

In this review, Prioleau and MacAlpine summarize recent advances in our understanding of how primary sequence, chromatin environment, and nuclear architecture contribute to the dynamic selection and activation of replication origins across diverse cell types and developmental stages.
Keywords: chromatin, DNA replication, epigenetics, nuclear architecture, origins of DNA replication
Abstract
For more than three decades, investigators have sought to identify the precise locations where DNA replication initiates in mammalian genomes. The development of molecular and biochemical approaches to identify start sites of DNA replication (origins) based on the presence of defining and characteristic replication intermediates at specific loci led to the identification of only a handful of mammalian replication origins. The limited number of identified origins prevented a comprehensive and exhaustive search for conserved genomic features that were capable of specifying origins of DNA replication. More recently, the adaptation of origin-mapping assays to genome-wide approaches has led to the identification of tens of thousands of replication origins throughout mammalian genomes, providing an unprecedented opportunity to identify both genetic and epigenetic features that define and regulate their distribution and utilization. Here we summarize recent advances in our understanding of how primary sequence, chromatin environment, and nuclear architecture contribute to the dynamic selection and activation of replication origins across diverse cell types and developmental stages.
Every cell cycle, tens of thousands of start sites of DNA replication must be established and coordinated to ensure the accurate and complete duplication of >3 billion base pairs (bp) of DNA in the human genome. Despite the critical role of the DNA replication program in ensuring the inheritance of genetic information, the selection and activation of replication origins is remarkably dynamic and plastic. Developmental and cell type-specific constraints on the DNA replication program dictate the number and distribution of origins throughout the genome. Extremes include increased origin density during the rapid cell divisions of early embryogenesis (Blumenthal et al. 1974; Hyrien et al. 1995) and tissue-specific amplicons that arise from the activation of select origins outside of the canonical cell cycle (Spradling 1981; Claycomb and Orr-Weaver 2005; Hannibal and Baker 2016). Primary sequence, epigenetic modifications, trans-acting factors, chromatin organization, and nuclear architecture all contribute to the selection and activation of mammalian replication origins. Mutations in key replication initiation factors may lead to severe developmental consequences (Bicknell et al. 2011; Kuo et al. 2012) or genomic instability and, potentially, tumorigenesis (Shima et al. 2007).
The location and distribution of replication origins throughout the genome define replicons, which are large sequence domains copied by the bidirectional movement of the replication fork away from an origin. More than half a century ago, the initial characterization of replicons using radiolabeled DNA fibers provided a rough accounting of the number of origins required to duplicate mammalian genomes (Cairns 1966). Depending on cell type, observed replicon sizes ranged from 30 to 450 kb, suggesting considerable variability in the distribution and utilization of replication origins (Berezney et al. 2000). Despite our knowledge with pinpoint accuracy of origin locations in prokaryotic and fungal genomes, researchers are just beginning to understand how cell type-to-cell type variability governs the density, distribution, and utilization of mammalian origins.
The replicon model proposed by Jacob et al. (1963) postulated that each replicon would be defined by a trans-acting initiator and a cis-acting replicator sequence representing the origin. This model elegantly accounted for the fact that not all pieces of genetic material were capable of autonomous replication but rather were propagated only when integrated into the context of a larger episome or chromosome that functioned as the replicon. Although the model was proposed to explain the replication of prokaryotic and phage genomes, Jacob et al. (1963) were remarkably clairvoyant when describing how their model may apply to more complex genomes:
“It is clear that chromosome reproduction in higher organisms is a very elaborate process requiring multiple control mechanisms which have to be coordinated. The analysis of these mechanisms as well as the exact role of the structural elements involved may turn out to be very difficult.”
Many of the basic tenets of the model have held up surprisingly well over time. Multiple conserved trans-acting factors function as the proposed initiator. The origin recognition complex (ORC) is a heterohexameric protein complex that binds DNA and is required to load Mcm2–7, the replicative helicase, onto the DNA (Bell and Kaguni 2013). In the G1 phase of the cell cycle, ORC and Cdc6 facilitate the cooperative loading of two Cdt1–Mcm2–7 complexes onto the DNA in a head-to-head orientation (Evrin et al. 2009; Remus et al. 2009). The loading of the Mcm2–7 helicase on the DNA completes the assembly of the prereplicative complex (pre-RC). Pre-RC assembly “licenses” the origin for potential activation in the subsequent S phase (Siddiqui et al. 2013). As cells progress into S phase, the activity of cyclin-dependent kinases (CDKs) and Dbf4-dependent kinases (DDKs) stimulates the recruitment of proteins, including Cdc45 and GINS, to the origin that are required to form an active Cdc45/Mcm2–7/GINS (CMG) helicase complex to initiate DNA unwinding, facilitate formation of the replisome, and prime DNA synthesis (Zegerman 2015). The activation of replication origins during S phase is in part regulated by limiting concentrations of key activating factors (Mantiero et al. 2011; Tanaka et al. 2011; Collart et al. 2013). Importantly, the separation of origin licensing (pre-RC assembly) and initiation into distinct cell cycle phases of G1 and S ensures that the genome is copied once and only once.
In contrast to the trans-activating initiator factors, the identification of conserved cis-acting replicator sequences that function as origins of DNA replication and define specific replicons has been hampered by the size and complexity of mammalian genomes. To date, only a handful of efficient origins that are activated across a large population of cells have been identified and characterized in depth by multiple groups using multiple approaches. Sequence analysis and dissection of these origins have failed, for the most part, to unveil a common and conserved cis-acting replicator element.
The recent and widespread adoption of genomic technologies has allowed for systematic and comprehensive genome-wide surveys to identify sites of DNA replication initiation (Cadoret et al. 2008; Sequeira-Mendes et al. 2009; Mesner et al. 2011; Petryk et al. 2016). Instead of cis-acting replicators being distributed across the genome at regular intervals in fixed locations, a much more complex and stochastic pattern of origin utilization has emerged. First, origins are not randomly distributed across the genome, as the density of mapped origins is highest in early-replicating chromosomal domains (Cadoret et al. 2008; Besnard et al. 2012). Second, there are many more potential origins established than there are activated initiation events during S phase; these excess potential origins serve as reserve origins during replicative stress (Ge et al. 2007; Ibarra et al. 2008). Third, there appears to be considerable cell-to-cell variability in the utilization of replication origins (Demczuk et al. 2012). Fourth, some primary sequence elements, including GC-rich sequences with the potential to form G quadruplexes, have been associated with origin function (Besnard et al. 2012; Cayrou et al. 2012); however, the low-complexity nature and broad distribution of these sequences suggest that they are not sufficient for origin activity. Finally, prior biochemical experiments (Vashee et al. 2003; Remus et al. 2004) and the recent crystal structure of ORC (Bleichert et al. 2015) revealed no obvious requirement or mechanisms for sequence specificity. Combined, these results indicate that the proposed replicator is most likely defined by a complex combination of sequence and chromatin-based features in higher eukaryotes. Here we focus on recent advances in the replication field to identify the distribution and usage of replication origins and the different mechanisms by which they are defined and regulated.
Identification of mammalian replication origins
Early work in mammalian cells used tritium labeling and fiber autoradiography to directly visualize the size distribution of the replicons resulting from bidirectional DNA replication (Edenberg and Huberman 1975). The density of the autoradiographic signal was proportional to the specific activity of the 3H thymidine; thus, replication fork movement away from the origin could be readily detected by pulse labeling with different specific activities of radioactive thymidine. A heterogeneous range of replicon sizes was observed from 30 to 450 kb per replicon, suggesting lower and upper bounds of ∼7000 and ∼100,000 for the number of replication origins in mammalian genomes (Berezney et al. 2000). The variance in the size distribution of replicons and thus the number of potential origins were thought to be due in part to cell type-specific determinants and foreshadowed the complexity and diversity of the mammalian DNA replication program.
The advent of molecular biology started a race to precisely identify the location and cis-acting features that define start sites of DNA replication in higher eukaryotes. Origins were identified by characteristic molecular intermediates of bidirectional DNA replication (Okazaki fragments, nascent strands, and bubbles) and plasmid-based genetic assays.
Genetic assays
In smaller eukaryotes like the budding yeast Saccharomyces cerevisiae, cis-acting replicator elements were first identified by their ability to ensure the long-term propagation and maintenance of episomal DNA. Functional screens identified hundreds of short sequence fragments, termed autonomously replicating sequence (ARS) elements, that were necessary for the faithful duplication and inheritance of the plasmid every cell cycle (Stinchcomb et al. 1979). Despite the success of ARS-based genetic screens in small eukaryotes (and prokaryotes), the assay has been a challenge to scale to higher eukaryotes, as the maintenance of a plasmid requires not only that it is replicated but that both copies are also segregated into each daughter cell following cell division. These challenges were overcome in part by using a modified version of the EBV replication origin that was functional for segregation, but not EBNA-mediated replication, to screen a large library of human DNA clones for replicator function (Krysan et al. 1989). Sequence length—not composition—was most strongly correlated with plasmid maintenance (Heinzel et al. 1991). Although these experiments argue that there is little sequence specificity required for replication initiation on a plasmid, it was also clear that sequence inserts derived from bacterial sources were less efficient at promoting plasmid replication. Thus, while no clear short stretches of primary sequences emerged, there did indeed appear to be encoded information specific to mammalian DNA that was important for efficient initiation of DNA replication in the context of a plasmid. Finally, given the complex nuclear organization of vertebrate genomes (Lieberman-Aiden et al. 2009; Dixon et al. 2012), it remains to be determined whether the episome/plasmid-based approaches in higher eukaryotes will adequately reflect and/or capture the higher-order chromosome organization and structure that are likely to be involved in the specification of initiation sites along chromosomes.
Identification of origins by characteristic molecular intermediates
Initiation of DNA replication and the semiconservative nature of the bidirectional replication forks result in the generation of specific molecular intermediates that are indicative of origin activation (Fig. 1). A number of experimental approaches have been developed to enrich and capture these replication intermediates in an effort to identify start sites of DNA replication across the mammalian genome. Replication intermediates include the generation of “bubbles” of localized regions of 2N DNA content as the forks move outward from the origin as well as two types of nascent strands. Okazaki fragments are short discontinuous nascent strands (∼150 bp) produced by lagging strand synthesis and primed by DNA polymerase α-mediated RNA synthesis. A consequence of bidirectional DNA replication is that there will be a sharp transition in the strandedness of Okazaki fragments at the origin. In contrast, short nascent strands are significantly longer than Okazaki fragments and result from RNA-primed leading strand synthesis at the origin.
Figure 1.

Mapping molecular intermediates of replication initiation. Schematic representation of replication intermediates and their detection by enrichment for short nascent strands (SNSs), replication bubbles, and Okazaki fragments. Three potential origins (two highly efficient and one inefficient) are depicted in a 25-kb region. For simplicity, initiation events are represented on the same molecule as an aggregate across the entire population; however, in reality, initiation events are stochastic and sparse, with the likelihood of more than one origin being activated in this region being very low. Restriction sites used for fragmentation for the bubble-seq method are indicated (restriction sites). Detection of replication bubbles is very sensitive but limited by the resolution of the restriction fragments and subject to false negatives when a restriction site is within the replication bubble. The distribution of Okazaki fragments and the DNA strand (Watson or Crick) to which they map provides information about replication fork direction (RFD); sharp transitions in strandedness mark origins and termination zones. However, this method cannot discriminate between a zone of initiation and a region containing several specific sites used in a stochastic manner. SNS mapping by treatment with λ-exonuclease provides increased resolution but can detect only the most efficient origins of DNA replication.
The 55-kb intergenic region between the convergently transcribed genes DHFR and 2BE2121 from Chinese hamster ovary (CHO) cells is perhaps the best-characterized mammalian replicon with regard to replication initiation (Hamlin et al. 2010). This locus has long served as the paradigm for the development of assays to detect mammalian replication origins in their chromosomal context. During the late 1980s and early 1990s, a complex and often conflicting view of origin usage emerged from this locus, which only served to highlight the strengths and weaknesses of each origin-mapping approach. Initial studies from the Hamlin laboratory (Heintz and Hamlin 1982) took advantage of the 800-fold tandem amplification of the DHFR locus in response to methotrexate to identify the “earliest labeling restriction fragments” (ELFs) from this region, which corresponded to two nonoverlapping fragments of 6 and 11 kb termed Ori-β and Ori-γ.
The location of Ori-β was subsequently refined to near-nucleotide resolution by the DePamphilis group (Burhans et al. 1990) by analyzing the distribution of Okazaki fragments. Alkaline hydrolysis of the RNA primer provided a 5′-OH for P32 end labeling of Okazaki fragments. The labeled Okazaki fragments were then hybridized to strand-specific sequences surrounding Ori-β, revealing a marked transition in strandedness that occurred within a 500-bp window. Together, these results suggested that 80% of the replication intermediates at the DHFR locus originated from a specific replicator element at Ori-β.
An alternative approach to identify origins was by the location of nascent strands—short molecular intermediates that arise from the elongating strand immediately following initiation of DNA replication (Vassilev and Johnson 1989). Nascent strands were enriched by pulse-labeling proliferating cells with BrdU, denaturing the duplex DNA, and enriching for BrdU-labeled nascent strands by immunoprecipitation. The BrdU-enriched DNA was subsequently size-fractionated by alkaline sucrose gradients and amplified to isolate the short (<1-kb) nascent strand replication intermediates. Similar to the Okazaki fragment studies, there was clear enrichment of nascent strands at Ori-β (Vassilev et al. 1990). A major advance in the nascent strand abundance assay used λ-exonuclease to selectively degrade small, broken DNA lacking a 5′ RNA–DNA junction indicative of polymerase α-mediated priming. (Bielinsky and Gerbi 1998).
In 1987, Brewer and Fangman (1987) developed an innovative two-dimensional (2D) gel electrophoresis approach to map and identify replication intermediates not by their molecular or biochemical properties but rather by their shape and structure. Specifically, linear DNA will migrate through an agarose gel with different kinetics than branched DNAs (replication fork) or those with 2N “bubbles” (origins). Strikingly, analysis of the 55-kb DHFR intergenic region identified “bubble” replication intermediates throughout the entire intergenic region (Vaughn et al. 1990). Thus, unlike the nascent strand abundance assay, which indicated that there were two preferred and well-defined replicators, the 2D gel electrophoresis data were consistent with the entire intergenic spacer functioning as a replication initiation zone, with low-frequency initiation events being observed everywhere except within the DHFR and 2BE2121 transcribed sequences.
These contrasting views ignited a firestorm of debate regarding the nature of origins: Do they arise from defined locations in the genome, or are there instead zones of potential initiation? In retrospect, these results are at least partially reconcilable given the scale of each approach, the breadth of the search space, the nature of the biochemical enrichment of replication intermediates, and the use of bulk population-based assays. Single-molecule DNA-combing assays, in which the incorporation of nucleotide analogs are visualized on single DNA fibers, also support the zone of initiation model at the DHFR locus (Lubelsky et al. 2011), but there are also examples of locus-specific origins being detected at other genomic locations (Anglana et al. 2003). However, despite providing a direct readout on how often an origin was used in a population, the combing assays were labor-intensive and difficult to scale horizontally (across the genome) and vertically (number of molecules detected) and thus often lacked sufficient statistical power.
Genome-wide surveys of replication origins
The development of genomic technologies to comprehensively survey and explore genome function, first by microarray-based approaches and more recently by next-generation sequencing, has significantly expanded the catalog of replication initiation events from several dozen to >100,000 across the entire genome (Cadoret et al. 2008; Sequeira-Mendes et al. 2009; Mesner et al. 2011; Besnard et al. 2012). Not surprisingly, the debate of specific replicators versus nonspecific initiation zones has continued from its humble beginnings at the DHFR locus into the genomic era. However, despite differences in resolution, the number of origins detected, and apparent concordance among and between assays, a general consensus is emerging that, while much of the genome is capable of supporting low levels of replication initiation events, preferred sites of initiation include regulatory elements (transcription start sites [TSSs] and enhancers).
Nascent strand abundance
Adaptation of short nascent strand (SNS) abundance assays to genome-wide approaches was relatively straightforward, and the first experiments used microarrays to survey ∼1% of human (HeLa) and mouse (mESC [mouse embryonic stem cell]) genomes for replication origins (Cadoret et al. 2008; Sequeira-Mendes et al. 2009). Size-selected nascent strands were enriched and purified from broken contaminating chromosomal fragments by treatment with excess λ-exonuclease before hybridization to microarrays. It should be noted that excess λ-exonuclease is important for the accurate identification of bona fide nascent strands, as the nuclease exhibits an intrinsic sequence bias that can lead to false positives (Foulk et al. 2015). Despite surveying only a limited portion of the genome, it was possible to estimate a population-based average interorigin distance of ∼60 kb (Cadoret et al. 2008). Notably, the distribution of replication origins was not random, as studies in both mouse (Sequeira-Mendes et al. 2009) and human cells (Cadoret et al. 2008) identified significant enrichment of nascent strands at DNase I-hypersensitive sites and gene regulatory elements. Further analysis of the sequence features associated with origins found that 35% of the nascent strands accumulated in the vicinity of CpG islands (CGIs) (Cadoret et al. 2008). This association was in agreement with a pioneering single-locus study that had previously implicated CGIs in vertebrate replication initiation (Delgado et al. 1998) and may suggest a common regulatory role for CGIs in both transcription and DNA replication. Although a third of origins were associated with CGIs, CGIs were not sufficient or necessary for origin function. These initial studies also established a clear correlation between origin density and GC content and identified large regions devoid of strong replication initiation sites (Cadoret et al. 2008), establishing a link between origin density and replication timing.
The microarray-based studies laid the foundations for comprehensive genome-wide studies using next-generation sequencing (Martin et al. 2011; Besnard et al. 2012; Picard et al. 2014; Cayrou et al. 2015). These comprehensive surveys of origin activity were able to identify >80,000 origins that were activated in a variety of human ESC and mESC lines. Nearly 35% of surveyed origins were shared across multiple cell lines (Besnard et al. 2012). These constitutive origins are among the strongest and are enriched for regulatory CGI elements. In contrast, origins specific to a single cell line accounted for only 10% of the identified origins. The ability to resolve origins to <1000 bp by the SNS assay provided an opportunity to identify sequence features that may be associated with the selection or activation of origins (discussed below).
Bubble trapping
The initiation of DNA replication within a linear fragment of DNA results in a 2N “bubble” replication intermediate. The reduced electrophoretic mobility of these bubble structures provided a mechanism to enrich for replication initiation events across the genome by trapping them in agarose plugs during gel electrophoresis (Mesner et al. 2006). Analyzing the 1% of the human genome targeted by ENCODE, Mesner et al. (2011) found evidence for replication initiation events across nearly 20% of the surveyed genome. As the bubble-trapping assay is dependent on prior restriction digest to linearize the chromosomes, it lacks the precision of the nascent strand abundance assay; however, it is more sensitive to detecting low-level initiation events over broad regions. Origin initiation events were distributed in both genic and intergenic regions but were significantly enriched for transcribed genes with activating chromatin marks at the promoter, which is consistent with the nascent strand abundance studies. Expansion of bubble trapping to the entire genome by next-generation sequencing linked origin activity and the density of bubble intermediates not with transcription units per se but rather with large domains of accessible DNase I-hypersensitive chromatin (Mesner et al. 2013).
Okazaki fragments
A comprehensive accounting of the strand-specific distribution of Okazaki fragments throughout the genome in theory can describe the precise location and efficiency of replication origins and the direction of replication fork movement within a replicon as well as identify termination zones. In a landmark study, Smith and Whitehouse (2012) provided the first genome-wide map of Okazaki fragments in the budding yeast S. cerevisiae. Okazaki fragments were enriched by inactivation of DNA ligase I and subsequently purified from asynchronous cells. The strand-specific nature of the Okazaki fragments was preserved during high-throughput sequencing, and origins of DNA replication were readily marked by the sharp transition in strandedness (Watson to Crick) surrounding an origin.
Application of Okazaki fragment mapping to the human genome reinforced the emerging paradigm that replication initiation in mammalian systems is a flexible system with very few highly efficient origins being used in the majority of cells (Petryk et al. 2016). Out of the entire human genome, only 66 origins could be identified by a sharp transition in Okazaki fragments that occurred within a 5-kb window, corresponding to <0.01% of the surveyed genome. Instead, much broader initiation zones were observed, with gradual transitions in Okazaki fragment strand distributions occurring over large domains with a mean size of 30 kb (6–150 kb). Unlike the SNS data, which identified discrete start sites of DNA replication throughout the genome, the Okazaki fragment analysis indicated that there were very few discrete sites of initiation used across the population of cells and that initiation events were distributed over broad zones. These seemingly conflicting results are at least partially reconcilable if start sites detected by SNSs represent an ensemble of all possible initiation events and if their stochastic usage throughout the population results in the broad zones of initiation detected by the Okazaki fragment analysis. Again, consistent with prior nascent strand- and bubble-trapping methods, CGIs and expressed genes were frequently colocalized with the borders of these initiation zones. Interestingly, the distribution of Okazaki fragments revealed that the directions of transcription and replication forks were frequently co-oriented, which may minimize the possibility of head-to-head polymerase collisions (Azvolinsky et al. 2009; Hoffman et al. 2015) and may explain the frequent colocalization of origins near TSSs.
Initiators to map potential origins
Chromatin immunoprecipitation coupled with microarrays or next-generation sequencing has been a powerful approach to map the binding sites of transcription factors and other DNA-binding proteins throughout genomes of most eukaryotes (Landt et al. 2012). ORC was one of the first DNA-binding complexes to be mapped by chromatin immunoprecipitation in S. cerevisiae (Wyrick et al. 2001). Despite the early successes in mapping ORC-binding sites in S. cerevisiae and Drosophila (Wyrick et al. 2001; MacAlpine et al. 2010), it has been a challenge to identify ORC-binding sites in mammalian systems. Despite the efforts of many laboratories, comprehensive maps of genome-wide ORC localization have become available only recently (Dellino et al. 2013; Miotto et al. 2016). No sequence motifs emerged from the analysis of the ORC-associated DNA, but rather ORC was found to be highly enriched in open DNase I-accessible chromatin at regulatory enhancers and promoters marked by activating histone chromatin modifications (Miotto et al. 2016). These data, together with biochemical data demonstrating a lack of sequence specificity for metazoan ORC (Vashee et al. 2003; Remus et al. 2004), suggest that ORC's association with DNA may be promiscuous and/or dependent on specific epigenetic features.
Concordance, controversy, and coalescence
The precise identification and localization of replication origins have been hotly contested subjects for nearly three decades (Hamlin et al. 2010). Despite the conservation of the eukaryotic trans-activating initiation factors, specific cis-activating replicator elements that are used in a majority of cells in a population have failed to emerge. The field has been mired in a quest for a precise accounting of the number and location of mammalian replication origins (Fig. 1). Nascent strand abundance assays, despite their nucleotide precision, enrich only for strong initiation events across a population of cells; bubble-trapping approaches are very sensitive at detecting initiation events but lack precision; Okazaki fragment mapping provides information about replication fork direction, but only a handful of robust origins with defined transitions in Okazaki fragments emerge from the overwhelming signal of passive replication. Except for a handful of loci, the majority of the genome appears to be passively replicated by the stochastic utilization of many potential and preferred initiation sites. Together, these data support a “flexible replicon” model (Cayrou et al. 2011) in which replicons are defined by not a single origin but rather multiple potential origins that are used in a stochastic fashion.
Identification of ORC-binding sites should provide a complete catalog of loci with the potential to function as origins of replication (Dellino et al. 2013; Miotto et al. 2016). ORC-binding sites share similarities with SNS-detected origins, including enrichment in early replicating domains and at gene regulatory elements. However, a large fraction of ORC-binding sites does not overlap with SNS-detected origins (Miotto et al. 2016), suggesting an uncoupling of replication initiation from the site of ORC binding. Recent in vitro and in vivo experiments in yeast suggest that transcription can slide the Mcm2–7 complex and displace the site of initiation (Gros et al. 2015). Transcription has also been shown to shape the distribution of the Mcm2–7 complex in a metazoan (Powell et al. 2015). These results suggest that Mcm2–7 redistribution during origin licensing may represent an additional mechanism to establish and shape the DNA replication program in a cell type-specific manner.
Given the complexity and plasticity of the mammalian DNA replication program and that almost any sequence has some potential to function as an origin, it will be beneficial going forward to develop comprehensive new technologies to study DNA replication at the single-molecule level (e.g., individual chromosomal fragments). This could be accomplished by single-cell adaptations of genomic techniques to profile DNA copy number (Knouse et al. 2014) and chromatin structure (Cusanovich et al. 2015) or by recent advances in single-molecule sequencing approaches. For example, nanopore sequencing translocates ssDNA through protein nanopores, and combinations of individual nucleotides are detected as discrete voltage changes across a membrane (Deamer and Branton 2002; Schneider and Dekker 2012). Analogous to DNA combing, it may be possible to pulse-label replicons with nucleotide analogs in order to identify and deconvolve replicated sequences by nanopore sequencing. In theory, this technology could overcome the current limitations of DNA combing (limited sample size and lack of sequence-level resolution) and provide robust estimates of the frequency, location, and distribution of initiation events.
Defining an origin
Sequence
A comprehensive catalog of tens of thousands of replication origins has provided an opportunity to revisit the role of primary sequence in defining replication origins. Although prior plasmid-based assays failed to identify a conserved cis-acting sequence (Heinzel et al. 1991), they did demonstrate that origin activity in human cells was more pronounced on mammalian DNA. Similarly, experiments at the human B-globin locus demonstrated that an 8-kb sequence was sufficient to promote DNA replication in an ectopic location (Aladjem et al. 1998). Computational analysis of sequences flanking origins mapped by SNS provided evidence that a specific structural feature of GC-rich DNA may be involved in the initiation of DNA replication (Cayrou et al. 2011, 2012; Besnard et al. 2012). Specifically, nascent strands accumulated ∼220 bp downstream from G-rich sequences with the potential of forming a noncanonical four-stranded helical structure termed the G quadruplex (G4). The basic structural unit is the G quartet, a square planar assembly of four Hoogsteen-bonded guanine bases. The signature motif predictive of G4 formation is four tracts of at least three guanines separated by other bases that can fold to form the secondary structure of the G4 (Rhodes and Lipps 2015). There are >370,000 predicted G4 motifs in the human genome (Huppert and Balasubramanian 2007), which is significantly more than the 80,000 identified origins, suggesting that G4s are not sufficient for origin activity.
Taking advantage of efficient homologous recombination in chicken DT40 cells (Winding and Berchtold 2001), investigators have been able to precisely insert an efficient origin comprising the chicken βA globin promoter into an ectopic location devoid of strong initiation events, which is replicated in mid-S phase (Hassan-Zadeh et al. 2012; Valton et al. 2014). Insertion of the chicken βA promoter origin was sufficient to produce short nascent strands, thus demonstrating its ability to function as an ectopic replicator; however, insertion of this replicator was insufficient to advance replication timing. Flanking the ectopic replicator with binding sites for the USF transcription factor induced a change in the local histone H3 acetylation and H3K4me2 and an advancement in replication timing that was dependent on the βA promoter origin. The synergistic impact of USF-binding and -activating chromatin marks on βA function may indicate a potential feed-forward mechanism that promotes the coordinated activation of neighboring cryptic origins to advance replication timing. The role of a prominent G4 motif in the βA promoter origin was also dissected genetically (Valton et al. 2014). Point mutations affecting the in vitro stability of G4 also impacted origin efficiency, thus providing evidence for a critical role of G4 formation in replication initiation. Not surprisingly, based on the asymmetric distribution of SNS intermediates surrounding potential G4 motifs from prior genome-wide studies (Cayrou et al. 2012), the orientation of the G4 motif within the βA promoter origin determined the position of replication initiation.
These findings highlight key questions concerning the mechanism by which specific sequences like G4s may impact origin function (Fig. 2). Potential models for G4 function include precisely positioning nucleosomes flanking the origin (Cayrou et al. 2015), which may facilitate pre-RC formation (Lipford and Bell 2001). Alternatively, G4 structures may be recognized by specific factors involved in the formation of a functional origin—both ORC and RecQL4 (human ortholog of Sld2) interact with G4 structures in vitro (Hoshina et al. 2013; Keller et al. 2014). G4 structures may also cooperate with other adjacent cis-acting sequence modules (Valton et al. 2014). For example, G4 structures may function to transiently pause the elongation of nascent strands that initiated within the immediate vicinity of the predicted structure. This is supported by their orientation-dependent function (Valton et al. 2014) and the polar accumulation of nascent strands 3′ from the G-rich sequence (Besnard et al. 2012; Cayrou et al. 2012). Replication of G4 structures is at least partially dependent on specialized polymerases and helicases. The Pif1 helicase unwinds G4 structures, and loss of Pif1 results in replication fork stalling (Lopes et al. 2011; Paeschke et al. 2011) and genomic instability (Paeschke et al. 2013). In addition, Rev1, a translesion polymerase, facilitates passage of the replication fork through G4 secondary structures, and loss of Rev1 activity uncouples canonical chromatin assembly from DNA synthesis, resulting in a failure to properly re-establish the epigenetic landscape and gene expression patterns (Schiavone et al. 2014). However, point mutations in the βA G4 structure disrupt not only SNS accumulation (as expected for a replication fork pause) but also the advancement in replication timing, which implies that the βA G4 has a bona fide role in initiation (Valton et al. 2014). Recent advances in genome editing will provide the tools to comprehensively and systematically assess the function of specific replicator sequences (including G4 structures) in contributing to origin efficiency and replication timing.
Figure 2.
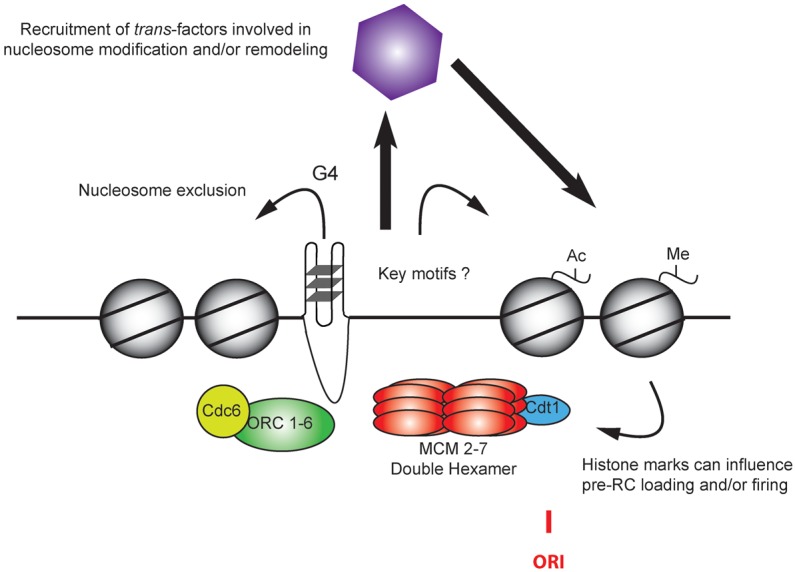
Defining a replication origin. Several features have been proposed to contribute to local origin activity. Nucleosome-free regions (NFRs) may be preferential sites for ORC binding, pre-RC formation, and/or activation. G4 motifs may also favor the formation of NFRs and/or recruitment of specific factors involved in origin licensing or activation. Cooperation with neighboring cis-regulatory sequences and chromatin modifications may also increase the capacity to form a NFR or contribute to the recruitment of trans-factors, influencing origin selection and activation. The site of initiation (ORI) is located ∼220 bp 3′ of the G4 structure, which may also function to transiently impede fork progression on the leading strand.
Epigenetic features
The plasticity of the mammalian DNA replication program is thought to be due in large part to epigenetic features that modulate the selection and activation of replication origins (Fig. 2; Table 1). For example, the activation of a predominant replication origin near the human β-globin locus is developmentally programmed to occur early in S phase in erythroid cells and in late S phase in nonerythroid cells (Kitsberg et al. 1993). This change in replication timing is correlated with differences in histone acetylation between erythroid and nonerythroid cells (Goren et al. 2008). Importantly, tethering different histone acetylases to the locus is sufficient to promote early replication in lymphoblastoid cells, and, conversely, the tethering of a histone deacetylase is sufficient to repress early replication in erythroid cells.
Table 1.
Genomic and epigenetic features associated with mammalian origin activity

The histone acetyltransferase (HAT) Hbo1 acetylates histone H4 on Lys5, Lys8, and/or Lys12. Hbo1 was initially identified via an interaction with Orc1 (Iizuka and Stillman 1999) and has been subsequently shown to interact with multiple pre-RC components (Burke et al. 2001; Iizuka et al. 2006), Hbo1 is recruited to origins via an interaction with Cdt1 (Miotto and Struhl 2008) and results in elevated H4ac levels local to the origin (Miotto and Struhl 2010). Although Hbo1 is essential for cellular proliferation in transformed culture cells, HBO1-null mutant Drosophila (McConnell et al. 2012) and primary mouse embryonic fibroblasts (MEFs) (Kueh et al. 2011) exhibit only mild defects in DNA replication and cellular proliferation, indicating that redundant chromatin-based mechanisms must exist to regulate origins. Hbo1 also interacts with BRPF3 to direct the preferential acetylation of H3K14 near TSSs (Feng et al. 2016). Depletion of BRPF3 induces replicative stress, decreases origin density, and reduces Cdc45 recruitment to chromatin. Altogether, these data suggest a role for Hbo1 in the control of both origin licensing and firing; however, the absence of this function can be compensated for by other mechanisms, including increased fork speed.
Methylation of histone H4K20 is also thought to influence the selection of replication origins across metazoan genomes. The bromo-adjacent homology (BAH) domain of Orc1 specifically interacts with H4K20me2 in higher eukaryotes, and mutating the Orc1-BAH domain results in an S-phase delay and reduced ORC occupancy at select origins (Kuo et al. 2012). The methylation of H4K20 is mediated by the methyltransferases PR-Set7 (H4K20me) and Suv4-20h1/2 (H4K20me2/3). PR-Set7 is cell cycle-regulated and is targeted for proteasome-mediated degradation in S phase (Abbas et al. 2010; Oda et al. 2010; Tardat et al. 2010). Deregulation of PR-Set7 activity via stabilization or overexpression results in rereplication and genomic instability presumably via inappropriate pre-RC assembly, as tethering of PR-Set7 is sufficient to promote Mcm2–7 helicase loading at select origins (Tardat et al. 2010). Efficient pre-RC assembly during mouse development is dependent on both PR-Set7 and Suv4-20h1/2 (Beck et al. 2012), suggesting that dimethylation of H4K20 is the critical methylation state for defining origins.
At the genome-wide scale, it is hard to envision H4K20me2 as a specificity factor for origins because H4K20me2 is the most abundant histone post-translational modification, accounting for >80% of the total H4 population (Yang et al. 2008). Thus, >95% of all nucleosomes will contain at least one H4 subunit dimethylated at Lys20. Despite the conservation of PR-Set7 and catastrophic developmental phenotypes resulting from the loss of PR-Set7 activity in both mammalian and Drosophila systems, recent studies using engineered histone arrays demonstrated that H4K20A mutants were viable in flies (McKay et al. 2015). These results indicate that H4K20 methylation is not strictly required for cell proliferation and instead may function to stabilize ORC on chromatin via the Orc1-BAH domain.
The transcription and DNA replication programs have long been thought to respond to similar chromatin cues, with chromatin environments that activate or repress transcription being associated with early- and late-replicating domains, respectively. Early studies from ENCODE (The ENCODE Project Consortium 2007) identified correlations between time of replication and specific chromatin marks over broad domains. The comprehensive mapping of replication origins throughout the genome provided an opportunity to identify specific and combinatorial patterns of chromatin modifications that may function to directly specify origin selection and activation at discrete chromosomal loci. A discriminant analysis of origins and chromatin landscape in human cells—aiming at pinpointing combinations of chromatin marks that characterize early-, mid-, and late-firing origins—found that early origins were characterized by their proximity to the combination of open chromatin marks (H3K9ac, H3K4me3, and H2AZ) and H4K20me1 (Picard et al. 2014). Similar results were observed in mESCs (Cayrou et al. 2015). H3K79me2 is also enriched at active genes and correlated with origin activity (Fu et al. 2013). Given the frequent colocalization of origins with TSSs and regulatory CGIs, it is not surprising that there is significant enrichment for chromatin signatures representative of promoters, enhancers, and active transcription.
More surprising was the correlation with polycomb-repressive chromatin found in human cell lines (Picard et al. 2014) and mESCs (Cayrou et al. 2015). Although not explicitly tested, these polycomb-repressive domains may represent a bivalent chromatin state enriched for both activating chromatin (H3K4me3) and polycomb-repressive chromatin (H3K27me), which are frequently found at developmentally regulated transcripts (Bernstein et al. 2006). The presence of bivalent chromatin at replication origins may provide a mechanism for the rapid developmentally programmed shifts in replication timing observed for select loci (e.g., β-globin).
The widespread availability of genome-wide DNA replication and chromatin data has identified many intriguing correlations that may have regulatory potential. However, it is important to stress that correlation does not equal causation. Complicating matters is the nonuniform distribution of replication origins across the genome. The frequent co-occurrence of replication origins with TSSs and regulatory elements makes it difficult to deconvolve the contribution of specific chromatin marks to the regulation of transcription, replication, or both. In addition, epigenetic writers (e.g., HATs and methyltransferases) are primarily thought of as modifying histone lysines but also frequently target lysines on nonhistone targets. Both HBO1 and PR-Set7 are known to directly modify nonhistone lysine targets, including critical cell cycle and pre-RC factors (Shi et al. 2007; Iizuka et al. 2008; Takawa et al. 2012); thus, the effects at a particular origin may be independent of the local chromatin environment.
Genome editing and the development of engineered histone arrays will facilitate a mechanistic understanding of how epigenetic features modulate the DNA replication program. CRISPR/Cas9 will allow for not only the rapid introduction of specific mutations into cis-acting sequences like CGIs and G4 structures but also the insertion of potential replicator sequences at ectopic locations. In addition, HATs and histone methyltransferases as well as histone deacetylases (HDACS) and histone demethylases can be directly targeted to specific loci by CRISPR/dCas9 fusion constructs with surgical precision (Thakore et al. 2016). Targeting of regulatory epigenetic writers and erasers to specific replicons will allow investigators to decipher the local epigenetic rules that regulate the DNA replication program while minimizing the potential for pleiotropic effects due to global dysregulation of transcription. Similarly, the development and use of engineered histone arrays (McKay et al. 2015) in mammalian systems will identify the specific role of key histone tail lysine residues in modulating the selection and activation of DNA replication origins.
Regulation of origins on the chromosome scale
In higher eukaryotes, large chromosomal domains replicate at characteristic times during S phase (Rhind and Gilbert 2013). The density, distribution, and activation of replication origins define this temporal DNA replication program. A hierarchy of interdependent and intradependent features contributes to the regulation of replication origins and the establishment of the DNA replication timing program (Fig. 3). These features include nuclear architecture, isochores, gene density, chromatin state, transcriptional activity, histone modifications, nucleosome positioning, primary sequence, and the availability of rate-limiting initiation factors. Together, they provide for a potentially massive combinatorial code by which to modulate and fine-tune the DNA replication program for different cell types and developmental stages.
Figure 3.
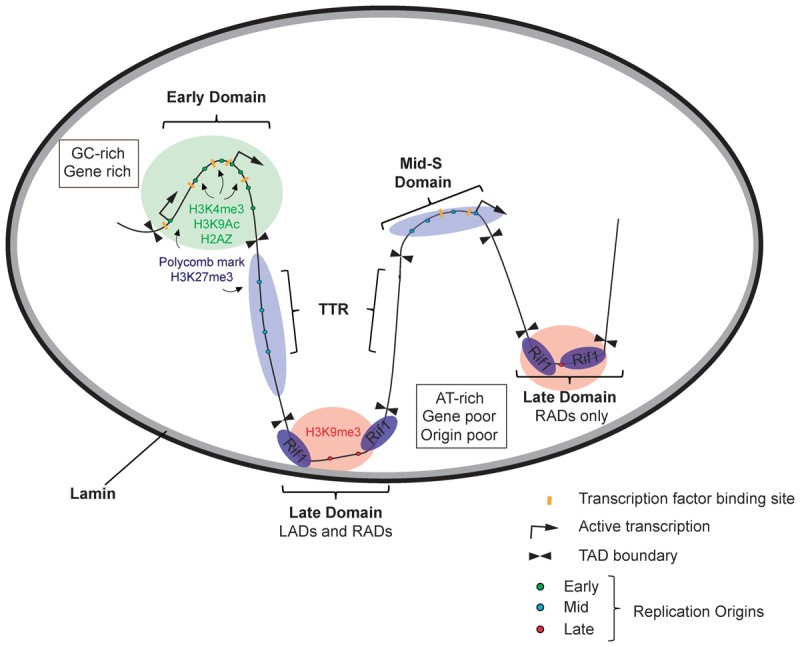
Hierarchy of temporal regulation. Active early, mid-, and late S-phase replication origins are represented as small green, blue, and red dots, respectively. Constant early-replicating domains are enriched for GC content, genes, regulatory elements (transcription factor-binding and enhancer sites), DNase I-hypersensitive sites, and origins of DNA replication. Early-replicating domains are found in the interior of the nucleus and are frequently associated with activating chromatin modifications and histone variants, including H3K4me3, H2AZ, and H3K9ac. Timing transition zones are also origin-rich and frequently associated with polycomb-repressive chromatin environments (H3K27me3). In contrast, late-replicating regions are AT-rich and gene-poor and exhibit a decreased density of origins. Late-replicating domains are associated with nuclear lamins and Rif1 (Rap1-interacting factor 1), which may lead to the formation of a microenvironment at the periphery of the nucleus or the nucleolus that restricts the activity of critical kinases (DDK and CDK) and/or limiting initiation factors.
Mammalian replication timing domains were first described using microarray-based approaches either at very low resolution (Woodfine et al. 2004) or targeting a chromosome arm (White et al. 2004) or select regions of the genome (Jeon et al. 2005). More recently, these approaches were extended genome-wide by tiling arrays (Hiratani et al. 2008) and next-generation sequencing (Hansen et al. 2010; Koren et al. 2014; Mukhopadhyay et al. 2014). The mammalian genome is segmented into broad regions with constant replication timing (early, mid, or late S phase) and narrow timing transition zones by which the replication timing switches from one domain to the next. The vast majority of protein-coding genes is located in early-replicating domains, suggesting that there is evolutionary pressure to ensure that protein-coding genes are duplicated early in S phase, as there is an increased accumulation of mutations in late-replicating domains (Stamatoyannopoulos et al. 2009; Koren et al. 2012).
Replication timing domains are remarkably conserved between different cell types (Ryba et al. 2011), individuals of the same species (Koren et al. 2014), and even different species (Ryba et al. 2010). Although still highly correlated on the megabase scale, at increased resolution, it is possible to find a significant number of smaller changes in replication timing that are developmentally regulated (Rivera-Mulia et al. 2015) or linked to the underlying genotype (Koren et al. 2014). For example, replication timing changes that occur during differentiation of mESCs typically involve the consolidation of smaller neighboring replication domains (Hiratani et al. 2008).
Constant replication timing domains are significantly larger than individual replicons, implying the coordinated activation of multiple replication origins across the domain. Indeed, early-replicating domains have a high density of efficient replication origins (Cadoret et al. 2008; Besnard et al. 2012). In contrast, late-replicating domains have far fewer origins detected by methods like SNS; however, that is not to imply that late-replicating regions are the consequence of being replicated from a replication fork emanating from a neighboring domain, as the distribution of Okazaki fragments is not consistent with the passage of a single fork (Petryk et al. 2016). Rather, it appears to be a cascade of stochastic and inefficient initiation events (Mesner et al. 2013) proceeding from the transition zone into the late domain (Petryk et al. 2016).
The time at which a locus replicates in S phase has long been associated with nuclear architecture (Li et al. 2001; Zhou et al. 2002). Late-replicating sequences are associated with the nuclear periphery and colocalized with lamin-associated domains (LADs) (Peric-Hupkes et al. 2010). More recently, genome-wide chromatin interaction maps generated via Hi-C (Lieberman-Aiden et al. 2009) revealed strong correlations between nuclear architecture and the DNA replication timing program (Ryba et al. 2010). Specifically, the boundaries of topologically associated domains (TADs) (Dixon et al. 2012) frequently demarcate replication timing domain boundaries (Pope et al. 2014). The mechanisms responsible for establishing and maintaining topological domains are being actively deciphered. The insulator element CTCF is enriched at the borders of TADs (Dixon et al. 2012), and ablation of a specific CTCF site at the Hox gene cluster permits an expansion of permissive chromatin and disruption of a TAD boundary (Narendra et al. 2015). Presumably, although it remains to be tested, the loss of the TAD boundary and expansion of open and active chromatin would result in an increased density of initiation events and an alteration in replication timing.
The conservation of replication timing domains suggests that they are hardwired into the genome at some fundamental level that is likely dependent on chromatin accessibility and nuclear architecture. Not surprisingly, it has proven difficult to globally disrupt the replication timing program. Recently, Rif1 (Rap1-interacting factor 1) has been identified as a global regulator of replication timing (Cornacchia et al. 2012; Hayano et al. 2012). Initially identified as a telomere-binding protein required for maintenance of telomere length in S. cerevisiae (Hardy et al. 1992), it was subsequently implicated in advancing the time of activation of late-replicating origins both at the telomeres and within the chromosome in Schizosaccharomyces pombe (Hayano et al. 2012). In mammalian systems, Rif1 is an essential factor required for organismal development. Depletion of Rif1 results in a consolidation of early- and late-replicating domains to be instead replicated in mid-S phase (or randomly throughout S phase) (Cornacchia et al. 2012). Rif1 localizes to late-replicating domains, many of which are concordant with LADs (Foti et al. 2016). Late-replicating regions of the genome marked only by Rif1 are most susceptible to advancing their time of replication in the absence of Rif1 function. However, in early-replicating regions of the genome, it is unclear what mediates the transition to later replication in the absence of Rif1. The change in timing could be direct due to alteration of interchromatin architecture or indirect via the increased competition for rate-limiting initiation factors (Yoshida et al. 2014).
While the molecular mechanisms that govern nuclear architecture and long-range chromatin interactions and how they impact the DNA replication timing program remain to be elucidated, it is worthwhile to step back and consider the replication program in the context of broader chromosomal features. In addition to topological domains, gene density and isochores of differing GC content are also excellent proxies for predicting replication timing. Early-replicating domains are enriched for GC content and gene regulatory elements, including promoters and enhancers. The high density of regulatory elements contributes to an open or accessible chromatin environment that is permissive for ORC binding (Dellino et al. 2013; Miotto et al. 2016). In Drosophila, ORC localizes to highly occupied target (HOT) regions of the genome where multiple DNA-binding factors colocalize (The modEncode Consortium et al. 2010). HOT regions are conserved in higher eukaryotes (Gerstein et al. 2010; The ENCODE Project Consortium 2012) and may represent a unique chromatin environment for the promiscuous and transient recruitment of regulatory DNA-binding factors. Other common regulatory features like CGIs contribute to the formation of G4 structures, which are implicitly associated with replication initiation events (Besnard et al. 2012; Cayrou et al. 2012). Finally, chromatin accessibility over the gene-dense early-replicating isochores facilitates intradomain chromatin interactions, which provide a localized environment for the coordinated activation of multiple origins throughout the domain. The domain-specific coordinated activation of replication origins is likely established in part by limiting concentrations of initiation factors (Mantiero et al. 2011; Tanaka et al. 2011) that serve to shape a precise replication timing program.
The mechanisms that govern how gene-poor and AT-rich isochores are copied late in S phase remain a mystery. The availability of open chromatin and nucleosome-free DNA for ORC recruitment is diminished and is reflected in the low density of identified ORC-binding sites and initiation events (Besnard et al. 2012; Dellino et al. 2013; Miotto et al. 2016). Despite the temporal delay in the activation of replication, these domains are copied for the most part with the same kinetics as early-replicating domains. That is to say that, at a single-molecule level, a similar number of active replicons contribute to the duplication of early- and late-replicating domains. However, there are exceptions, like developmentally regulated chromosome fragile sites (CFSs). For example, the CFS FRA3B is late-replicating in both lymphoblastoid and fibroblast-derived human cell lines (Letessier et al. 2011). However, in lymphoblastoid cells, there is an absence of initiation events within the 700-kb domain encompassing the fragile site, thereby rendering the domain dependent on replication forks emanating from outside the locus. Intriguingly, genomic instability at these developmentally regulated fragile sites may also be dependent on transcription of ultralong genes (>500 kb) (Wilson et al. 2015). It remains to be determined whether the breaks and resulting genomic instability are due to collisions between transcription and replication forks (Helmrich et al. 2011) or the generation of RNA:DNA hybrid R loops (Stirling et al. 2012) or, alternatively, whether cell type-specific transcription through these loci serves to displace the Mcm2–7 helicase complex from the template DNA (Gros et al. 2015; Powell et al. 2015), further reducing potential initiation events.
The past year, 2015, has seen remarkable advances in the DNA replication field. Decades of biochemical studies have culminated in what can only be described as finding the holy grail—multiple times. Detailed atomic- and near-atomic-resolution structures were obtained by X-ray crystallography and cryo-electron microscopy for ORC (Bleichert et al. 2015) and the Mcm2–7 complex (Li et al. 2015), respectively. In addition, the Diffley laboratory (Yeeles et al. 2015) has reconstituted licensed DNA replication in vitro using 42 polypeptides, providing insight into the precise order of events leading to initiation and defining the minimal complement of factors required for regulated eukaryotic DNA replication. Future in vitro experiments using purified proteins from mammalian systems will lead the way forward to explicitly test the minimal cis- and trans-acting factors required for regulated initiation from a defined locus. However, it will be difficult, if not impossible, to capture the full complexity of the mechanisms and nuclear features that regulate chromosomal replication in vivo. Analogous to the varied mechanisms by which transcription is regulated at individual genes, it is likely that diverse chromatin-mediated mechanisms will exist to regulate individual origins to ensure the faithful inheritance of genetic information across cell types and developmental stages.
Acknowledgments
We thank our laboratories for critical comments and suggestions. We are also indebted to reviewers for their insightful comments and suggestions. This work was supported by grants from the National Institutes of Health (R01-GM104097) and the American Cancer Society (RSG-11-048-01-DMC) to D.M.M., and the Association pour la Recherche sur le Cancer (Equipe Labellisée), the Agence Nationale pour la Recherche (ANR-15-CE12-0004-01), and the IdEx Paris Sorbonne to M.-N.P. M.-N.P is supported by the Institut National de la Santé et de la Recherche Médicale.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.285114.116.
References
- Abbas T, Shibata E, Park J, Jha S, Karnani N, Dutta A. 2010. CRL4(Cdt2) regulates cell proliferation and histone gene expression by targeting PR-Set7/Set8 for degradation. Mol Cell 40: 9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aladjem MI, Rodewald LW, Kolman JL, Wahl GM. 1998. Genetic dissection of a mammalian replicator in the human β-globin locus. Science 281: 1005–1009. [DOI] [PubMed] [Google Scholar]
- Anglana M, Apiou F, Bensimon A, Debatisse M. 2003. Dynamics of DNA replication in mammalian somatic cells: nucleotide pool modulates origin choice and interorigin spacing. Cell 114: 385–394. [DOI] [PubMed] [Google Scholar]
- Azvolinsky A, Giresi PG, Lieb JD, Zakian VA. 2009. Highly transcribed RNA polymerase II genes are impediments to replication fork progression in Saccharomyces cerevisiae. Mol Cell 34: 722–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck DB, Burton A, Oda H, Ziegler-Birling C, Torres-Padilla ME, Reinberg D. 2012. The role of PR-Set7 in replication licensing depends on Suv4-20h. Genes Dev 26: 2580–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell SP, Kaguni JM. 2013. Helicase loading at chromosomal origins of replication. Cold Spring Harb Perspect Biol 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezney R, Dubey DD, Huberman JA. 2000. Heterogeneity of eukaryotic replicons, replicon clusters, and replication foci. Chromosoma 108: 471–484. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. 2006. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125: 315–326. [DOI] [PubMed] [Google Scholar]
- Besnard E, Babled A, Lapasset L, Milhavet O, Parrinello H, Dantec C, Marin J-M, Lemaitre J-M. 2012. Unraveling cell type-specific and reprogrammable human replication origin signatures associated with G-quadruplex consensus motifs. Nat Struct Mol Biol 19: 837–844. [DOI] [PubMed] [Google Scholar]
- Bicknell LS, Bongers EMHF, Leitch A, Brown S, Schoots J, Harley ME, Aftimos S, Al-Aama JY, Bober M, Brown PAJ, et al. 2011. Mutations in the pre-replication complex cause Meier-Gorlin syndrome. Nat Genet 43: 356–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielinsky AK, Gerbi SA. 1998. Discrete start sites for DNA synthesis in the yeast ARS1 origin. Science 279: 95–98. [DOI] [PubMed] [Google Scholar]
- Bleichert F, Botchan MR, Berger JM. 2015. Crystal structure of the eukaryotic origin recognition complex. Nature 519: 321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenthal AB, Kriegstein HJ, Hogness DS. 1974. The units of DNA replication in Drosophila melanogaster chromosomes. Cold Spring Harb Symp Quant Biol 38: 205–223. [DOI] [PubMed] [Google Scholar]
- Brewer BJ, Fangman WL. 1987. The localization of replication origins on ARS plasmids in S. cerevisiae. Cell 51: 463–471. [DOI] [PubMed] [Google Scholar]
- Burhans WC, Vassilev LT, Caddle MS, Heintz NH, DePamphilis ML. 1990. Identification of an origin of bidirectional DNA replication in mammalian chromosomes. Cell 62: 955–965. [DOI] [PubMed] [Google Scholar]
- Burke TW, Cook JG, Asano M, Nevins JR. 2001. Replication factors MCM2 and ORC1 interact with the histone acetyltransferase HBO1. J Biol Chem 276: 15397–15408. [DOI] [PubMed] [Google Scholar]
- Cadoret J-C, Meisch F, Hassan-Zadeh V, Luyten I, Guillet C, Duret L, Quesneville H, Prioleau M-N. 2008. Genome-wide studies highlight indirect links between human replication origins and gene regulation. Proc Natl Acad Sci 105: 15837–15842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns J. 1966. Autoradiography of HeLa cell DNA. J Mol Biol 15: 372–373. [DOI] [PubMed] [Google Scholar]
- Cayrou C, Coulombe P, Vigneron A, Stanojcic S, Ganier O, Peiffer I, Rivals E, Puy A, Laurent-Chabalier S, Desprat R, et al. 2011. Genome-scale analysis of metazoan replication origins reveals their organization in specific but flexible sites defined by conserved features. Genome Res 21: 1438–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayrou C, Coulombe P, Puy A, Rialle S, Kaplan N, Segal E, Méchali M. 2012. New insights into replication origin characteristics in metazoans. Cell Cycle 11: 658–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayrou C, Ballester B, Peiffer I, Fenouil R, Coulombe P, Andrau J-C, van Helden J, Méchali M. 2015. The chromatin environment shapes DNA replication origin organization and defines origin classes. Genome Res 25: 1873–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claycomb JM, Orr-Weaver TL. 2005. Developmental gene amplification: insights into DNA replication and gene expression. Trends Genet 21: 149–162. [DOI] [PubMed] [Google Scholar]
- Collart C, Allen GE, Bradshaw CR, Smith JC, Zegerman P. 2013. Titration of four replication factors is essential for the Xenopus laevis midblastula transition. Science 341: 893–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornacchia D, Dileep V, Quivy JP, Foti R, Tili F, Santarella-Mellwig R, Antony C, Almouzni G, Gilbert DM, Buonomo SB. 2012. Mouse Rif1 is a key regulator of the replication-timing programme in mammalian cells. EMBO J 31: 3678–3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusanovich DA, Daza R, Adey A, Pliner HA, Christiansen L, Gunderson KL, Steemers FJ, Trapnell C, Shendure J. 2015. Epigenetics. Multiplex single-cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 348: 910–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deamer DW, Branton D. 2002. Characterization of nucleic acids by nanopore analysis. Acc Chem Res 35: 817–825. [DOI] [PubMed] [Google Scholar]
- Delgado S, Gómez M, Bird A, Antequera F. 1998. Initiation of DNA replication at CpG islands in mammalian chromosomes. EMBO J 17: 2426–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dellino GI, Cittaro D, Piccioni R, Luzi L, Banfi S, Segalla S, Cesaroni M, Mendoza-Maldonado R, Giacca M, Pelicci PG. 2013. Genome-wide mapping of human DNA-replication origins: levels of transcription at ORC1 sites regulate origin selection and replication timing. Genome Res 23: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demczuk A, Gauthier MG, Veras I, Kosiyatrakul S, Schildkraut CL, Busslinger M, Bechhoefer J, Norio P. 2012. Regulation of DNA replication within the immunoglobulin heavy-chain locus during B cell commitment. PLoS Biol 10: e1001360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. 2012. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485: 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edenberg HJ, Huberman JA. 1975. Eukaryotic chromosome replication. Annu Rev Genet 9: 245–284. [DOI] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. 2007. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447: 799–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evrin C, Clarke P, Zech J, Lurz R, Sun J, Uhle S, Li H, Stillman B, Speck C. 2009. A double-hexameric MCM2–7 complex is loaded onto origin DNA during licensing of eukaryotic DNA replication. Proc Natl Acad Sci 106: 20240–20245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Vlassis A, Roques C, Lalonde M-E, González-Aguilera C, Lambert J-P, Lee S-B, Zhao X, Alabert C, Johansen JV, et al. 2016. BRPF3-HBO1 regulates replication origin activation and histone H3K14 acetylation. EMBO J 35: 176–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foti R, Gnan S, Cornacchia D, Dileep V, Bulut-Karslioglu A, Diehl S, Buness A, Klein FA, Huber W, Johnstone E, et al. 2016. Nuclear architecture organized by Rif1 underpins the replication-timing program. Mol Cell 61: 260–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foulk MS, Urban JM, Casella C, Gerbi SA. 2015. Characterizing and controlling intrinsic biases of λ exonuclease in nascent strand sequencing reveals phasing between nucleosomes and G-quadruplex motifs around a subset of human replication origins. Genome Res 25: 725–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu H, Maunakea AK, Martin MM, Huang L, Zhang Y, Ryan M, Kim R, Lin CM, Zhao K, Aladjem MI. 2013. Methylation of histone H3 on lysine 79 associates with a group of replication origins and helps limit DNA replication once per cell cycle. PLoS Genet 9: e1003542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge XQ, Jackson DA, Blow JJ. 2007. Dormant origins licensed by excess Mcm2-7 are required for human cells to survive replicative stress. Genes Dev 21: 3331–3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein MB, Lu ZJ, Van Nostrand EL, Cheng C, Arshinoff BI, Liu T, Yip KY, Robilotto R, Rechtsteiner A, Ikegami K, et al. 2010. Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science 330: 1775–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goren A, Tabib A, Hecht M, Cedar H. 2008. DNA replication timing of the human β-globin domain is controlled by histone modification at the origin. Genes Dev 22: 1319–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gros J, Kumar C, Lynch G, Yadav T, Whitehouse I, Remus D. 2015. Post-licensing specification of eukaryotic replication origins by facilitated Mcm2-7 sliding along DNA. Mol Cell 60: 797–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamlin JL, Mesner LD, Dijkwel PA. 2010. A winding road to origin discovery. Chromosome Res 18: 45–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannibal RL, Baker JC. 2016. Selective amplification of the genome surrounding key placental genes in trophoblast giant cells. Curr Biol 26: 230–236. [DOI] [PubMed] [Google Scholar]
- Hansen RS, Thomas S, Sandstrom R, Canfield TK, Thurman RE, Weaver M, Dorschner MO, Gartler SM, Stamatoyannopoulos JA. 2010. Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc Natl Acad Sci 107: 139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy CF, Sussel L, Shore D. 1992. A RAP1-interacting protein involved in transcriptional silencing and telomere length regulation. Genes Dev 6: 801–814. [DOI] [PubMed] [Google Scholar]
- Hassan-Zadeh V, Chilaka S, Cadoret J-C, Ma MK-W, Boggetto N, West AG, Prioleau M-N. 2012. USF binding sequences from the HS4 insulator element impose early replication timing on a vertebrate replicator. PLoS Biol 10: e1001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayano M, Kanoh Y, Matsumoto S, Renard-Guillet C, Shirahige K, Masai H. 2012. Rif1 is a global regulator of timing of replication origin firing in fission yeast. Genes Dev 26: 137–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintz NH, Hamlin JL. 1982. An amplified chromosomal sequence that includes the gene for dihydrofolate reductase initiates replication within specific restriction fragments. Proc Natl Acad Sci 79: 4083–4087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzel SS, Krysan PJ, Tran CT, Calos MP. 1991. Autonomous DNA replication in human cells is affected by the size and the source of the DNA. Mol Cell Biol 11: 2263–2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmrich A, Ballarino M, Tora L. 2011. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol Cell 44: 966–977. [DOI] [PubMed] [Google Scholar]
- Hiratani I, Ryba T, Itoh M, Yokochi T, Schwaiger M, Chang C-W, Lyou Y, Townes TM, Schübeler D, Gilbert DM. 2008. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol 6: e245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman EA, McCulley A, Haarer B, Arnak R, Feng W. 2015. Break-seq reveals hydroxyurea-induced chromosome fragility as a result of unscheduled conflict between DNA replication and transcription. Genome Res 25: 402–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshina S, Yura K, Teranishi H, Kiyasu N, Tominaga A, Kadoma H, Nakatsuka A, Kunichika T, Obuse C, Waga S. 2013. Human origin recognition complex binds preferentially to G-quadruplex-preferable RNA and single-stranded DNA. J Biol Chem 288: 30161–30171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppert JL, Balasubramanian S. 2007. G-quadruplexes in promoters throughout the human genome. Nucleic Acids Res 35: 406–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyrien O, Maric C, Méchali M. 1995. Transition in specification of embryonic metazoan DNA replication origins. Science 270: 994–997. [DOI] [PubMed] [Google Scholar]
- Ibarra A, Schwob E, Méndez J. 2008. Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication. Proc Natl Acad Sci 105: 8956–8961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iizuka M, Stillman B. 1999. Histone acetyltransferase HBO1 interacts with the ORC1 subunit of the human initiator protein. J Biol Chem 274: 23027–23034. [DOI] [PubMed] [Google Scholar]
- Iizuka M, Matsui T, Takisawa H, Smith MM. 2006. Regulation of replication licensing by acetyltransferase Hbo1. Mol Cell Biol 26: 1098–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iizuka M, Sarmento OF, Sekiya T, Scrable H, Allis CD, Smith MM. 2008. Hbo1 Links p53-dependent stress signaling to DNA replication licensing. Mol Cell Biol 28: 140–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob F, Brenner S, Cuzin F. 1963. On the regulation of DNA replication in bacteria. Cold Spring Harb Symp Quant Biol 28: 329–348. [Google Scholar]
- Jeon Y, Bekiranov S, Karnani N, Kapranov P, Ghosh S, MacAlpine D, Lee C, Hwang DS, Gingeras TR, Dutta A. 2005. Temporal profile of replication of human chromosomes. Proc Natl Acad Sci 102: 6419–6424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller H, Kiosze K, Sachsenweger J, Haumann S, Ohlenschläger O, Nuutinen T, Syväoja JE, Görlach M, Grosse F, Pospiech H. 2014. The intrinsically disordered amino-terminal region of human RecQL4: multiple DNA-binding domains confer annealing, strand exchange and G4 DNA binding. Nucleic Acids Res 42: 12614–12627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitsberg D, Selig S, Keshet I, Cedar H. 1993. Replication structure of the human β-globin gene domain. Nature 366: 588–590. [DOI] [PubMed] [Google Scholar]
- Knouse KA, Wu J, Whittaker CA, Amon A. 2014. Single cell sequencing reveals low levels of aneuploidy across mammalian tissues. Proc Natl Acad Sci 111: 13409–13414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren A, Polak P, Nemesh J, Michaelson JJ, Sebat J, Sunyaev SR, McCarroll SA. 2012. Differential relationship of DNA replication timing to different forms of human mutation and variation. Am J Hum Genet 91: 1033–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren A, Handsaker RE, Kamitaki N, Karlić R, Ghosh S, Polak P, Eggan K, McCarroll SA. 2014. Genetic variation in human DNA replication timing. Cell 159: 1015–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krysan PJ, Haase SB, Calos MP. 1989. Isolation of human sequences that replicate autonomously in human cells. Mol Cell Biol 9: 1026–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kueh AJ, Dixon MP, Voss AK, Thomas T. 2011. HBO1 is required for H3K14 acetylation and normal transcriptional activity during embryonic development. Mol Cell Biol 31: 845–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo AJ, Song J, Cheung P, Ishibe-Murakami S, Yamazoe S, Chen JK, Patel DJ, Gozani O. 2012. The BAH domain of ORC1 links H4K20me2 to DNA replication licensing and Meier-Gorlin syndrome. Nature 484: 115–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Bernstein BE, Bickel P, Brown JB, Cayting P, et al. 2012. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res 22: 1813–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letessier A, Millot GA, Koundrioukoff S, Lachages AM, Vogt N, Hansen RS, Malfoy B, Brison O, Debatisse M. 2011. Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature 470: 120–123. [DOI] [PubMed] [Google Scholar]
- Li F, Chen J, Izumi M, Butler MC, Keezer SM, Gilbert DM. 2001. The replication timing program of the Chinese hamster β-globin locus is established coincident with its repositioning near peripheral heterochromatin in early G1 phase. J Cell Biol 154: 283–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Zhai Y, Zhang Y, Li W, Yang M, Lei J, Tye B-K, Gao N. 2015. Structure of the eukaryotic MCM complex at 3.8 Å. Nature 524: 186–191. [DOI] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. 2009. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326: 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipford JR, Bell SP. 2001. Nucleosomes positioned by ORC facilitate the initiation of DNA replication. Mol Cell 7: 21–30. [DOI] [PubMed] [Google Scholar]
- Lombraña R, Almeida R, Revuelta I, Madeira S, Herranz G, Saiz N, Bastolla U, Gómez M. 2013. High-resolution analysis of DNA synthesis start sites and nucleosome architecture at efficient mammalian replication origins. EMBO J 32: 2631–2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes J, Piazza A, Bermejo R, Kriegsman B, Colosio A, Teulade-Fichou M-P, Foiani M, Nicolas A. 2011. G-quadruplex-induced instability during leading-strand replication. EMBO J 30: 4033–4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubelsky Y, Sasaki T, Kuipers MA, Lucas I, Le Beau MM, Carignon S, Debatisse M, Prinz JA, Dennis JH, Gilbert DM. 2011. Pre-replication complex proteins assemble at regions of low nucleosome occupancy within the Chinese hamster dihydrofolate reductase initiation zone. Nucleic Acids Res 39: 3141–3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAlpine HK, Gordân R, Powell SK, Hartemink AJ, MacAlpine DM. 2010. Drosophila ORC localizes to open chromatin and marks sites of cohesin complex loading. Genome Res 20: 201–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantiero D, Mackenzie A, Donaldson A, Zegerman P. 2011. Limiting replication initiation factors execute the temporal programme of origin firing in budding yeast. EMBO J 30: 4805–4814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin MM, Ryan M, Kim R, Zakas AL, Fu H, Lin CM, Reinhold WC, Davis SR, Bilke S, Liu H, et al. 2011. Genome-wide depletion of replication initiation events in highly transcribed regions. Genome Res 21: 1822–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell KH, Dixon M, Calvi BR. 2012. The histone acetyltransferases CBP and Chameau integrate developmental and DNA replication programs in Drosophila ovarian follicle cells. Development 139: 3880–3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay DJ, Klusza S, Penke TJR, Meers MP, Curry KP, McDaniel SL, Malek PY, Cooper SW, Tatomer DC, Lieb JD, et al. 2015. Interrogating the function of metazoan histones using engineered gene clusters. Dev Cell 32: 373–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesner LD, Crawford EL, Hamlin JL. 2006. Isolating apparently pure libraries of replication origins from complex genomes. Mol Cell 21: 719–726. [DOI] [PubMed] [Google Scholar]
- Mesner LD, Valsakumar V, Karnani N, Dutta A, Hamlin JL, Bekiranov S. 2011. Bubble-chip analysis of human origin distributions demonstrates on a genomic scale significant clustering into zones and significant association with transcription. Genome Res 21: 377–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesner LD, Valsakumar V, Cieslik M, Pickin R, Hamlin JL, Bekiranov S. 2013. Bubble-seq analysis of the human genome reveals distinct chromatin-mediated mechanisms for regulating early- and late-firing origins. Genome Res 23: 1774–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miotto B, Struhl K. 2008. HBO1 histone acetylase is a coactivator of the replication licensing factor Cdt1. Genes Dev 22: 2633–2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miotto B, Struhl K. 2010. HBO1 histone acetylase activity is essential for DNA replication licensing and inhibited by Geminin. Mol Cell 37: 57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miotto B, Ji Z, Struhl K. 2016. Selectivity of ORC binding sites and the relation to replication timing, fragile sites, and deletions in cancers. Proc Natl Acad Sci 10.1073/pnas.1609060113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The modEncode Consortium, Roy S, Ernst J, Kharchenko PV, Kheradpour P, Negre N, Eaton ML, Landolin JM, Bristow CA, Ma L, et al. 2010. Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science 330: 1787–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay R, Lajugie J, Fourel N, Selzer A, Schizas M, Bartholdy B, Mar J, Lin CM, Martin MM, Ryan M, et al. 2014. Allele-specific genome-wide profiling in human primary erythroblasts reveal replication program organization. PLoS Genet 10: e1004319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra V, Rocha PP, An D, Raviram R, Skok JA, Mazzoni EO, Reinberg D. 2015. CTCF establishes discrete functional chromatin domains at the Hox clusters during differentiation. Science 347: 1017–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda H, Hubner MR, Beck DB, Vermeulen M, Hurwitz J, Spector DL, Reinberg D. 2010. Regulation of the histone H4 monomethylase PR-Set7 by CRL4(Cdt2)-mediated PCNA-dependent degradation during DNA damage. Mol Cell 40: 364–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paeschke K, Capra JA, Zakian VA. 2011. DNA replication through G-quadruplex motifs is promoted by the Saccharomyces cerevisiae Pif1 DNA helicase. Cell 145: 678–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paeschke K, Bochman ML, Garcia PD, Cejka P, Friedman KL, Kowalczykowski SC, Zakian VA. 2013. Pif1 family helicases suppress genome instability at G-quadruplex motifs. Nature 497: 458–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peric-Hupkes D, Meuleman W, Pagie L, Bruggeman SWM, Solovei I, Brugman W, Gräf S, Flicek P, Kerkhoven RM, van Lohuizen M, et al. 2010. Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol Cell 38: 603–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petryk N, Kahli M, d'Aubenton-Carafa Y, Jaszczyszyn Y, Shen Y, Silvain M, Thermes C, Chen C-L, Hyrien O. 2016. Replication landscape of the human genome. Nat Commun 7: 10208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard F, Cadoret J-C, Audit B, Arneodo A, Alberti A, Battail C, Duret L, Prioleau M-N. 2014. The spatiotemporal program of DNA replication is associated with specific combinations of chromatin marks in human cells. PLoS Genet 10: e1004282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope BD, Ryba T, Dileep V, Yue F, Wu W, Denas O, Vera DL, Wang Y, Hansen RS, Canfield TK, et al. 2014. Topologically associating domains are stable units of replication-timing regulation. Nature 515: 402–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell SK, MacAlpine HK, Prinz JA, Li Y, Belsky JA, MacAlpine DM. 2015. Dynamic loading and redistribution of the Mcm2-7 helicase complex through the cell cycle. EMBO J 34: 531–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remus D, Beall EL, Botchan MR. 2004. DNA topology, not DNA sequence, is a critical determinant for Drosophila ORC-DNA binding. EMBO J 23: 897–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remus D, Beuron F, Tolun G, Griffith JD, Morris EP, Diffley JFX. 2009. Concerted loading of Mcm2-7 double hexamers around DNA during DNA replication origin licensing. Cell 139: 719–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhind N, Gilbert DM. 2013. DNA replication timing. Cold Spring Harb Perspect Biol 5: a010132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes D, Lipps HJ. 2015. G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res 43: 8627–8637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera-Mulia JC, Buckley Q, Sasaki T, Zimmerman J, Didier RA, Nazor K, Loring JF, Lian Z, Weissman S, Robins AJ, et al. 2015. Dynamic changes in replication timing and gene expression during lineage specification of human pluripotent stem cells. Genome Res 25: 1091–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rondinelli B, Schwerer H, Antonini E, Gaviraghi M, Lupi A, Frenquelli M, Cittaro D, Segalla S, Lemaitre J-M, Tonon G. 2015. H3K4me3 demethylation by the histone demethylase KDM5C/JARID1C promotes DNA replication origin firing. Nucleic Acids Res 43: 2560–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryba T, Hiratani I, Lu J, Itoh M, Kulik M, Zhang J, Schulz TC, Robins AJ, Dalton S, Gilbert DM. 2010. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Res 20: 761–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryba T, Hiratani I, Sasaki T, Battaglia D, Kulik M, Zhang J, Dalton S, Gilbert DM. 2011. Replication timing: a fingerprint for cell identity and pluripotency. PLoS Comput Biol 7: e1002225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiavone D, Guilbaud G, Murat P, Papadopoulou C, Sarkies P, Prioleau M-N, Balasubramanian S, Sale JE. 2014. Determinants of G quadruplex-induced epigenetic instability in REV1-deficient cells. EMBO J 33: 2507–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider GF, Dekker C. 2012. DNA sequencing with nanopores. Nat Biotechnol 30: 326–328. [DOI] [PubMed] [Google Scholar]
- Sequeira-Mendes J, Díaz-Uriarte R, Apedaile A, Huntley D, Brockdorff N, Gómez M. 2009. Transcription initiation activity sets replication origin efficiency in mammalian cells. PLoS Genet 5: e1000446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Kachirskaia I, Yamaguchi H, West LE, Wen H, Wang EW, Dutta S, Appella E, Gozani O. 2007. Modulation of p53 function by SET8-mediated methylation at lysine 382. Mol Cell 27: 636–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shima N, Alcaraz A, Liachko I, Buske TR, Andrews CA, Munroe RJ, Hartford SA, Tye BK, Schimenti JC. 2007. A viable allele of Mcm4 causes chromosome instability and mammary adenocarcinomas in mice. Nat Genet 39: 93–98. [DOI] [PubMed] [Google Scholar]
- Siddiqui K, On KF, Diffley JFX. 2013. Regulating DNA replication in eukarya. Cold Spring Harb Perspect Biol 5: a012930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DJ, Whitehouse I. 2012. Intrinsic coupling of lagging-strand synthesis to chromatin assembly. Nature 483: 434–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spradling AC. 1981. The organization and amplification of two chromosomal domains containing Drosophila chorion genes. Cell 27: 193–201. [DOI] [PubMed] [Google Scholar]
- Stamatoyannopoulos JA, Adzhubei I, Thurman RE, Kryukov GV, Mirkin SM, Sunyaev SR. 2009. Human mutation rate associated with DNA replication timing. Nat Genet 41: 393–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stinchcomb DT, Struhl K, Davis RW. 1979. Isolation and characterisation of a yeast chromosomal replicator. Nature 282: 39–43. [DOI] [PubMed] [Google Scholar]
- Stirling PC, Chan YA, Minaker SW, Aristizabal MJ, Barrett I, Sipahimalani P, Kobor MS, Hieter P. 2012. R-loop-mediated genome instability in mRNA cleavage and polyadenylation mutants. Genes Dev 26: 163–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takawa M, Cho HS, Hayami S, Toyokawa G, Kogure M, Yamane Y, Iwai Y, Maejima K, Ueda K, Masuda A, et al. 2012. Histone lysine methyltransferase SETD8 promotes carcinogenesis by deregulating PCNA expression. Cancer Res 72: 3217–3227. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Nakato R, Katou Y, Shirahige K, Araki H. 2011. Origin association of Sld3, Sld7, and Cdc45 proteins is a key step for determination of origin-firing timing. Curr Biol 21: 2055–2063. [DOI] [PubMed] [Google Scholar]
- Tardat M, Brustel J, Kirsh O, Lefevbre C, Callanan M, Sardet C, Julien E. 2010. The histone H4 Lys 20 methyltransferase PR-Set7 regulates replication origins in mammalian cells. Nat Cell Biol 12: 1086–1093. [DOI] [PubMed] [Google Scholar]
- Thakore PI, Black JB, Hilton IB, Gersbach CA. 2016. Editing the epigenome: technologies for programmable transcription and epigenetic modulation. Nat Methods 13: 127–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valton A-L, Hassan-Zadeh V, Lema I, Boggetto N, Alberti P, Saintomé C, Riou J-F, Prioleau M-N. 2014. G4 motifs affect origin positioning and efficiency in two vertebrate replicators. EMBO J 33: 732–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vashee S, Cvetic C, Lu W, Simancek P, Kelly TJ, Walter JC. 2003. Sequence-independent DNA binding and replication initiation by the human origin recognition complex. Genes Dev 17: 1894–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev L, Johnson EM. 1989. Mapping initiation sites of DNA replication in vivo using polymerase chain reaction amplification of nascent strand segments. Nucleic Acids Res 17: 7693–7705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev LT, Burhans WC, DePamphilis ML. 1990. Mapping an origin of DNA replication at a single-copy locus in exponentially proliferating mammalian cells. Mol Cell Biol 10: 4685–4689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughn JP, Dijkwel PA, Hamlin JL. 1990. Replication initiates in a broad zone in the amplified CHO dihydrofolate reductase domain. Cell 61: 1075–1087. [DOI] [PubMed] [Google Scholar]
- West AG, Huang S, Gaszner M, Litt MD, Felsenfeld G. 2004. Recruitment of histone modifications by USF proteins at a vertebrate barrier element. Mol Cell 16: 453–463. [DOI] [PubMed] [Google Scholar]
- White EJ, Emanuelsson O, Scalzo D, Royce T, Kosak S, Oakeley EJ, Weissman S, Gerstein M, Groudine M, Snyder M, et al. 2004. DNA replication-timing analysis of human chromosome 22 at high resolution and different developmental states. Proc Natl Acad Sci 101: 17771–17776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson TE, Arlt MF, Park SH, Rajendran S, Paulsen M, Ljungman M, Glover TW. 2015. Large transcription units unify copy number variants and common fragile sites arising under replication stress. Genome Res 25: 189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winding P, Berchtold MW. 2001. The chicken B cell line DT40: a novel tool for gene disruption experiments. J Immunol Methods 249: 1–16. [DOI] [PubMed] [Google Scholar]
- Woodfine K, Fiegler H, Beare DM, Collins JE, McCann OT, Young BD, Debernardi S, Mott R, Dunham I, Carter NP. 2004. Replication timing of the human genome. Hum Mol Genet 13: 191–202. [DOI] [PubMed] [Google Scholar]
- Wyrick JJ, Aparicio JG, Chen T, Barnett JD, Jennings EG, Young RA, Bell SP, Aparicio OM. 2001. Genome-wide distribution of ORC and MCM proteins in S. cerevisiae: high-resolution mapping of replication origins. Science 294: 2357–2360. [DOI] [PubMed] [Google Scholar]
- Yang H, Pesavento JJ, Starnes TW, Cryderman DE, Wallrath LL, Kelleher NL, Mizzen CA. 2008. Preferential dimethylation of histone H4 lysine 20 by Suv4-20. J Biol Chem 283: 12085–12092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeeles JTP, Deegan TD, Janska A, Early A, Diffley JFX. 2015. Regulated eukaryotic DNA replication origin firing with purified proteins. Nature 519: 431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Bacal J, Desmarais D, Padioleau I, Tsaponina O, Chabes A, Pantesco V, Dubois E, Parrinello H, Skrzypczak M, et al. 2014. The histone deacetylases sir2 and rpd3 act on ribosomal DNA to control the replication program in budding yeast. Mol Cell 54: 691–697. [DOI] [PubMed] [Google Scholar]
- Zegerman P. 2015. Evolutionary conservation of the CDK targets in eukaryotic DNA replication initiation. Chromosoma 124: 309–321. [DOI] [PubMed] [Google Scholar]
- Zhou J, Ermakova OV, Riblet R, Birshtein BK, Schildkraut CL. 2002. Replication and subnuclear location dynamics of the immunoglobulin heavy-chain locus in B-lineage cells. Mol Cell Biol 22: 4876–4889. [DOI] [PMC free article] [PubMed] [Google Scholar]