

Abstract
In the past, the growth hormone (GH) – insulin-like growth factor-I (IGF-I) axis was thought to be the central system regulating childhood growth and therefore responsible for short stature and tall stature. However, recent findings have revealed that the GH-IGF-I axis is just one of many regulatory systems that control chondrogenesis in the growth plate, the biological process that drives height gain. Consequently, normal growth in children depends not only on GH and IGF-I but on multiple hormones, paracrine factors, extracellular matrix molecules, and intracellular proteins that regulate growth plate chondrocytes. Mutations in genes encoding many of these local proteins cause short stature or tall stature. Similarly genome-wide association studies have revealed that the normal variation in height appears to be due largely to genes outside the GH-IGF-I axis that affect growth at the growth plate through a wide variety of mechanisms. These findings point to a new conceptual framework for understanding short and tall stature, which is centered not on two particular hormones but rather on the growth plate, the structure responsible for height gain.
Introduction
For decades, the conceptual framework for understanding short stature and tall stature has been centered on the growth hormone (GH) – insulin-like growth factor-I (IGF-I) axis. Children grew taller, it was thought, primarily because the pituitary gland produces GH, which stimulates the liver to produce IGF-I, which makes children grow in height. This mindset was quite understandable historically because the role of the GH-IGF-I axis in longitudinal bone growth was discovered by the 1950s,1 and the measurement of these two circulating factors, GH and IGF-I, was readily accomplished by radioimmunoassay by the 1960s and 1970s respectively,2,3 and also because treatment with GH has been the main therapeutic approach available to treat short stature. Based on this paradigm, short stature has sometimes been divided into defects within the GH-IGF-I system versus those outside the axis.4 This thinking also tended to dominate our speculations about the causes of idiopathic short stature; ISS might be due to either (1) secondary IGF deficiency (due to subtle disorders of GH secretion), (2) primary IGF deficiency (low serum IGF-I with a normal GH secretion), (3) IGF resistance and (4) “other causes”.5, 6 Similarly, it has been suggested that the normal variation in height in the general population is due to subtle modulation of the GH – IGF-I axis.7,8
However, unambiguous defects in the GH-IGF-I axis can only be identified in a small minority of children with short stature.9 In some of these children, the underlying molecular defects have been identified, including mutations causing GH deficiency (including GH1, GHRHR), GH insensitivity/primary IGF-I deficiency (including GHR, IGFI, IGFALS, STAT5B) and IGF insensitivity (IGF1R).10 However, these genetic abnormalities are rare. Far more children fail GH stimulation tests, but this failure appears primarily to result from poor test specificity.11,12 Similarly, many short children have IGF-I levels in the lower part of the normal range or even below the normal range. This condition has been labeled primary IGF-I deficiency, but it is unclear how many of these children have low IGF-I levels secondary to poor nutritional intake,13 for example due to diminished appetite,14 subtle chronic disease,15 other primary disorders16 or simply due to a delay in the physiological increase in IGF-I that occurs with age and puberty, since many short children have delayed maturation of other physiological processes.13 Thus, the vast majority of short children do not have a well-substantiated defect in the GH-IGF-I axis.9
GH and IGF-I stimulate linear growth (gain in stature) in children by acting on the growth plate (Figure 1). The growth plate is a thin layer of cartilage that is found in most bones outside the skull and face, including the long bones and vertebrae. In the growth plate, chondrocytes proliferate, hypertrophy, and secrete cartilage extracellular matrix (Figure 1). These processes generate new cartilage tissue, which is subsequently remodeled into bone tissue.17,18 The net result is that new bone is progressively created at the growth plate, causing bones to grow longer and children to grow taller. GH acts on the growth plate to stimulate new bone formation both through circulating IGF-I and also locally, in part through local IGF-I production.1,18
Figure 1.
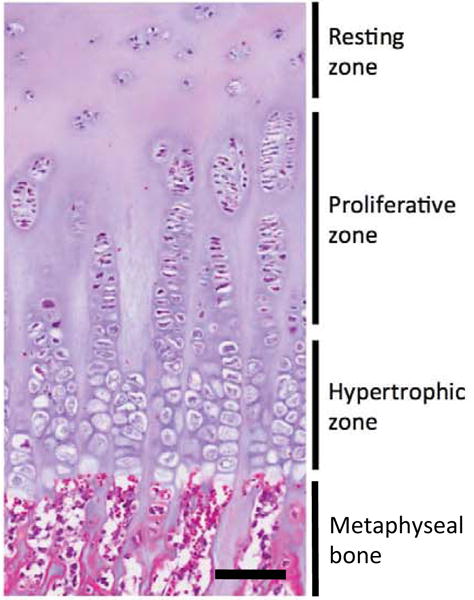
Human growth plate histology from an 11-year old boy. The growth plate comprises three histologically and functionally distinct zones; the resting, proliferative, and hypertrophic zones. Bar represents 100 μm.
However, recent findings, in both basic and clinical studies, have revealed that the GH – IGF-I axis is just one of many regulatory systems that control chondrogenesis in the growth plate and therefore regulate linear growth in children (Figure 2). Consequently, normal growth in children requires not just normal concentrations of GH and IGF-I but also normal production and action of multiple other hormones, paracrine factors, and extracellular matrix molecules, as well as normal function of multiple intracellular processes required for chondrocyte proliferation, hypertrophy, and extracellular matrix production. Recent studies have identified many new genes that, when mutated, cause short stature or tall stature, the large majority of which do not participate in the GH-IGF-I system. Similarly normal variation in height appears to be due largely to genes outside the GH-IGF-I axis that affect growth at the growth plate through a wide variety of mechanisms.19 These new findings point to a new conceptual framework for understanding short and tall stature, a framework which is centered not on two particular hormones but rather on the growth plate itself, the structure responsible for height gain.
Figure 2.
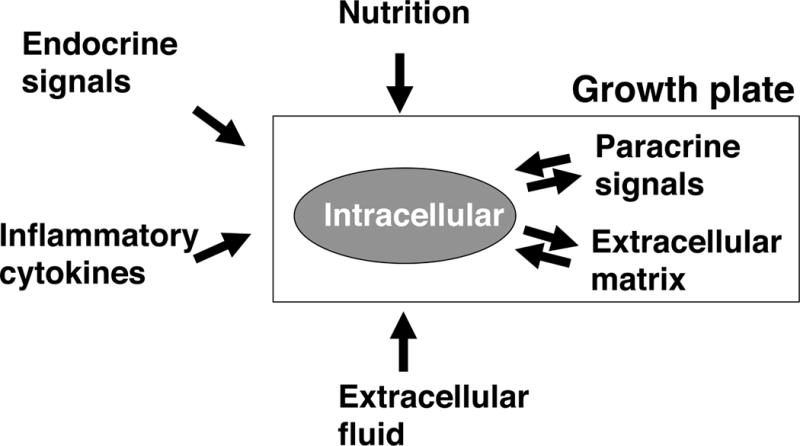
Schematic diagram depicting the regulation of growth plate function. Growth plate chondrocyte (grey oval) proliferation and differentiation are regulated by many factors, including nutritional, endocrine, inflammatory cytokines, extracellular fluid (e.g. oxygen, pH), paracrine, extracellular matrix, and intracellular mechanisms. Not depicted are the interactions among many of these systems; for example, nutritional intake strongly affects endocrine regulators of the growth plate.
Normal growth requires normal signaling through many pathways at the growth plate
In the past, the study of childhood growth was severely limited by available experimental methods. Endocrine factors could be measured by immunoassay in the circulation, but there were few available methods to study, for example, paracrine factors that act locally in the growth plate without entering the circulation, or to study intracellular molecular pathways that regulate chondrocyte proliferation and differentiation. However, in the past few decades a wide variety of new experimental approaches have yielded an explosion of information about the function of the growth plate. For example, knockout of many genes not previously known to be important in the growth plate have produced phenotypes involving skeletal growth at the growth plate, thus opening up unexpected new areas of growth plate physiology. In parallel, new molecular genetic techniques used in clinical research have identified genetic abnormalities causing short stature, many of which occur in genes involved, not in the GH-IGF-I axis, but in other, often local, pathways necessary for normal growth plate function. Together, the basic biology and clinical genetic studies have synergistically expanded our view of childhood growth physiology and pathophysiology.
Multiple hormones and cytokines directly regulate growth plate function
In addition to GH and IGF-I, multiple other hormones regulate linear growth, including thyroid hormone, glucocorticoids, estrogens, and androgens. There is evidence that each of these endocrine factors regulate growth in part by a direct action on the growth plate. For example, infusion of dexamethasone, a synthetic glucocorticoid, directly into the growth plate causes local slowing of growth in that growth plate and addition of dexamethasone to culture medium slows growth of cultured fetal metatarsal bones.20,21 Clinically, treatment with GH can partially compensate for a low dose of glucocorticoid, improving linear growth, but has little effect at high concentrations of glucocorticoids.22 Similarly, there is evidence for direct effects of thyroid hormone,23,24 androgen,25,26 and estrogen.27,28 In addition, there is interaction among these hormonal systems. For example, glucocorticoids have complex effects on GH production29 and inhibit thyroid hormone production.30
Estrogen has complex effects on the growth plate, not only altering growth rate, but also accelerating loss of progenitor cells in the resting zone and thereby accelerating the developmental program of growth plate senescence, causing earlier cessation of growth.31,32 Consequently, inactivating mutations in the estrogen receptor ERα33,34 or in aromatase,35 the enzyme that converts androgens to estrogens, cause the program of growth plate senescence to progress more slowly and thereby allow prolonged linear growth beyond adolescence and therefore adult tall stature. Aromatase inhibitors produce similar effects and thus are under investigation as a treatment for short stature in boys.36,37 Some of the estrogen that modulates growth physiologically may be locally produced by growth plate chondrocytes which express aromatase and other steroid-metabolizing enzymes.28,38
Proinflammatory cytokines are endogenously produced by growth plate chondrocytes and may act intrinsically to modulate longitudinal bone growth.39 Furthermore, the growth plate is targeted by extrinsic factors including cortisol and proinflammatory cytokines, both induced by stress and chronic inflammation.40 Some of these proinflammatory cytokines negatively regulate growth plate function.41 There is evidence that tumor necrosis factor-α, interleukin-1β, and interleukin-6 act directly on growth plate cartilage to suppress bone growth.39,42,43 These cytokines may act in synergy further potentiating their negative effects on growth plate cartilage function.43
Longitudinal bone growth is also regulated by nutritional intake, mediated in part through a complex endocrine network that includes leptin, IGF-I, sex steroids, thyroid hormone, and glucocorticoids.18 As a result, undernourished children have impaired linear growth (despite elevated GH levels) whereas obese children have normal or mildly accelerated growth (despite low GH levels).44
Growth plate function can also be affected by physical mechanisms. Even relatively low doses of ionizing radiation, such as 10 Gy, can impair longitudinal growth.45 Mechanical compression across the growth plate also impairs bone elongation46 in part due to decreased enlargement of hypertrophic chondrocytes.47 It has been suggested that the inhibitory effects of compression on the growth plate contributes to progression of scoliosis and tibia vara (Blount’s disease).46 Similarly, physical compression is used clinically to correct limb length inequalities and angular deformities.48 The effects of dynamic load variation due to exercise in children has not been well established.49
Multiple paracrine factors in the growth plate regulate linear growth
One area in which our understanding has advanced enormously involves the role of paracrine signals in the growth plate. Paracrine factors are secreted by growth plate chondrocytes, or sometimes cells in the surrounding perichondrium, and act locally on chondrocytes to regulate proliferation and differentiation. In vitro and in vivo approaches have uncovered a host of paracrine factors necessary for normal growth plate function, including multiple fibroblast growth factors (FGFs),50–53 bone morphogenetic proteins (BMPs),54–56 WNTs,57,58 the parathyroid hormone-related protein (PTHrP) and Indian hedgehog (IHH) pathway,17 and the C-type natriuretic peptide (CNP)-NPR2 pathway.59 Mutations in genes involved in these paracrine signaling systems can severely impair bone growth in both mice and humans.
For example, fibroblast growth factor receptor-3 (FGFR3) acts as a negative regulator of growth plate chondrogenesis. Consequently, activating mutations in FGF receptor-3 impair bone elongation in patients with achondroplasia, hypochondroplasia and thanatophoric dysplasia. A recent report has identified an activating FGFR3 mutation in a family with autosomal dominant proportionate short stature60. Conversely, heterozygous61 and homozygous62 inactivating mutations in FGFR3 have been reported in individuals with tall stature. At the growth plate, FGF signaling through FGFR3 leads to activation of several pathways including the MAPK and JAK/STAT pathways and negatively regulates growth by decreasing proliferation in the proliferative zone, decreasing matrix production, accelerating the onset of hypertrophic differentiation, and decreasing the size of the hypertrophic chondrocytes53,63–65. These effects are at least in part due to interaction with other paracrine factors including downregulation of IHH expression, as well as interactions with CNP and BMP signaling.65–67
The PTHrP and IHH paracrine system also plays a critical role in the regulation of growth plate function. These two paracrine factors form a negative feedback loop within the growth plate that regulates chondrocyte hypertrophy and proliferation.17 Consequently, mutations in the genes for IHH, PTHrP, and PTH1R cause specific skeletal dysplasias. For example, heterozygous mutations in PTHLH, the gene that encodes PTHrP, cause short stature and short fingers in individuals with brachydactyly, type E2.68
Another paracrine factor of importance in the growth plate is CNP. This peptide was named based on its structural similarity to atrial natriuretic peptide (ANP), but has a very different physiological role, serving instead as a local, positive regulator of growth plate function.69–71 Consequently, homozygous inactivating mutations in NPR2, the receptor for CNP, cause a severe skeletal dysplasia, termed acromesomelic dysplasia, Moroteaux type.72 Interestingly, heterozygous mutations cause a milder phenotype, presenting as short stature without clear signs of a skeletal dysplasia.16 Recent studies suggest that approximately 2% of children presenting with idiopathic short stature have mutations in this gene.73–75 Conversely, overexpression of CNP76,77 or activating mutations in NPR278,79 cause tall stature. Binding of CNP to NPR2 stimulates the receptor guanylyl cyclase activity thereby increasing synthesis of cGMP, activating the type II cGMP-dependent protein kinase.80 Therefore, mice deficient in that protein kinase also show severely impaired growth plate function.81 This signaling system leads to inhibition of the MAPK pathway, thus antagonizing FGFR signaling82, providing an explanation for the beneficial effects of CNP in a mouse model of achondroplasia.83
Bone morphogenetic proteins (BMPs), also known as growth and differentiation factors (GDFs), belong to the transforming growth factor-beta (TGFβ) superfamily of paracrine factors. The BMPs were originally discovered as the component of demineralized bone matrix that is able to induce ectopic bone formation and later found to regulate a multitude of processes in skeletal development, including spatial regulation of proliferation and differentiation in the growth plate.59 Consistently, inactivating mutations in the genes for several BMPs, their receptors, and antagonists cause skeletal dysplasias, including brachydactyly A2 (BMP2 or BMPR1B), brachydactyly A1 and C (GDF5), chondrodysplasia, Grebe type (GDF5), Klippel-Feil syndrome 1 (GDF6), proximal symphalangism 1A (NOGGIN). Local modulation by antagonists and sequestration by extracellular matrix molecules are crucial for local regulation of BMP and TGFβ signaling during development and growth. Consequently, excessive TGFβ signaling has been identified as an important pathogenic mechanism for Marfan’s syndrome,84 which includes disproportionate overgrowth and aortic dilation,85 caused by mutations in the fibrillin 1 (FBN1) gene. Angiotensin II receptor blockers act as TGFβ antagonists and are therefore being evaluated in clinical studies for prevention of aortic dilatation in Marfan’s syndrome.86
Cartilage extracellular matrix is crucial for linear growth
Chondrocytes secrete a unique extracellular matrix containing specific collagens, non-collagenous proteins, and proteoglycans, which also are vital to normal growth plate function. This extracellular matrix provides the compressible, resilient structural properties of cartilage and also interacts with signaling molecules to regulate growth plate chondrogenesis.87 As a result, mutations in genes that encode matrix proteins and proteoglycans often interfere with growth plate function.88 For example, mutations in COL10A1, the gene encoding collagen type X, causes a skeletal dysplasia termed metaphyseal chondrodysplasia, Schmid type.
Mutations in the gene ACAN, encoding aggrecan, a major proteoglycan component of the cartilage extracellular matrix, also affect linear growth. Homozygous mutations cause a severe skeletal dysplasia, spondyloepimetaphyseal dysplasia aggrecan type.89 Heterozygous mutations can present as a milder skeletal dysplasia, spondyloepiphyseal dysplasia type Kimberley90 or as short stature without an evident radiographic skeletal dysplasia, which can either be disproportionate91 or proportionate.92 The short stature is typically associated with an advanced bone age and early cessation of growth.92 In some patients, this disorder affects not only the growth plate cartilage but also the articular cartilage, causing osteochondritis dissecans and early-onset osteoarthritis.91,92 In animal models, ACAN deficiency causes growth plate dysfunction, including decreased chondrocyte proliferation and accelerated hypertrophic chondrocyte differentiation associated with disrupted IHH, FGF, and BMP signaling.93,94
There is evidence that other non-collagenous matrix proteins also interact with paracrine signals. For example biglycan and decorin modulate growth and bone formation by interactions with TGFβ as evidenced by postnatal growth retardation and osteoporosis of biglycan deficient mice.95 Similarly, excessive TGFβ signaling has been identified as an important pathogenic mechanism in Marfan’s syndrome, which includes disproportionate overgrowth, aortic dilation, and is caused by heterozygous mutations in the fibrillin 1 (FBN1) gene.84,85 Interestingly, FBN1 mutations also cause short stature in Weill-Marchesani syndrome, geleophysic dysplasia, and acromicric dysplasia.96
Intracellular pathways regulate growth plate function and linear growth
A variety of intracellular pathways that play important roles in growth plate chondrogenesis have also been discovered. For example, transcription factors Sox5, 6, and 9 are critical regulators of chondrocyte differentiation.97 As one would expect, mutations affecting key intracellular pathways cause growth disorders in humans. For example, inactivating mutations in SOX9 cause a severe skeletal dysplasia, campomelic dysplasia.97
As another example, homozygous inactivating mutations in SHOX, another transcription factor expressed in the growth plate,98 cause Langer mesomelic dysplasia, which includes severe defects in bone growth. Heterozygous inactivating mutations, or deletions of SHOX or its enhancer regions, cause a milder skeletal dysplasia, Leri-Weill dyschondrosteosis or can present clinically as idiopathic short stature, with body proportions that are mildly affected or sometimes within the normal range.99 SHOX mutations account for 2–15% of individuals presenting with idiopathic short stature, depending on the study.100 Conversely, increased copies of SHOX are associated with tall stature in individuals with Klinefelter syndrome and other type of sex chromosome aneuploidy.101
Another pathway important for skeletal growth due to its effect on cellular proliferation and differentiation of growth plate chondrocytes is the Ras/Mitogen activated protein kinase (MAPK) signaling pathway. This pathway integrates signals from several growth factors including FGFs, CNP, and EGF.102 Ras, a small GTPase, signals through MAPK cascades to phosphorylate numerous cytoplasmic and nuclear proteins, regulating cell proliferation and differentiation.103 Activation of this pathway results in a number of overlapping syndromes including Noonan, LEOPARD, Costello, cardio-facio-cutaneous, and neurofibromatosis-Noonan syndrome, all characterized by neurocutaneous manifestations but also postnatal growth failure of varying degree.104,105 In contrast, Sotos syndrome (characterized by tall stature) is associated with decreased activity of the Ras/MAPK pathway.106
Skeletal growth is also regulated by Nuclear Factor kappa B (NF-κB), a group of seven transcription factors, including p65 (RelA), c-Rel, RelB, p50/p105 (NF-κB1), and p52/p100 (NF-κB2). In growth plate chondrocytes, NF-κB p65 helps mediate the stimulatory effects of GH and IGF-1 on chondrogenesis.107 In humans, heterozygous loss of function mutations in IκBα, an essential component of the NF-κB pathway, result in GH insensitivity and growth failure as well as ectodermal dysplasia and immunodeficiency.108
Mutations in genes encoding proteins involved in fundamental cellular processes can produce severe global growth deficiencies, termed primordial dwarfisms, which affect not just the growth plate but multiple other tissues and typically impair both pre- and postnatal growth.109 For example, 3M syndrome, which includes severe intrauterine growth retardation and postnatal short stature, is caused by defects in one of three genes: CUL7,110 OBSL1,111 and CCDC8.112 The products of these three genes form a complex that plays a critical role in maintaining microtubule integrity with defects leading to aberrant cell division.113 Similarly, mutations in the centrosomal protein, pericentrin, cause microcephalic osteodysplastic primordial dwarfism (MOPD) type II114 while other centrosomal proteins are implicated in Seckel syndrome. Mutations in the DNA origin recognition complex underlie Meier-Gorlin Syndrome,115,116 and defects in DNA damage repair underlie growth disorders such as Cockayne Syndrome and Bloom Syndrome.
Interestingly, tall stature can be caused by mutations in genes that control epigenetic modifications, including DNA and histone methylation, and thereby chromatin formation and gene expression. Heterozygous mutations in DNA methyltransferase 3A (DNMT3A) cause tall stature, a distinctive facial appearance, and intellectual disability.117 Similarily, heterozygous mutations in EZH2, an enzyme that specifically methylates lysine residue 27 of histone 3 (H3K27) which is associated with transcriptional repression, causes Weaver syndrome, characterized by pre- and postnatal overgrowth, and a markedly advanced bone age.118 In addition, most cases of Sotos syndrome are caused by mutations in NSD1, which acts as a methyltransferase to methylate histone H3 lysine 36 (H3K36) and other substrates and also interacts with nuclear hormone receptors, thereby regulating transcription of target genes.106
Copy number variation and short stature
Recent evidence suggests that approximately 10% of patients with idiopathic short stature carry a disease-causing copy number variation (CNV).119–121 The phenotype of subjects with CNVs includes both growth failure of prenatal onset and postnatal onset, both proportionate and disproportionate short stature, and both syndromic and nonsyndromic short stature.120,121 However, in individual subjects, it is often difficult to know whether the growth failure was due to the CNV, and therefore additional studies are needed to define better the prevalence and phenotypes of CNV-associated short stature.
Normal variation in adult height
It has long been known that the normal variation in human stature has a large genetic component and shows a polygenic inheritance pattern. Only recently have the specific genes involved begun to be elucidated. A large meta-analysis of genome-wide association studies identified 423 loci that contribute to normal adult stature variation.122 Presumably each locus contains at least one gene in which common polymorphisms affect human linear growth. Although the precise gene within each locus that is responsible for the effect typically cannot be pinpointed with certainty, there is evidence that a large number of the causative genes are expressed in and function in the growth plate.122,123 These implicated genes include multiple genes involved in paracrine signaling by the PTHrP-IHH feedback loop, BMPs, FGFs, WNTs/β-catenin, and CNP. These loci also contain a much smaller number of genes known to participate in the GH-IGF-I axis, including GH1, which encodes GH, GHSR, which encodes the GH secretagogue receptor, and IGF1R, which encodes the principal receptor for IGF-I. It is likely that many children with short stature, particularly those with mild short stature with a pedigree suggesting a polygenic inheritance, have inherited multiple polymorphisms which negatively modulate linear growth associated with short stature. Interestingly, a recent study in tall Europeans reported that common sequence variants are also associated with tall stature.124
Current findings require a broader conceptual framework for short stature
The findings discussed above necessitate a new framework with which to conceptualize short stature. The categorization of idiopathic short stature into subtypes of GH deficiency, GH insensitivity, IGF-I deficiency and IGF-I insensitivity, which seemed reasonable when we could only measure hormones and knew little about the causes of short stature, now is not nearly broad enough. Using this system would require us to jam all the myriad abnormalities involving intracellular, extracellular matrix, and paracrine signaling defects into the category “IGF-I insensitivity.” Instead, the large picture emerging from recent findings places GH and IGF-I as important factors for growth but as only two of many factors that influence growth plate function and therefore human growth.
A broader conceptual framework can now be formulated that is centered, not on the GH-IGF-I axis, but on the growth plate (Figure 2), the biological structure that is responsible for linear growth. Short stature is caused by growth plate dysfunction that can result either from a primary defect, that is, a disorder intrinsic to the growth plate, or a secondary defect, in which the growth plate is adversely affected by a disorder elsewhere in the body (Table 1, Figure 2). Primary defects may involve: 1) Paracrine signaling systems in the growth plate, 2) Cartilage extracellular matrix molecules, and 3) Intracellular pathways in growth plate chondrocytes. In secondary disorders, growth plate chondrocytes can be adversely affected through a variety of mechanisms, including abnormal: 1) Nutritional effects (mediated in part by endocrine signals), 2) Endocrine signaling, 3) Inflammatory cytokines, 4) Extracellular fluid (such as acidosis), and 5) Physical factors (such as radiation). This scheme provides a conceptual framework based on the underlying biological mechanisms. For some disorders, including many dysmorphic syndromes, constitutional delay of growth, and idiopathic short stature, the mechanism responsible for growth plate dysfunction remains unknown. A classification scheme proposed by the European Society for Paediatric Endocrinology, which also divides the causes of short stature into primary and secondary defects, is less mechanism-based but useful for practical clinical purposes125. Although growth disorders can be environmental or polygenic in etiology, many growth disorders arise from single gene defects (Table 2).
Table 1.
Etiology of short stature, organised by mechanism of growth plate dysfunction
| Intrinsic to growth plate |
| Paracrine |
|
| Cartilage extracellular matrix |
|
| Intracellular |
|
| Extrinsic to growth plate |
| Nutritional intake |
|
| Endocrine |
|
| Inflammatory cytokines |
|
| Extracellular fluid |
|
| Physical factors |
|
This list provides examples of disorders affecting linear growth but is not exhaustive.
Table 2.
Examples of single gene defects that cause disorders of childhood growth.
| Intrinsic to growth plate | |||||
|---|---|---|---|---|---|
| Mechanism | Gene | Effect on protein | Disorder | Effect on linear growth | Inheritance |
| Paracrine | |||||
| CNP signaling | NPR2 | Loss of function | ISS | Short stature | AD* |
| Loss of function | Moroteaux acromesomelic dysplasia | Short stature | AR* | ||
| Gain of function | Overgrowth with or without skeletal deformities | Tall stature | AD* | ||
| FGF signaling | FGFR3 | Gain of function | Hypochondroplasia Achondroplasia Thanatophoric dysplasia ISS |
Short stature | AD |
| FGFR3 | Loss of function | CATSHL syndrome | Tall stature | AD | |
| PTHrP signaling | GNAS | Loss of function | Albright hereditary osteodystrophy | Short stature | AD |
| PTH1R | Loss of function | Blomstrand chondrodysplasia | Short stature | AD | |
| Gain of function | Jansen metaphyseal dysplasia | Short stature | AD | ||
| Cartilage extracellular matrix | |||||
| Extracellular matrix structure/function | ACAN | Abnormal structure | ISS | Short stature | AD* |
| Spondyloepiphyseal dysplasia type Kimberley | Short stature | AD* | |||
| Spondyloepimetaphyseal dysplasia aggrecan type | Extreme short stature | AR* | |||
| COL2A1 | Abnormal structure | Multiple skeletal dysplasias | Short stature | AD | |
| COL10A1 | Abnormal structure | Metaphyseal chondrodysplasia, Schmid type | Short stature | AD | |
| COMP | Abnormal structure | Pseudoachondroplasia | Short stature | AD | |
| FBN1 | Abnormal structure | Marfan syndrome | Tall stature | AD | |
| Intracellular | |||||
| Transcription factors | SOX9 | Loss of function | Campomelic dysplasia | Short stature | AD |
| SHOX | Loss of function | ISS | Short stature | AD* | |
| Loss of function | Leri–Weill dyschondrosteosis | Short stature | AD* | ||
| Loss of function | Langer mesomelic dysplasia (homozygous) | Extreme short stature | AR* | ||
| Microtubule function | CUL7 | Loss of function | 3M syndrome 1 | Short stature | AR |
| OBSL1 | Loss of function | 3M syndrome 2 | Short stature | AR | |
| RAS–MAPK signaling | PTPN11 KRAS Others |
Gain of function | Noonan syndrome | Short stature | AD |
| Epigenetic defects | NSD1 | Loss of function | Sotos syndrome | Tall stature | AD |
| EZH2 | Uncertain | Weaver syndrome | Tall stature | AD | |
| DNMT3A | Loss of function | DNMT3A overgrowth syndrome | Tall stature | AD | |
| Extrinsic to growth plate | |||||
| Endocrine | |||||
| GH signaling | GH1 | Loss of function | Isolated GH deficiency | Short stature | AR, AD |
| GHRHR | Loss of function | Isolated GH deficiency | Short stature | AR | |
| POU1F1 PROP1 LHX3 others |
Loss of function | Combined pituitary hormone deficiency | Short stature | AR | |
| GHR | Loss of function | GH insensitivity syndrome | Short stature | AR | |
| STAT5B | Loss of function | Growth hormone insensitivity with immune dysfunction | Short stature | AR | |
| IGF-1 signaling | IGF1 | Loss of function | IGF-I deficiency | Short stature | AD, AR |
| IGF1R | Loss of function | IGF resistance | Short stature | AD, AR | |
| ALS | Loss of function | ALS deficiency | Short stature | AR | |
| Thyroid hormone signaling | TSHR PAX8 NKX2-1 FOXE1 NKX2-5 |
Loss of function | Thyroid dysgenesis | Short stature | AD, AR |
| SLC5A5 TP DUOX2, SLC26A4 TG IYD/DEHAL1 |
Loss of function | Thyroid dyshormonogenesis | Short stature | AR | |
| THRB | Loss of function | Thyroid hormone resistance | Short stature | AD, AR | |
| THRA | Loss of function | Thyroid hormone resistance | Short stature | AD | |
| Glucocorticoid signaling | PRKAR1A | Loss of function | Carney complex (including glucocorticoid excess) | Short stature | AD |
| MC2R | Loss of function | Familial glucocorticoid deficiency | Tall stature | AR | |
| Estrogen signaling | ESR1 (estrogen receptor) | Loss of function | Estrogen resistance | Tall stature | AR |
| CYP19A1 (aromatase) | Loss of function | Estrogen deficiency | Tall stature | AR | |
Notes: This list provides examples of single gene defects affecting linear growth but is not exhaustive. Abbreviations: ISS, idiopathic short stature; AD, autosomal dominant; AR, autosomal recessive.
Mutations in these genes can be considered semidominant in that heterozygous mutations produce a mild phenotype while homozygous mutations a more severe phenotype.
Skeletal dysplasias, isolated short stature, stature within the normal range, and tall stature represent a spectrum at the molecular level
Previously, skeletal dysplasias and idiopathic short stature were considered to be largely distinct entities. However, recent findings indicate that defects in the same genes, such as SHOX, NPR2, ACAN, and FGFR3 can present clinically either as a skeletal dysplasia or as idiopathic short stature. The more severe phenotype tends to occur when the gene involved is critical for growth plate function, the mutation severely alters protein function, and/or the mutation occurs in the homozygous state, whereas the milder phenotype, short stature with normal bone morphology, tends to occur when the gene involved is less critical for growth plate function, the mutation only partially disrupts protein function, and/or when the mutation occurs in the heterozygous state. (Figure 3). Furthermore genome-wide association studies suggest that stature in the lower part of the normal range can also be considered part of this spectrum since it appears to be due in part to polymorphisms in the same genes in which mutations cause skeletal dysplasia, such as COL10A1 (responsible for metaphyseal chondrodysplasia, Schmid type), ACAN (spondyloepimetaphyseal dysplasia, aggrecan type), and HOXD13 (Brachydactyly, type E).123 Thus polymorphisms, which have a mild effect on protein function or expression, tend to cause stature in the lower part of the normal range, mildly deleterious and/or heterozygous mutations tend to present as idiopathic short stature or a mild skeletal dysplasia, and strongly deleterious and/or homozygous mutations tend to cause a more severe skeletal dysplasia (Figure 3).
Figure 3.
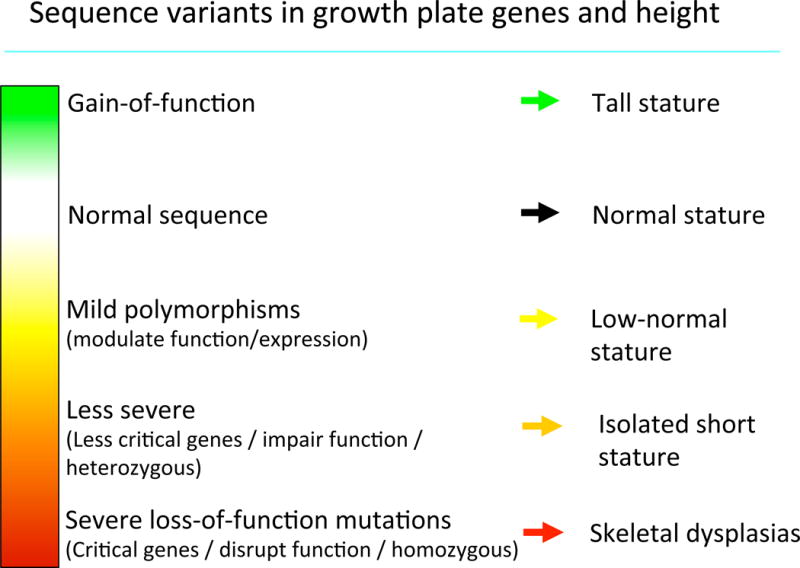
Diagram depicting the phenotypic spectrum that can be caused by sequence variants in genes that regulate growth plate chondrogenesis. The spectrum shown here applies to genes that promote longitudinal bone growth, such as NPR2. For genes that inhibit longitudinal bone growth, such as FGFR3, the spectrum is reversed in that gain-of-function mutations cause short stature while loss-of-function causes tall stature.
In some cases, tall stature can involve the same genes as those for short stature, but in tall stature, the mutation causing tall stature has the opposite functional effect on the gene product (Figure 3). For example, as discussed above, homozygous inactivating mutation in NPR2 cause a skeletal dysplasia with severe short stature, heterozygous inactivating mutations present as a milder skeletal dysplasia or idiopathic short stature, and activating mutations in NPR2 cause tall stature.78,79 Another example mentioned previously is FGFR3, in which activating mutations cause proportionate short stature, hypochondroplasia, achondroplasia, and thanatophoric dysplasia, whereas inactivating mutations have been reported to cause tall stature.61,62 Similarly, loss of function mutations or decreased copy number of SHOX cause short stature whereas increased copy number, including that of Klinefelter syndrome, is associated with tall stature.
The temporal and spatial distribution of gene expression and function affect the phenotype
Some genetic defects, such as activating mutations in FGFR3 causing achondroplasia, present with short stature at birth, whereas other genetic defects, such as heterozygous inactivating mutations in SHOX that cause Leri-Weill dyschondrosteosis, often result in a normal birth length with short stature developing at a later age. In some cases, this difference may be due to the severity of the growth plate abnormality. For example, severe activating mutations in FGFR3 associated with thanaphoric dysplasia and achondroplasia cause growth failure of prenatal onset,126,127 whereas a milder mutation causing hypochondroplasia often produces a birth length within the normal range with short stature developing later. However, in other cases, the temporal onset, pre- vs. postnatal, may depend on whether the gene is important in the fetal growth plate, which is morphologically and functionally somewhat different from the growth plate in childhood.
As described above, some defects in genes required for growth plate function produce proportionate short stature whereas others produce disproportionate short stature. Presumably, disproportionate short stature indicates that the gene product involved has a more critical role in some growth plates than in others. Many genetic defects tend to affect growth plates in the long bones more than growth plates in the vertebrae, causing a disproportionate short stature with a greater reduction in leg and arm lengths than in trunk length. We speculate that this common pattern may reflect the fact that the growth plates in the long bones function at a much greater pace than each of the individual growth plates in vertebrae and thus may be more susceptible to dysregulation. On the other hand, some disorders, such as brachyolmia, tend to affect the spine more than the long bones, causing disproportionate short stature with a short trunk. Brachyolmia is genetically heterogeneous, including mutations in PAPSS2 which encodes a sulfotransferase, required for sulfation of a variety of molecules, including cartilage glycosaminoglycans and DHEA.128
Mutations in some genes impair development and/or function not only of the growth plate but also non-skeletal structures, causing associated congenital anomalies, that is, syndromic short stature. For example, Noonan syndrome, which is caused by mutations in genes involved in the Ras/MAPK pathway such as PTPN11, often cause short stature associated with distinctive facies, cardiac and renal anomalies, developmental delay, and/or coagulation defects.129 However, mutations in PTPN11 have also been reported in patients who presented with short stature and were not recognized to have Noonan syndrome prior to sequencing.130
Growth hormone therapy for short stature
Although most short stature is caused by defects outside the GH-IGF-I system, treatment with recombinant GH is still somewhat effective in accelerating linear growth in many children with short stature. For example, GH treatment increases the linear growth rate of children with SHOX deficiency,131 Noonan syndrome,132 and idiopathic short stature.133,134 Indeed, endogenous growth hormone excess due to a pituitary adenoma can cause remarkable tall stature in otherwise normal children. These findings indicate that the stimulatory effect of GH on growth plate chondrocyte proliferation and hypertrophy, which is partly mediated by increased IGF-I,135–137 can, in many cases, non-specifically accelerate linear growth and thereby partially compensate for unrelated molecular defects affecting the growth plate.
Conclusions
For decades, the dominant conceptual framework for understanding short and tall stature was centered on the GH-IGF-I axis. However, recent findings in basic molecular and cellular biology and in clinical genetics have uncovered a vast array of other regulatory systems that control skeletal growth and an accompanying vast array of genetic defects outside the GH-IGF-I axis that can cause disorders of linear growth. As a result, the traditional view of short or tall stature that is centered on the GH-IGF-I axis is now far too narrow to encompass the ever-growing catalogue of defects that cause abnormal linear growth. A much broader conceptual framework can be based on the simple concept that linear growth disorders necessarily are due to dysfunction of the skeletal growth plate, the structure responsible for bone elongation and therefore overall body size. Consequently, short stature can more generally be conceptualized as a primary or secondary disorder of the growth plate chondrocytes. A related concept that has emerged is that sequence variants in genes that affect growth plate function can produce a phenotypic spectrum that ranges from a severe skeletal dysplasia to disproportionate or proportionate short stature, to normal variation in height, to tall stature. It is likely that high-throughput sequencing approaches will continue to expand the list of genetic defects that can cause growth plate dysfunction and disorders of linear growth. The clinical application of these broad sequencing approaches will allow the physician to identify the etiology of growth failure from among the myriad possibilities. We can therefore anticipate that the number of children who receive the unhelpful diagnosis of idiopathic short stature will continue to diminish. With these advances, we can look forward to treatment approaches that are tailored to the specific genetic cause of the disorder.
Footnotes
Declaration of interests
FDL, and JMW have no interests to declare. JB is listed as a co-inventor on a patent application by the National Institutes of Health for targeted treatment of cartilage disorders. LS has received speakers’ honoraria and/or research support from Novo Nordisk, Pfizer, Ferring, and Merck Serono and has submitted a patent application for novel peptides to treat bone or cartilage disorders and other diseases. AD has been a faculty speaker at continuing medical education symposia sponsored by Novo Nordisk, Sandoz, and Ipsen. MP has received research support from Novo Nordisk, Pfizer, Teva, personal fees from Novo Nordisk, and is a director of NG Solutions Ltd. ON has received an ESPE research fellowship sponsored by Novo Nordisk and speaker’s honorarium from Lilly.
References
- 1.Daughaday WH. Growth hormone axis overview–somatomedin hypothesis. Pediatr Nephrol. 2000;14(7):537–40. doi: 10.1007/s004670000334. [DOI] [PubMed] [Google Scholar]
- 2.Furlanetto RW, Underwood LE, Van Wyk JJ, D’Ercole AJ. Estimation of somatomedin-C levels in normals and patients with pituitary disease by radioimmunoassay. J Clin Invest. 1977;60(3):648–57. doi: 10.1172/JCI108816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roth J, Glick SM, Yalow RS, Berson SA. The Influence of Blood Glucose on the Plasma Concentration of Growth Hormone. Diabetes. 1964;13:355–61. doi: 10.2337/diab.13.4.355. [DOI] [PubMed] [Google Scholar]
- 4.Melmed S, Williams RH, Larsen PR, Kronenberg H. Williams textbook of endocrinology. 12th. Philadelphia: Elsevier/Saunders; 2011. [Google Scholar]
- 5.Rosenfeld RG. The molecular basis of idiopathic short stature. Growth Horm IGF Res. 2005;15(Suppl A):S3–5. doi: 10.1016/j.ghir.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 6.Wit JM, Clayton PE, Rogol AD, Savage MO, Saenger PH, Cohen P. Idiopathic short stature: definition, epidemiology, and diagnostic evaluation. Growth hormone & IGF research: official journal of the Growth Hormone Research Society and the International IGF Research Society. 2008;18(2):89–110. doi: 10.1016/j.ghir.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 7.Codner E, Mericq MV, Maheshwari HG, et al. Relationship between serum growth hormone binding protein levels and height in young men. Journal of pediatric endocrinology & metabolism: JPEM. 2000;13(7):887–92. doi: 10.1515/jpem.2000.13.7.887. [DOI] [PubMed] [Google Scholar]
- 8.Gill MS, Thalange NK, Foster PJ, et al. Regular fluctuations in growth hormone (GH) release determine normal human growth. Growth hormone & IGF research: official journal of the Growth Hormone Research Society and the International IGF Research Society. 1999;9(2):114–22. doi: 10.1054/ghir.1999.0095. [DOI] [PubMed] [Google Scholar]
- 9.Sisley S, Trujillo MV, Khoury J, Backeljauw P. Low incidence of pathology detection and high cost of screening in the evaluation of asymptomatic short children. The Journal of pediatrics. 2013;163(4):1045–51. doi: 10.1016/j.jpeds.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dauber A, Rosenfeld RG, Hirschhorn JN. Genetic Evaluation of Short Stature. The Journal of clinical endocrinology and metabolism. 2014:jc20141506. doi: 10.1210/jc.2014-1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stanley TL, Levitsky LL, Grinspoon SK, Misra M. Effect of body mass index on peak growth hormone response to provocative testing in children with short stature. The Journal of clinical endocrinology and metabolism. 2009;94(12):4875–81. doi: 10.1210/jc.2009-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marin G, Domene HM, Barnes KM, Blackwell BJ, Cassorla FG, Cutler GB., Jr The effects of estrogen priming and puberty on the growth hormone response to standardized treadmill exercise and arginine-insulin in normal girls and boys. The Journal of clinical endocrinology and metabolism. 1994;79(2):537–41. doi: 10.1210/jcem.79.2.8045974. [DOI] [PubMed] [Google Scholar]
- 13.Rosenbloom AL. Idiopathic short stature: conundrums of definition and treatment. International journal of pediatric endocrinology. 2009;2009:470378. doi: 10.1155/2009/470378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wudy SA, Hagemann S, Dempfle A, et al. Children with idiopathic short stature are poor eaters and have decreased body mass index. Pediatrics. 2005;116(1):e52–7. doi: 10.1542/peds.2004-1684. [DOI] [PubMed] [Google Scholar]
- 15.Rosenbloom AL. Is there a role for recombinant insulin-like growth factor-I in the treatment of idiopathic short stature? Lancet. 2006;368(9535):612–6. doi: 10.1016/S0140-6736(06)69205-2. [DOI] [PubMed] [Google Scholar]
- 16.Olney RC, Bukulmez H, Bartels CF, et al. Heterozygous mutations in natriuretic peptide receptor-B (NPR2) are associated with short stature. J Clin Endocrinol Metab. 2006;91(4):1229–32. doi: 10.1210/jc.2005-1949. [DOI] [PubMed] [Google Scholar]
- 17.Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423(6937):332–6. doi: 10.1038/nature01657. [DOI] [PubMed] [Google Scholar]
- 18.Nilsson O, Marino R, De Luca F, Phillip M, Baron J. Endocrine regulation of the growth plate. Horm Res. 2005;64(4):157–65. doi: 10.1159/000088791. [DOI] [PubMed] [Google Scholar]
- 19.Lango Allen H, Estrada K, Lettre G, et al. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature. 2010;467(7317):832–8. doi: 10.1038/nature09410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mushtaq T, Bijman P, Ahmed SF, Farquharson C. Insulin-like growth factor-I augments chondrocyte hypertrophy and reverses glucocorticoid-mediated growth retardation in fetal mice metatarsal cultures. Endocrinology. 2004;145(5):2478–86. doi: 10.1210/en.2003-1435. [DOI] [PubMed] [Google Scholar]
- 21.Baron J, Huang Z, Oerter KE, Bacher JD, Cutler GB., Jr Dexamethasone acts locally to inhibit longitudinal bone growth in rabbits. Am J Physiol. 1992;263(3 Pt 1):E489–92. doi: 10.1152/ajpendo.1992.263.3.E489. [DOI] [PubMed] [Google Scholar]
- 22.Rivkees SA, Danon M, Herrin J. Prednisone dose limitation of growth hormone treatment of steroid-induced growth failure. The Journal of pediatrics. 1994;125(2):322–5. doi: 10.1016/s0022-3476(94)70219-5. [DOI] [PubMed] [Google Scholar]
- 23.Wang L, Shao YY, Ballock RT. Thyroid hormone interacts with the Wnt/beta-catenin signaling pathway in the terminal differentiation of growth plate chondrocytes. J Bone Miner Res. 2007;22(12):1988–95. doi: 10.1359/jbmr.070806. [DOI] [PubMed] [Google Scholar]
- 24.Barnard JC, Williams AJ, Rabier B, et al. Thyroid hormones regulate fibroblast growth factor receptor signaling during chondrogenesis. Endocrinology. 2005;146(12):5568–80. doi: 10.1210/en.2005-0762. [DOI] [PubMed] [Google Scholar]
- 25.Raz P, Nasatzky E, Boyan BD, Ornoy A, Schwartz Z. Sexual dimorphism of growth plate prehypertrophic and hypertrophic chondrocytes in response to testosterone requires metabolism to dihydrotestosterone (DHT) by steroid 5-alpha reductase type 1. Journal of cellular biochemistry. 2005;95(1):108–19. doi: 10.1002/jcb.20298. [DOI] [PubMed] [Google Scholar]
- 26.Ren SG, Malozowski S, Sanchez P, Sweet DE, Loriaux DL, Cassorla F. Direct administration of testosterone increases rat tibial epiphyseal growth plate width. Acta endocrinologica. 1989;121(3):401–5. doi: 10.1530/acta.0.1210401. [DOI] [PubMed] [Google Scholar]
- 27.Borjesson AE, Lagerquist MK, Liu C, et al. The role of estrogen receptor alpha in growth plate cartilage for longitudinal bone growth. J Bone Miner Res. 2010;25(12):2690–700. doi: 10.1002/jbmr.156. [DOI] [PubMed] [Google Scholar]
- 28.Chagin AS, Chrysis D, Takigawa M, Ritzen EM, Savendahl L. Locally produced estrogen promotes fetal rat metatarsal bone growth; an effect mediated through increased chondrocyte proliferation and decreased apoptosis. J Endocrinol. 2006;188(2):193–203. doi: 10.1677/joe.1.06364. [DOI] [PubMed] [Google Scholar]
- 29.Mazziotti G, Giustina A. Glucocorticoids and the regulation of growth hormone secretion. Nature reviews Endocrinology. 2013;9(5):265–76. doi: 10.1038/nrendo.2013.5. [DOI] [PubMed] [Google Scholar]
- 30.Benker G, Raida M, Olbricht T, Wagner R, Reinhardt W, Reinwein D. TSH secretion in Cushing’s syndrome: relation to glucocorticoid excess, diabetes, goitre, and the ‘sick euthyroid syndrome’. Clinical endocrinology. 1990;33(6):777–86. doi: 10.1111/j.1365-2265.1990.tb03915.x. [DOI] [PubMed] [Google Scholar]
- 31.Nilsson O, Weise M, Landman EB, Meyers JL, Barnes KM, Baron J. Evidence that estrogen hastens epiphyseal fusion and cessation of longitudinal bone growth by irreversibly depleting the number of resting zone progenitor cells in female rabbits. Endocrinology. 2014;155(8):2892–9. doi: 10.1210/en.2013-2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weise M, De-Levi S, Barnes KM, Gafni RI, Abad V, Baron J. Effects of estrogen on growth plate senescence and epiphyseal fusion. Proc Natl Acad Sci U S A. 2001;98(12):6871–6. doi: 10.1073/pnas.121180498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Quaynor SD, Stradtman EW, Jr, Kim HG, et al. Delayed puberty and estrogen resistance in a woman with estrogen receptor alpha variant. The New England journal of medicine. 2013;369(2):164–71. doi: 10.1056/NEJMoa1303611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith EP, Boyd J, Frank GR, et al. Estrogen resistance caused by a mutation in the estrogen-receptor gene in a man. The New England journal of medicine. 1994;331(16):1056–61. doi: 10.1056/NEJM199410203311604. [DOI] [PubMed] [Google Scholar]
- 35.Morishima A, Grumbach MM, Simpson ER, Fisher C, Qin K. Aromatase deficiency in male and female siblings caused by a novel mutation and the physiological role of estrogens. The Journal of clinical endocrinology and metabolism. 1995;80(12):3689–98. doi: 10.1210/jcem.80.12.8530621. [DOI] [PubMed] [Google Scholar]
- 36.Dunkel L. Update on the role of aromatase inhibitors in growth disorders. Horm Res. 2009;71(Suppl 1):57–63. doi: 10.1159/000178040. [DOI] [PubMed] [Google Scholar]
- 37.Wit JM, Hero M, Nunez SB. Aromatase inhibitors in pediatrics. Nature reviews Endocrinology. 2012;8(3):135–47. doi: 10.1038/nrendo.2011.161. [DOI] [PubMed] [Google Scholar]
- 38.van der Eerden BC, Lowik CW, Wit JM, Karperien M. Expression of estrogen receptors and enzymes involved in sex steroid metabolism in the rat tibia during sexual maturation. J Endocrinol. 2004;180(3):457–67. doi: 10.1677/joe.0.1800457. [DOI] [PubMed] [Google Scholar]
- 39.Fernandez-Vojvodich P, Palmblad K, Karimian E, Andersson U, Savendahl L. Pro-inflammatory cytokines produced by growth plate chondrocytes may act locally to modulate longitudinal bone growth. Hormone research in paediatrics. 2012;77(3):180–7. doi: 10.1159/000337569. [DOI] [PubMed] [Google Scholar]
- 40.Savendahl L. Endocrinology The Effect of Acute and Chronic Stress on Growth. Sci Signal. 2012;5(247) doi: 10.1126/scisignal.2003484. [DOI] [PubMed] [Google Scholar]
- 41.Sederquist B, Fernandez-Vojvodich P, Zaman F, Savendahl L. Impact of inflammatory cytokines on longitudinal bone growth. Journal of molecular endocrinology. 2014 doi: 10.1530/JME-14-0006. [DOI] [PubMed] [Google Scholar]
- 42.MacRae VE, Farquharson C, Ahmed SF. The restricted potential for recovery of growth plate chondrogenesis and longitudinal bone growth following exposure to pro-inflammatory cytokines. J Endocrinol. 2006;189(2):319–28. doi: 10.1677/joe.1.06609. [DOI] [PubMed] [Google Scholar]
- 43.Martensson K, Chrysis D, Savendahl L. Interleukin-1beta and TNF-alpha act in synergy to inhibit longitudinal growth in fetal rat metatarsal bones. J Bone Miner Res. 2004;19(11):1805–12. doi: 10.1359/JBMR.040805. [DOI] [PubMed] [Google Scholar]
- 44.Phillip M, Moran O, Lazar L. Growth without growth hormone. Journal of pediatric endocrinology & metabolism: JPEM. 2002;15(Suppl 5):1267–72. [PubMed] [Google Scholar]
- 45.Couto-Silva AC, Trivin C, Esperou H, et al. Final height and gonad function after total body irradiation during childhood. Bone Marrow Transplant. 2006;38(6):427–32. doi: 10.1038/sj.bmt.1705455. [DOI] [PubMed] [Google Scholar]
- 46.Stokes IA, Aronsson DD, Dimock AN, Cortright V, Beck S. Endochondral growth in growth plates of three species at two anatomical locations modulated by mechanical compression and tension. J Orthop Res. 2006;24(6):1327–34. doi: 10.1002/jor.20189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stokes IA, Mente PL, Iatridis JC, Farnum CE, Aronsson DD. Enlargement of growth plate chondrocytes modulated by sustained mechanical loading. J Bone Joint Surg Am. 2002;84-A(10):1842–8. doi: 10.2106/00004623-200210000-00016. [DOI] [PubMed] [Google Scholar]
- 48.Lykissas MG, Jain VV, Manickam V, Nathan S, Eismann EA, McCarthy JJ. Guided growth for the treatment of limb length discrepancy: a comparative study of the three most commonly used surgical techniques. J Pediatr Orthop B. 2013;22(4):311–7. doi: 10.1097/BPB.0b013e32836132f0. [DOI] [PubMed] [Google Scholar]
- 49.Caine D, Howe W, Ross W, Bergman G. Does repetitive physical loading inhibit radial growth in female gymnasts? Clin J Sport Med. 1997;7(4):302–8. doi: 10.1097/00042752-199710000-00007. [DOI] [PubMed] [Google Scholar]
- 50.Hung IH, Yu K, Lavine KJ, Ornitz DM. FGF9 regulates early hypertrophic chondrocyte differentiation and skeletal vascularization in the developing stylopod. Dev Biol. 2007;307(2):300–13. doi: 10.1016/j.ydbio.2007.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lazarus JE, Hegde A, Andrade AC, Nilsson O, Baron J. Fibroblast growth factor expression in the postnatal growth plate. Bone. 2007;40(3):577–86. doi: 10.1016/j.bone.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 52.Liu Z, Lavine KJ, Hung IH, Ornitz DM. FGF18 is required for early chondrocyte proliferation, hypertrophy and vascular invasion of the growth plate. Dev Biol. 2007;302(1):80–91. doi: 10.1016/j.ydbio.2006.08.071. [DOI] [PubMed] [Google Scholar]
- 53.Mancilla EE, De Luca F, Uyeda JA, Czerwiec FS, Baron J. Effects of fibroblast growth factor-2 on longitudinal bone growth. Endocrinology. 1998;139(6):2900–4. doi: 10.1210/endo.139.6.6032. [DOI] [PubMed] [Google Scholar]
- 54.De Luca F, Barnes KM, Uyeda JA, et al. Regulation of growth plate chondrogenesis by bone morphogenetic protein-2. Endocrinology. 2001;142(1):430–6. doi: 10.1210/endo.142.1.7901. [DOI] [PubMed] [Google Scholar]
- 55.Nilsson O, Parker EA, Hegde A, Chau M, Barnes KM, Baron J. Gradients in bone morphogenetic protein-related gene expression across the growth plate. J Endocrinol. 2007;193(1):75–84. doi: 10.1677/joe.1.07099. [DOI] [PubMed] [Google Scholar]
- 56.Pogue R, Lyons K. BMP signaling in the cartilage growth plate. Curr Top Dev Biol. 2006;76:1–48. doi: 10.1016/S0070-2153(06)76001-X. [DOI] [PubMed] [Google Scholar]
- 57.Andrade AC, Nilsson O, Barnes KM, Baron J. Wnt gene expression in the post-natal growth plate: regulation with chondrocyte differentiation. Bone. 2007;40(5):1361–9. doi: 10.1016/j.bone.2007.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kuss P, Kraft K, Stumm J, et al. Regulation of cell polarity in the cartilage growth plate and perichondrium of metacarpal elements by HOXD13 and WNT5A. Dev Biol. 2014;385(1):83–93. doi: 10.1016/j.ydbio.2013.10.013. [DOI] [PubMed] [Google Scholar]
- 59.Lui JC, Nilsson O, Baron J. Recent insights into the regulation of the growth plate. Journal of molecular endocrinology. 2014 doi: 10.1530/JME-14-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kant SG, Cervenkova I, Balek L, et al. A novel variant of FGFR3 causes proportionate short stature. Eur J Endocrinol. 2015;172:763–70. doi: 10.1530/EJE-14-0945. [DOI] [PubMed] [Google Scholar]
- 61.Toydemir RM, Brassington AE, Bayrak-Toydemir P, et al. A novel mutation in FGFR3 causes camptodactyly, tall stature, and hearing loss (CATSHL) syndrome. American journal of human genetics. 2006;79(5):935–41. doi: 10.1086/508433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Makrythanasis P, Temtamy S, Aglan MS, Otaify GA, Hamamy H, Antonarakis SE. A Novel Homozygous Mutation in FGFR3 Causes Tall Stature, Severe Lateral Tibial Deviation, Scoliosis, Hearing Impairment, Camptodactyly, and Arachnodactyly. Human mutation. 2014;35(8):959–63. doi: 10.1002/humu.22597. [DOI] [PubMed] [Google Scholar]
- 63.Baron J, Klein KO, Yanovski JA, et al. Induction of growth plate cartilage ossification by basic fibroblast growth factor. Endocrinology. 1994;135(6):2790–3. doi: 10.1210/endo.135.6.7988472. [DOI] [PubMed] [Google Scholar]
- 64.Chen L, Adar R, Yang X, et al. Gly369Cys mutation in mouse FGFR3 causes achondroplasia by affecting both chondrogenesis and osteogenesis. J Clin Invest. 1999;104(11):1517–25. doi: 10.1172/JCI6690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Minina E, Kreschel C, Naski MC, Ornitz DM, Vortkamp A. Interaction of FGF, Ihh/Pthlh, and BMP signaling integrates chondrocyte proliferation and hypertrophic differentiation. Developmental cell. 2002;3(3):439–49. doi: 10.1016/s1534-5807(02)00261-7. [DOI] [PubMed] [Google Scholar]
- 66.Foldynova-Trantirkova S, Wilcox WR, Krejci P. Sixteen years and counting: the current understanding of fibroblast growth factor receptor 3 (FGFR3) signaling in skeletal dysplasias. Human mutation. 2012;33(1):29–41. doi: 10.1002/humu.21636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xie Y, Zhou S, Chen H, Du X, Chen L. Recent research on the growth plate: Advances in fibroblast growth factor signaling in growth plate development and disorders. J Mol Endocrinol. 2014;53(1):T11–34. doi: 10.1530/JME-14-0012. [DOI] [PubMed] [Google Scholar]
- 68.Klopocki E, Hennig BP, Dathe K, et al. Deletion and point mutations of PTHLH cause brachydactyly type E. Am J Hum Genet. 2010;86(3):434–9. doi: 10.1016/j.ajhg.2010.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chusho H, Tamura N, Ogawa Y, et al. Dwarfism and early death in mice lacking C-type natriuretic peptide. Proc Natl Acad Sci U S A. 2001;98(7):4016–21. doi: 10.1073/pnas.071389098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mericq V, Uyeda JA, Barnes KM, De Luca F, Baron J. Regulation of fetal rat bone growth by C-type natriuretic peptide and cGMP. Pediatr Res. 2000;47(2):189–93. doi: 10.1203/00006450-200002000-00007. [DOI] [PubMed] [Google Scholar]
- 71.Pejchalova K, Krejci P, Wilcox WR. C-natriuretic peptide: an important regulator of cartilage. Molecular genetics and metabolism. 2007;92(3):210–5. doi: 10.1016/j.ymgme.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 72.Bartels CF, Bukulmez H, Padayatti P, et al. Mutations in the transmembrane natriuretic peptide receptor NPR-B impair skeletal growth and cause acromesomelic dysplasia, type Maroteaux. American journal of human genetics. 2004;75(1):27–34. doi: 10.1086/422013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Amano N, Mukai T, Ito Y, et al. Identification and functional characterization of two novel NPR2 mutations in Japanese patients with short stature. The Journal of clinical endocrinology and metabolism. 2014;99(4):E713–8. doi: 10.1210/jc.2013-3525. [DOI] [PubMed] [Google Scholar]
- 74.Vasques GA, Amano N, Docko AJ, et al. Heterozygous mutations in natriuretic peptide receptor-B (NPR2) gene as a cause of short stature in patients initially classified as idiopathic short stature. The Journal of clinical endocrinology and metabolism. 2013;98(10):E1636–44. doi: 10.1210/jc.2013-2142. [DOI] [PubMed] [Google Scholar]
- 75.Wang SR, Jacobsen CM, Carmichael H, et al. Heterozygous Mutations in Natriuretic Peptide Receptor-B (NPR2) Gene as a Cause of Short Stature. Human mutation. 2015;36(4):474–81. doi: 10.1002/humu.22773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bocciardi R, Giorda R, Buttgereit J, et al. Overexpression of the C-type natriuretic peptide (CNP) is associated with overgrowth and bone anomalies in an individual with balanced t(2;7) translocation. Human mutation. 2007;28(7):724–31. doi: 10.1002/humu.20511. [DOI] [PubMed] [Google Scholar]
- 77.Moncla A, Missirian C, Cacciagli P, et al. A cluster of translocation breakpoints in 2q37 is associated with overexpression of NPPC in patients with a similar overgrowth phenotype. Human mutation. 2007;28(12):1183–8. doi: 10.1002/humu.20611. [DOI] [PubMed] [Google Scholar]
- 78.Hannema SE, van Duyvenvoorde HA, Premsler T, et al. An activating mutation in the kinase homology domain of the natriuretic peptide receptor-2 causes extremely tall stature without skeletal deformities. J Clin Endocrinol Metab. 2013;98(12):E1988–98. doi: 10.1210/jc.2013-2358. [DOI] [PubMed] [Google Scholar]
- 79.Miura K, Kim OH, Lee HR, et al. Overgrowth syndrome associated with a gain-of-function mutation of the natriuretic peptide receptor 2 (NPR2) gene. American journal of medical genetics Part A. 2014;164A(1):156–63. doi: 10.1002/ajmg.a.36218. [DOI] [PubMed] [Google Scholar]
- 80.Teixeira CC, Agoston H, Beier F. Nitric oxide, C-type natriuretic peptide and cGMP as regulators of endochondral ossification. Dev Biol. 2008;319(2):171–8. doi: 10.1016/j.ydbio.2008.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Miyazawa T, Ogawa Y, Chusho H, et al. Cyclic GMP-dependent protein kinase II plays a critical role in C-type natriuretic peptide-mediated endochondral ossification. Endocrinology. 2002;143(9):3604–10. doi: 10.1210/en.2002-220307. [DOI] [PubMed] [Google Scholar]
- 82.Krejci P, Masri B, Fontaine V, et al. Interaction of fibroblast growth factor and C-natriuretic peptide signaling in regulation of chondrocyte proliferation and extracellular matrix homeostasis. J Cell Sci. 2005;118(Pt 21):5089–100. doi: 10.1242/jcs.02618. [DOI] [PubMed] [Google Scholar]
- 83.Lorget F, Kaci N, Peng J, et al. Evaluation of the therapeutic potential of a CNP analog in a Fgfr3 mouse model recapitulating achondroplasia. Am J Hum Genet. 2012;91(6):1108–14. doi: 10.1016/j.ajhg.2012.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Neptune ER, Frischmeyer PA, Arking DE, et al. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nature genetics. 2003;33(3):407–11. doi: 10.1038/ng1116. [DOI] [PubMed] [Google Scholar]
- 85.Dietz HC, Pyeritz RE. Mutations in the human gene for fibrillin-1 (FBN1) in the Marfan syndrome and related disorders. Human molecular genetics. 1995;4:1799–809. doi: 10.1093/hmg/4.suppl_1.1799. Spec No. [DOI] [PubMed] [Google Scholar]
- 86.Brooke BS, Habashi JP, Judge DP, Patel N, Loeys B, Dietz HC., 3rd Angiotensin II blockade and aortic-root dilation in Marfan’s syndrome. The New England journal of medicine. 2008;358(26):2787–95. doi: 10.1056/NEJMoa0706585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Koziel L, Kunath M, Kelly OG, Vortkamp A. Ext1-dependent heparan sulfate regulates the range of Ihh signaling during endochondral ossification. Developmental cell. 2004;6(6):801–13. doi: 10.1016/j.devcel.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 88.Jochmann K, Bachvarova V, Vortkamp A. Reprint of: Heparan sulfate as a regulator of endochondral ossification and osteochondroma development. Matrix Biol. 2014;35:239–47. doi: 10.1016/j.matbio.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 89.Tompson SW, Merriman B, Funari VA, et al. A recessive skeletal dysplasia, SEMD aggrecan type, results from a missense mutation affecting the C-type lectin domain of aggrecan. American journal of human genetics. 2009;84(1):72–9. doi: 10.1016/j.ajhg.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gleghorn L, Ramesar R, Beighton P, Wallis G. A mutation in the variable repeat region of the aggrecan gene (AGC1) causes a form of spondyloepiphyseal dysplasia associated with severe, premature osteoarthritis. American journal of human genetics. 2005;77(3):484–90. doi: 10.1086/444401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Stattin EL, Wiklund F, Lindblom K, et al. A missense mutation in the aggrecan C-type lectin domain disrupts extracellular matrix interactions and causes dominant familial osteochondritis dissecans. American journal of human genetics. 2010;86(2):126–37. doi: 10.1016/j.ajhg.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nilsson O, Guo MH, Dunbar N, et al. Short stature, accelerated bone maturation, and early growth cessation due to heterozygous aggrecan mutations. The Journal of clinical endocrinology and metabolism. 2014;99:E1510–8. doi: 10.1210/jc.2014-1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lauing KL, Cortes M, Domowicz MS, Henry JG, Baria AT, Schwartz NB. Aggrecan is required for growth plate cytoarchitecture and differentiation. Dev Biol. 2014;396(2):224–36. doi: 10.1016/j.ydbio.2014.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Watanabe H, Yamada Y. Chondrodysplasia of gene knockout mice for aggrecan and link protein. Glycoconj J. 2002;19(4–5):269–73. doi: 10.1023/A:1025344332099. [DOI] [PubMed] [Google Scholar]
- 95.Xu T, Bianco P, Fisher LW, et al. Targeted disruption of the biglycan gene leads to an osteoporosis-like phenotype in mice. Nature genetics. 1998;20(1):78–82. doi: 10.1038/1746. [DOI] [PubMed] [Google Scholar]
- 96.Cain SA, McGovern A, Baldwin AK, Baldock C, Kielty CM. Fibrillin-1 mutations causing Weill-Marchesani syndrome and acromicric and geleophysic dysplasias disrupt heparan sulfate interactions. PLoS One. 2012;7(11):e48634. doi: 10.1371/journal.pone.0048634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Akiyama H, Lefebvre V. Unraveling the transcriptional regulatory machinery in chondrogenesis. Journal of bone and mineral metabolism. 2011;29(4):390–5. doi: 10.1007/s00774-011-0273-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Marchini A, Marttila T, Winter A, et al. The short stature homeodomain protein SHOX induces cellular growth arrest and apoptosis and is expressed in human growth plate chondrocytes. J Biol Chem. 2004;279(35):37103–14. doi: 10.1074/jbc.M307006200. [DOI] [PubMed] [Google Scholar]
- 99.Malaquias AC, Scalco RC, Fontenele EG, et al. The sitting height/height ratio for age in healthy and short individuals and its potential role in selecting short children for SHOX analysis. Hormone research in paediatrics. 2013;80(6):449–56. doi: 10.1159/000355411. [DOI] [PubMed] [Google Scholar]
- 100.Binder G. Short stature due to SHOX deficiency: genotype, phenotype, and therapy. Hormone research in paediatrics. 2011;75(2):81–9. doi: 10.1159/000324105. [DOI] [PubMed] [Google Scholar]
- 101.Ottesen AM, Aksglaede L, Garn I, et al. Increased number of sex chromosomes affects height in a nonlinear fashion: a study of 305 patients with sex chromosome aneuploidy. American journal of medical genetics Part A. 2010;152A(5):1206–12. doi: 10.1002/ajmg.a.33334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yasoda A, Komatsu Y, Chusho H, et al. Overexpression of CNP in chondrocytes rescues achondroplasia through a MAPK-dependent pathway. Nat Med. 2004;10(1):80–6. doi: 10.1038/nm971. [DOI] [PubMed] [Google Scholar]
- 103.Cseh B, Doma E, Baccarini M. “RAF” neighborhood: Protein-protein interaction in the Raf/Mek/Erk pathway. FEBS letters. 2014;588(15):2398–406. doi: 10.1016/j.febslet.2014.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lee BH, Kim JM, Jin HY, Kim GH, Choi JH, Yoo HW. Spectrum of mutations in Noonan syndrome and their correlation with phenotypes. The Journal of pediatrics. 2011;159(6):1029–35. doi: 10.1016/j.jpeds.2011.05.024. [DOI] [PubMed] [Google Scholar]
- 105.Stevenson DA, Yang FC. The musculoskeletal phenotype of the RASopathies. American journal of medical genetics Part C, Seminars in medical genetics. 2011;157C(2):90–103. doi: 10.1002/ajmg.c.30296. [DOI] [PubMed] [Google Scholar]
- 106.Visser R, Kant SG, Wit JM, Breuning MH. Overgrowth syndromes:from classical to new. Pediatric endocrinology reviews: PER. 2009;6(3):375–94. [PubMed] [Google Scholar]
- 107.Wu S, Fadoju D, Rezvani G, De Luca F. Stimulatory effects of insulin-like growth factor-I on growth plate chondrogenesis are mediated by nuclear factor-kappaB p65. J Biol Chem. 2008;283(49):34037–44. doi: 10.1074/jbc.M803754200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wu S, Walenkamp MJ, Lankester A, Bidlingmaier M, Wit JM, De Luca F. Growth hormone and insulin-like growth factor I insensitivity of fibroblasts isolated from a patient with an I{kappa}B{alpha} mutation. The Journal of clinical endocrinology and metabolism. 2010;95(3):1220–8. doi: 10.1210/jc.2009-1662. [DOI] [PubMed] [Google Scholar]
- 109.Klingseisen A, Jackson AP. Mechanisms and pathways of growth failure in primordial dwarfism. Genes & development. 2011;25(19):2011–24. doi: 10.1101/gad.169037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Huber C, Dias-Santagata D, Glaser A, et al. Identification of mutations in CUL7 in 3-M syndrome. Nature genetics. 2005;37(10):1119–24. doi: 10.1038/ng1628. [DOI] [PubMed] [Google Scholar]
- 111.Hanson D, Murray PG, Sud A, et al. The primordial growth disorder 3-M syndrome connects ubiquitination to the cytoskeletal adaptor OBSL1. American journal of human genetics. 2009;84(6):801–6. doi: 10.1016/j.ajhg.2009.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hanson D, Murray PG, O’Sullivan J, et al. Exome sequencing identifies CCDC8 mutations in 3-M syndrome, suggesting that CCDC8 contributes in a pathway with CUL7 and OBSL1 to control human growth. American journal of human genetics. 2011;89(1):148–53. doi: 10.1016/j.ajhg.2011.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yan J, Yan F, Li Z, et al. The 3M complex maintains microtubule and genome integrity. Molecular cell. 2014;54(5):791–804. doi: 10.1016/j.molcel.2014.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rauch A, Thiel CT, Schindler D, et al. Mutations in the pericentrin (PCNT) gene cause primordial dwarfism. Science. 2008;319(5864):816–9. doi: 10.1126/science.1151174. [DOI] [PubMed] [Google Scholar]
- 115.Bicknell LS, Bongers EM, Leitch A, et al. Mutations in the pre-replication complex cause Meier-Gorlin syndrome. Nature genetics. 2011;43(4):356–9. doi: 10.1038/ng.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Guernsey DL, Matsuoka M, Jiang H, et al. Mutations in origin recognition complex gene ORC4 cause Meier-Gorlin syndrome. Nature genetics. 2011;43(4):360–4. doi: 10.1038/ng.777. [DOI] [PubMed] [Google Scholar]
- 117.Tatton-Brown K, Seal S, Ruark E, et al. Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nat Genet. 2014;46(4):385–8. doi: 10.1038/ng.2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gibson WT, Hood RL, Zhan SH, et al. Mutations in EZH2 cause Weaver syndrome. American journal of human genetics. 2012;90(1):110–8. doi: 10.1016/j.ajhg.2011.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Canton AP, Costa SS, Rodrigues TC, et al. Genome-wide screening of copy number variants in children born small for gestational age reveals several candidate genes involved in growth pathways. Eur J Endocrinol. 2014;171(2):253–62. doi: 10.1530/EJE-14-0232. [DOI] [PubMed] [Google Scholar]
- 120.van Duyvenvoorde HA, Lui JC, Kant SG, et al. Copy number variants in patients with short stature. Eur J Hum Genet. 2014;22(5):602–9. doi: 10.1038/ejhg.2013.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zahnleiter D, Uebe S, Ekici AB, et al. Rare copy number variants are a common cause of short stature. PLoS genetics. 2013;9(3):e1003365. doi: 10.1371/journal.pgen.1003365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wood AR, Esko T, Yang J, et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nature genetics. 2014;46(11):1173–86. doi: 10.1038/ng.3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lui JC, Nilsson O, Chan Y, et al. Synthesizing genome-wide association studies and expression microarray reveals novel genes that act in the human growth plate to modulate height. Hum Mol Genet. 2012;21(23):5193–201. doi: 10.1093/hmg/dds347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Liu F, Hendriks AE, Ralf A, et al. Common DNA variants predict tall stature in Europeans. Human genetics. 2014;133(5):587–97. doi: 10.1007/s00439-013-1394-0. [DOI] [PubMed] [Google Scholar]
- 125.Wit JM, Ranke MB, Kelnar CJH. The ESPE classification of paediatric endocrine diagnoses – Foreword. Hormone Research. 2007;68(Suppl 2):1–120. [Google Scholar]
- 126.Chitty LS, Griffin DR, Meaney C, et al. New aids for the non-invasive prenatal diagnosis of achondroplasia: dysmorphic features, charts of fetal size and molecular confirmation using cell-free fetal DNA in maternal plasma. Ultrasound in obstetrics & gynecology: the official journal of the International Society of Ultrasound in Obstetrics and Gynecology. 2011;37(3):283–9. doi: 10.1002/uog.8893. [DOI] [PubMed] [Google Scholar]
- 127.Chitty LS, Khalil A, Barrett AN, Pajkrt E, Griffin DR, Cole TJ. Safe, accurate, prenatal diagnosis of thanatophoric dysplasia using ultrasound and free fetal DNA. Prenatal diagnosis. 2013;33(5):416–23. doi: 10.1002/pd.4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ahmad M, Faiyaz Ul Haque M, Ahmad W, et al. Distinct, autosomal recessive form of spondyloepimetaphyseal dysplasia segregating in an inbred Pakistani kindred. American journal of medical genetics. 1998;78(5):468–73. [PubMed] [Google Scholar]
- 129.Roberts AE, Allanson JE, Tartaglia M, Gelb BD. Noonan syndrome. Lancet. 2013;381(9863):333–42. doi: 10.1016/S0140-6736(12)61023-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wang SR, Carmichael H, Andrew SF, et al. Large-scale pooled next-generation sequencing of 1077 genes to identify genetic causes of short stature. The Journal of clinical endocrinology and metabolism. 2013;98(8):E1428–37. doi: 10.1210/jc.2013-1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Blum WF, Crowe BJ, Quigley CA, et al. Growth hormone is effective in treatment of short stature associated with short stature homeobox-containing gene deficiency: Two-year results of a randomized, controlled, multicenter trial. The Journal of clinical endocrinology and metabolism. 2007;92(1):219–28. doi: 10.1210/jc.2006-1409. [DOI] [PubMed] [Google Scholar]
- 132.Noordam C, Van der Burgt I, Sengers RC, Delemarre-van de Waal HA, Otten BJ. Growth hormone treatment in children with Noonan’s syndrome: four year results of a partly controlled trial. Acta Paediatr. 2001;90(8):889–94. [PubMed] [Google Scholar]
- 133.Albertsson-Wikland K, Aronson AS, Gustafsson J, et al. Dose-dependent effect of growth hormone on final height in children with short stature without growth hormone deficiency. The Journal of clinical endocrinology and metabolism. 2008;93(11):4342–50. doi: 10.1210/jc.2008-0707. [DOI] [PubMed] [Google Scholar]
- 134.Leschek EW, Rose SR, Yanovski JA, et al. Effect of growth hormone treatment on adult height in peripubertal children with idiopathic short stature: a randomized, double-blind, placebo-controlled trial. J Clin Endocrinol Metab. 2004;89(7):3140–8. doi: 10.1210/jc.2003-031457. [DOI] [PubMed] [Google Scholar]
- 135.Hunziker EB, Wagner J, Zapf J. Differential effects of insulin-like growth factor I and growth hormone on developmental stages of rat growth plate chondrocytes in vivo. J Clin Invest. 1994;93(3):1078–86. doi: 10.1172/JCI117058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Nilsson O, Mitchum RD, Jr, Schrier L, et al. Growth plate senescence is associated with loss of DNA methylation. J Endocrinol. 2005;186(1):241–9. doi: 10.1677/joe.1.06016. [DOI] [PubMed] [Google Scholar]
- 137.Wang J, Zhou J, Cheng CM, Kopchick JJ, Bondy CA. Evidence supporting dual, IGF-I-independent and IGF-I-dependent, roles for GH in promoting longitudinal bone growth. J Endocrinol. 2004;180(2):247–55. doi: 10.1677/joe.0.1800247. [DOI] [PubMed] [Google Scholar]