

Abstract
Recently, mutations in the zinc finger MYND-type containing 11 (ZMYND11) gene were identified in patients with autism spectrum disorders, intellectual disability, aggression, and complex neuropsychiatric features, supporting that this gene is implicated in 10p15.3 microdeletion syndrome. We report a novel de novo variant in the ZMYND11 gene (p.Ser421Asn) in a patient with a complex neurodevelopmental phenotype. The patient is a 24-yr-old Caucasian/Filipino female with seizures, global developmental delay, sensorineural hearing loss, hypotonia, dysmorphic features, and other features including a happy disposition and ataxic gait similar to Angelman syndrome. In addition, this patient had uncommon features including eosinophilic esophagitis and multiple, severe allergies not described in similar ZMYND11 cases. This new case further supports the association of ZMYND11 with autistic-like phenotypes and suggests that ZMYND11 should be included in the list of potentially causative candidate genes in cases with complex neurodevelopmental phenotypes.
Keywords: absence seizures, absent speech, asthma, atonic seizures, broad-based gait, central hypoxia, clinodactyly of the 5th finger, congenital sensorineural hearing impairment, esophagitis, gait ataxia, gastroesophageal reflux, microcephaly, neurogenic bladder, prominent epicanthal folds, seasonal allergy, severe global developmental delay
INTRODUCTION
10p15.3 microdeletion syndrome has been associated with autism spectrum disorders and intellectual disabilities. It is only recently that zinc finger MYND-type containing 11 (ZMYND11) has been proposed as a candidate gene for this syndrome characterized by patients with overlapping symptoms, including complex cognitive, behavioral, and developmental difficulties. Coe et al. (2014) identified five different truncating variants in ZMYND11 in patients presenting with the symptoms described above. Descipio et al. described a cohort of 19 patients with submicroscopic deletions involving 10p15.3 and cognitive/behavioral problems, speech delay and language disorder, motor delay, craniofacial dysmorphism, hypotonia, brain anomalies, and seizures. In 18 of these patients, the deletion included ZMYND11 (Descipio et al. 2012). Here, we present a female patient with a de novo missense mutation in the ZMYND11 gene (Chr10:294310G>A; p.Ser421Asn), with a clinical phenotype (global developmental delay, seizures, hypotonia, and hearing loss) comparable with the 10p15.3 microdeletion cases.
RESULTS
Clinical Presentation and Family History
A 24-yr-old Caucasian/Filipino female, born to nonconsanguineous, healthy parents, was referred to The Translational Genomics Research Institute's (TGen's) Center for Rare Childhood Disorders. Her clinical features include severe global developmental delay, hypotonia, intractable epilepsy, mild-to-moderate bilateral sensorineural hearing loss, and an ataxic gait. She also has dysmorphic features consisting of prominent jaw, wide mouth, broad nasal root and bulbous nasal tip, deep set eyes, small feet (7.7 in), microcephaly (occipitofrontal circumference [OFC] 51.5 cm), epicanthal folds, and clinodactyly of the fifth finger (Fig. 1). Other unique features included a neurogenic bladder, extrinsic asthma, severe eosinophilic esophagitis, esophageal reflux, multiple food, medication, and environmental allergies, atopic dermatitis, and allergic urticaria.
Figure 1.

Patient phenotypic assessment depicting: (A) facial dysmorphism, (B) clinodactyly of the fifth finger, (C) small feet (7.7 in), and (D) epicanthal folds.
The patient was born to a 21-yr-old G1P0 Caucasian mother. She had routine prenatal care including two unremarkable ultrasounds. The pregnancy was complicated by preterm labor at 7 mo and mild hypertension during the last 3–4 wk of pregnancy that were both treated with bed rest. The child was born full-term by an uncomplicated, induced vaginal delivery weighing 8 lb, 4 oz and measuring 20.5 in. She was monitored in the neonatal intensive care unit and required oxygen for 48 h because of mild respiratory distress. She also had mild jaundice but did not require phototherapy; she was discharged home on day 4 with no neonatal problems. By 4 mo of age, the parents noted developmental delay and hypotonia.
Seizures began at 9 mo of age and remain intractable, despite treatment with several antiepileptic drugs (including carbamazepine, gabapentin, lamotrigine, phenobarbital, phenytoin, tiagabine, divalproex, clonazepam, topiramate, and levetiracetam), a ketogenic diet, and vagal nerve stimulation. A magnetic resonance imaging (MRI) scan of the brain performed at 10 mo showed some nonspecific cortical abnormalities. At 9 mo, she was also diagnosed with mild-to-moderate bilateral sensorineural hearing impairment, which showed no further progression with age. There is a positive family history of congenital bilateral hearing impairment from the mother's paternal grandfather and the father's maternal uncle and aunt. It is unclear whether this has an independent genetic cause. Ophthalmological examination was normal except for myopia. Cardiology evaluation revealed a benign innocent murmur. Muscle biopsy, urine organic acids, plasma amino acids, creatine kinase, and urine mucopolysaccharides were normal.
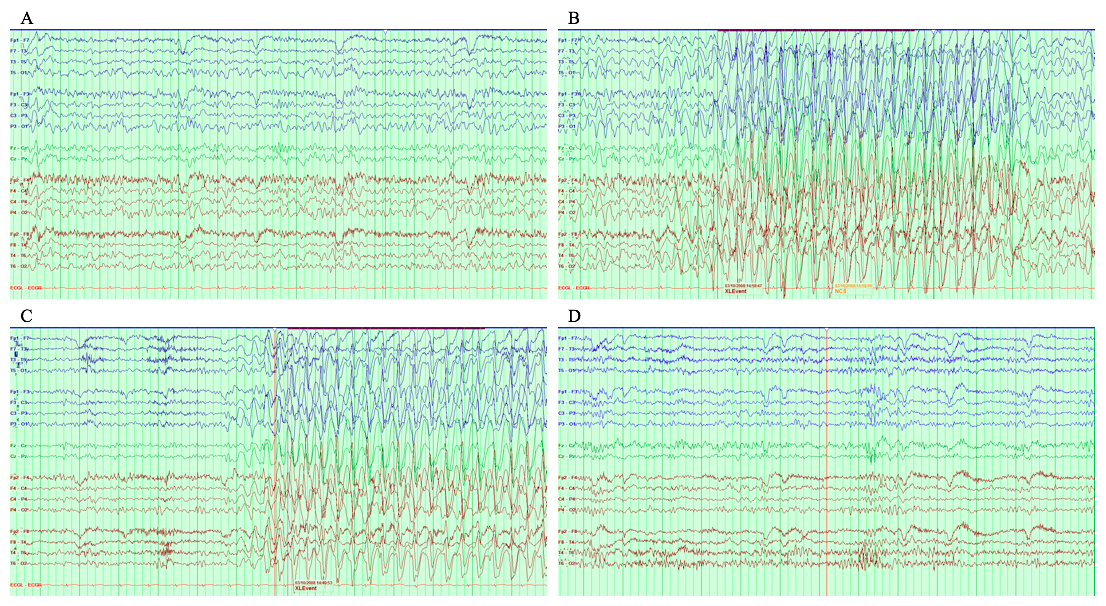
At 8 yr of age, a brain MRI showed cerebral atrophy and delayed myelination without evidence of focal abnormality (Fig. 2). At 15 yr, a vagal nerve stimulator was implanted, which significantly improved control of her atonic (drop) and tonic–clonic seizures. The first electroencephalogram (EEG) study completed at 16.5 yr of age showed epileptiform discharges. Another EEG study was done at 21.5 yr of age, which showed background disorganization (Supplemental Fig. 1). She continues to suffer from daily myoclonic jerks and absence seizures, with occasional tonic–clonic seizures usually triggered by intercurrent illnesses.
Figure 2.

Magnetic resonance imaging (MRI) images indicate that the ventricles and cerebral sulci are moderately prominent for the patient's age and show abnormal myelination with peripheral myelination less than expected. The radiology report impression concludes cerebral atrophy and delayed myelination without evidence of focal abnormality.
Her developmental history is significant for being globally delayed, making slow progress in all areas except for speech. There is no history of regression. She rolled over at 9 mo, sat at 18 mo, began crawling at 24 mo, and started walking between 4 and 5 yr. Currently, she wears ankle foot orthoses and can walk with assistance for short distances, but is ataxic with a wide-based, festinating gait. Over the past 2 years, she has developed stereotypical movements with an athetotic quality. She is nonverbal but makes sounds. She is able to understand to some degree and can communicate using a few signs. She is very social, smiles almost constantly, and has a happy disposition with some hyperactivity. Her appearance is similar to children with Angelman syndrome. She requires constant care with all of her activities of daily living and occasionally requires catheterization because of her neurogenic bladder.
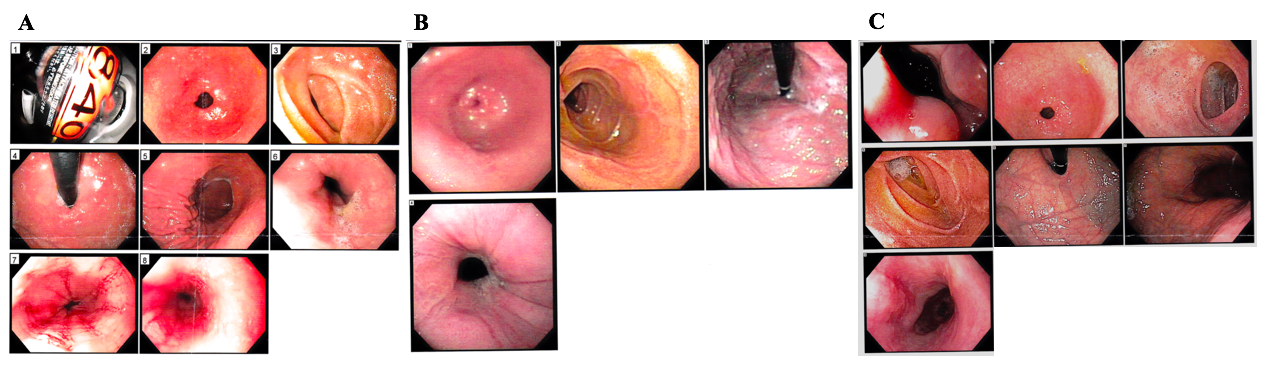
In addition, the patient suffers from severe eosinophilic esophagitis (Supplemental Fig. 2), gastroesophageal reflux, extrinsic asthma, atopic dermatitis, and multiple environmental, drug, and food allergies. She also suffers from chronic otitis media, chronic sinusitis, and recurrent lower respiratory infections. Her food, medication, and environmental allergies can lead to various immune responses, including eczema and asthma exacerbations, rhinitis, urticaria, and anaphylaxis. She has 25 reported food allergies and 27 reported environmental allergies. Her inability to vary her diet, sensitivity to medications, and multiple allergies contribute to further difficulties in managing this patient.
Previous Genetic/Diagnostic Testing
Baylor College of Medicine performed the Chromosomal Microarray analysis using the CMA Oligo V7.1 method. The results from this method were reported normal. The CMA Oligo V7.0 method was tested as well; however, the DNA was of insufficient quality to determine any significant results. FISH (fluorescence in situ hybridization) testing for Angelman syndrome/Prader–Willi syndrome (AS/PWS) was negative. UBE3A sequence analysis was negative. Polymerase chain reaction (PCR) showed that the patient does not demonstrate uniparental disomy on Chr15 (AS/PWS). Urinalysis results were normal. There was no evidence of proteinuria ruling out renal Fanconi syndrome.
Genomic Analysis
Exome sequencing of genomic DNA of the patient and family members led to the identification of a de novo variant, ENSP00000381053.3:p.Ser421Asn (Chr10:294310G>A), in the ZMYND11 gene that was not present in the dbSNP142, the ExAC (Exome Aggregation Consortium) 65k exome database, nor an in-house exome data set of approximately 2600 exomes (Table 1). The coverage depth for this variant in the proband is 92× (Supplemental Table 1). Both parents and an unaffected sibling do not carry this variant. This variant is located between the PWWP and MYND domains, highly conserved among several species (Fig. 3A), and predicted to be damaging (Combined Annotation-Dependent Depletion [CADD] score: 28.5) (Kircher et al. 2014). In addition to the damaging CADD score, other tools predicted this variant to be damaging and highly conserved. The PhyloP score was 0.98 out of 1.0, indicating high conservation between species (Siepel et al. 2006). The LRT (likelihood ratio test) predicts the significantly conserved amino acids between 31 different species (Chun and Fay 2009). This tool called a deleterious nonsynonymous variant in ZMYND11. PolyPhen-2 and MutationTaster predicted the variant to be damaging as well (Schwarz et al. 2010; Adzhubei et al. 2013). However, the SIFT (Scoring Sorting Intolerant from Tolerant) score predicted this variant to be tolerated (Kumar et al. 2009). ExAC shows that there are 217.2 expected missense mutations and only 90 observed with a z-score of 4.22. Additionally, there are 28.6 expected loss-of-function mutations and zero observed. The probability of loss-of-function intolerance (pLI) score for ZMYND11 is 1.0, meaning that this gene is intolerable to loss-of-function variation. Phosphorylation of Ser421 has been reported during mitosis (Dephoure et al. 2008). Other variants in the ZMYND11 gene have been linked to autism spectrum disorders, autosomal-dominant mental retardation, global developmental delay, and seizures (Supplemental Table 2). The patient, father, and unaffected sister carry a reported likely pathogenic variant in GJB2 (rs72474224; NM_004004.5:c.109G>A; NP_003995.2:p.Val37Ile). A second mutation in GJB2 was not identified in the patient by exome sequencing despite that the coding region was sufficiently covered (>20×) (Landrum et al. 2014). In other hearing loss genes, only variants of unknown significance that do not fit any inheritance models were found.
Table 1.
Variant table
| Gene | Chr | dbSNP | DNA ref (HG19/GRC37) | Protein ref | Proband GT | Father GT | Mother GT | Unaffected sib GT | Variant type | CADD |
|---|---|---|---|---|---|---|---|---|---|---|
| ZMYND11 | 10 | NA | g.294310G>A | ENSP00000381053.3:p.Ser421Asn | G|A | G|G | G|G | G|G | NSC | 28.5 |
| Coverage depth | 92 | 98 | 132 | 103 |
dbSNP, Databse for Short Genetic Variations; GT, genotype; NSC, missense variant, CADD, Combined Annotation-Dependent Depletion score; NA, not applicable.
Figure 3.

(A) High conservation of the ZMYND11 region across multiple species. (B) Reported variants of ZMYND11. The variant described in this report is in red.
DISCUSSION
ZMYND11, also called BS69, specifically recognizes H3K36me3 on H3.3 (histone 3, subtype 3) and is speculated to regulate RNA polymerase II during the elongation step of transcription. It has been proposed that ZMYND11 is important for the “fine-tuning” of gene expression rather than acting as an essential regulator. ZMYND11 knockdown moderately affects gene expression (Wen et al. 2014). Additionally, ZMYND11 has been demonstrated to have an inhibitory role in neuronal and muscle differentiation processes; however, the detailed mechanisms remain unknown (Yu et al. 2009).
Using a support vector machine technique that measures the change in free energy of point mutations, it was predicted that the p.Ser421Asn variant would lead to an increase in protein stability (Cheng et al. 2006). However, the serine at the 421 position has been reported to be phosphorylated during mitosis (Dephoure et al. 2008). Phosphorylation can play a large role in protein–protein interaction as well as signaling. Therefore, the substitution of serine at this position with asparagine, which is not a side chain commonly known to be phosphorylated posttranscriptionally, can potentially have a damaging effect on protein function and activity. ZMYND11 (or BS69) expression has been primarily found in the kidney, but lower levels have also been found in the lung, brain, spleen, thymus, and testis by northern blot analysis (Hateboer et al. 1995). This is further supported in the GeneCards database that reports mRNA is ubiquitously expressed; however, protein expression seems to be localized to lymphocytes, retinal cells, hepatocytes, and epithelial cells of the kidney. There is a positive correlation between these tissues and the clinical features described in this patient. The nervous system, immune system, and excretory system are areas where this patient is severely affected.
Coe et al. published cases of loss-of-function mutations in ZMYND11 in patients with clinical phenotypes similar to the case presented here. The case most similar to our patient is that of a 32-yr-old male with a different ZMYND11 variant (Chr10(GRCh37):g.294294_294295del), but a comparable phenotype including hypotonia, lack of speech, epilepsy, a movement disorder (choreoathetosis), and facial dysmorphisms including asymmetric skull, deep-set eyes, hypertelorism, an overfolded upper helix left ear, and a long philtrum. Other cases were generally characterized by developmental delay, epilepsy, hypotonia, and autism spectrum disorders (Coe et al. 2014). Another similar, single case was recently described in a boy with severe syndromic intellectual disability with a similar de novo mutation in ZMYND11 (c.1798C>T; p.Arg600Trp; NM_006624) (Cobben et al. 2014). Figure 3B shows all previously reported variants of ZMYND11 and Supplemental Table 2 presents the full phenotypic comparison with all other reported ZMYND11 variants. Sensorineural hearing loss has been reported in 10p15.3 microdeletion syndrome but is not commonly described in these patients (Vargiami et al. 2014). It is unclear whether the bilateral sensorineural hearing loss in this patient is due to an independent genetic cause other than ZMYND11. A positive family history was reported; yet, no clear genetic cause could be identified in this patient in any of the nonsyndromic hearing genes (http://hereditaryhearingloss.org/).
Our patient also suffers from distinct symptoms, including the presence of eosinophilic esophagitis and severe allergies. ZMYND11 is involved in the negative regulation of NF-κB, by direct interaction with TRAF3 (Ikeda et al. 2010). NF-κB is a primary transcription factor responsible for regulating the inflammatory responses of the immune system and diverse physiological processes, such as immunity, inflammation, proliferation, and apoptosis (Tak et al. 2001). In addition, NF-κB has been shown to be involved in hypereosinophilic syndromes and allergies (Ather et al. 2011). Dysregulation of the NF-κB pathway is involved in many diseases, particularly ones associated with chronic inflammation, immunodeficiency, or cancer.
A mutation in ZMYND11 could result in dysregulation of NF-κB. Without this negative feedback regulation, the NF-κB-dependent pathway during eosinophil activation is disrupted and results in inflammation because of an overactivation of eosinophils (Bouffi et al. 2013). This mechanism can potentially explain the eosinophilic esophagitis and severe allergy problems seen in this patient. Although it is not uncommon to see comorbidities of allergies, autoimmunity, asthma, and gastrointestinal issues with autism spectrum disorder (ASD) patients, none of the previously reported patients with a ZMYND11 variant have presented with these specific symptoms (Zerbo et al. 2015). This may allude to the importance of the specific Ser421 residue of ZMYND11 in NF-κB signaling, but further studies are needed to understand this relationship.
Previous examples have shown the importance of the role of ZMYND11 in autism spectrum disorders and syndromic intellectual disabilities. However, the complexity of this de novo missense mutation is unique in its association with eosinophilic esophagitis and severe allergies as well as specific similarities, including the patient's neurological phenotype, happy disposition, and gait, that show similarity to Angelman syndrome (Supplemental Movie). The discovery of this genotype–phenotype relationship is significant to further characterize ZMYND11 as it relates to pathogenic variants.
METHODS
Exome Sequencing
DNA was extracted from blood obtained from the parents, proband, and an unaffected sibling. Exome libraries were prepared with the SureSelectXT2 Human All Exon V4 Kit (Agilent Technologies), followed by 100-bp paired-end sequencing on a HiSeq2000 instrument (Illumina Inc.) by GeneDX (GeneDX). Filtered reads were aligned to the human genome (Hg19/GRC37) using the Burrows–Wheeler transform (BWA-MEM) (Li and Durbin 2009). Reads were sorted and PCR duplicates were removed using Picard (Li et al. 2009). Base quality recalibration and indel realignment were performed using the Genome Analysis Toolkit (GATK) (McKenna et al. 2010). Variants were called jointly with HaplotypeCaller and recalibrated with GATK, annotated with SnpEff, and selected (SnpSift) for protein-coding events (Cingolani et al. 2012). Prediction scores were loaded from dbNSFP (Database for Nonsynonymous SNPs' Functional Predictions) and used for filtering (Supplemental Table 3). Autosomal-recessive, autosomal-dominant, and X-linked inheritance models were considered when analyzing the data. Relatedness and gender of the four individuals were checked with Plink v1.07.
ADDITIONAL INFORMATION
Data Deposition and Access
Exome data of individuals have been added to the Database of Genotypes and Phenotypes (dbGaP; http://www.ncbi.nlm.nih.gov/gap) under project phs000816.v1.p1. This variant has been reported to ClinVar (htpp://www.ncbi.nlm.nih.gov/clinvar/) under accession number SCV000267194. The raw sequence data of the father (C4RCD0190_D1_1_1417643), mother (C4RCD0190_M1_2_1417642), patient (C4RCD0190_C1_2_1417641), and unaffected sibling (C4RCD0190_S1_1_1417644) were submitted to the Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) with the respective accession ID numbers SRS1411269, SRS1411266, SRS1411267, and SRS1411268.
Ethics Statement
The participating family provided written consent and was enrolled into the Center for Rare Childhood Disorders Program at The Translational Genomics Research Institute (TGen). The study protocol and consent procedure were approved by the Western Institutional Review Board (study number 20120789).
Acknowledgments
We thank the family for participating in this study and also all the other families within TGen's Center for Rare Childhood Disorders. We thank Bradley Coe and Evan Eichler for sharing their insight into this ZMYND11 variation. A.M.M. was supported by the Helios Education Foundation through the Helios Scholars at TGen summer internship program in biomedical research at the TGen in Phoenix, Arizona. Finally, this manuscript is also dedicated to the memory of our dear friend and colleague Jason Corneveaux. His contributions to our laboratory were innumerable and significant, and he will be sorely missed.
Author Contributions
A.M.M., A.L.S., S.S., A.M.C., C.B., A.L.C., S.R., and I.S. were involved in data analysis. J.J.C., R.F.R., I.S.P., M.D.B., and M.R. composed or performed the bioinformatics pipeline. A.M.M., N.B., K.R., and V.N. examined the clinical phenotype of the patient. A.M.M., N.B., and I.S. drafted the paper. All authors where involved in data interpretation and read, revised, and approved the final manuscript.
Competing Interest Statement
The authors have declared no competing interest.
Referees
Gholson Lyon
Anonymous
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
REFERENCES
- Adzhubei I, Jordan DM, Sunyaev SR. 2013. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet 7: Unit 7.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ather JL, Hodgkins SR, Janssen-Heininger YMW, Poynter ME. 2011. Airway epithelial NF-κB activation promotes allergic sensitization to an innocuous inhaled antigen. Am J Respir Cell Mol Biol 44: 631–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouffi C, Rochman M, Zust C, Stucke E, Kartashov A, Fulkerson P. 2013. IL-33 markedly activates murine eosinophils by an NFκB-dependent mechanism differentially dependent upon an IL-4-driven autoinflammatory loop. J Immunol 191: 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, Randall A, Baldi P. 2006. Prediction of protein stability changes for single-site mutations using support vector machines. Proteins 62: 1125–1132. [DOI] [PubMed] [Google Scholar]
- Chun S, Fay JC. 2009. Identification of deleterious mutations within three human genomes. Genome Res 19: 1553–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. 2012. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w 1118; iso-2; iso-3. Fly (Austin) 6: 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobben JM, Weiss MM, van Dijk FS, De Reuver R, de Kruiff C, Pondaag W, Hennekam RC, Yntema HG. 2014. A de novo mutation in ZMYND11, a candidate gene for 10p15.3 deletion syndrome, is associated with syndromic intellectual disability. Eur J Med Genet 57: 636–638. [DOI] [PubMed] [Google Scholar]
- Coe BP, Witherspoon K, Rosenfeld JA, van Bon BWM, Vulto-van Silfhout AT, Bosco P, Friend KL, Baker C, Buono S, Vissers LELM, et al. 2014. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat Genet 46: 1063–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dephoure N, Zhou C, Villén J, Beausoleil SA, Bakalarski CE, Elledge SJ, Gygi SP. 2008. A quantitative atlas of mitotic phosphorylation. Proc Natl Acad Sci 105: 10762–10767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Descipio C, Conlin L, Rosenfeld J, Tepperberg J, Pasion R, Patel A, McDonald MT, Aradhya S, Ho D, Goldstein J, et al. 2012. Subtelomeric deletion of chromosome 10p15.3: clinical findings and molecular cytogenetic characterization. Am J Med Genet A 158A: 2152–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hateboer G, Gennissen A, Ramos YF, Kerkhoven RM, Sonntag-Buck V, Stunnenberg HG, Bernards R. 1995. BS69, a novel adenovirus E1A-associated protein that inhibits E1A transactivation. EMBO J 14: 3159–3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda O, Miyasaka Y, Yoshida R, Mizushima A, Oritani K, Sekine Y, Kuroda M, Yasui T, Fujimuro M, Muromoto R, et al. 2010. BS69 cooperates with TRAF3 in the regulation of Epstein–Barr virus–derived LMP1/CTAR1-induced NF-κB activation. FEBS Lett 584: 865–872. [DOI] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. 2014. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46: 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng P. 2009. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4: 1073–1081. [DOI] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, Maglott DR. 2014. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 42: D980–D985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; Subgroup 1000 Genome Project Data Processing. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. 2010. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 7: 575–576. [DOI] [PubMed] [Google Scholar]
- Siepel A, Pollard KS, Haussler D. 2006. New methods for detecting lineage-specific selection. In Lecture notes in computer science (including subseries lecture notes in artificial intelligence and lecture notes in bioinformatics), Vol. 3909, pp. 190–205. Springer, Berlin. [Google Scholar]
- Tak PP, Firestein GS, Tak PP, Firestein GS. 2001. NF-κB: a key role in inflammatory diseases. J Clin Invest 107: 7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargiami E, Ververi A, Kyriazi M, Papathanasiou E, Gioula G, Gerou S, Al-Mutawa H, Kambouris M, Zafeiriou DI. 2014. Severe clinical presentation in monozygotic twins with 10p15.3 microdeletion syndrome. Am J Med Genet A 164: 764–768. [DOI] [PubMed] [Google Scholar]
- Wen H, Li Y, Li H, Shi X. 2014. ZMYND11: an H3.3-specific reader of H3K36me3. Cell Cycle 13: 2153–2154. [DOI] [PubMed] [Google Scholar]
- Yu B, Shao Y, Zhang C, Chen Y, Zhong Q, Zhang J, Yang H, Zhang W, Wan J. 2009. BS69 undergoes SUMO modification and plays an inhibitory role in muscle and neuronal differentiation. Exp Cell Res 315: 3543–3553. [DOI] [PubMed] [Google Scholar]
- Zerbo O, Leong A, Barcellos L, Bernal P, Fireman B, Croen LA. 2015. Immune mediated conditions in autism spectrum disorders. Brain Behav Immun 46: 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}