

Abstract
Recent research has highlighted the importance of the two-way interaction between the nervous and immune systems. This interaction is particularly important in the bowel because of the unique properties of this organ. The lumen of the gut is lined by a very large but remarkably thin surface that separates the body from the enteric microbiome. Immune defenses against microbial invasion are thus well developed, and neuroimmune interactions are important in regulating and integrating these defenses. Important concepts in the phylogeny of neuroimmunity, enteric neuronal and glial regulation of immunity, changes that occur in the enteric nervous system during inflammation, the fundamental role of serotonin (5-HT) in enteric neuroimmune mechanisms, and future perspectives are reviewed.
Inflammation and Neuroimmunity in the Bowel
Inflammatory bowel diseases (IBD) are chronic intestinal inflammatory conditions that affect approximately one million individuals in the USA [1]. The prevalence of IBD is increasing nationally and internationally. Despite significant advances in understanding the pathophysiology of IBD, its pathogenesis remains unknown and IBD continues to cause a great deal of morbidity.
The intestine is essential for life. It is the site of nutrient ingestion and also of where these nutrients, many of which are macromolecules, are digested into small molecules that the gut can absorb. Within the bowel, moreover, there is a resident microbiome, which is a net plus in value [2] but also a danger that must be restricted to the enteric lumen and prevented from invading [3]. The intestinal lumen is external to the body. The thin barrier that separates the lumen from the internal milieu of the body, which in most regions of the gut is only one cell thick, allows essential nutrients to be absorbed but prevents the absorption of toxins and invasion by commensal and pathogenic organisms. Microbial translocation across the intestinal lining triggers inflammation, which helps to control infection [3], although inflammation is also capable of inflicting a great deal of collateral damage to the intestinal wall. Managing digestion, absorption, and protection from microbial invasion is complicated and requires considerable organization, integration, and rapid direction of resources to sites where they are most needed. These resources include secretion of enzymes, ions, mucous, and water, the complex motor patterns that the intestinal smooth muscle generates, and a variety of hormones. Signaling through the nervous system evolved to accomplish organization, integration, and rapid direction of resources. In the bowel, the details of the management of these functions have been delegated to the enteric nervous system (ENS), which can, uniquely, function independently of CNS control [4]. The brain, of course, powerfully influences the ENS through sympathetic and parasympathetic nerves, but the bowel and its microbial content, possibly through the ENS, also reciprocally affect the brain. The intestinal microbiota can influence satiety, metabolism, and even mood [2,5–7]; moreover, electrical stimulation of the vagus nerves, which mimics gut–brain signaling, improves memory [8] and has been employed to treat depression [9]. One of the most important functions of the intestinal innervation, however, is to modulate intestinal inflammation [10].
The roles of individual neurotransmitters/neuromodulators in intestinal inflammation have recently been reviewed [11] and those of tachykinins, in particular, have been extensively summarized [12]. Instead of a comprehensive examination of the pro- and anti-inflammatory activities of every transmitter in the gut, we focus on the phylogeny of neuronal regulation of intestinal inflammation, bidirectional neuroimmune interactions in the regulation and consequences of intestinal inflammation, as well as the central roles that serotonin plays as a signaling molecule in triggering, enhancing, and countering inflammation.
Phylogeny of Neuroimmune Interactions
Survival demands that every animal distinguishes between the nutrients it ingests from invaders and toxins. This ability is so fundamental that it probably arose early in evolution. Defenses against invasion include innate immune mechanisms such as the secretion of cytokines and phagocytic activity. These mechanisms appear to have preceded the evolution of adaptive immunity, which added memory, sophistication, and regulation to the layers of defense. Secreted signaling molecules function in intercellular communication, a vital component of adaptive immunity. Many of these molecules are also found in primitive organisms and thus have been retained in evolution [13]. Some molecules have been re-purposed as additional cells and systems joined the defensive community and reactions increased in specificity and speed. In the mammalian bowel, endocrine and neurocrine signaling helps to coordinate and regulate innate and adaptive immune mechanisms of defense. Macrophages and their dendritic cell relatives have now transcended their primitive phagocytic ancestors and become nexi of intercellular communication. These cells interact with the lymphocytic mediators of adaptive immune responses, and both macrophages and lymphocytes respond to hormonal and neuronal regulation [13]. Neurotransmission evolved from phylogenetically older methods of signaling. Hormones and paracrine messengers are employed for intercellular signaling in animals, such as sponges, that lack a nervous system. Environmental stimuli cause specialized cells in the epidermis and the intestinal lining of primitive multicellular organisms to secrete signaling metabolites. The hormones and metabolites that are used for signaling in animals without a nervous system diffuse throughout the organism to be recognized by target cells with appropriate receptors. Ultimately, endocrine cells and neurons separated from the epithelium but both continued to secrete signaling molecules. Endocrine cells secrete into the circulation while neurons project to individual target cells or even, at synapses, to regions of target cells [14]. Early in evolution, multicellularity led to the development of paracrine signaling, which involves the secretion of diffusible molecules that act in the neighborhood of the secreting cell. Nevertheless, even paracrine signaling enables multiple cells to cooperate for the common defense of the organism. Later, the acquisition of endocrine signaling enabled distant cells to be recruited, with specificity achieved through molecular recognition in a ligand–receptor relationship. Eventually, the exquisite anatomical specificity and speed of neuronal conduction were added to the ligand–receptor relationship to vastly enhance the adaptability and integration of responses.
The evolution of the defensive response is reflected in the mechanisms that are operative in the mammalian bowel. Paracrine, endocrine, and neurocrine signaling are all utilized; early mechanisms thus were not supplanted as newer ones evolved. The newer mechanisms have been added to, and may modulate or regulate the primitive original responses that have been retained as effectors of inflammation. Sponges, for example, contain phagocytic amoeboid cells (archeocytes) that are pluripotent stem cells [15]. These cells contribute not only to nutrient uptake but also to defense of the whole organism. Some of these primitive phagocytes secrete soluble signaling molecules, including cytokines and nitric oxide (NO), that are retained as endocrine and/or neurocrine messengers in higher animals.
The mammalian gut faces a stiff microbial challenge. It must contend with bombardment by a vast number of potential pathogenic bacteria, fungi, and viruses, while simultaneously maintaining homeostasis of a resident community of commensal organisms [16]. The epithelial surface of the intestine produces a diversity of antimicrobial peptides that help the bowel to meet its microbial challenges [17]. Paneth and other cells secrete α- and β-defensins, C-type lectins of the REG3 family, secretory phospholipase A2, and lysozyme when bacterial products activate receptors these cells express. The management of the secretion of these antimicrobial peptides is subject to endocrine and neurocrine regulation [16]. The gut thus has a powerful antimicrobial apparatus. Its safe and efficient operation requires the coordination and integration that the nervous system uniquely provides.
Enteric Neuronal and Glial Regulation of Immunity
It has increasingly become evident that interactions between the enteric nervous system (ENS) and the immune system play important roles in the modulation of inflammation [18]. These interactions involve the action of neurotransmitters, neuromodulators, and cytokines that carry signals, often bidirectionally, between enteric neurons and immune cells [19,20]. Disappointingly, neurons do not form traditional synapses with immune cells, which complicates the use of anatomical tools to investigate neuroimmune interactions; however, axons and immune cells do form ‘close associations’ that are often interpreted to be the anatomical substrate of functional interactions [21,22]. In the case of neurons and macrophages, the bidirectional link is dependent on macrophage secretion of bone morphogenetic protein 2 (BMP2) and neuronal secretion of colony stimulating factor 1 [22]. This muscularis macrophage–ENS communication is responsive to intestinal microbiota and also may enhance interactions between the ENS and the immune system of the bowel. Once ‘close associations’ have been established, nerves can swiftly modulate inflammation; for example, sympathetic nerves to the gut quickly protect tissues from bacteria-induced inflammatory damage [23]. Norepinephrine from the sympathetic axons does this by activating β2-adrenoceptors on muscularis macrophages. Transmitters that neurons secrete can positively (e.g., tachykinins/NK1 receptors) or negatively (e.g., acetylcholine/α7 nicotinic receptors) affect the severity of inflammatory responses of the bowel, and thus neurons may not merely be passive bystanders in intestinal inflammation and IBD [10]. Certainly, the activity of neurons during inflammation has profound consequences for gastrointestinal (GI) secretion and motility [24].
Inflammation may have important consequences for neurons and their relationship to enteric glia. It has become evident that enteric glia, which together with neurons comprise the ENS, are not simply the ‘glue’ that holds neurons and neurites of the ENS together, but are active participants in neuroimmune regulation and other neuro effect or mechanisms [25,26]. Enteric glia, for example, may help to maintain the intestinal barrier and exert neuroprotective effects. Neurons very much need protection when an intestinal inflammatory response is in high dudgeon. Intestinal inflammation can be destructive to enteric neurons, which may be killed during the reaction, in an act of ultimate betrayal by their glial partners [27,28]. During inflammation, purines stimulate P2X7 receptors, which activate pannexin-1 channels in neurons. Neurons then secrete purines (perhaps ATP) that stimulate P2Y1 receptors on glia. These receptors activate glia, which are then activated and induced to synthesize NO, which opens connexin-43 hemichannels in their plasma membrane that allow even more purine to be released. The glial purine then stimulates neuronal P2X7 receptors, which execute the neurons.
The idea that glia can act in a nurturing, protective fashion for the neurons they ensheath would seem to be at odds with the concept that they play the role of inflammation-induced executioner. One finding certainly suggesting that glia are anti-inflammatory was the observation that acyclovir-driven glial ablation in transgenic mice that express herpes simplex thymidine kinase in cells that express the glial marker, glial fibrillary acidic protein (GFAP), initiates a fulminant and lethal inflammation of the ileum [29]. Cells that are not glia, however, may express GFAP. In the CNS, neuronal stem cells express GFAP [30], and the GFAP-expressing cells of the gut that give rise to neurons may thus be stem cells [31]. Toxic products may also diffuse from acyclovir-ablated glia to affect other types of cell; moreover, many enteric glia do not express GFAP [32]. Enteric glia, furthermore, are not homogenous. Instead they exhibit a remarkable diversity, differing in the various layers of the gut wall, and possibly also along its length [26,33,34]. Enteric glial ablation, furthermore, which does not target GFAP, does not cause intestinal inflammation [35]. It is thus possible that it is not the glial ablation that occurs in response to acyclovir in GFAP∷ thymidine kinase mice that causes inflammation; that is, the inflammation may not be cell-autonomous. A more comprehensive marker for enteric glia than GFAP is proteolipid protein-1 (PLP1) [32], which together with S-100β appears to be found in almost all enteric glia throughout the length of the bowel in each of its layers. Glial ablation in mice that express diphtheria toxin driven from the Plp1 promoter is not accompanied by intestinal inflammation [36]. The ability of enteric glia to prevent intestinal inflammation is therefore, at a minimum, questionable. On the other hand, no matter whether during inflammation enteric glia protect or kill their associated neurons, evidence suggests that enteric neurons can develop from Sox10-expressing enteric glial precursor cells and that intestinal inflammation stimulates glia to generate new neurons [37]. Enteric glia also continue to be generated and multiply during life. New enteric glia appear to be generated primarily in the myenteric plexus, but the nascent glial cells then have the ability to migrate, under the attractive guidance of luminal microbiota, to the enteric mucosa [38]. Enteric glia, therefore, probably play a dual role in intestinal inflammation–not only as a grim reaper that kills neurons but also as a savior that, through neurogenesis, undoes the harm that inflammation causes to the ENS.
Changes in the ENS During Intestinal Inflammation
Changes reported in the ENS in patients with IBD include an increase in numbers of enteric neurons, altered neurotransmitter synthesis, content, and release, changes in glial cell numbers, and myenteric ganglionitis [39]. Inflammation also causes multiple changes in the intrinsic motor circuits of the intestine, including neuronal hyperexcitability, increased synaptic facilitation, and decreased descending inhibitory neuromuscular transmission that appears to be related to an attenuation of purinergic transmission [24, 40–46]. These changes are associated with a resultant loss of myenteric neurons_and can ultimately lead to long-lasting disruptions in colonic motor activity [24,47]. There are also data to suggest, however, that inflammation-induced neuroplasticity can increase the number of enteric neurons [48] and contribute to disrupted motility or, conversely, that a pre-existing excess of enteric neurons may actually predispose individuals to intestinal inflammatory disease [10].
Dysfunction of the bowel is found in non-inflamed regions, as well as in the portions of the gut that display obvious inflammation [49]. This dysfunction is associated with long-lasting changes in enteric neural circuits. ENS dysfunction caused by inflammation induces secretion, as well as motility, to become abnormal, and even in non-inflamed portions of the intestine. In animals with trinitrobenzene sulfonic acid (TNBS)-induced colitis, non-cholinergic secretion was decreased significantly in the ileum, even though inflammation could not be detected in ileal tissue [49]. This secretory change was associated with a dramatic reduction of slow excitatory synaptic transmission in VIP-expressing secretomotor neurons of the submucosal plexus. In addition, ileal submucosal cholinergic neurons were more excitable, and action potentials in intrinsic primary afferent neurons (IPANs) were broader than in the absence of colitis. Although nothing can yet be said about the mechanism by which inflammation in the colon disturbs the ENS in the ileum, it is clear that altered neuronal function is profound and widespread in the bowel even when inflammation appears to be confined. As a result, global intestinal dysfunction can be expected from inflammation as the regulatory apparatus of the bowel inappropriately adapts to an inflammatory insult. It is possible that neurons cause inflammation to spread to the small intestine as a result of even highly localized lesions in the colon. When a small region of colon is cauterized, proinflammatory cytokine secretion increases with an accompanying decrease in alanine absorption as far proximally as the duodenum [50]. A long-lived hyperexcitability characterizes small intestinal intrinsic primary afferent neurons after TNBS-induced colitis that persists for weeks after resolution of the colitis [51]. A similar excitation persists after acute infection of the bowel [52] and may contribute to postinfectious IBS [52,53].
Serotonin
One of the most fundamental paracrine/neurocrine messengers found in the epithelium of the mammalian bowel is 5-HT. This signaling molecule is abundant in the bowel of cyclostomes, teleosts, amphibians, and reptiles [54,55]. 5-HT-producing enterochromaffin (EC) cells are located in the epithelial lining of the gut of almost all vertebrates [56] and, where they are not found, the mucosa receives a serotonergic innervation from intrinsic enteric neurons [57]. The EC cells that secrete 5-HT are the most numerous of the enteroendocrine cells of the gut [58] and, in contrast to other types of enteroendocrine cell, which are more limited in location, EC cells are found throughout the bowel, from stomach through colon [59].
The biosynthesis of 5-HT depends on the hydroxylation of tryptophan, which in the gut is catalyzed by tryptophan hydroxylase 1 (TPH1) and TPH2. Each is a separate gene product. TPH1 is the rate-limiting enzyme for 5-HT biosynthesis in EC cells and, in rats and mice, mast cells, while TPH2 is the relevant isoform in neurons [60–63]. 5-HT signals through members of seven groups of 5-HT receptor, all but one of which, the ligand-gated ion channel 5-HT3 receptor [64], are G protein-coupled. At least 15 individual 5-HT receptor subtypes have been identified in the bowel [65]. Enteric neurons express 5-HT1A [66], 5-HT3 [67], 5-HT4 [68,69], and 5-HT7 receptors [70,71], which participate in the regulation of GI motility, while immune effector cells express the 5-HT2A, 2B, 2C, 5-HT3, 5-HT4, and 5-HT7 receptor subtypes [72].
After 5-HT stimulates one of its receptors, 5-HT must be removed rapidly to prevent excessive activation and/or receptor desensitization. 5-HT inactivation requires transmembrane transport because 5-HT cannot be catabolized extracellularly; moreover, 5-HT is charged at a physiological pH and thus poorly traverses plasma-membrane lipid bilayers. The inactivation of 5-HT thus requires the mediation of a selective plasmalemmal, sodium-dependent serotonin transporter (SERT) [73]. Inhibition or deletion of SERT amplifies 5-HT-mediated responses, while increases in SERT activity diminish responses to 5-HT [74]. Within the gut, neurons and enterocytes both express SERT [75]. Platelets, which are not intrinsic to the bowel, circulate through it and also are SERT-expressing [76]. Platelet uptake of 5-HT thus contributes to terminating its enteric activity. Neuronal SERT is important in regulating serotonergic neurotransmission while mucosal and platelet SERT modulate the paracrine serotonergic signaling of EC cell-derived 5-HT. Platelets may also be important in enabling EC cell-derived 5-HT to act in an endocrine fashion by transporting 5-HT to distant targets such as liver [77] and bone [78]. Following reuptake, 5-HT can be transported into vesicles [79] and reused, or it can be catabolized by a variety of intracellular enzymes including monoamine oxidase (MAO) [80,81].
5-HT influences the activity of many of the effector cells that participate in adaptive or innate immune responses in the gut. Macrophages and T cells have even been reported to produce small amounts of 5-HT [82], while rat and mouse mast cells synthesize, store, secrete, and take up 5-HT [83]. 5-HT has been found to modulate chemotaxis, leukocyte activation, proliferation, cytokine secretion, anergy, and apoptosis in immunocytes [82,84]. 5-HT is also vasoactive and plays a role in cell-mediated immunity by facilitating trafficking of lymphocytes through post-capillary venules [85–87]. In addition, 5-HT accumulates in sympathetic nerve terminals in lymphoid tissue and, once in sympathetic terminal axons, enters synaptic vesicles and is co-released along with norepinephrine [88,89]. The sympathetic innervation in gut-associated lymphoid tissue (GALT) may thus also be a source of the 5-HT that regulates intestinal immunity.
Experiments with animal models have indicated that 5-HT plays important roles in intestinal inflammation. Initial gain-of-function studies with mice lacking SERT (SERTKO mice) suggested that 5-HT is proinflammatory. The severity of TNBS-induced colitis and the colitis that occurs spontaneously in mice lacking IL-10 is each increased significantly when combined with the deletion of SERT [90]. Later work established that the severity of inflammation is significantly diminished in mice lacking TPH1 (TPH1KO mice) owing to the selective ablation of mucosal 5-HT [72,91]. The idea emerged from this work that 5-HT released from EC cells enhances inflammation through an action on 5-HT7 receptors that dendritic cells express [72,92,93] (Figure 1). Unfortunately, as compelling as this idea seems to be, strong evidence has been advanced for an equally compelling but contrary hypothesis. Both sides of the argument agree that dendritic cells in the intestine express the 5-HT7 receptor; however, the contrary evidence suggests that the dendritic cell 5-HT7 receptor is anti-inflammatory, not proinflammatory [94]. A 5-HT7 antagonist, SB-269970, and the deletion of 5-HT7 receptors were found to increase the severity of inflammation, and stimulation of the 5-HT7 receptor exerted anti-inflammatory effects. A difference between the studies is that the proinflammatory side [93] employed a dose of SB-269970 that was 2500-fold higher than that utilized by the anti-inflammatory advocates [94]. Conceivably, the higher dose of SB-269970 exerted non-specific effects. Clearly, 5-HT from EC cells cannot drive inflammation through the 5-HT7 receptors of dendritic cells if stimulation of these receptors opposes inflammation. The proinflammatory response to EC cell 5-HT thus remains to be explained. It is clear that 5-HT can raise intracellular cAMP levels in mature dendritic cells, but dendritic cells express many more 5-HT receptors than only 5-HT7 [95].
Figure 1. Interactions That Modulate Intestinal Inflammation.
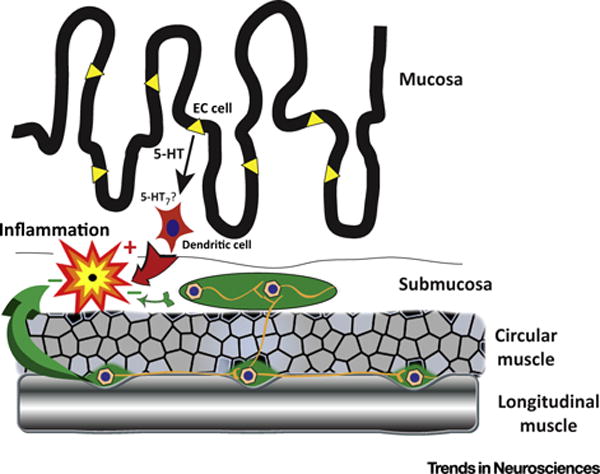
5-HT is stored in enterochromaffin (EC; yellow) cells of the gastrointestinal mucosa. The secretion of 5-HT from EC cells is proinflammatory. 5-HT has been postulated to stimulate 5-HT7 receptors, which dendritic cells (red) express and, in turn, drive immunity and inflammation. Contrary evidence, however, suggests that the 5-HT7 mediated effect on dendritic cells is inhibitory. Enteric neurons located in ganglia (green) of the myenteric plexus, between the circular and longitudinal layers of smooth muscle, also produce 5-HT. Terminals of these neurons project to the submucosal plexus (green). The neuronal pool of 5-HT is anti-inflammatory and, through 5-HT4 receptor stimulation, exerts neuroprotective effects and promotes neurogenesis to help enable the enteric nervous system to survive the detrimental effects of inflammation.
Whatever the explanation of the proinflammatory effects of 5-HT turns out to be after inflammation has been initiated, the participation of 5-HT gathers force in a positive feedback loop. Inflammation leads to the downregulation of SERT, which in turn makes 5-HT more effective [96,97]. Inflammation also causes the numbers of EC cells to increase, which presumably leads to still more 5-HT secretion [98,99]. This initiates a self-perpetuating cycle because intestinal inflammation causes downregulation of SERT that is accompanied by an increase in EC cell number which can result in higher levels of available 5-HT owing to decreased 5-HT inactivation and/or larger amounts 5-HT released [96,97,99]. Conversely, the selective pharmacological inhibition of mucosal but not neuronal TPH, similarly to deletion of TPH1, decreases the severity of experimental inflammation [63].
The clinical relevance of the proinflammatory effects of 5-HT is not yet totally clear. Serum serotonin levels have been found to correlate with the severity of pouchitis, which suggests that the secretion of 5-HT from EC cells is chronically elevated in patients who have received colectomies for Crohn’s disease or ulcerative colitis [100]. SERT transcription is decreased in individuals with UC, which should enhance responses to secreted 5-HT [101]. The gene encoding SERT, SLC6A4, moreover, displays a promoter polymorphism, and the LL variant has been associated with microscopic colitis, suggesting that SLC6A4 might be one the genes participating in generating intestinal inflammation [102]. The effects of mechanical forces and adenosine receptors that drive 5-HT secretion from EC cells have also been found to be amplified in IBD, providing a mechanism for the chronically increased release of this proinflammatory messenger [103]. The interleukin IL-1β and bacterial lipopolysaccharide (LPS) were each also observed to provoke greater secretion of 5-HT from EC cells isolated from the mucosa of individuals with Crohn’s disease than from those of control subjects [104]. The use of selective antagonists proved that IL-1β and bacterial LPS each act specifically on their cognate receptors. The enhanced responsiveness of EC cells to bacterial products and a cytokine during IBD is consistent with the idea that there is self-enhancing, autocatalytic secretion of 5-HT in IBD. A phenomenon of this type could therefore contribute to the pathogenesis of IBD and/or to the severity of GI symptoms. One interesting observation is that, during experimental inflammation in mice and in human patients with IBD, there is a strong upregulation of 5-HT7 receptor-expressing dendritic cells in the lamina propria [94]. Whether the stimulation of these receptors is driving inflammation or modulating it, however, cannot be determined until the above-discussed conflict over the effect of 5-HT7 stimulation of dendritic cells is resolved.
In contrast to mucosal 5-HT, neuronal 5-HT appears to be anti-inflammatory. Although deletion of TPH1, which is expressed in EC cells, decreases the severity of inflammation, deletion of TPH2, which is expressed in neurons, makes inflammation far more severe [63]. It is plausible that this effect is due to 5-HT-mediated neuroprotection [105–107]. This action of 5-HT is 5-HT4-dependent. 5-HT4 receptors promote neurogenesis as well as neuroprotection [48,105,108]; therefore, serotonin-induced neurogenesis may also contribute to the ability of the ENS to survive bouts of intestinal inflammation. Because intestinal inflammation is itself destructive of enteric neurons, 5-HT-mediated neuroprotection may be an essential shield that evolved to protect the bowel when the intestine mounts an inflammatory response to defend itself from infection or when inflammation occurs abnormally as a component of a GI disorder [109]. The previously discussed increase in enteric neurons after inflammation or in association with IBD may be a reflection of 5-HT-mediated neuroprotection and/or neurogenesis.
Concluding Remarks and Future Perspectives
Most earlier investigations of IBD, whether of pathogenesis or therapy, have, understandably, concentrated on innate and adaptive immunity [110]. The neuronal component has not yet been exploited for insight into causation, contributions to severity, or especially to therapy. Neuronal contributions to the dysfunction of IBD are, however, as reviewed above, significant. Neurons have the ability to modulate immunity, and thus the severity of the inflammatory process and inflammation, in turn, exerts a powerful effect on the ENS. In fact, given the unique ability of the gut to control its own behavior, and the central role that the ENS plays in that activity, it is reasonable to expect that the bidirectional interactions of immunoeffectors and the ENS is responsible, if not for IBD, then at least for some of the GI discomfort patients with IBD suffer. A great deal of this discomfort is the result of disrupted patterns of GI motility; evidence suggests that, in ulcerative colitis, total bowel transit is slow and stasis occurs in the proximal colon, although rectosigmoid transit is increased and retrograde contractions disappear [111], giving rise to diarrhea [112]. Studies in animal models of inflammation suggest that inflammation inhibits calcium-activated potassium channels in intrinsic primary afferent neurons, which makes these cells overly excitable [24,113]. The strength of synapses between interneurons is also increased, while inhibitory purinergic neuromuscular transmission is diminished [45] as a result of the oxidative stress associated with inflammation [46]. These effects create greatly enhanced activity in the ENS that creates what has been dubbed an enteric ‘attention deficit disorder’ [24]. The ability of the inflamed bowel to generate peristaltic reflexes along its proximodistal length is interrupted. Many small-molecule drugs have been developed over the years to modulate the output of the nervous system and, although such compounds have been employed effectively in the treatment of IBS, for example serotonergic agonists and antagonists [114], little is known about the therapeutic efficacy of neuroactive compounds in IBD.
The emerging idea that the ENS is involved in the regulation of more activities than merely the motility and secretion of the gut is likely to provide insights into many disorders that are now classified as idiopathic. Inflammatory or other causes of enteric dysfunction, for example, often accompany diseases that are thought to be entirely or primarily neuronal. An illustration is autism spectrum disorder (ASD) in which GI disease is fourfold more common than in the general population [115]. Hyperfunction of SERT has been found in a subset of patients with ASD and, when the human gene encoding the hyperfunctional SERT (SERT Ala56) is expressed in mice, the animals exhibit behaviors reminiscent of ASD [116]. The ENS of the SERT Ala56 mice, moreover, is hypoplastic, GI motility is slow, and intestinal barrier function is compromised [74]. The disorder of the gut in SERT Ala56 mice has been traced to an inability of 5-HT to stimulate its receptors adequately (the abnormal SERT clears 5-HT too quickly) and the crucial receptor for enteric neurogenesis was found to be 5-HT4. As a consequence, administration of a 5-HT4 agonist throughout development completely prevents the SERT Ala56-associated GI disturbances. These observations illustrate the powerful effects, for better or worse, that altered neuronal function exerts on the structure of the gut and its subsequent behavior. They also illustrate the great, but as yet not fully exploited, therapeutic potential of neuroactive compounds. With respect to IBD, it is known that inflammation is destructive of enteric neurons; therefore it is possible that 5-HT4-promoted neuroprotection or stimulation of adult neurogenesis [48,105,106,108] would be helpful in treating patients with IBD. The large stockpile of neuroactive drugs in clinical use may thus hold a few gems that, once explored, might help to alleviate the suffering of patients with IBD and other GI ailments. The future in this area may thus hold many twists and turns that cannot today be imagined. The expansion of the IBD universe to include the contributions of the ENS provides a new field for exploration and expansion of knowledge.
Trends.
Despite recent advances, the pathogenesis of inflammatory bowel disease (IBD) remains unknown.
Neuroimmune interactions contribute to the pathophysiology of intestinal inflammation, and enteric neuronal dysfunction during IBD causes considerable morbidity.
Neurotransmitters, neuromodulators, and cytokines participate in neuroimmune signaling, which is often bidirectional and may involve enteric glia.
5-HT is a paracrine, endocrine, and neurocrine signaling molecule present in the nervous system and/or mucosal epithelium of the gut of all vertebrates.
Mucosal 5-HT activates innate and adaptive immune responses, which protect against microbial invasion, but may also damage enteric neurons. Enteric neuronal 5-HT is neuroprotective, stimulates neurogenesis, and is anti-inflammatory. Pharmacological alteration of serotonergic mechanisms may be therapeutically beneficial in IBD.
Outstanding Questions.
How does mucosal 5-HT drive intestinal inflammation intestinal inflammation? What role(s) do 5HT7 receptors play and do they stimulate or inhibit dendritic cells? In addition to dendritic cells, which other effectors of immunity does 5-HT regulate?
Is the proinflammatory response to mucosal 5-HT of pathogenic significance in IBD?
Polymorphisms in SLC6A4, which encodes the 5-HT transporter, have been linked to psychiatric disorders and irritable bowel syndrome; do they also predispose to IBD?
What are the specific enteric neuronal mechanisms that predispose or contribute to the severity of IBD?
Can small-molecule neuroactive drugs be used in the treatment of IBD?
Do the neuroimmune interactions that occur during IBS affect the CNS and alter mood and behavior?
Is GI dysfunction an intrinsic constituent of primary neuronal diseases, such as ASD?
Acknowledgments
Supported by National Institutes of Health grants NS15547, DK093786, and by the Einhorn Charitable Trust.
References
- 1.Ananthakrishnan AN. Epidemiology and risk factors for IBD. Nat Rev Gastroenterol Hepatol. 2015;12:205–217. doi: 10.1038/nrgastro.2015.34. [DOI] [PubMed] [Google Scholar]
- 2.Goyal MS, et al. Feeding the brain and nurturing the mind: linking nutrition and the gut microbiota to brain development. Proc Natl Acad Sci USA. 2015;112:14105–14112. doi: 10.1073/pnas.1511465112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sanchez de Medina F, et al. Intestinal inflammation and mucosal barrier function. Inflamm Bowel Dis. 2014;20:2394–2404. doi: 10.1097/MIB.0000000000000204. [DOI] [PubMed] [Google Scholar]
- 4.Furness JB. The enteric nervous system: normal functions and enteric neuropathies. Neurogastroenterol Motil. 2008;20(Suppl 1):32–38. doi: 10.1111/j.1365-2982.2008.01094.x. [DOI] [PubMed] [Google Scholar]
- 5.Mayer EA, et al. Gut microbes and the brain: paradigm shift in neuroscience. J Neurosci. 2014;34:15490–15496. doi: 10.1523/JNEUROSCI.3299-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sampson TR, Mazmanian SK. Control of brain development, function, and behavior by the microbiome. Cell Host Microbe. 2015;17:565–576. doi: 10.1016/j.chom.2015.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Forsythe P, et al. Vagal pathways for microbiome–brain–gut axis communication. Adv Exp Med Biol. 2014;817:115–133. doi: 10.1007/978-1-4939-0897-4_5. [DOI] [PubMed] [Google Scholar]
- 8.Clark KB, et al. Enhanced recognition memory following vagus nerve stimulation in human subjects. Nat Neurosci. 1999;2:94–98. doi: 10.1038/4600. [DOI] [PubMed] [Google Scholar]
- 9.Rush AJ, et al. Vagus nerve stimulation (VNS) for treatment-resistant depressions: a multicenter study. Biol Psychiatry. 2000;47:276–286. doi: 10.1016/s0006-3223(99)00304-2. [DOI] [PubMed] [Google Scholar]
- 10.Margolis KG, et al. Enteric neuronal density contributes to the severity of intestinal inflammation. Gastroenterology. 2011;141:588–598. doi: 10.1053/j.gastro.2011.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Costes LM, et al. Neural networks in intestinal immunoregulation. Organogenesis. 2013;9:216–223. doi: 10.4161/org.25646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steinhoff MS, et al. Tachykinins and their receptors: contributions to physiological control and the mechanisms of disease. Physiol Rev. 2014;94:265–301. doi: 10.1152/physrev.00031.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ottaviani E, et al. Common evolutionary origin of the immune and neuroendocrine systems: from morphological and functional evidence to in silico approaches. Trends Immunol. 2007;28:497–502. doi: 10.1016/j.it.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 14.Hartenstein V. The neuroendocrine system of invertebrates: a developmental and evolutionary perspective. J Endocrinol. 2006;190:555–570. doi: 10.1677/joe.1.06964. [DOI] [PubMed] [Google Scholar]
- 15.Funayama N. The stem cell system in demosponges: suggested involvement of two types of cells: archeocytes (active stem cells) and choanocytes (food-entrapping flagellated cells) Dev Genes Evol. 2013;223:23–38. doi: 10.1007/s00427-012-0417-5. [DOI] [PubMed] [Google Scholar]
- 16.Kedees MH, et al. Functional activity of murine intestinal mucosal cells is regulated by the glucagon-like peptide-1 receptor. Peptides. 2013;48:36–44. doi: 10.1016/j.peptides.2013.07.022. [DOI] [PubMed] [Google Scholar]
- 17.Mukherjee S, Hooper LV. Antimicrobial defense of the intestine. Immunity. 2015;42:28–39. doi: 10.1016/j.immuni.2014.12.028. [DOI] [PubMed] [Google Scholar]
- 18.Margolis KG, Gershon MD. Neuropeptides and inflammatory bowel disease. Curr Opin Gastroenterol. 2009;25:503–511. doi: 10.1097/MOG.0b013e328331b69e. [DOI] [PubMed] [Google Scholar]
- 19.Buhner S, Schemann M. Mast cell–nerve axis with a focus on the human gut. Biochim Biophys Acta. 2012;1822:85–92. doi: 10.1016/j.bbadis.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 20.Verheijden S, et al. Neuron-macrophage crosstalk in the intestine: a ‘microglia’ perspective. Front Cell Neurosci. 2015;9:403. doi: 10.3389/fncel.2015.00403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cailotto C, et al. Neuro-anatomical evidence indicating indirect modulation of macrophages by vagal efferents in the intestine but not in the spleen. PLoS ONE. 2014;9:e87785. doi: 10.1371/journal.pone.0087785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muller PA, et al. Crosstalk between muscularis macrophages and enteric neurons regulates gastrointestinal motility. Cell. 2014;158:300–313. doi: 10.1016/j.cell.2014.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gabanyi I, et al. Neuro-immune interactions drive tissue programming in intestinal macrophages. Cell. 2016;164:378–391. doi: 10.1016/j.cell.2015.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mawe GM. Colitis-induced neuroplasticity disrupts motility in the inflamed and post-inflamed colon. J Clin Invest. 2015;125:949–955. doi: 10.1172/JCI76306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neunlist M, et al. Enteric glial cells: recent developments and future directions. Gastroenterology. 2014;147:1230–1237. doi: 10.1053/j.gastro.2014.09.040. [DOI] [PubMed] [Google Scholar]
- 26.Sharkey KA. Emerging roles for enteric glia in gastrointestinal disorders. J Clin Invest. 2015;125:918–925. doi: 10.1172/JCI76303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brown IA, et al. Enteric glia mediate neuron death in colitis through purinergic pathways that require connexin-43 and nitric oxide. Cell Mol Gastroenterol Hepatol. 2016;2:77–91. doi: 10.1016/j.jcmgh.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gulbransen BD, et al. Activation of neuronal P2X7 receptor–pannexin-1 mediates death of enteric neurons during colitis. Nat Med. 2012;18:600–604. doi: 10.1038/nm.2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bush TG, et al. Fulminant jejuno-ileitis following ablation of enteric glia in adult transgenic mice. Cell. 1998;93:189–201. doi: 10.1016/s0092-8674(00)81571-8. [DOI] [PubMed] [Google Scholar]
- 30.Kempermann G, et al. Neurogenesis in the adult hippo-campus. Cold Spring Harb Perspect Biol. 2015;7:a018812. doi: 10.1101/cshperspect.a018812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gershon MD. Behind an enteric neuron there may lie a glial cell. J Clin Invest. 2011;121:3386–3389. doi: 10.1172/JCI59573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rao M, et al. Enteric glia express proteolipid protein 1 and are a transcriptionally unique population of glia in the mammalian nervous system. Glia. 2015;63:2040–2057. doi: 10.1002/glia.22876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boesmans W, et al. Heterogeneity and phenotypic plasticity of glial cells in the mammalian enteric nervous system. Glia. 2015;63:229–241. doi: 10.1002/glia.22746. [DOI] [PubMed] [Google Scholar]
- 34.Gulbransen BD, Sharkey KA. Novel functional roles for enteric glia in the gastrointestinal tract. Nat Rev Gastroenterol Hepatol. 2012;9:625–632. doi: 10.1038/nrgastro.2012.138. [DOI] [PubMed] [Google Scholar]
- 35.Nasser Y, et al. Role of enteric glia in intestinal physiology: effects of the gliotoxin fluorocitrate on motor and secretory function. Am J Physiol Gastrointest Liver Physiol. 2006;291:G912–G927. doi: 10.1152/ajpgi.00067.2006. [DOI] [PubMed] [Google Scholar]
- 36.Rao M. Defining the role of glia in the enteric nervous system. J Pediatr Gastroenterol Nutr. 2015;61:000–000. [Google Scholar]
- 37.Laranjeira C, et al. Glial cells in the mouse enteric nervous system can undergo neurogenesis in response to injury. J Clin invest. 2011;121:3412–3424. doi: 10.1172/JCI58200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kabouridis PS, et al. Microbiota controls the homeostasis of glial cells in the gut lamina propria. Neuron. 2015;85:289–295. doi: 10.1016/j.neuron.2014.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bernardini N, et al. Immunohistochemical analysis of myenteric ganglia and interstitial cells of Cajal in ulcerative colitis. J Cell Mol Med. 2012;16:318–327. doi: 10.1111/j.1582-4934.2011.01298.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Furness JB, et al. Intrinsic primary afferent neurons and nerve circuits within the intestine. Prog Neurobiol. 2004;72:143–164. doi: 10.1016/j.pneurobio.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 41.Linden DR, et al. Enhanced excitability of myenteric AH neurones in the inflamed guinea-pig distal colon. J Physiol. 2003;547:589–601. doi: 10.1113/jphysiol.2002.035147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krauter EM, et al. Synaptic plasticity in myenteric neurons of the guinea-pig distal colon: presynaptic mechanisms of inflammation-induced synaptic facilitation. J Physiol. 2007;581:787–800. doi: 10.1113/jphysiol.2007.128082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lomax AE, et al. Synaptic facilitation and enhanced neuronal excitability in the submucosal plexus during experimental colitis in guinea-pig. J Physiol. 2005;564:863–875. doi: 10.1113/jphysiol.2005.084285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roberts JA, et al. The roles of purinergic signaling during gastrointestinal inflammation. Curr Opin Pharmacol. 2012;12:659–666. doi: 10.1016/j.coph.2012.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Strong DS, et al. Purinergic neuromuscular transmission is selectively attenuated in ulcerated regions of inflamed guinea pig distal colon. J Physiol. 2010;588:847–859. doi: 10.1113/jphysiol.2009.185082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roberts JA, et al. Oxidative stress disrupts purinergic neuromuscular transmission in the inflamed colon. J Physiol. 2013;591:3725–3737. doi: 10.1113/jphysiol.2013.254136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Linden DR, et al. Indiscriminate loss of myenteric neurones in the TNBS-inflamed guinea-pig distal colon. Neurogastroenterol Motil. 2005;17:751–760. doi: 10.1111/j.1365-2982.2005.00703.x. [DOI] [PubMed] [Google Scholar]
- 48.Belkind-Gerson J, et al. Colitis induces enteric neurogenesis through a 5-HT4-dependent mechanism. Inflamm Bowel Dis. 2015;21:870–878. doi: 10.1097/MIB.0000000000000326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hons IM, et al. Alterations to enteric neural signaling underlie secretory abnormalities of the ileum in experimental colitis in the guinea pig. Am J Physiol Gastrointest Liver Physiol. 2009;296:G717–G726. doi: 10.1152/ajpgi.90472.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barada K, et al. Electrocautery-induced localized colonic injury elicits increased levels of pro-inflammatory cytokines in small bowel and decreases jejunal alanine absorption. Cytokine. 2015;71:109–118. doi: 10.1016/j.cyto.2014.08.009. [DOI] [PubMed] [Google Scholar]
- 51.Linden DR. Enhanced excitability of guinea pig ileum myenteric AH neurons during and following recovery from chemical colitis. Neurosci Lett. 2013;545:91–95. doi: 10.1016/j.neulet.2013.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cocciolillo S, Collins SM. The long-term functional consequences of acute infectious diarrhea. Curr Opin Gastroenterol. 2016;32:1–6. doi: 10.1097/MOG.0000000000000233. [DOI] [PubMed] [Google Scholar]
- 53.Spiller R, Garsed K. Postinfectious irritable bowel syndrome. Gastroenterology. 2009;136:1979–1988. doi: 10.1053/j.gastro.2009.02.074. [DOI] [PubMed] [Google Scholar]
- 54.Goodrich JT, et al. Phylogeny of enteric serotonergic neurons. J Comp Neurol. 1980;190:15–28. doi: 10.1002/cne.901900103. [DOI] [PubMed] [Google Scholar]
- 55.Trandaburu T, Trandaburu I. Serotonin (5-hydroxy-tryptamine, 5-HT) immunoreactive endocrine and neural elements in the chromaffin enteropancreatic system of amphibians and reptiles. Acta Histochem. 2007;109:237–247. doi: 10.1016/j.acthis.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 56.Grosell M, et al. Fish Physiology: The Multifunctional Gut of Fish. Elsevier Science; 2010. [Google Scholar]
- 57.Anderson C, Campbell G. Immunohistochemical study of 5-HT-containing neurons in the teleost intestine: relationship to the presence of enterochromaffin cells. Cell Tissue Res. 1988;254:553–559. doi: 10.1007/BF00226505. [DOI] [PubMed] [Google Scholar]
- 58.Ahlman H, Nilsson The gut as the largest endocrine organ in the body. Ann Oncol. 2001;12(Suppl 2):S63–S68. doi: 10.1093/annonc/12.suppl_2.s63. [DOI] [PubMed] [Google Scholar]
- 59.Latorre R, et al. Enteroendocrine cells: a review of their role in brain-gut communication. Neurogastroenterol Motil. 2016;28:620–630. doi: 10.1111/nmo.12754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Walther DJ, et al. Synthesis of serotonin by a second tryptophan hydroxylase isoform. Science. 2003;299:76. doi: 10.1126/science.1078197. [DOI] [PubMed] [Google Scholar]
- 61.Li Z, et al. Essential roles of enteric neuronal serotonin in gastrointestinal motility and the development/survival of enteric dopaminergic neurons. J Neurosci. 2011;31:8998–9009. doi: 10.1523/JNEUROSCI.6684-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cote F, et al. Disruption of the nonneuronal tph1 gene demonstrates the importance of peripheral serotonin in cardiac function. Proc Natl Acad Sci USA. 2003;100:13525–13530. doi: 10.1073/pnas.2233056100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Margolis KG, et al. Pharmacological reduction of mucosal but not neuronal serotonin opposes inflammation in mouse intestine. Gut. 2014;63:928–937. doi: 10.1136/gutjnl-2013-304901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Derkach V, et al. 5-HT3 receptors are membrane ion channels. Nature. 1989;339:706–709. doi: 10.1038/339706a0. [DOI] [PubMed] [Google Scholar]
- 65.Stasi C, et al. Serotonin receptors and their role in the pathophysiology and therapy of irritable bowel syndrome. Tech Coloproctol. 2014;18:613–621. doi: 10.1007/s10151-013-1106-8. [DOI] [PubMed] [Google Scholar]
- 66.Kirchgessner AL, et al. Identification of cells that express 5-HT1A receptors in the nervous systems of the bowel and pancreas. J Comp Neurol. 1996;364:439–455. doi: 10.1002/(SICI)1096-9861(19960115)364:3<439::AID-CNE5>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 67.Glatzle J, et al. Expression of 5-HT3 receptors in the rat gastrointestinal tract. Gastroenterology. 2002;123:217–226. doi: 10.1053/gast.2002.34245. [DOI] [PubMed] [Google Scholar]
- 68.Liu M, et al. Expression and function of 5-HT4 receptors in the mouse enteric nervous system. Am J Physiol Gastrointest Liver Physiol. 2005;289:G1148–G1163. doi: 10.1152/ajpgi.00245.2005. [DOI] [PubMed] [Google Scholar]
- 69.Poole DP, et al. Identification of neurons that express 5-hydroxytryptamine4 receptors in intestine. Cell Tissue Res. 2006;325:413–422. doi: 10.1007/s00441-006-0181-9. [DOI] [PubMed] [Google Scholar]
- 70.Tonini M, et al. 5-HT7 receptors modulate peristalsis and accommodation in the guinea pig ileum. Gastroenterology. 2005;129:1557–1566. doi: 10.1053/j.gastro.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 71.Yaakob NS, et al. Distribution of 5-HT3, 5-HT4, and 5-HT7 receptors along the human colon. J Neurogastroenterol Motil. 2015;21:361–369. doi: 10.5056/jnm14157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shajib MS, Khan WI. The role of serotonin and its receptors in activation of immune responses and inflammation. Acta physiologica. 2015;213:561–574. doi: 10.1111/apha.12430. [DOI] [PubMed] [Google Scholar]
- 73.Blakely RD, et al. Molecular physiology of norepinephrine and serotonin transporters. J Exp Biol. 1994;196:263–281. doi: 10.1242/jeb.196.1.263. [DOI] [PubMed] [Google Scholar]
- 74.Margolis KG, et al. Serotonin transporter variant drives preventable gastrointestinal abnormalities in development and function. J Clin Invest. 2016 doi: 10.1172/JCI84877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gershon MD. Review article: serotonin receptors and transporters–roles in normal and abnormal gastrointestinal motility. Aliment Pharmacol Ther. 2004;20(Suppl 7):3–14. doi: 10.1111/j.1365-2036.2004.02180.x. [DOI] [PubMed] [Google Scholar]
- 76.Beikmann BS, et al. Serotonin uptake is largely mediated by platelets versus lymphocytes in peripheral blood cells. ACS chem Neurosci. 2013;4:161–170. doi: 10.1021/cn300146w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lesurtel M, et al. Platelet-derived serotonin mediates liver regeneration. Science. 2006;312:104–107. doi: 10.1126/science.1123842. [DOI] [PubMed] [Google Scholar]
- 78.Kode A, et al. Lrp5 regulation of bone mass and serotonin synthesis in the gut. Nat Med. 2014;20:1228–1229. doi: 10.1038/nm.3698. [DOI] [PubMed] [Google Scholar]
- 79.Weihe E, et al. Localization of vesicular monoamine transporter isoforms (VMAT1 and VMAT2) to endocrine cells and neurons in rat. J Mol Neurosci. 1994;5:149–164. doi: 10.1007/BF02736730. [DOI] [PubMed] [Google Scholar]
- 80.Gershon MD, Ross LL. Radioisotopic studies of the binding, exchange, and distribution of 5-hydroxytrypta-mine synthesized from its radioactive precursor. J Physiol. 1966;186:451–476. doi: 10.1113/jphysiol.1966.sp008046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gershon MD, et al. Type-specific localization of mono-amine oxidase in the enteric nervous system: relatonship to 5-hydroxytryptamine, neuropeptides, and sympathetic nerves. J Comp Neurol. 1990;301:191–213. doi: 10.1002/cne.903010205. [DOI] [PubMed] [Google Scholar]
- 82.Baganz NL, Blakely RD. A dialogue between the immune system and brain, spoken in the language of serotonin. ACS Chem Neurosci. 2013;4:48–63. doi: 10.1021/cn300186b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Manning BM, et al. Single-cell analysis of mast cell degranulation induced by airway smooth muscle-secreted chemokines. Biochim Biophys Acta. 2015;1850:1862–1868. doi: 10.1016/j.bbagen.2015.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Arreola R, et al. Immunomodulatory effects mediated by serotonin. J Immunol Res. 2015;2015:354957. doi: 10.1155/2015/354957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Askenase PW, et al. Serotonin initiation of delayed-type hypersensitivity: mediation by a primitive Thy-1+ antigen-specific clone or by specific monoclonal IgE antibody. Skin Pharmacol. 1991;4(Suppl 1):25–42. doi: 10.1159/000210981. [DOI] [PubMed] [Google Scholar]
- 86.Askenase PW, et al. T cell-dependent mast cell degranulation and release of serotonin in murine delayed-type hypersensitivity. J Exp Med. 1980;152:1358–1374. doi: 10.1084/jem.152.5.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gershon RK, et al. Requirement for vasoactive amines for production of delayed-type hypersensitvity skin reactions. J Exp Med. 1975;142:732–747. doi: 10.1084/jem.142.3.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Thoa NB, et al. The accumulation of carbon 14-serotonin in the guinea-pig vas deferens. J Pharmacol Exp Ther. 1969;169:68–73. [PubMed] [Google Scholar]
- 89.Saito A, Lee TJ. Serotonin as an alternative transmitter in sympathetic nerves of large cerebral arteries of the rabbit. Circ Res. 1987;60:220–228. doi: 10.1161/01.res.60.2.220. [DOI] [PubMed] [Google Scholar]
- 90.Bischoff SC, et al. Role of serotonin in intestinal inflammation: knockout of serotonin reuptake transporter exacerbates 2,4,6-trinitrobenzene sulfonic acid colitis in mice. Am J Physiol Gastrointest Liver Physiol. 2009;296:G685–G695. doi: 10.1152/ajpgi.90685.2008. [DOI] [PubMed] [Google Scholar]
- 91.Ghia JE, et al. Serotonin has a key role in pathogenesis of experimental colitis. Gastroenterology. 2009;137:1649–1660. doi: 10.1053/j.gastro.2009.08.041. [DOI] [PubMed] [Google Scholar]
- 92.Li N, et al. Serotonin activates dendritic cell function in the context of gut inflammation. Am J Pathol. 2011;178:662–671. doi: 10.1016/j.ajpath.2010.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kim JJ, et al. Targeted inhibition of serotonin type 7 (5-HT7) receptor function modulates immune responses and reduces the severity of intestinal inflammation. J Immunol. 2013;190:4795–4804. doi: 10.4049/jimmunol.1201887. [DOI] [PubMed] [Google Scholar]
- 94.Guseva D, et al. Serotonin 5-HT7 receptor is critically involved in acute and chronic inflammation of the gastrointestinal tract. Inflamm Bowel Dis. 2014;20:1516–1529. doi: 10.1097/MIB.0000000000000150. [DOI] [PubMed] [Google Scholar]
- 95.Idzko M, et al. The serotoninergic receptors of human dendritic cells: identification and coupling to cytokine release. J Immunol. 2004;172:6011–6019. doi: 10.4049/jimmunol.172.10.6011. [DOI] [PubMed] [Google Scholar]
- 96.Linden DR, et al. Serotonin availability is increased in mucosa of guinea pigs with TNBS-induced colitis. Am J Physiol Gastrointest Liver Physiol. 2003;285:G207–G216. doi: 10.1152/ajpgi.00488.2002. [DOI] [PubMed] [Google Scholar]
- 97.Linden DR, et al. Serotonin transporter function and expression are reduced in mice with TNBS-induced colitis. Neurogastroenterol Motil. 2005;17:565–574. doi: 10.1111/j.1365-2982.2005.00673.x. [DOI] [PubMed] [Google Scholar]
- 98.Spiller RC, et al. Increased rectal mucosal enteroendocrine cells, T lymphocytes, and increased gut permeability following acute Campylobacter enteritis and in post-dysenteric irritable bowel syndrome. Gut. 2000;47:804–811. doi: 10.1136/gut.47.6.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.O’Hara JR, et al. Enteroendocrine cells and 5-HT availability are altered in mucosa of guinea pigs with TNBS ileitis. Am J Physiol Gastrointest Liver Physiol. 2004;287:G998–G1007. doi: 10.1152/ajpgi.00090.2004. [DOI] [PubMed] [Google Scholar]
- 100.Wang Y, et al. Correlation between serum serotonin and endoscopy inflammation scores in patients with ileal pouches. J Crohns Colitis. 2013;7:e133–e142. doi: 10.1016/j.crohns.2012.07.028. [DOI] [PubMed] [Google Scholar]
- 101.Coates MD, et al. Molecular defects in mucosal serotonin content and decreased serotonin reuptake transporter in ulcerative colitis and IBS. Gastroentrology. 2004;126:1657–1664. doi: 10.1053/j.gastro.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 102.Sikander A, et al. Association of serotonin transporter promoter polymorphism (5-HTTLPR) with microscopic colitis and ulcerative colitis. Dig Dis Sci. 2015;60:887–894. doi: 10.1007/s10620-014-3482-y. [DOI] [PubMed] [Google Scholar]
- 103.Chin A, et al. The role of mechanical forces and adenosine in the regulation of intestinal enterochromaffin cell serotonin secretion. Am J Physiol Gastrointest Liver Physiol. 2012;302:G397–G405. doi: 10.1152/ajpgi.00087.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kidd M, et al. IL1beta- and LPS-induced serotonin secretion is increased in EC cells derived from Crohn’s disease. Neurogastroenterol Motil. 2009;21:439–450. doi: 10.1111/j.1365-2982.2008.01210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Liu MT, et al. 5-HT4 receptor-mediated neuroprotection and neurogenesis in the enteric nervous system of adult mice. J Neurosci. 2009;29:9683–9699. doi: 10.1523/JNEUROSCI.1145-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bianco F, et al. Prucalopride exerts neuroprotection in human enteric neurons. Am J Physiol Gastrointest Liver Physiol. 2016;310:G768–G775. doi: 10.1152/ajpgi.00036.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gershon MD. 5-HT4-mediated neuroprotection: a new therapeutic modality on the way? Am J Physiol Gastrointest Liver Physiol. 2016;310:G766–G767. doi: 10.1152/ajpgi.00120.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Goto K, et al. Activation of 5-HT4 receptors facilitates neurogenesis from transplanted neural stem cells in the anastomotic ileum. J physiol Sci. 2016;66:67–76. doi: 10.1007/s12576-015-0396-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gershon MD. Serotonin is a sword and a shield of the bowel: serotonin plays offense and defense. Trans Am Clin Climato Assoc. 2012;123:268–280. [PMC free article] [PubMed] [Google Scholar]
- 110.Manuc TE, et al. Recent insights into the molecular pathogenesis of Crohn’s disease: a review of emerging therapeutic targets. Clin Exp Gastroenterol. 2016;9:59–70. doi: 10.2147/CEG.S53381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bassotti G, et al. Twenty-four-hour manometric study of colonic propulsive activity in patients with diarrhea due to inflammatory (ulcerative colitis) and non-inflammatory (irritable bowel syndrome) conditions. Int J Colorectal Dis. 2004;19:493–497. doi: 10.1007/s00384-004-0604-6. [DOI] [PubMed] [Google Scholar]
- 112.Rao SS, et al. Studies on the mechanism of bowel disturbance in ulcerative colitis. Gastroenterology. 1987;93:934–940. doi: 10.1016/0016-5085(87)90554-3. [DOI] [PubMed] [Google Scholar]
- 113.Linden DR, et al. Cyclooxygenase-2 contributes to dysmotility and enhanced excitability of myenteric AH neurones in the inflamed guinea pig distal colon. J Physiol. 2004;557:191–205. doi: 10.1113/jphysiol.2004.062174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hornby PJ. Drug discovery approaches to irritable bowel syndrome. Expert Opin Drug Discov. 2015;10:809–824. doi: 10.1517/17460441.2015.1049528. [DOI] [PubMed] [Google Scholar]
- 115.McElhanon BO, et al. Gastrointestinal symptoms in autism spectrum disorder: a meta-analysis. Pediatrics. 2014;133:872–883. doi: 10.1542/peds.2013-3995. [DOI] [PubMed] [Google Scholar]
- 116.Veenstra-VanderWeele J, et al. Autism gene variant causes hyperserotonemia, serotonin receptor hypersensitivity, social impairment and repetitive behavior. Proc Natl Acad Sci USA. 2012;109:5469–5474. doi: 10.1073/pnas.1112345109. [DOI] [PMC free article] [PubMed] [Google Scholar]