) was used to test whether differences in expression levels were statistically significant between the case and control groups as implemented in R. The K-S test was chosen (over the F test comparison of means) because of the non-normality of the expression level distributions among miRNAs. Our statistical significance threshold allowing for multiple testing correction was determined using the False Discovery Rate (FDR) procedure of Benjamini-Hochberg [12]. Based on the number of miRNA elements, this threshold was set at 1.2 × 10−4. We also considered a relaxed (or nominal) significance threshold of 0.05. In addition to using statistical significance thresholds for miRNA selection, we also included a count threshold of >800 to exclude miRNAs that had very low expression levels and were unlikely to be replicated with the less-sensitive RT-qPCR. The K-S test was also used to determine significant differential miRNA and SOCS6 expression from the RT-qPCR relative expression data.
Correlation to patient characteristics
The Pearson correlation coefficient was calculated using RT-qPCR data for MS samples (n = 24) and patient characteristics: EDSS, age, disease duration, and progression duration. A correlation coefficient (r value) >±0.5 was considered strong, ±0.3–0.49 moderate, and <±0.29 weak.
Gene target prediction
miRSystem integrates seven different target gene prediction algorithms and contains experimentally validated data on miRNA:mRNA interactions [13]. This integration system was used to identify genes that may be targeted by more than one of our identified dysregulated miRNAs.
Analysis of SOCS6 expression
Five hundred nanograms of total RNA was reverse transcribed using high-capacity cDNA reverse transcription kits (Applied Biosystems, Thermo Fisher Scientific, USA) in 21 SPMS and 21 HC samples. qPCR was performed using an exon-spanning TaqMan probe for SOCS6 (ref: Hs00377781_m1). Expression of SOCS6 was determined as relative expression to the housekeeping genes GAPDH (ref: 4326317E) and β-actin (ref: 4326215E) using a ViiA 7 (Applied Biosystems, Thermo Fisher Scientific, USA).
Results
We used NGS to establish miRNA expression profiles in CD4+ T cells from a cohort of 12 SPMS and 12 HC samples. RT-qPCR was then employed to validate differences in miRNA expression in the NGS cohort as well as a replication cohort of 12 SPMS and 10 HC samples (total 24 SPMS and 22 HC).
NGS
We observed three statistically significant miRNAs (miR-451a, miR-1246, and miR-144-5p) at the FDR-corrected threshold (Additional file 1: Figure S1), which probably reflects the modest sample size. These miRNAs were very lowly expressed (<100 reads per sample), and we were unable to confirm this dysregulation with RT-qPCR. We also observed 42 miRNAs at the nominal significance threshold (97 % of these were down-regulated). Of these 42 miRNAs, only 10 met our secondary criteria of having a read count >800: miR-21-5p (p = 0.031), miR-23a-3p (p = 0.007), miR-26b-5p (p = 0.031), miR-27a-3p (p = 0.031), miR-27b-3p (p = 0.031), miR-29b-3p (p = 0.007), miR-30e-5p (p = 0.031), miR-142-3p (p = 0.031), miR-155-5p (p = 0.031), and miR-221-3p (p = 0.031). Each of these miRNAs was found to be down-regulated in SPMS as summarized in Fig. 1 and was forwarded for replication testing in an independent cohort.
Fig. 1.
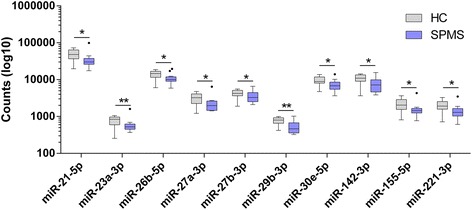
Tukey boxplot demonstrating the ten most significantly dysregulated microRNAs identified using NGS. Data is presented as log10 of the read count and clearly exhibits the down-regulation of miRNAs in SPMS (purple) compared to HC (gray). Whiskers represent data within 1.5 interquartile range (IQR) of the upper and lower quartile. Data points outside of the 1.5 IQR are represented by black dots. *p < 0.05, **p < 0.01
RT-qPCR
To confirm our NGS findings, the top ten most dysregulated miRNAs were selected for further analysis in 24 SPMS and 22 HC samples using RT-qPCR (including the 12 SPMS and 12 HC samples that underwent NGS analysis). Of these ten miRNAs, RT-qPCR confirmed significant down-regulation of miR-21-5p (p = 0.0048), miR-26b-5p (p = 0.007) miR-29b-3p (p = 0.00001), miR-142-3p (p = 0.05), and miR-155-5p (p = 0.001) in SPMS CD4+ T cells (Fig. 2). These five miRNAs were confirmed in the original NGS cohort, the replication cohort, and the combined cohort. This provides statistically significant evidence of replication, indicating that these five miRNAs are very unlikely to be false positives. A trend of down-regulation of miRNA in SPMS samples was still observed across all ten miRNAs.
Fig. 2.
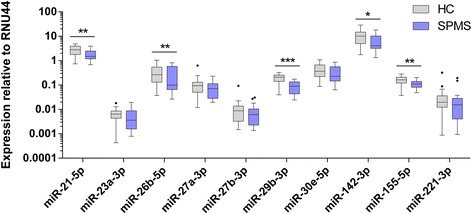
Tukey boxplot of top ten miRNAs expression (relative to RNU44) using RT-qPCR. Significant down-regulation of miR-21-5p, miR-26b-3p, miR-29b-3p, miR-142-3p, and miR-155-5p in SPMS was confirmed. Whiskers represent data within 1.5 interquartile range (IQR) of the upper and lower quartile. Data points outside of the 1.5 IQR are represented by black dots. p < 0.05, **p < 0.01, ***p < 0.001
Comparison of methods
Concordance of differential expression can vary between quantitation methods [14]. To determine the magnitude of fold-change in SPMS vs. HC, we compared RT-qPCR and NGS results and found no change in the degree of decreased expression between NGS and RT-qPCR methods in the miRNAs confirmed by RT-qPCR (Fig. 3).
Fig. 3.
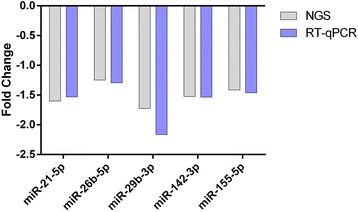
Comparison of miRNA fold-change between NGS and RT-qPCR. Magnitude of change is consistent between NGS and RT-qPCR methods
Correlation to patient characteristics
No strong correlations between miRNA expression and patient characteristics were identified (Table 2). However, moderate positive correlation between EDSS and miR-21-5p, miR-26b-5p, and miR-29b-3p was seen. Further positive correlation was also found between disease duration and miR-21-5p and miR-155-5p. All miRNAs demonstrated weak correlation to patient age and progression duration.
Table 2.
Correlation coefficients calculated from RT-qPCR data against patient characteristics
| miR-21-5p | miR-26b-5p | miR-29b-3p | miR-142-3p | miR-155-5p | |
|---|---|---|---|---|---|
| EDSS | 0.34 | 0.42 | 0.41 | 0.28 | 0.26 |
| Age (HC) | 0.22 | 0.17 | 0.31 | 0.21 | −0.08 |
| Age (SPMS) | −0.07 | −0.17 | −0.17 | −0.30 | −0.01 |
| Disease duration | 0.49 | 0.15 | 0.23 | −0.08 | 0.49 |
| Progression duration | 0.12 | 0.12 | 0.11 | −0.07 | 0.17 |
Correlation of miRNA expression and age of HC has also been calculated as a reference point for age of patients. Moderate correlations are in bold text.
Target prediction
miRNA fold-change was <2 for all miRNAs. It is therefore unlikely that any one particular miRNA is causing a significant effect on gene expression alone. It is more likely to be a combination of multiple miRNAs targeting a few specific genes. Furthermore, as RT-qPCR is a less-sensitive methodology than NGS, and the trend of down-regulation is still observed (though not significant) in the other miRNAs, all ten miRNAs were cross-analyzed for potential gene targets. miRSystem was used to identify genes that have multiple target genes in common, both in the five confirmed miRNAs and all ten miRNAs identified by NGS. One gene, bromodomain and WD repeat domain containing 1 (BRWD1), is targeted by all five confirmed miRNAs. No genes are targeted by all ten miRNAs; however, eight genes are targeted by eight of the miRNAs (Table 3).
Table 3.
Genes identified by miRSystem targeted by eight of the ten microRNAs
| miR-21-5p | miR-23a-3p | miR-26b-5p | miR-27a-3p | miR-27b-3p | miR-29b-3p | miR-30e-5p | miR-142-3p | miR-155-5p | miR-221-3p | |
|---|---|---|---|---|---|---|---|---|---|---|
| ACVR2B | V | V | V | V | V | V | V | V | ||
| ZBTB41 | V | V | V | V | V | V | V | V | ||
| BRWD1 | V | V | V | V | V | V | V | V | ||
| CAMTA1 | V | V | V | V | V | V | V | V | ||
| CFL2 | V | V | V | V | V | V | V | V | ||
| SOCS6 | V | V | V | V | V | V | V | V | ||
| MIER3 | V | V | V | V | V | V | V | V | ||
| KLF12 | V | V | V | V | V | V | V | V |
Verified targeting miRNAs are identified with a “V”
These genes are involved in transmembrane ligand binding, regulation of actin filaments, or are transcription factors. However, only one gene is specifically linked to immune cell function, SOCS6 (suppressor of cytokine signaling 6). This gene has previously been reported to negatively regulate T cell activation by promoting ubiquitin-dependent proteolysis [15] and was consequently selected for further investigated.
SOCS6 expression
Gene expression analysis using RT-qPCR was conducted to determine whether SOCS6 is up-regulated in SPMS CD4+ T cells in direct negative correlation to the miRNA expression (Fig. 4). Both the preliminary and validation cohorts were analyzed, and SOCS6 expression is increased in SPMS compared to HC. Normalization against GAPDH and β-actin generated the same results (data for β-actin not shown).
Fig. 4.
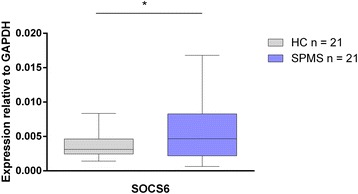
Expression of SOCS6 relative to GAPDH. Up-regulation of SOCS6 in SPMS is significant though widely distributed (*p = 0.042)
Discussion
This is the first study in MS to utilize NGS for miRNA expression profiling in the CD4+ T cells of SPMS patients. We found 42 miRNAs that are dysregulated in the CD4+ T cells of SPMS patients as compared to controls: 97 % of which were down-regulated. TaqMan assays confirmed five of these miRNAs (miR-21-5p, miR-26b-5p, miR-29b-3p, miR-142-3p, and miR-155-5p) to be down-regulated in SPMS. Each of these miRNAs (excluding miR-26b) has been reported on previously in MS though not necessarily in SPMS or CD4+ T cells. Lindberg et al. [11] identified seven miRNAs dysregulated in CD4+ T cells from RRMS patients but did not identify dysregulation in any of the five miRNA in this study. Thus, down-regulation of these miRNAs may be exclusive to SPMS.
Here, we report a decrease in miR-155-5p expression in MS. miR-155-5p has a pro-inflammatory role in MS and is up-regulated in a number of tissues. Studies of postmortem brain tissue find a gradient of miR-155-5p expression that peaks in active lesions [16] and associated neurovascular units [17] and decreases through chronic lesions and normal appearing white matter to a low baseline in healthy control (non-MS) white matter [17, 18]. This increased expression of miR-155 has been associated with the suppression of CD47 in active lesions that creates a permissive environment for myelin phagocytosis [16]; focal adhesion and cell-cell complex molecules in the blood-brain barrier, thus increasing permeability [17] and; AKR1C1 and AKR1C2, essential for biosynthesis of allopregnanolone (a neuroprotective steroid) [18].
Interestingly, a study of miR-155 in the EAE mouse model found that miR-155 expression in CD4+ T cells increases during EAE and that miR-155−/− mice had an attenuation of EAE [19]. Specifically, Th17 cells lacking miR-155-5p are unable to cause EAE [20]. miR-155-5p is required for normal immune function [21], and together, these studies confirm that the significant role miR-155-5p over-expression plays in the inflammatory process of MS. In contrast, our finding of miR-155-5p down-regulation may be exclusive to SPMS patients and/or CD4+ T cells and is consistent with SPMS as a non-inflammatory mediated disease.
miR-155-5p and miR-142-3p have been identified as dysregulated in RRMS PBMCs [22], and a recent study on autologous hematopoietic stem cell transplant (AHSCT) also found co-dysregulation of miR-155-5p and miR-142-3p [23]. Contrary to our results, Arruda et al. found these miRNAs to be up-regulated in MS patient CD4+ T cells before treatment (cohort was 75 % SPMS). However, AHSCT is most effective in active MS disease, and six of the 19 SPMS patients enrolled in the Arruda et al. study presented with gadolinium-enhancing lesions in the year approaching the treatment indicating inflammatory activity. Further, the average disease duration in the Arruda et al. study was 8.1 years, as opposed to 25.6 (primary cohort) or 18.6 (replication cohort) years in our study. Our data is corroborated further by NGS expression analysis, which is a more sensitive measure of expression changes.
In a study of potential biomarkers in Alzheimer’s disease (AD), miR-26b-5p was shown to be down-regulated in the serum and CSF of AD patients when compared to patients with inflammatory neurological diseases [24], supporting the predominantly neurodegenerative pathology of SPMS. Over-expression of miR-29b insystemic lupus erythematosus (SLE) has been linked to hypomethylation of DNA in CD4+ T cells [25]. While there are currently no studies on DNA methylation in SPMS, it would be interesting to see if the down-regulation of miR-29b that we have identified here in CD4+ T cells is associated with genome-wide hypermethylation in SPMS.
Increased miR-21-5p promotes differentiation of Th17 cells in the EAE mouse model, and miR-21-5p knock-out mice are resistant to EAE [26]. Fenoglio et al. found increased miR-21-5p expression in RRMS (active relapse phase) PMBCs compared to controls, though no difference in SPMS. Again, this may be attributed to the relatively small sample size (n = 6) [27].
Also of interest, we previously reported miR-20a-5p down-regulation in the whole blood of all MS subtypes [28]. This miRNA was one of the 42 dysregulated miRNAs identified by NGS and is significantly down-regulated in SPMS compared to HC. However, it narrowly missed the 800 read cut-off for qPCR confirmation. miR-20a-5p is also predicted to target SOCS6.
Eight of the top ten dysregulated miRNAs were predicted to target SOCS6 using MirSystem. Consistent with this, increased expression of SOCS6 in the SPMS cohort is in direct negative correlation with the miRNA expression profiles, strongly indicating a mRNA:miRNA relationship. To our knowledge, this is the first study to identify SOCS6 as a gene of interest in MS. It is a highly conserved gene with very low expression levels in healthy thymus and brain tissues and is down-regulated in gastric, colorectal, and pancreatic cancers [29–32]. In colorectal cancer, methylation changes have been ruled out as the mechanism of down-regulation [31]; therefore, down-regulation may be due to altered miRNA expression. MiR-424-5p is responsible for the down-regulation of SOCS6 in pancreatic cancer [32]; however, we found no differences in miR-424-5p expression between SPMS and HC in this study.
The function of SOCS6 as a negative regulator of T cell activation [15] and its observed over-expression in SPMS CD4+ T cells supports the notion of reduced immune activity in SPMS. Very little is known about SOCS6, and more studies are required to determine if it may be a novel therapeutic target.
This is the first study to use NGS miRNA profiling to assess miRNA expression in the CD4+ T cells of SPMS patients. Future studies should focus on using the same technique in treatment naïve RRMS patients to determine if this is a SPMS exclusive trend and remove the confounding factor of treatment effects. Furthermore, miRNA expression profiles of other cell subtypes should be investigated, as whole blood analysis is likely masking significant changes in individual cell subsets. Ideally, all of our patients would have had inactive SPMS; however, as SPMS is a difficult disease stage to define and collect, we have included some active SPMS patients in this study. In this study, we chose to focus on CD4+ T cells as they are thought to be the main cell infiltrates. Our previous studies also show that CD4+ T cells exhibit significant changes in methylation profiles in RRMS [33, 34].
Conclusions
Here, we have shown a general down-regulation of miRNAs in CD4+ T cells compared to HC, with five miRNAs confirmed as significant in two independent assays. This indicates that miRNA expression may be over-normalizing in SPMS CD4+ T cells. SOCS6 is a predicted target of the majority of these miRNAs and, consistent with this, we found SOCS6 to be up-regulated in this cohort. These are novel findings that point towards a diminished role for CD4+ T cells in SPMS and add further evidence for SPMS being a neurodegenerative disease stage, not an inflammation-driven one.
Acknowledgements
We would like to thank the MS patients and clinical team at the John Hunter Hospital MS clinic who participated in this study. We also acknowledge the Analytical Biomolecular Research Facility at the University of Newcastle for the flow cytometry support and the Diamantina Institute at the University of Queensland for the NGS services.
Funding
This study was supported by the John Hunter Hospital Charitable Trust and the Bloomfield Group Foundation. KAS, VEM, and RAL are supported by fellowships from Multiple Sclerosis Research Australia. KAS is also funded by the Trish MS Research Foundation postgraduate scholarship. VEM is supported by a postdoctoral fellowship from the Canadian Institutes of Health Research.
Availability of data and material
Data available on request and subject to ethics committee approval.
Authors’ contributions
KAS, MCB, and RAL wrote the main manuscript text. KAS, VEM, SA, and NG performed the experiments. MCB and RAL analyzed the NGS data. RJS, LT, and JLS supervised the study. All authors reviewed the manuscript. All authors read and approved the final manuscript.
Competing interests
JL-S receives non-direct funding as well as honoraria for presentations and membership on advisory boards from Genzyme, Biogen, Bayer Health Care, Merck, Teva, and Novartis, Australia.
The authors, KAS, MCB, RAL, VEM, SA, NG, RJS, and LT, declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
The Hunter New England Health Research Ethics Committee and University of Newcastle Ethics committee approved this study (05/04/13.09 and H-505-0607, respectively), and methods were carried out in accordance with institutional guidelines on human subject experiments. Written and informed consent was obtained from all patient and control subjects.
Abbreviations
- AD
Alzheimer’s disease
- AHSCT
Autologous hematopoietic stem cell transplant
- CNS
Central nervous system
- DNA
Deoxyribonucleic acid
- EAE
Experimental autoimmune encephalitis
- EDSS
Expanded disability status scale
- FDR
False discovery rate
- GA
Glatiramer acetate
- HC
Healthy controls
- K-S test
Kolmogorov-Smirnov test
- miRNA
MicroRNA
- MS
Multiple sclerosis
- NGS
Next-generation sequencing
- PBMC
Peripheral blood mononuclear cells
- RNA
Ribonucleic acid
- RRMS
Relapsing remitting MS
- RT-qPCR
Reverse transcription quantitative polymerase chain reaction
- SD
Standard deviation
- SOCS6
Suppressor of cytokine signaling 6
- SPMS
Secondary progressive MS
Additional file
{kind=link}
Volcano plot of differentially expressed miRNAs identified with NGS. The FDR-corrected significance threshold is demarked with a green line at p < 1.2 × 10−4. Three miRNAs were identified at the threshold. Mean read counts were low in all three miRNAs: miR-451a (SPMS mean = 76.3, HC mean = 18.9), miR-1246 (SPMS mean = 94.9, HC mean = 51.9), and miR-144-5p (SPMS mean = 15.1, HC mean = 5.5). Differential expression could not be replicated with RT-qPCR. (PNG 57 kb)
References
- 1.Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372:1502–1517. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- 2.Broux B, Stinissen P, Hellings N. Which immune cells matter? The immunopathogenesis of multiple sclerosis. Crit Rev Immunol. 2013;33:283–306. doi: 10.1615/CritRevImmunol.2013007453. [DOI] [PubMed] [Google Scholar]
- 3.Segal BM. Stage-specific immune dysregulation in multiple sclerosis. J Interferon Cytokine Res. 2014;34:633–640. doi: 10.1089/jir.2014.0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Raphael I, Webb J, Stuve O, Haskins W, Forsthuber T. Body fluid biomarkers in multiple sclerosis: how far we have come and how they could affect the clinic now and in the future. Expert Rev Clin Immunol. 2015;11:69–91. doi: 10.1586/1744666X.2015.991315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keller A, Leidinger P, Steinmeyer F, Stahler C, Franke A, Hemmrich-Stanisak G, Kappel A, Wright I, Dorr J, Paul F, et al. Comprehensive analysis of microRNA profiles in multiple sclerosis including next-generation sequencing. Mult Scler. 2013;20:295–303. doi: 10.1177/1352458513496343. [DOI] [PubMed] [Google Scholar]
- 6.Keller A, Leidinger P, Meese E, Haas J, Backes C, Rasche L, Behrens JR, Pfuhl C, Wakonig K, Giess RM, et al. Next-generation sequencing identifies altered whole blood microRNAs in neuromyelitis optica spectrum disorder which may permit discrimination from multiple sclerosis. J Neuroinflammation. 2015;12:196. doi: 10.1186/s12974-015-0418-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Felice B, Mondola P, Sasso A, Orefice G, Bresciamorra V, Vacca G, Biffali E, Borra M, Pannone R. Small non-coding RNA signature in multiple sclerosis patients after treatment with interferon-beta. BMC Med Genomics. 2014;7:26. doi: 10.1186/1755-8794-7-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Polman CH, Reingold SC, Banwell B, Clanet M, Cohen JA, Filippi M, Fujihara K, Havrdova E, Hutchinson M, Kappos L, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. 2011;69:292–302. doi: 10.1002/ana.22366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014;42:D68–73. doi: 10.1093/nar/gkt1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lindberg RL, Hoffmann F, Mehling M, Kuhle J, Kappos L. Altered expression of miR-17-5p in CD4+ lymphocytes of relapsing-remitting multiple sclerosis patients. Eur J Immunol. 2010;40:888–898. doi: 10.1002/eji.200940032. [DOI] [PubMed] [Google Scholar]
- 12.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol. 1995;57:289–300. [Google Scholar]
- 13.Lu T-P, Lee C-Y, Tsai M-H, Chiu Y-C, Hsiao CK, Lai L-C, Chuang EY. miRSystem: an integrated system for characterizing enriched functions and pathways of microRNA targets. PLoS ONE. 2012;7:e42390. doi: 10.1371/journal.pone.0042390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mestdagh P, Hartmann N, Baeriswyl L, Andreasen D, Bernard N, Chen C, Cheo D, D'Andrade P, DeMayo M, Dennis L, et al. Evaluation of quantitative miRNA expression platforms in the microRNA quality control (miRQC) study. Nat Methods. 2014;11:809–815. doi: 10.1038/nmeth.3014. [DOI] [PubMed] [Google Scholar]
- 15.Choi YB, Son M, Park M, Shin J, Yun Y. SOCS-6 negatively regulates T cell activation through targeting p56lck to proteasomal degradation. J Biol Chem. 2010;285:7271–7280. doi: 10.1074/jbc.M109.073726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Junker A, Krumbholz M, Eisele S, Mohan H, Augstein F, Bittner R, Lassmann H, Wekerle H, Hohlfeld R, Meinl E. MicroRNA profiling of multiple sclerosis lesions identifies modulators of the regulatory protein CD47. Brain. 2009;132:3342–3352. doi: 10.1093/brain/awp300. [DOI] [PubMed] [Google Scholar]
- 17.Lopez-Ramirez MA, Wu D, Pryce G, Simpson JE, Reijerkerk A, King-Robson J, Kay O, de Vries HE, Hirst MC, Sharrack B, et al. MicroRNA-155 negatively affects blood-brain barrier function during neuroinflammation. FASEB J. 2014;28(6):2551–65. doi: 10.1096/fj.13-248880. [DOI] [PubMed] [Google Scholar]
- 18.Noorbakhsh F, Ellestad KK, Maingat F, Warren KG, Han MH, Steinman L, Baker GB, Power C. Impaired neurosteroid synthesis in multiple sclerosis. Brain. 2011;134:2703–2721. doi: 10.1093/brain/awr200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murugaiyan G, Beynon V, Mittal A, Joller N, Weiner HL. Silencing microRNA-155 ameliorates experimental autoimmune encephalomyelitis. J Immunol. 2011;187:2213–2221. doi: 10.4049/jimmunol.1003952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu R, Huffaker TB, Kagele DA, Runtsch MC, Bake E, Chaudhuri AA, Round JL, O'Connell RM. MicroRNA-155 confers encephalogenic potential to Th17 cells by promoting effector gene expression. J Immunol. 2013;190:5972–5980. doi: 10.4049/jimmunol.1300351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, van Dongen S, Grocock RJ, Das PP, Miska EA, et al. Requirement of bic/microRNA-155 for normal immune function. Science. 2007;316:608–611. doi: 10.1126/science.1139253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Waschbisch A, Atiya M, Linker RA, Potapov S, Schwab S, Derfuss T. Glatiramer acetate treatment normalizes deregulated microRNA expression in relapsing remitting multiple sclerosis. PLoS One. 2011;6:e24604. doi: 10.1371/journal.pone.0024604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arruda LC, Lorenzi JC, Sousa AP, Zanette DL, Palma PV, Panepucci RA, Brum DS, Barreira AA, Covas DT, Simoes BP, et al. Autologous hematopoietic SCT normalizes miR-16, -155 and -142-3p expression in multiple sclerosis patients. Bone Marrow Transplant. 2015;50:380–389. doi: 10.1038/bmt.2014.277. [DOI] [PubMed] [Google Scholar]
- 24.Galimberti D, Villa C, Fenoglio C, Serpente M, Ghezzi L, Cioffi SM, Arighi A, Fumagalli G, Scarpini E. Circulating miRNAs as potential biomarkers in Alzheimer’s disease. J Alzheimers Dis. 2014;42:1261–1267. doi: 10.3233/JAD-140756. [DOI] [PubMed] [Google Scholar]
- 25.Qin H, Zhu X, Liang J, Wu J, Yang Y, Wang S, Shi W, Xu J. MicroRNA-29b contributes to DNA hypomethylation of CD4+ T cells in systemic lupus erythematosus by indirectly targeting DNA methyltransferase 1. J Dermatol Sci. 2013;69:61–67. doi: 10.1016/j.jdermsci.2012.10.011. [DOI] [PubMed] [Google Scholar]
- 26.Murugaiyan G, da Cunha AP, Ajay AK, Joller N, Garo LP, Kumaradevan S, Yosef N, Vaidya VS, Weiner HL. MicroRNA-21 promotes Th17 differentiation and mediates experimental autoimmune encephalomyelitis. J Clin Invest. 2015;125:1069–1080. doi: 10.1172/JCI74347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fenoglio C, Cantoni C, De Riz M, Ridolfi E, Cortini F, Serpente M, Villa C, Comi C, Monaco F, Mellesi L, et al. Expression and genetic analysis of miRNAs involved in CD4+ cell activation in patients with multiple sclerosis. Neurosci Lett. 2011;504:9–12. doi: 10.1016/j.neulet.2011.08.021. [DOI] [PubMed] [Google Scholar]
- 28.Cox MB, Cairns MJ, Gandhi KS, Carroll AP, Moscovis S, Stewart GJ, Broadley S, Scott RJ, Booth DR, Lechner-Scott J. Consortium ANMSG: MicroRNAs miR-17 and miR-20a inhibit T cell activation genes and are under-expressed in MS whole blood. PLoS One. 2010;5:e12132. doi: 10.1371/journal.pone.0012132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lai RH, Wang MJ, Yang SH, Chen JY. Genomic organization and functional characterization of the promoter for the human suppressor of cytokine signaling 6 gene. Gene. 2009;448:64–73. doi: 10.1016/j.gene.2009.07.022. [DOI] [PubMed] [Google Scholar]
- 30.Lai RH, Hsiao YW, Wang MJ, Lin HY, Wu CW, Chi CW, Li AF, Jou YS, Chen JY. SOCS6, down-regulated in gastric cancer, inhibits cell proliferation and colony formation. Cancer Lett. 2010;288:75–85. doi: 10.1016/j.canlet.2009.06.025. [DOI] [PubMed] [Google Scholar]
- 31.Letellier E, Schmitz M, Baig K, Beaume N, Schwartz C, Frasquilho S, Antunes L, Marcon N, Nazarov PV, Vallar L, et al. Identification of SOCS2 and SOCS6 as biomarkers in human colorectal cancer. Br J Cancer. 2014;111:726–735. doi: 10.1038/bjc.2014.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu K, Hu G, He X, Zhou P, Li J, He B, Sun W. MicroRNA-424-5p suppresses the expression of SOCS6 in pancreatic cancer. Pathol Oncol Res. 2013;19:739–748. doi: 10.1007/s12253-013-9637-x. [DOI] [PubMed] [Google Scholar]
- 33.Graves M, Benton M, Lea R, Boyle M, Tajouri L, Macartney-Coxson D, Scott R, Lechner-Scott J. Methylation differences at the HLA-DRB1 locus in CD4+ T-Cells are associated with multiple sclerosis. Mult Scler. 2014;20:1033–1041. doi: 10.1177/1352458513516529. [DOI] [PubMed] [Google Scholar]
- 34.Maltby VE, Graves MC, Lea RA, Benton MC, Sanders KA, Tajouri L, Scott RJ, Lechner-Scott J. Genome-wide DNA methylation profiling of CD8+ T cells shows a distinct epigenetic signature to CD4+ T cells in multiple sclerosis patients. Clin Epigenetics. 2015;7:118. doi: 10.1186/s13148-015-0152-7. [DOI] [PMC free article] [PubMed] [Google Scholar]