

Abstract
The integration of genome-scale studies such as whole-exome sequencing (WES) into the clinical care of children with cancer has the potential to provide insight into the genetic basis of an individual's cancer with implications for clinical management. This report describes the results of clinical tumor and germline WES for a patient with a rare tumor diagnosis, rosette-forming glioneuronal tumor of the fourth ventricle (RGNT). Three pathogenic gene alterations with implications for clinical care were identified: somatic activating hotspot mutations in FGFR1 (p.N546K) and PIK3CA (p.H1047R) and a germline pathogenic variant in PTPN11 (p.N308S) diagnostic for Noonan syndrome. The molecular landscape of RGNT is not well-described, but these data are consistent with prior observations regarding the importance of the interconnected MAPK and PI3K/AKT/mTOR signaling pathways in this rare tumor. The co-occurrence of FGFR1, PIK3CA, and PTPN11 alterations provides further evidence for consideration of RGNT as a distinct molecular entity from pediatric low-grade gliomas and suggests potential therapeutic strategies for this patient in the event of tumor recurrence as novel agents targeting these pathways enter pediatric clinical trials. Although RGNT has not been definitively linked with cancer predisposition syndromes, two prior cases have been reported in patients with RASopathies (Noonan syndrome and neurofibromatosis type 1 [NF1]), providing an additional link between these tumors and the mitogen-activated protein kinase (MAPK) signaling pathway. In summary, this case provides an example of the potential for genome-scale sequencing technologies to provide insight into the biology of rare tumors and yield both tumor and germline results of potential relevance to patient care.
Keywords: neoplasm of the central nervous system
INTRODUCTION
Rosette-forming glioneuronal tumor of the fourth ventricle (RGNT) is a rare central nervous system (CNS) tumor that primarily affects older children and young adults. Although RGNTs have an indolent natural history and can be cured with complete resection, no proven therapies exist for the treatment of partially resected, progressive, or recurrent tumors (Ellezam et al. 2012; Gessi et al. 2014). RGNT was recently included in the World Health Organization (WHO) classification system as a Grade I glioneuronal tumor, with a characteristic biphasic histologic appearance consisting of neurocytic rosettes and a piloid astrocytic component (Louis et al. 2007; Rosenblum 2007; Adesina 2010), but little is known about the biologic basis of these tumors. Targeted molecular analyses in small cohorts of RGNT have revealed a lack of the recurrent BRAF and IDH1/2 alterations found in low-grade gliomas (LGGs) (Bidinotto et al. 2015), with somatic mutations identified in FGFR1 (Gessi et al. 2014) and PIK3CA (Ellezam et al. 2012) in a subset of cases (Gessi et al. 2011, 2012; Solis et al. 2011), linking the mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K) signaling pathways to RGNT pathogenesis. No definitive link between RGNT and cancer predisposition syndromes is known, but two cases of RGNT (Sherman et al. 2009; Karafin et al. 2011) have been reported in children with the RASopathies Noonan syndrome and neurofibromatosis type 1 (NF1) (Scheithauer et al. 2009).
In this report, we describe the results of clinical tumor and germline whole-exome sequencing (WES) for a child with RGNT. This analysis revealed three key genetic alterations with potential implications for clinical care: somatic activating hotspot mutations in FGFR1 and PIK3CA and a pathogenic germline PTPN11 variant diagnostic for Noonan syndrome. The co-occurrence of these three mutations in a patient with RGNT confirms previous observations regarding the molecular pathways altered in this rare tumor and suggests possible therapeutic strategies in the event of tumor recurrence. More generally, this case provides an example of the diagnostic value of genome-scale testing for patients with rare tumors such as RGNT and highlights the importance of integrating both tumor and germline testing for childhood cancer patients (Zhang et al. 2015; Parsons et al. 2016).
RESULTS
Clinical Presentation
The patient is a 12-yr-old African–American female who presented to Texas Children's Cancer Center for evaluation of papilledema that was incidentally discovered on a yearly optometric examination. Persistent mild headaches, intermittent vomiting, and a mildly ataxic gait were reported in retrospect. The child had a complex medical and social history, including premature delivery between 32 and 36 wk of gestation and a maternal history of HIV infection and substance abuse. She was adopted shortly after birth and showed failure to thrive, developmental delay (speaking a few words at 2 yr of age and first walking at 2.5 yr), and spastic diplegic cerebral palsy. Her height and weight had been consistently measured below the fifth percentile for age since infancy. Structural cardiac anomalies (mild supravalvular pulmonary stenosis, small perimembranous ventricular septal defect, and coronary arterial dilation) were diagnosed in early childhood and medically managed without surgical intervention. The patient had not previously been evaluated by a geneticist.
Physical examination was notable for short stature (below fifth percentile). Examination of the head and neck demonstrated hypertelorism with downslanting palpebral fissures, a short nose with depressed nasal root, deep philtrum, and low-set ears. Auscultation revealed a grade 2/6 ejection murmur and systolic ejection click. No rashes, macules, or patches were identified. On neurologic assessment, mild ataxia was noted, but examination of cranial nerves, coordination, sensation, muscle strength, and deep tendon reflexes was within normal limits.
Magnetic resonance imaging (MRI) of the brain and spine demonstrated a large complex T2 hyper-/hypointense lobular heterogeneously enhancing mass filling the expanded fourth ventricle and protruding through its outlets with associated obstructive hydrocephalus (Fig. 1A,B). A small enhancing metastatic nodule was noted in the inferior recess of the third ventricle (Fig. 1B). Post contrast spine MRI at presentation demonstrated T1 hyperintense substance within the terminal thecal sac, reflecting hemorrhage and/or drop metastasis; this subsequently improved on follow-up scans. A suboccipital craniotomy was performed and near total resection of the primary tumor was achieved. The patient did not receive adjuvant chemotherapy or radiation therapy. Following recovery from surgery, she underwent inpatient physical and occupational therapy and multidisciplinary management of her comorbid conditions.
Figure 1.
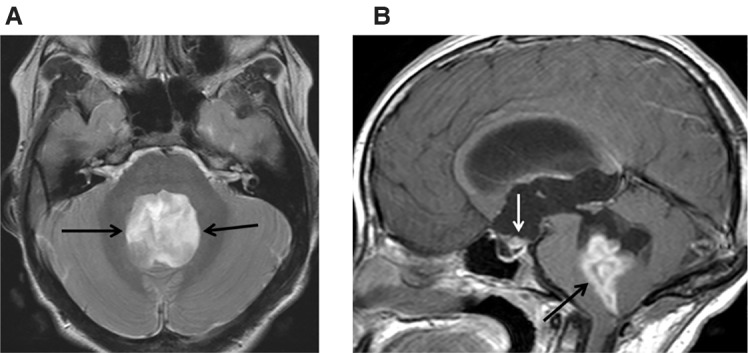
Brain MRI of mass at presentation. (A) Axial T2 and (B) post gadolinium T1 sagittal. The intraventricular tumor expands and obstructs the fourth ventricle, shows mixed T2 hyper-/hypointense signal (A, black arrows) and enhances heterogeneously (B, black arrow). There is also a small enhancing metastatic nodule at the inferior third ventricular recess (B, white arrow).
Pathologic review revealed a RGNT with mixed architecture consisting of rosette-forming neurocytic cells with synaptophysin-positive cores (Fig. 2A,B), sheets of oligo-like cells with delicate capillary network (Fig. 2C), and piloid cells with gliofibrillary processes (Fig. 2D). An infarct-type necrosis with peri-infarct arcade of vascular proliferation, as well as regional calcification, was noted. Routine clinical molecular testing was negative for somatic alterations in BRAF (V600E point mutation and KIAA1549-BRAF fusion) and hotspot mutations in IDH1 and IDH2 by pyrosequencing.
Figure 2.

Immunohistopathology demonstrates (A) neurocytic rosettes with (B) synaptophysin-positive cores, (C) sheets of oligo-like neurocytes with associated delicate vascular network, and (D) regions with intermixed piloid gliofibrillary architecture. The histologic features are those of a rosette-forming glioneuronal tumor. Magnification: (A,B) 400×; (C,D) 200×.
The patient is now 16 mo out from neurosurgical resection, with stable residual tumor noted on follow-up MRI studies. Neuro-ophthalmologic examination has also been stable with right internuclear ophthalmoplegia. She has made a gradual overall recovery with physical and occupational therapy and is followed by the cardiology and genetics services.
Family History
The patient has five full and three half biologic siblings ranging in age between 3 and 23 yr, all in good health and without clinical features of Noonan syndrome. A family history of cancer in adulthood was reported in two maternal uncles (throat cancer; pancreatic cancer) and the maternal grandfather (colorectal cancer). Multiple paternal relatives reportedly died of cancers of unknown types. Additional details of the family history were limited by the patient's history of adoption.
Genomic Analysis
The patient was enrolled in the Baylor College of Medicine institutional review board (IRB)-approved BASIC3 (Baylor Advancing Sequencing into Childhood Cancer Care) study, a National Human Genome Research Institute (NHGRI)- and National Cancer Institute (NCI)-funded Clinical Sequencing Exploratory Research project seeking to investigate the impact of WES on the care of pediatric oncology patients (Scollon et al. 2014). As part of this study, WES in a College of American Pathologists (CAP)- and Clinical Laboratory Improvement Amendment (CLIA)-certified laboratory was performed on paired frozen tumor and blood samples as previously described (Parsons et al. 2016).
Analysis of the tumor WES data after subtraction of germline variants observed in the blood sample revealed four somatic (tumor-specific) mutations (Table 1), including mutations in the FGFR1 and PIK3CA oncogenes. The exon 12 missense FGFR1 mutation identified (c.1638C>A; p.N546K) occurs within the kinase domain of the protein and is the most frequent somatic FGFR1 mutation reported to date, having been found in both low- and high-grade gliomas and glioneuronal tumors (Gessi et al. 2014; Forbes et al. 2015). It has been shown to activate MAPK and PI3K/AKT/mTOR signaling pathways (Turner and Grose 2010; Zhang et al. 2013b). The c.3140A>G missense mutation (p.H1047R) identified in exon 21 of PIK3CA is the most frequently observed PIK3CA hotspot alteration in human cancers, including high-grade gliomas and glioneuronal tumors (Ellezam et al. 2012), and has also been demonstrated to result in constitutive activation of the PI3K/AKT/mTOR pathway (Bader et al. 2005; Engelman 2009; Wu et al. 2014; Thorpe et al. 2015). Tumor WES also revealed novel somatic mutations in PPP1R1A and RNF216, genes that have been reported to be rarely mutated in cancer (Forbes et al. 2015) and are not known to contribute to cancer pathogenesis.
Table 1.
Somatic mutations identified by tumor whole-exome sequencing
| Gene | Genomic coordinates (hg19) | HGVS cDNA | HGVS protein | Variant type | COSMIC ID | Variant allele fraction |
|---|---|---|---|---|---|---|
| FGFR1 | Chr 8: 38274849 | NM_023110: c.1638C>A | p.N546K | Missense | COSM19176 | 0.29 |
| PIK3CA | Chr 3: 178952085 | NM_006218: c.3140A>G | p.H1047R | Missense | COSM775 | 0.33 |
| PPP1R1A | Chr 12: 54975791 | NM_006741: c.372_373 delinsCA | p.E124_S125 delinsDT | Dinucleotide substitution | N/A | 0.38 |
| RNF216 | Chr 7: 5662795 | NM_207116: c.2297G>A | p.R766H | Missense | N/A | 0.21 |
HGVS, Human Genome Variation Society; COSMIC, Catalogue of Somatic Mutations in Cancer; N/A, not applicable.
Germline WES revealed a c.923A>G (p.N308S) PTPN11 pathogenic variant (Table 2), a well-described alteration in patients with Noonan syndrome (Tartaglia et al. 2002). The germline WES report also included nine variants of uncertain significance in genes related to cancer or intellectual disability (Supplemental Table S1) and seven pathogenic variants in genes associated with rare autosomal-recessive Mendelian disorders (Supplemental Table S2). Parental blood samples were unavailable for analysis.
Table 2.
Pathogenic germline variants in disease genes related to clinical phenotype
| Gene | Genomic coordinates (hg19) | HGVS cDNA | HGVS protein | Variant type | SIFT/PolyPhen-2 predicted effect | dbSNP/dbVarID | Genotype |
|---|---|---|---|---|---|---|---|
| PTPN11 | Chr 12: 112915524 | NM_002834: c.923A>G | p.N308S | Missense | Tolerated/benign | rs121918455 | Heterozygous |
HGVS, Human Genome Variation Society; SIFT, Sorting Tolerant from Intolerant; dbSNP, Database for Short Genetic Variations.
DISCUSSION
Our patient showed clinical features of Noonan syndrome, a RASopathy characterized by distinctive facial features more prominent in infancy and childhood, short stature, pectus abnormality, cryptorchidism, congenital heart defects, and increased risk of bleeding (Romano et al. 2010; Niemeyer 2014). A genetic diagnosis of Noonan syndrome had not previously been made until germline WES identified a known pathogenic variant in PTPN11 (c.923A>G, p.N308S) (Tartaglia et al. 2002). Approximately half of Noonan syndrome cases can be attributed to pathogenic variants in the PTPN11 gene (Tartaglia et al. 2002), which encodes the protein tyrosine phosphatase SHP2, a cell-signaling molecule in the MAPK signaling pathway (Fig. 3; Keilhack et al. 2005; Niemeyer 2014). Notably, SIFT and PolyPhen-2 algorithms predicted this alteration as tolerated or benign, highlighting the limitations of existing prediction models for variant pathogenicity and the continued need for a thorough clinical assessment of patients harboring variants.
Figure 3.

Overlapping RAS/MAPK and PIK3CA/AKT/mTOR signaling pathways and potential therapeutic targets. Pathogenic variants identified in this patient are FGFR1, PTPN11, and PIK3CA. FGFR, fibroblast growth factor receptor; FGF, fibroblast growth factor; PI3K, phosphoinositide-3 kinase; mTOR, mammalian target of rapamycin; NF1, neurofibromatosis type 1; BRAF, B-Raf proto-oncogene, serine/threonine kinase; MEK, mitogen-activated protein kinase/ERK kinase; ERK, extracellular-signal-regulated kinase.
Patients with Noonan syndrome require multidisciplinary medical care for management of a wide range of associated comorbidities. Although patients with a PTPN11 pathogenic variant are at increased risk of developing a spectrum of cancers, most commonly hematologic malignancies such as juvenile myelomonocytic leukemia (Romano et al. 2010), no specific surveillance program exists (Jongmans et al. 2011). Less commonly, CNS tumors including oligodendroglioma and other low-grade glial tumors have been associated with PTPN11 Noonan syndrome (Smpokou et al. 2015), including a single case of RGNT (Sherman et al. 2009). A second case of RGNT in a patient with a clinical diagnosis of Noonan syndrome, but unknown genetic etiology has also been described (Karafin et al. 2011).
The somatic mutations identified in our patient with RGNT suggest potential targets for molecular therapeutics in the event of tumor recurrence. Although clinical data for this rare tumor are limited, surgical resection and adjuvant radiation therapy would be considered a standard approach (Zhang et al. 2013a). However, should these modalities be contraindicated or insufficient, the presence of activating somatic FGFR1 and PIK3CA mutations raises the possibility of targeted inhibition of the MAPK and PI3K/AKT/mTOR signaling pathways as an enticing albeit unproven intervention (Fig. 3). Activating FGFR1 mutations, amplifications, and translocations have been reported in a variety of adult and pediatric cancers (Liang et al. 2013) and FGFR inhibitors are in clinical trial development for adult malignancies (Kim et al. 2011; Andre et al. 2013; Angevin et al. 2013; Soria et al. 2014; Cabanillas et al. 2015; Schlumberger et al. 2016) but not yet tested in children. Similarly, mutations and copy number alterations in a number of genes in the PI3K/AKT/mTOR pathway (most prominently PIK3CA, PTEN, TSC1, and TSC2) are recurrent events in numerous cancer types (Samuels and Ericson 2006; Thorpe et al. 2015). Agents targeting multiple nodes in the PI3K/AKT/mTOR pathway are in clinical development, including PI3K inhibitors, AKT inhibitors, mTOR inhibitors, TORC1/2 inhibitors, and PI3K/mTOR inhibitors (Weigelt and Downward 2012; Rodon et al. 2013; Fruman and Rommel 2014) and have begun to be evaluated in pediatric cancer patients. Aberrations of the PI3K/AKT/mTOR pathway have been suggested as a negative prognostic factor in the small number of RGNT cases described in the literature; of four tumors with somatic PIK3CA mutations, two recurred (Ellezam et al. 2012; Gessi et al. 2014). Somatic PIK3CA alterations have been associated with clinically aggressive features in LGGs, such as in pilocytic astrocytomas with uniquely anaplastic histology (Rodriguez et al. 2011). Given the small number of RGNTs subjected to genetic analysis, the prognostic significance of these specific mutations in RGNT is currently unclear.
Although comprehensive data regarding the mutational landscape of RGNT are lacking, results from the few cases sequenced to date provide clues into the genetic and biological basis of these tumors. RGNTs share histologic similarity to pediatric LGGs, tumors defined by genetic alterations in the MAPK signaling pathway (Zhang et al. 2013b), and our data confirm previous reports of frequent MAPK pathway alterations in RGNT. However, the genes mutated in LGGs and RGNTs appear somewhat distinct: for example, missense mutations in FGFR1 appear frequently in RGNTs (Gessi et al. 2014) but are rare events in pediatric LGGs (Zhang et al. 2013b), whereas the characteristic BRAF fusion seen in pilocytic astrocytomas has not been detected in RGNTs (Gessi et al. 2012). Concurrent FGFR1 and PTPN11 mutations, as found in our patient, have been identified in rare BRAF wild-type midline pilocytic astrocytomas and may represent an alternate mechanism of MAPK pathway activation or potentially have a potentiating effect on tumorigenesis (Zhang et al. 2013b).
Interestingly, concurrent somatic mutations were identified in FGFR1 and PIK3CA, which code for gene products involved in distinct but closely communicating molecular signaling pathways. We report variant allele fractions of 29% (FGFR1) and 33% (PIK3CA) in this case, implying that a majority of sequenced cells (58% and 66%, respectively) carry each mutation (presuming heterozygous mutations in these oncogenes) and that the mutations therefore co-occur in tumor cells as opposed to being separate subclonal events. The identification of mutations in both MAPK and PI3K/AKT/mTOR signaling (Fig. 3) distinguishes RGNTs from the “single pathway” MAPK-driven LGGs. Interestingly, concurrent mutations in these same pathways have been reported in a variety of other cancer types (Janku et al. 2011), including a recurrent RGNT in an adult patient (Gessi et al. 2014). Previous targeted sequencing studies have revealed PIK3CA mutations, which are common in high-grade gliomas but not LGGs, in three of four RGNTs analyzed (Ellezam et al. 2012; Cachia et al. 2014). Functional studies describing the interplay of these alterations would be of biologic and therapeutic interest.
The identification of both germline and somatic mutations with potential implications for clinical care in this case highlights the potential benefit of integrating both tumor and germline testing in the evaluation of childhood cancer patients. Although most clinical tumor sequencing (primarily consisting of mutation panels including selected cancer genes and variants) is performed without concurrent analysis of a matched normal tissue sample, recent studies have highlighted the importance of paired tumor/germline analysis to decrease false-positive somatic mutation calls and distinguish germline variants from somatic mutations (Jones et al. 2015; Raymond et al. 2016). Given an observed frequency of pathogenic germline cancer susceptibility variants in 8%–10% of childhood cancer patients (Mody et al. 2015; Zhang et al. 2015; Parsons et al. 2016), clarification of whether individual mutations are somatic or germline is of particular relevance in the pediatric setting. Our patient provides a pertinent example: the specific germline PTPN11 (p.N308S) pathogenic variant identified in this child with Noonan syndrome has been previously reported as a somatic event in several cases of acute lymphoblastoid leukemia (Zhang et al. 2011; Roberts et al. 2014).
In summary, this case of a child with RGNT and Noonan syndrome demonstrates the potential of genome-scale sequencing technologies such as WES to provide insight into the biology of rare tumors and yield both tumor and germline results of potential relevance for patient care. The identification of three pathogenic mutations (FGFR1, PIK3CA, and PTPN11) converging on MAPK and PI3K/AKT/mTOR signaling provides further evidence for the importance of these pathways in RGNT and offers possible therapeutic strategies in the event of tumor recurrence. Clinical trials evaluating agents that target these molecular pathways will be necessary to demonstrate the benefit of these drugs and genotype-directed precision oncology strategies for childhood cancer patients.
METHODS
WES of peripheral blood and tumor biopsy samples was performed in the CLIA-certified Genetics Laboratories at Baylor College of Medicine including library construction, exome capture by VCRome version 2.1 (target size 35.45 Mb) and 2 × 100-bp paired-end sequencing on an Illumina HiSeq instrument as previously described (Yang et al. 2013; Parsons et al. 2016). The tumor/germline matched pair was sequenced on a single lane of a HiSeq 2500 with mean coverage of 197× (blood) and 223× (tumor) and a target base coverage of 20× at 98% (blood and tumor). Germline and tumor WES reports were completed in 14 and 20 wk, respectively. The reported FGFR1, PIK3CA, and PTPN11 variants were confirmed by Sanger sequencing.
ADDITIONAL INFORMATION
Data Deposition and Access
Tumor and germline WES data will be deposited to the National Center for Biotechnology Information (NCBI) Database of Genotypes and Phenotypes (dbGaP; http://www.ncbi.nlm.nih.gov/gap) with the remainder of the BASIC3 patient cohort data upon study completion, under accession number phs001026.v1.p1. The tumor and germline variants have been submitted into ClinVar (Landrum et al. 2014) (http://www.ncbi.nlm.nih.gov/clinvar/) under accession numbers SCV000292258–SCV000292262.
Ethics Statement
The BASIC3 study protocol (H-30755) was approved by the Baylor College of Medicine IRB, which is also the IRB for Texas Children's Hospital, the study's clinical site. Written informed consent for study enrollment was obtained by a trained study project manager or genetic counselor as previously described (Scollon et al. 2014).
Acknowledgments
The authors are grateful to the patient and her family as well as the clinical and research staff at Texas Children's Cancer Center for their participation in the study.
Author Contributions
Patient clinical care was provided by F.Y.L., A.J., S.B., and A.M.A. Study design, participant recruitment, consent, and support were provided by F.Y.L., K.B., A.B., S.S., R.R.-M., M.M.C., A.R., S.E.P., and D.W.P. Sequence data analysis and interpretation were performed by R.P., A.R., S.E.P., and D.W.P., F.Y.L., K.B., A.R., S.E.P., D.W.P. wrote the initial draft of the manuscript. All authors contributed to reviewing and editing the final draft.
Funding
The BASIC3 study is a Clinical Sequencing Exploratory Research (CSER) program project supported by NHGRI/NCI 1U01HG006485. This work was also supported by a Sontag Foundation Distinguished Scientist Award (D.W.P.) and a Stand Up To Cancer St Baldrick's Pediatric Dream Team Translational Research Grant (SU2CAACR-DT1113); Stand Up To Cancer is a program of the Entertainment Industry Foundation administered by the American Association for Cancer. F.Y.L. is a Kurt Groten Family Research Scholar.
Competing Interest Statement
The authors have declared no competing interest.
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
REFERENCES
- Adesina AM, Tihan T, Fuller CE, Poussaint TY. 2010. Atlas of pediatric brain tumors, pp. 205–208. Springer, New York. [Google Scholar]
- Andre F, Bachelot T, Campone M, Dalenc F, Perez-Garcia JM, Hurvitz SA, Turner N, Rugo H, Smith JW, Deudon S, et al. 2013. Targeting FGFR with dovitinib (TKI258): preclinical and clinical data in breast cancer. Clin Cancer Res 19: 3693–3702. [DOI] [PubMed] [Google Scholar]
- Angevin E, Lopez-Martin JA, Lin CC, Gschwend JE, Harzstark A, Castellano D, Soria JC, Sen P, Chang J, Shi M, et al. 2013. Phase I study of dovitinib (TKI258), an oral FGFR, VEGFR, and PDGFR inhibitor, in advanced or metastatic renal cell carcinoma. Clin Cancer Res 19: 1257–1268. [DOI] [PubMed] [Google Scholar]
- Bader AG, Kang S, Zhao L, Vogt PK. 2005. Oncogenic PI3K deregulates transcription and translation. Nat Rev Cancer 5: 921–929. [DOI] [PubMed] [Google Scholar]
- Bidinotto LT, Scapulatempo-Neto C, Mackay A, de Almeida GC, Scheithauer BW, Berardinelli GN, Torrieri R, Clara CA, Feltrin LT, Viana-Pereira M, et al. 2015. Molecular profiling of a rare rosette-forming glioneuronal tumor arising in the spinal cord. PLoS One 10: e0137690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabanillas ME, Schlumberger M, Jarzab B, Martins RG, Pacini F, Robinson B, McCaffrey JC, Shah MH, Bodenner DL, Topliss D, et al. 2015. A phase 2 trial of lenvatinib (E7080) in advanced, progressive, radioiodine-refractory, differentiated thyroid cancer: a clinical outcomes and biomarker assessment. Cancer 121: 2749–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cachia D, Prado MP, Theeler B, Hamilton J, McCutcheon I, Fuller GN. 2014. Synchronous rosette-forming glioneuronal tumor and diffuse astrocytoma with molecular characterization: a case report. Clin Neuropathol 33: 407–411. [DOI] [PubMed] [Google Scholar]
- Ellezam B, Theeler BJ, Luthra R, Adesina AM, Aldape KD, Gilbert MR. 2012. Recurrent PIK3CA mutations in rosette-forming glioneuronal tumor. Acta Neuropathol 123: 285–287. [DOI] [PubMed] [Google Scholar]
- Engelman JA. 2009. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer 9: 550–562. [DOI] [PubMed] [Google Scholar]
- Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S, et al. 2015. COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res 43: D805–D811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruman DA, Rommel C. 2014. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov 13: 140–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gessi M, Waha A, Setty P, Waha A, Pietsch T. 2011. Analysis of KIAA1549-BRAF fusion status in a case of rosette-forming glioneuronal tumor of the fourth ventricle (RGNT). Neuropathology 31: 654–657. [DOI] [PubMed] [Google Scholar]
- Gessi M, Lambert SR, Lauriola L, Waha A, Collins VP, Pietsch T. 2012. Absence of KIAA1549-BRAF fusion in rosette-forming glioneuronal tumors of the fourth ventricle (RGNT). J Neurooncol 110: 21–25. [DOI] [PubMed] [Google Scholar]
- Gessi M, Moneim YA, Hammes J, Goschzik T, Scholz M, Denkhaus D, Waha A, Pietsch T. 2014. FGFR1 mutations in Rosette-forming glioneuronal tumors of the fourth ventricle. J Neuropathol Exp Neurol 73: 580–584. [DOI] [PubMed] [Google Scholar]
- Janku F, Lee JJ, Tsimberidou AM, Hong DS, Naing A, Falchook GS, Fu S, Luthra R, Garrido-Laguna I, Kurzrock R. 2011. PIK3CA mutations frequently coexist with RAS and BRAF mutations in patients with advanced cancers. PLoS One 6: e22769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Anagnostou V, Lytle K, Parpart-Li S, Nesselbush M, Riley DR, Shukla M, Chesnick B, Kadan M, Papp E, et al. 2015. Personalized genomic analyses for cancer mutation discovery and interpretation. Sci Transl Med 7: 283ra253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jongmans MC, van der Burgt I, Hoogerbrugge PM, Noordam K, Yntema HG, Nillesen WM, Kuiper RP, Ligtenberg MJ, van Kessel AG, van Krieken JH, et al. 2011. Cancer risk in patients with Noonan syndrome carrying a PTPN11 mutation. Eur J Hum Genet 19: 870–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karafin M, Jallo GI, Ayars M, Eberhart CG, Rodriguez FJ. 2011. Rosette forming glioneuronal tumor in association with Noonan syndrome: pathobiological implications. Clin Neuropathol 30: 297–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keilhack H, David FS, McGregor M, Cantley LC, Neel BG. 2005. Diverse biochemical properties of Shp2 mutants. Implications for disease phenotypes. J Biol Chem 280: 30984–30993. [DOI] [PubMed] [Google Scholar]
- Kim KB, Chesney J, Robinson D, Gardner H, Shi MM, Kirkwood JM. 2011. Phase I/II and pharmacodynamic study of dovitinib (TKI258), an inhibitor of fibroblast growth factor receptors and VEGF receptors, in patients with advanced melanoma. Clin Cancer Res 17: 7451–7461. [DOI] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, Maglott DR. 2014. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 42: D980–D985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang G, Chen G, Wei X, Zhao Y, Li X. 2013. Small molecule inhibition of fibroblast growth factor receptors in cancer. Cytokine Growth Factor Rev 24: 467–475. [DOI] [PubMed] [Google Scholar]
- Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P. 2007. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114: 97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mody RJ, Wu YM, Lonigro RJ, Cao X, Roychowdhury S, Vats P, Frank KM, Prensner JR, Asangani I, Palanisamy N, et al. 2015. Integrative clinical sequencing in the management of refractory or relapsed cancer in youth. JAMA 314: 913–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemeyer CM. 2014. RAS diseases in children. Haematologica 99: 1653–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons D, Roy A, Yang Y, Wang T, Scollon S, Bergstrom K, Kerstein RA, Gutierrez S, Petersen AK, Bavle A, et al. 2016. Diagnostic yield of clinical tumor and germline whole-exome sequencing for children with solid tumors. JAMA Oncol 10.1001/jamaoncol.2015.5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond VM, Gray SW, Roychowdhury S, Joffe S, Chinnaiyan AM, Parsons DW, Plon SE; Clinical Sequencing Exploratory Research Consortium Tumor Working Group. 2016. Germline findings in tumor-only sequencing: points to consider for clinicians and laboratories. J Natl Cancer Inst 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang YL, Pei D, McCastlain K, Ding L, Lu C, Song G, et al. 2014. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med 371: 1005–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodon J, Dienstmann R, Serra V, Tabernero J. 2013. Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol 10: 143–153. [DOI] [PubMed] [Google Scholar]
- Rodriguez EF, Scheithauer BW, Giannini C, Rynearson A, Cen L, Hoesley B, Gilmer-Flynn H, Sarkaria JN, Jenkins S, Long J, et al. 2011. PI3K/AKT pathway alterations are associated with clinically aggressive and histologically anaplastic subsets of pilocytic astrocytoma. Acta Neuropathol 121: 407–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano AA, Allanson JE, Dahlgren J, Gelb BD, Hall B, Pierpont ME, Roberts AE, Robinson W, Takemoto CM, Noonan JA. 2010. Noonan syndrome: clinical features, diagnosis, and management guidelines. Pediatrics 126: 746–759. [DOI] [PubMed] [Google Scholar]
- Rosenblum MK. 2007. The 2007 WHO Classification of Nervous System Tumors: newly recognized members of the mixed glioneuronal group. Brain Pathol 17: 308–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels Y, Ericson K. 2006. Oncogenic PI3K and its role in cancer. Curr Opin Oncol 18: 77–82. [DOI] [PubMed] [Google Scholar]
- Scheithauer BW, Silva AI, Ketterling RP, Pula JH, Lininger JF, Krinock MJ. 2009. Rosette-forming glioneuronal tumor: report of a chiasmal-optic nerve example in neurofibromatosis type 1: special pathology report. Neurosurgery 64: E771–E772; discussion E772. [DOI] [PubMed] [Google Scholar]
- Schlumberger M, Jarzab B, Cabanillas ME, Robinson B, Pacini F, Ball DW, McCaffrey J, Newbold K, Allison R, Martins RG, et al. 2016. A phase II trial of the multitargeted tyrosine kinase inhibitor lenvatinib (E7080) in advanced medullary thyroid cancer. Clin Cancer Res 22: 44–53. [DOI] [PubMed] [Google Scholar]
- Scollon S, Bergstrom K, Kerstein RA, Wang T, Hilsenbeck SG, Ramamurthy U, Gibbs RA, Eng CM, Chintagumpala MM, Berg SL, et al. 2014. Obtaining informed consent for clinical tumor and germline exome sequencing of newly diagnosed childhood cancer patients. Genome Med 6: 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman CB, Ali-Nazir A, Gonzales-Gomez I, Finlay JL, Dhall G. 2009. Primary mixed glioneuronal tumor of the central nervous system in a patient with Noonan syndrome: a case report and review of the literature. J Pediatr Hematol Oncol 31: 61–64. [DOI] [PubMed] [Google Scholar]
- Smpokou P, Zand DJ, Rosenbaum KN, Summar ML. 2015. Malignancy in Noonan syndrome and related disorders. Clin Genet 88: 516–522. [DOI] [PubMed] [Google Scholar]
- Solis OE, Mehta RI, Lai A, Mehta RI, Farchoukh LO, Green RM, Cheng JC, Natarajan S, Vinters HV, Cloughesy T, et al. 2011. Rosette-forming glioneuronal tumor: a pineal region case with IDH1 and IDH2 mutation analyses and literature review of 43 cases. J Neurooncol 102: 477–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soria JC, DeBraud F, Bahleda R, Adamo B, Andre F, Dienstmann R, Delmonte A, Cereda R, Isaacson J, Litten J, et al. 2014. Phase I/IIa study evaluating the safety, efficacy, pharmacokinetics, and pharmacodynamics of lucitanib in advanced solid tumors. Ann Oncol 25: 2244–2251. [DOI] [PubMed] [Google Scholar]
- Tartaglia M, Kalidas K, Shaw A, Song X, Musat DL, van der Burgt I, Brunner HG, Bertola DR, Crosby A, Ion A, et al. 2002. PTPN11 mutations in Noonan syndrome: molecular spectrum, genotype-phenotype correlation, and phenotypic heterogeneity. Am J Hum Genet 70: 1555–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorpe LM, Yuzugullu H, Zhao JJ. 2015. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer 15: 7–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner N, Grose R. 2010. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer 10: 116–129. [DOI] [PubMed] [Google Scholar]
- Weigelt B, Downward J. 2012. Genomic determinants of PI3K pathway inhibitor response in cancer. Front Oncol 2: 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Renuse S, Sahasrabuddhe NA, Zahari MS, Chaerkady R, Kim MS, Nirujogi RS, Mohseni M, Kumar P, Raju R, et al. 2014. Activation of diverse signalling pathways by oncogenic PIK3CA mutations. Nat Commun 5: 4961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, Braxton A, Beuten J, Xia F, Niu Z, et al. 2013. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med 369: 1502–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Mullighan CG, Harvey RC, Wu G, Chen X, Edmonson M, Buetow KH, Carroll WL, Chen IM, Devidas M, et al. 2011. Key pathways are frequently mutated in high-risk childhood acute lymphoblastic leukemia: a report from the Children's Oncology Group. Blood 118: 3080–3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Babu R, McLendon RE, Friedman AH, Adamson C. 2013a. A comprehensive analysis of 41 patients with rosette-forming glioneuronal tumors of the fourth ventricle. J Clin Neurosci 20: 335–341. [DOI] [PubMed] [Google Scholar]
- Zhang J, Wu G, Miller CP, Tatevossian RG, Dalton JD, Tang B, Orisme W, Punchihewa C, Parker M, Qaddoumi I, et al. 2013b. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet 45: 602–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Walsh MF, Wu G, Edmonson MN, Gruber TA, Easton J, Hedges D, Ma X, Zhou X, Yergeau DA, et al. 2015. Germline mutations in predisposition genes in pediatric cancer. N Engl J Med 373: 2336–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.