

Abstract
Osteosarcoma is the most common primary bone cancer. It can be cured by aggressive surgery and chemotherapy, but outcomes for metastatic or chemoresistant disease remain dismal. Cancer sequencing studies have shown that the p53 pathway is dysregulated in nearly every case, often by translocation; however, no studies of osteosarcoma evolution or intratumor heterogeneity have been done to date. We studied a patient with chemoresistant, metastatic disease over the course of 3 years. We performed exome sequencing on germline DNA and DNA collected from tumor at three separate time points. We compared variant calls and variant allele frequencies between different samples. We identified subclonal mutations in several different genes in the primary tumor sample and found that one particular subclone dominated subsequent tumor samples at relapse. This clone was marked by a novel TP53-KPNA3 translocation and loss of the opposite-strand wild-type TP53 allele. Future research must focus on the functional significance of such clones and strategies to eliminate them.
Keywords: neoplasm of the skeletal system, osteosarcoma
INTRODUCTION
Osteosarcoma is the most common primary bone cancer and primarily affects adolescents and young adults (Mirabello et al. 2009). Anatomically, osteosarcoma most commonly affects metaphyseal regions of the long bones (distal femur, proximal tibia, and proximal humerus). Peak incidence occurs in adolescence and is associated with the adolescent growth spurt. Aggressive surgery alone cures a fraction of affected patients. With the addition of neoadjuvant chemotherapy with methotrexate, doxorubicin, and cisplatin, patients with nonmetastatic disease at diagnosis currently have >70% long-term relapse-free survival (Mirabello et al. 2009). However, despite many attempts to incorporate other active agents into this therapeutic backbone, outcomes have not improved in the last three decades. Additionally, patients with metastatic or relapsed disease continue to have dismal outcomes. As such, identifying and targeting the biological drivers of metastatic and relapsed osteosarcoma remains important in developing further improvements in therapy.
Osteosarcoma is linked to germline predisposition syndromes, most strongly hereditary retinoblastoma (Hansen et al. 1985) and Li–Fraumeni syndrome (McIntyre et al. 1994). In addition, osteosarcoma predisposition has also been linked to germline mutations in RECQ helicases, which causes Rothmund–Thomson syndrome (Kitao et al. 1999), and SQSTM1, which predisposes to Paget's disease of the bone (Laurin et al. 2002). More recently, genome-wide association studies have identified two risk loci near the GRM4 locus (Savage et al. 2013). The only known environmental risk factor for developing osteosarcoma is exposure to ionizing radiation.
A recent next-generation sequencing study of 34 primary pediatric osteosarcoma samples identified recurrent somatic mutations impairing p53 activity in nearly every sample (Chen et al. 2014). The most striking mutations are translocations involving the TP53 gene itself, particularly translocations involving the first intron. Such a rearrangement has even been reported in the germline of one family with clinical Li–Fraumeni syndrome but without mutation in the TP53 coding sequence (Ribi et al. 2015). Other genomic aberrations affecting the p53 pathway include genomic loss of TP53 and modification of the MDM2 locus. Recurrent somatic mutations were also found in RB1, ATRX, and DLG2. Another next-generation sequencing study also identified frequent copy number changes affecting genes in the PI3K–mTOR pathway (Perry et al. 2014). However, none of these studies investigated genomic drivers of chemoresistance.
Genomically, osteosarcoma may be more similar to adult-type cancers than other solid tumors that affect young people. Unlike most pediatric cancers, osteosarcoma displays a high rate of somatic mutation and even displays foci of kataegis, a specific type of localized hypermutation previously identified in breast (Nik-Zainal et al. 2012), pancreatic, lung, and other types of cancer (Alexandrov et al. 2013), but not previously identified in other pediatric cancers. In addition, unlike most other pediatric cancer subtypes that respond well to multiagent chemotherapy, osteosarcoma remains primarily a surgical disease where grossly unresectable disease is incurable with chemotherapy. In many adult cancer types, including lung (de Bruin et al. 2014; Zhang et al. 2014), kidney (Gerlinger et al. 2012), and prostate cancer (Gundem et al. 2015; Hong et al. 2015), deep sequencing has been used to study dynamics of intratumor heterogeneity and to understand how these cancers change as they metastasize and develop chemoresistance. In some cases, a small fraction of the tumor already harboring a resistance-conferring mutation at diagnosis can become the dominant clone at the time of relapse. In other cases, under the selective pressure of chemotherapy, acquisition of new mutations drives the relapse clone (Ding et al. 2012). Occasionally, polyclonal seeding can lead to metastasis and resistance (Gundem et al. 2015).
Herein, we describe genetic changes detected in longitudinally collected samples from an aggressive, multiply relapsed metastatic osteosarcoma patient. These samples were taken over the course of several years and through multiple courses of therapy. We performed whole-exome sequencing from formalin-fixed, paraffin-embedded (FFPE) tumor samples to study the evolution of this cancer during this time. Single-nucleotide variants (SNVs), insertions or deletions (indels), and structural variants (SVs) were called. Strikingly, we identified a novel translocation involving TP53 that accumulated in frequency over time. Unlike previously reported translocations, this one affected the 3′ untranslated region (3′ UTR) of TP53, revealing a novel mechanism for dysregulating p53. Together, these results give fresh insight into the genetic changes that occur as an osteosarcoma evolves, relapses, and metastasizes. To our knowledge, no other study has investigated the subclonal dynamics of osteosarcoma from diagnosis to death from multiple relapses.
RESULTS
Clinical Presentation and Family History
The patient was a 25-yr-old man diagnosed with conventional, osteoblastic-type high-grade osteosarcoma of the right fibula (Fig. 1). He received two rounds of preoperative chemotherapy with cisplatin and doxorubicin before his primary tumor resection and received two more rounds postoperatively. However, 8 months after finishing chemotherapy, he was found to have tumor recurrences in his fibula and lung. After two rounds of salvage chemotherapy with methotrexate, ifosfamide, and etoposide, a secondary resection was performed. He continued to receive chemotherapy, with ifosfamide, etoposide, and cisplatin. Nevertheless, he continued to develop more metastatic disease in his lungs, axilla, scapula, and chest wall, and a tertiary tumor sample was taken at this time that continued to show viable tumor. Despite further attempts at salvage with docetaxel, gemcitabine, and radiation, he ultimately died of his disease.
Figure 1.

Time line of events in the patient's history, in months after diagnosis. Diagnosis of initial disease or relapse displayed above the time line; interventions displayed below the time line. The patient was initially treated with neoadjuvant and adjuvant chemotherapy sandwiched around a large resection of the primary site of disease. This was followed by several relapses that were treated with the surgeries and chemotherapy regimens as indicated. Please see text for further details. Chemotherapy regimens: AP, cisplatin and doxorubicin (adriamycin); MEI, methotrexate, etoposide, and ifosfamide; IVP, ifosfamide, etoposide, and cisplatin; GD, gemcitabine and docetaxel.
Genomic Analyses
Germline tissue (blood) and primary, secondary, and tertiary tumor samples were submitted for whole-exome sequencing. Alignment statistics are shown in Supplemental Table S1. More than 45 million reads were obtained from each sample, and 95% of these were mapped. This resulted in mean coverage depth of 40×–57× across all targeted regions.
Detection of Nonsynonymous Variants
In all, there were a total of 118 nonsynonymous variants in the primary tumor sample, 191 variants in the secondary tumor sample, and 290 variants in the tertiary tumor sample (Fig. 2). A large number of the variants overlapped, especially between the secondary and tertiary samples. The number of variants detected in each sample increased over time, which may reflect accumulating mutations in response to chemotherapy. These variants are listed in Supplemental Table S2. Despite the fact that mean coverage depth was only 40×–57× and that the blood sample was processed differently than the tumor samples (which were FFPE), the vast majority of variants detected in germline tissue were also detected in the tumor samples (Supplemental Fig. S1).
Figure 2.
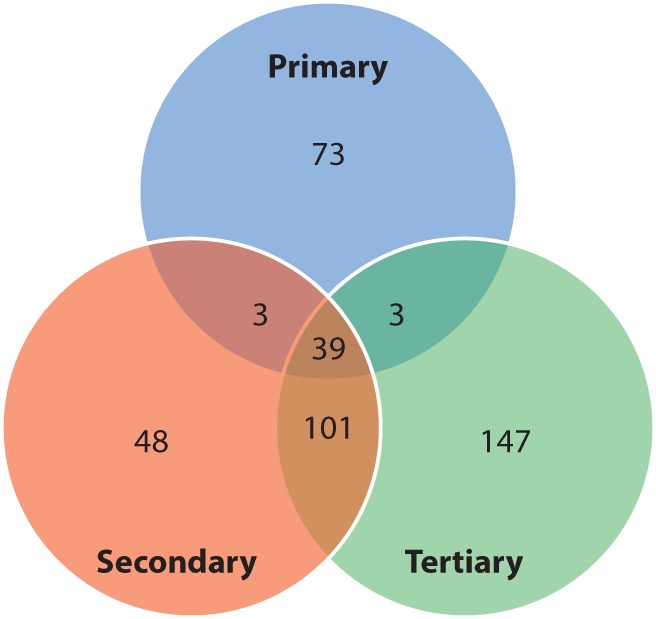
Venn diagram of nonsynonymous variants identified in each sample. MuTect was used to call variants in each sample that did not appear in the germline sequencing sample. See Supplemental Table S2 for all variants.
In addition, indels were detected using Pindel (Supplemental Table S3), and copy number variation was computed using ExomeCNV (Supplemental Table S4; Supplemental File S1).
Tumor Evolution
Next we used these variant calls to examine the evolutionary relationships among these samples by constructing a maximum parsimony phylogenetic tree and performing a principal coordinate analysis in an unbiased manner. As shown in Figure 3A,B, these calculations concurred with the time relationship between these samples. The primary tumor sample diverged from the germline (blood) sample, and the secondary and tertiary samples represented a further step in tumor evolution away from the primary sample.
Figure 3.

Intersample relationships. (A) Phylogenetic tree. The Maximum Parsimony tree was obtained using the Max-mini branch-and-bound algorithm. The branch lengths were calculated using the average pathway method. Evolutionary analyses were conducted in MEGA6. (B) Principal coordinate analysis. The x- and y-axes represent the first and second coordinates, respectively. In this case, the first and second principal coordinates are genetic variation and somatic mutation, respectively. (C) Heatmap displaying total root-mean-square deviation of each sample's variant allele frequencies from each other.
In other tumor types, studies of intratumor heterogeneity have shown that chemotherapeutic resistance may arise from the outgrowth of a subclonal population that was present at diagnosis. However, this has not previously been studied in osteosarcoma. Thus, we next examined how these tumor samples related to one another to identify how subclonal populations shifted over the course of time. We calculated relationships between samples by calculating the total root-mean-square deviation (RMSD) of each sample's variant allele frequencies (Fig. 3C). When compared with the reference genome, this again shows a clear progression from germline to primary, secondary, and tertiary tumor samples corresponding to their relation in time. As these relationships are calculated from variant allele frequencies, this implies the progression of a subclonal population in the primary tumor sample that becomes the dominant clone in the tertiary sample.
Translocation Involving Chromosomes 13 and 17
Previous osteosarcoma sequencing studies have shown somatic events affecting the TP53 pathway in nearly every osteosarcoma sample studied; the most common TP53-related events in osteosarcoma are translocations involving the TP53 gene itself (Chen et al. 2014; Perry et al. 2014; Ribi et al. 2015). Thus we next applied a soft-clipping-based detection method called sample heterogeneity estimation and assembly by reference (SHEAR) (Landman et al. 2014) and a custom script we named “soft-clipping and realignment detects structural variant frequency” (SCARF; see Methods for further detail) to identify fusion events in our exome sequencing results (Supplemental Table S5). Strikingly, in all three tumor specimens, we identified a t(13;17) translocation that fuses the 3′ UTR of TP53 to the first intron of KPNA3 (Fig. 4A; Supplemental File S2). Because this translocation involves an intron, our standard exome-sequencing analysis pipeline did not detect it. Most translocations in osteosarcoma involve the first intron of TP53; translocations involving the 3′ UTR of TP53 have not previously been reported.
Figure 4.

TP53-KPNA3 translocation. (A) A schematic showing translocation between the last exon of TP53 and the first intron of KPNA3 as identified by whole-exome sequencing. The genomic coordinates are based on hg19. (B) Sanger sequencing validation of the translocation breakpoint in secondary and tertiary tumor samples.
To validate this finding, we performed Sanger sequencing on the three tumor samples. We sequenced an amplicon produced with one primer aligning to Chromosome 13 and a second to Chromosome 17 (Fig. 4B). We were unable to amplify the translocation from the primary tumor sample, but sequencing from the secondary and tertiary tumor samples showed the translocation breakpoint concurring with the exome sequencing.
When we examined the frequency of reads supporting this translocation in our three tumor samples, we noticed a striking accumulation of these reads in later tumor samples. The ratio of reads supporting the translocation to reads supporting the reference TP53 sequence increased from 0:65 in the germline sample to 10:62, 42:2, and 45:4 in the primary, secondary, and tertiary samples, respectively (Fig. 5). The increasing proportion of reads supporting the translocation again supports the hypothesis that this translocation marks a subclonal population that was present in the primary tumor sample but became the dominant clone in subsequent postchemotherapy samples.
Figure 5.

Reads in each sample supporting the reference TP53 exon versus the TP53-KPNA3 translocation breakpoint. Genomic coordinates based on hg19. Duplicates included.
The number of reads corresponding to the translocation may be an underestimate of the actual predominance of the translocation, as exome-captured reads would be enriched for sequences containing exonic TP53 sequences. In support of this, we did not detect any reads aligning to the KPNA3 intron alone, unconjugated to the TP53 exon.
In other osteosarcomas, translocations involving TP53 usually co-occur with loss of the wild-type allele (Chen et al. 2014). In our case, because so few of the reads align to the reference sequence, it appears that the predominant clone has also undergone loss of the wild-type TP53 allele. Accordingly, fluorescence in situ hybridization (FISH) for TP53 in the secondary and tertiary tumor samples appeared to show loss of one copy of TP53 in many cells (Supplemental Fig. S2).
Finally, to determine the effect of this translocation on TP53 protein levels, we performed immunohistochemistry (IHC) for TP53 on samples from biopsy, primary, secondary, and tertiary archival tumor samples (Supplemental Fig. S3). There was no detectable TP53 protein in any of the four samples by IHC, implying that the translocation results in functional TP53 loss.
DISCUSSION
Unlike many other malignancies of childhood or young adulthood, osteosarcoma outcomes have not improved for several decades (Mirabello et al. 2009). Although chemotherapy may treat microscopic residual disease, grossly unresected disease rarely responds to chemotherapy. The next breakthroughs in osteosarcoma therapy will need to overcome this intrinsic chemoresistance. In other types of cancer, studies of cancer evolution have unveiled the tumor to be polyclonal at diagnosis, but under the selective pressure of chemotherapy, one or a few therapy-resistant clones emerge to cause relapse (Ding et al. 2012). In retrospect, the mutations present in the relapse clone are often detectable from the initial tumor sample at a subclonal level. For example, one study of prostate cancer found an enrichment of TP53 mutations in metastatic subclones (Hong et al. 2015).
Here, for the first time, we show a similar pattern in osteosarcoma. Next-generation sequencing efforts in osteosarcoma have revealed variations affecting TP53 and related genes in nearly every case. In this study we identify a subclonal TP53 translocation in the initial tumor sample that became the dominant clone in subsequent chemotherapy-resistant samples. These results suggest that although other mutations, such as those affecting the PI3K–mTOR pathway (Perry et al. 2014; Grignani et al. 2015), may contribute to osteosarcoma pathogenesis, overcoming therapy resistance may require therapy directed at p53-deficient osteosarcoma.
Genomic instability and p53 inactivation are hallmarks of many cancer types, but SVs involving the TP53 locus have heretofore been limited to two cancer types: prostate cancer and osteosarcoma. This led Chen et al. to speculate that in osteosarcoma, deletion or translocation of p53 may actually be a result of genomic instability rather than the driver event (Chen et al. 2014). In our case, the TP53-KPNA3 translocation was present at only subclonal frequency in the diagnostic specimen, which suggests that it was actually a late event in osteosarcoma formation.
The TP53-KPNA3 translocation described here was not the only genetic aberration to accumulate in frequency over time. Several other variants of unclear significance also accumulated in the dominant clone (Supplemental Table S2). Unlike TP53 translocations, none of these SNVs is in genes previously reported to be recurrently mutated in osteosarcoma. Although we cannot exclude the possibility that these mutations also contribute to the emergence of this subclone, the recurrence of TP53 translocations in other osteosarcomas suggests that the TP53 translocation is the key genomic feature of the dominant recurrent subclone.
To our knowledge, translocations involving the TP53 3′ UTR, such as the TP53-KPNA3 translocation we identify here, have not previously been characterized, in either osteosarcoma or any other type of cancer. However, this subclonal translocation was not detected by our routine exome-sequencing analysis pipeline (detecting genomic variants present in only coding regions). In fact, it required an algorithm using soft-clipped reads to accurately detect and estimate frequencies of structural variations with precise breakpoints at the nucleotide level across these tumors. Similar translocations, involving noncoding regions such as introns and 5′ or 3′ UTRs, have likely been missed in previous studies. Although FFPE tissues may produce false positives when calling SNVs because of C>T artifacts, they are not known to produce false translocations. Current sequencing data and analysis pipelines may require custom modification to include introns and intergenic regions and detect such translocations in cancer-related genes from FFPE-based sequencing projects.
In osteosarcoma, translocation involving one allele of TP53 commonly co-occurs with loss of the opposite allele, and this appears to be especially true in samples derived from osteosarcoma metastases (Miller et al. 1990; Chen et al. 2014). Cancer cells deficient for P53 are resistant to DNA damage–triggered apoptosis in response to chemotherapy and radiation (Lowe et al. 1993, 1994). However, this cannot be rescued by exogenous expression of the TP53 open reading frame (ORF) alone (Breen et al. 2007), suggesting that regulatory elements surrounding the endogenous TP53 locus are also required for the p53 response. In fact, several RNA-binding proteins (Rosenstierne et al. 2008), including HuR (Mazan-Mamczarz et al. 2003), RNPC1 (Zhang et al. 2011), and Wig-1 (Vilborg et al. 2009), bind to conserved elements in the TP53 3′ UTR and are responsible for up-regulating expression in the setting of DNA damage. Alternatively, the loss of these regulatory elements may affect transcript stability through cellular processes such as nonsense-mediated decay. In addition a region of the 5′ UTR has been shown to base-pair with the 3′ UTR, and this base-pairing is also required for the induction of p53 following DNA damage (Chen and Kastan 2010). For these reasons, this translocation, which separates the TP53 coding sequence from its 3′ UTR, in combination with loss of the wild-type allele, is likely to result in a loss of a cell's capacity to induce p53 in response to DNA damage such as by standard chemotherapy. This may explain the emergence of this clone after chemotherapy.
METHODS
Patient Samples and Whole-Exome Sequencing
Blood and tumor samples were taken from a patient treated at Yonsei Cancer Center, Yonsei University College of Medicine, Seoul, Republic of Korea.
For the germline sequencing, DNA was extracted from blood using LaboPass Blood Miniprep Kit (Cosmogenetech, Ltd.). For tumor samples, FFPE blocks with a preponderance of tumor cells were decalcified, and DNA was extracted using RecoverAll Total Nucleic Acid Isolation Kit (Thermo Fisher Scientific). Library preparation and exome capture were done using the SureSelect Library Prep Kit and the SureSelect Automated Hybridization Kit (Agilent Technologies, Inc.). Whole-exome sequencing was performed on one lane of paired-end, 100-bp reads on Illumina HiSeq for each sample.
Quality control was performed using the NGS QC Toolkit 2.3.3 (Patel and Jain 2012). Sequences were aligned to the hg19 reference genome using bwa-0.7.10 (Li and Durbin 2009). Duplicates were marked and discarded using Picard 1.34. Genome Analysis Toolkit (GATK) 3.4 was used to recalibrate base quality score, perform local realignment around indels, and perform pooled multisample genotyping (McKenna et al. 2010; DePristo et al. 2011). SNVs were identified using MuTect 1.1.4 (Cibulskis et al. 2013); indels were identified using Pindel 0.2.5 (Ye et al. 2009); and copy number aberrations were identified using ExomeCNV 1.4 (Sathirapongsasuti et al. 2011).
Detection of SNVs at Subclonal Frequency
SNVs present at subclonal frequency were detected using SHEAR (Landman et al. 2014). The relationship between samples was calculated by RMSD. The RMSD value between two samples, s1 and s2, was calculated by employing the equation:
where L is a set of all sequenced loci with read depth >10 and VAF(l,s) is the variant (nonreference) allele frequency on the locus l of the sample s. The custom Perl script used in the calculation is downloadable at https://github.com/compbioUTSW/RMSD_of_VAF and is also available in the Supplemental Material (Supplemental File S3).
Detection of Structural Variants at Subclonal Frequency
Finally, we sought to detect novel SVs, including those present at subclonal frequency. To detect SVs using our whole-exome sequencing, we ran SHEAR as above to detect and estimate the frequency of structural variation within a heterogeneous individual tumor (Landman et al. 2014). We found the subclonal TP53–KPNA3 fusion across primary, secondary, and tertiary tumors with different estimated frequencies. To further investigate the TP53–KPNA3 fusion event with exact breakpoints at nucleotide precision and its frequencies across tumors, we produced a custom script, which we have named “soft-clipping and realignment detects structural variant frequency” (SCARF) (Supplemental File S4).
Our custom script takes advantage of soft-clipped reads—that is, reads in which one end of a paired-end read is perfectly aligned, but the opposite end is partially aligned to a nearby region. The unaligned, soft-clipped subsequences are usually ignored. We collected soft-clipped sequences in which (a) the soft-clipping position was identical in at least two reads; (b) the aligned portion of the subsequence covered at least 10 bases; and (c) the mismatch ratio in the aligned portion was <0.1. The soft-clipped subsequences were then merged into one contig and aligned to the reference genome using BLAT (BLAST-like alignment tool; Kent 2002) to discover the translocation partner. The BLAT search required that the previously aligned portion of these soft-clipped reads not align to the translocation partner to ensure uniqueness. Frequency of the translocation was estimated as the number of reads supporting the translocation divided by the total number of reads supporting the translocation or mapping to a 200-bp window around the breakpoint from either partner in the translocation. The script used to detect such SVs, along with a toy example for demonstration purposes, is downloadable at https://github.com/compbioUTSW/SCARF (Supplemental File S4).
Sanger Sequencing
Validation of the t(13;17) translocation was performed using one primer mapping to Chromosome 13 (CTGCATGTCAATCTACAATCACC) and one mapping to Chromosome 17 (GGCCCACTTCACCGTACTAA). The resulting 205-bp amplicon was gel-extracted and sequenced from both ends by Sanger sequencing.
Fluorescence In Situ Hybridization
Fluorescence in situ hybridization for TP53 was performed using the Vysis LSI TP53/CEP 17 FISH probe kit (Abbott Molecular) following standard protocols.
p53 Immunohistochemistry
IHC for p53 (dilution 1:100, clone D07; Novocastra) was performed on FFPE archival blocks following standard protocols using a Ventana automatic immunostainer (Ventana, Benchmark).
ADDITIONAL INFORMATION
Data Deposition and Access
Raw sequencing data have been uploaded to the European Genome-phenome Archive (EGA; https://www.ebi.ac.uk/ega/home) under study accession EGAS00001001805, and the t(13;17) translocation sequence has been submitted to GenBank (http://www.ncbi.nlm.nih.gov/genbank/) under accession KX396266.
Ethics Statement
All sequencing was performed under the supervision and approval of the Institutional Review Board (IRB) at Severance Hospital, Yonsei University College of Medicine, Republic of Korea, according to the Declaration of Helsinki principles. Written informed consent was received from the participant before performing this study.
Acknowledgments
K.S.C. is a Damon Runyon-Sohn Pediatric Fellow supported by the Damon Runyon Cancer Research Foundation (DRSG-4P-13). Contributions by J.K. were funded by CPRIT grant RP150596. S.Y.R.’s research was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (HI13C2096). T.H.H. was supported by the Korea Health Technology R&D Project through the Korea Health Industry Development Institute, funded by the Ministry of Health & Welfare (HI13C2162), Republic of Korea.
Author Contributions
W.S.K., H.C.C., S.Y.R., and T.H.H. designed research studies. W.S.K., S.H.K., and J.K. conducted experiments and acquired and/or analyzed data. K.S.C., J.K., and T.H.H. analyzed exome sequencing data, and K.S.C., J.K., W.S.K., H.C.C., S.Y.R., and T.H.H. interpreted sequencing results. K.S.C., J.K., W.S.K., H.C.C., S.Y.R., and T.H.H. wrote the manuscript.
Competing Interest Statement
The authors have declared no competing interest.
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
REFERENCES
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, et al. 2013. Signatures of mutational processes in human cancer. Nature 500: 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breen L, Heenan M, Amberger-Murphy V, Clynes M. 2007. Investigation of the role of p53 in chemotherapy resistance of lung cancer cell lines. Anticancer Res 27: 1361–1364. [PubMed] [Google Scholar]
- Chen J, Kastan MB. 2010. 5′-3′-UTR interactions regulate p53 mRNA translation and provide a target for modulating p53 induction after DNA damage. Genes Dev 24: 2146–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Bahrami A, Pappo A, Easton J, Dalton J, Hedlund E, Ellison D, Shurtleff S, Wu G, Wei L, et al. 2014. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep 7: 104–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. 2013. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 31: 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruin EC, McGranahan N, Mitter R, Salm M, Wedge DC, Yates L, Jamal-Hanjani M, Shafi S, Murugaesu N, Rowan AJ, et al. 2014. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science 346: 251–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43: 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, Ritchey JK, Young MA, Lamprecht T, McLellan MD, et al. 2012. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 481: 506–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, et al. 2012. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 366: 883–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grignani G, Palmerini E, Ferraresi V, D'Ambrosio L, Bertulli R, Asaftei SD, Tamburini A, Pignochino Y, Sangiolo D, Marchesi E, et al. 2015. Sorafenib and everolimus for patients with unresectable high-grade osteosarcoma progressing after standard treatment: a non-randomised phase 2 clinical trial. Lancet Oncol 16: 98–107. [DOI] [PubMed] [Google Scholar]
- Gundem G, Van Loo P, Kremeyer B, Alexandrov LB, Tubio JM, Papaemmanuil E, Brewer DS, Kallio HM, Hognas G, Annala M, et al. 2015. The evolutionary history of lethal metastatic prostate cancer. Nature 520: 353–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen MF, Koufos A, Gallie BL, Phillips RA, Fodstad O, Brøgger A, Gedde-Dahl T, Cavenee WK. 1985. Osteosarcoma and retinoblastoma: a shared chromosomal mechanism revealing recessive predisposition. Proc Natl Acad Sci 82: 6216–6220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong MK, Macintyre G, Wedge DC, Van Loo P, Patel K, Lunke S, Alexandrov LB, Sloggett C, Cmero M, Marass F, et al. 2015. Tracking the origins and drivers of subclonal metastatic expansion in prostate cancer. Nat Commun 6: 6605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ. 2002. BLAT—the BLAST-like alignment tool. Genome Res 12: 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitao S, Shimamoto A, Goto M, Miller RW, Smithson WA, Lindor NM, Furuichi Y. 1999. Mutations in RECQL4 cause a subset of cases of Rothmund–Thomson syndrome. Nat Genet 22: 82–84. [DOI] [PubMed] [Google Scholar]
- Landman SR, Hwang TH, Silverstein KA, Li Y, Dehm SM, Steinbach M, Kumar V. 2014. SHEAR: sample heterogeneity estimation and assembly by reference. BMC Genomics 15: 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurin N, Brown JP, Morissette J, Raymond V. 2002. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am J Hum Genet 70: 1582–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe SW, Ruley HE, Jacks T, Housman DE. 1993. p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell 74: 957–967. [DOI] [PubMed] [Google Scholar]
- Lowe SW, Bodis S, McClatchey A, Remington L, Ruley HE, Fisher DE, Housman DE, Jacks T. 1994. p53 status and the efficacy of cancer therapy in vivo. Science 266: 807–810. [DOI] [PubMed] [Google Scholar]
- Mazan-Mamczarz K, Galban S, Lopez de Silanes I, Martindale JL, Atasoy U, Keene JD, Gorospe M. 2003. RNA-binding protein HuR enhances p53 translation in response to ultraviolet light irradiation. Proc Natl Acad Sci 100: 8354–8359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre JF, Smith-Sorensen B, Friend SH, Kassell J, Borresen AL, Yan YX, Russo C, Sato J, Barbier N, Miser J. 1994. Germline mutations of the p53 tumor suppressor gene in children with osteosarcoma. J Clin Oncol 12: 925–930. [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CW, Aslo A, Tsay C, Slamon D, Ishizaki K, Toguchida J, Yamamuro T, Lampkin B, Koeffler HP. 1990. Frequency and structure of p53 rearrangements in human osteosarcoma. Cancer Res 50: 7950–7954. [PubMed] [Google Scholar]
- Mirabello L, Troisi RJ, Savage SA. 2009. Osteosarcoma incidence and survival rates from 1973 to 2004: data from the Surveillance, Epidemiology, and End Results Program. Cancer 115: 1531–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik-Zainal S, Alexandrov Ludmil B, Wedge David C, Van Loo P, Greenman Christopher D, Raine K, Jones D, Hinton J, Marshall J, Stebbings Lucy A, et al. 2012. Mutational processes molding the genomes of 21 breast cancers. Cell 149: 979–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel RK, Jain M. 2012. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS One 7: e30619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry JA, Kiezun A, Tonzi P, Van Allen EM, Carter SL, Baca SC, Cowley GS, Bhatt AS, Rheinbay E, Pedamallu CS, et al. 2014. Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc Natl Acad Sci 111: E5564–E5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard. http://broadinstitute.github.io/picard/.
- Ribi S, Baumhoer D, Lee K, Edison, Teo AS, Madan B, Zhang K, Kohlmann WK, Yao F, Lee WH, et al. 2015. TP53 intron 1 hotspot rearrangements are specific to sporadic osteosarcoma and can cause Li–Fraumeni syndrome. Oncotarget 6: 7727–7740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenstierne MW, Vinther J, Mittler G, Larsen L, Mann M, Norrild B. 2008. Conserved CPEs in the p53 3′ untranslated region influence mRNA stability and protein synthesis. Anticancer Res 28: 2553–2559. [PubMed] [Google Scholar]
- Sathirapongsasuti JF, Lee H, Horst BA, Brunner G, Cochran AJ, Binder S, Quackenbush J, Nelson SF. 2011. Exome sequencing-based copy-number variation and loss of heterozygosity detection: ExomeCNV. Bioinformatics 27: 2648–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage SA, Mirabello L, Wang Z, Gastier-Foster JM, Gorlick R, Khanna C, Flanagan AM, Tirabosco R, Andrulis IL, Wunder JS, et al. 2013. Genome-wide association study identifies two susceptibility loci for osteosarcoma. Nat Genet 45: 799–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilborg A, Glahder JA, Wilhelm MT, Bersani C, Corcoran M, Mahmoudi S, Rosenstierne M, Grander D, Farnebo M, Norrild B, et al. 2009. The p53 target Wig-1 regulates p53 mRNA stability through an AU-rich element. Proc Natl Acad Sci 106: 15756–15761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. 2009. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25: 2865–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Cho SJ, Shu L, Yan W, Guerrero T, Kent M, Skorupski K, Chen H, Chen X. 2011. Translational repression of p53 by RNPC1, a p53 target overexpressed in lymphomas. Genes Dev 25: 1528–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Fujimoto J, Zhang J, Wedge DC, Song X, Zhang J, Seth S, Chow C-W, Cao Y, Gumbs C, et al. 2014. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science 346: 256–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.