

Abstract
Microglia play a central role in the pathogenesis of HIV-associated dementia not only by acting as conduits of viral entry but also as reservoirs for productive and latent virus infection, and as producers of neurotoxins. Interaction between CX3CL1 (fractalkine) and FKN receptor (CX3CR1) is highly functional in the brain, and is known to regulate a complex network of paracrine and autocrine interactions between neurons and microglia. The purpose of the present study was to determine what extent HIV-1 Tat protein causes the alteration of CX3CR1 expression and to investigate the regulatory mechanism for CX3CR1 expression. Here we showed that exposure of primary microglia and BV2 cells to exogenous Tat protein resulted in down-regulation of CX3CR1 expression at both the mRNA and protein levels, with a concomitant induction of proinflammatory responses. Next, we further showed that NF-κB activation by Tat treatment negatively regulated CX3CR1 expression. Since a YY1 binding site ~10kb upstream of CX3CR1 promoter was predicted in rats, mice and humans, the classical NF-κB-YY1 regulatory pathway was considered. Our findings indicated that Tat repressed CX3CR1 expression via NF-κB-YY1 regulatory pathway. To gain insight into the effect of Tat on CX3CL1-CX3CR1 communication, calcium mobilization, MAPK activation and microglial migration, respectively, were tested in microglial cells after successive treatment with Tat and CX3CL1. The results suggested that Tat disrupted the responses of microglia to CX3CL1. Taken together, these results demonstrate that HIV-1 Tat protein suppresses CX3CR1 expression in microglia via NF-κB-YY1 pathway and attenuates CX3CL1-induced functional response of microglia.
Introduction
Over the past three decades, acquired immune deficiency syndrome (AIDS) caused by the human immunodeficiency virus type-1 (HIV-1) has remained the one of the most serious infectious disease challenges facing public health on a global level. In the era of combined active antiretroviral therapy (cART), HIV infection has evolved from being a death sentence to a more manageable chronic condition, with the advantage of successful virus suppression and increased longevity of the infected individuals. However, lack of penetration of some of the antiretroviral regimens, persistent inflammation and co-morbid conditions has resulted in increased prevalence of complications correlated with HIV-1-associated neurological disorders (HAND). HAND encompasses a diverse spectrum of clinical manifestations involving cognitive, motor and behavioral symptomatology [1]. Both viral and cellular factors play key roles in amplification of the pathological cascade in the CNS, which in part, involves impaired function and activation state of the resident microglia [2].
In the CNS, microglia are the primary target cells promoting productive HIV-1 replication and this process in turn, is accompanied by uptake and release of toxic viral proteins such as the transactivator Tat and the envelope gp120 [3]. HIV Tat is a virally encoded regulatory protein that promotes virus replication, can be released by HIV-infected cells into the extracellular space, cerebrospinal fluid (CSF) and sera and be taken up by neighboring uninfected cells [4,5]. Previous studies have demonstrated that treatment of microglial cells with Tat resulted in increased microglial activation with the induction of generation of superoxide, tumor necrosis factor-α (TNF-α), phagocytosis and nitrite release, which in turn, cumulatively resulted in exacerbated pathology associated with HAND [6–9].
The G protein-coupled receptor CX3CR1 is expressed primarily by microglia in the CNS [10]. The specific ligand for CX3CR1 is the chemokine CX3CL1, which is expressed in the neurons, as a transmembrane protein and binds to CX3CR1 on microglia with a high affinity. CX3CL1/CX3CR1 axis establishes a unique communication system between neurons and microglia in the brain [10–12]. CX3CL1 binds to CX3CR1 in a membranes-bound form or as a shed/secreted form after proteolysis by ADAM10 and ADAM17 metalloproteases, respectively [13]. This pattern of cross talk plays an important role in microglial activation and neuroinflammation [10–12]. Changes in expression of CX3CR1 and impaired CX3CL1/CX3CR1 interaction are known to play critical roles in CX3CL1-dependent cellular functions. For example, IL-15 has been reported to negatively regulate the expression of CX3CR1 in natural killer (NK) cells and peripheral blood mononuclear cells (PBMC), resulting in decreased responsiveness of these cells to CX3CL1 [14,15]. Similarly, RANKL (receptor activator of nuclear factor-κB ligand) has been shown to reduce the expression of CX3CR1 in RAW 264.7 cells through the PI3K/Akt signaling pathway, resulting in decreased CX3CL1-induced cellular responses [16]. It has also been shown that in the CNS LPS downregulates expression of CX3CR1 in microglia thereby suppressing the functional response of cells to CX3CL1. Furthermore, deficiency of CX3CR1 is known to result in protracted microglial activation in CX3CR1−/− mice [17–19]. Additionally, morphine and HIV-1 Tat has been shown to cumulatively downregulate CX3CR1 expression, thereby implicating that deficits in CX3CL1/CX3CR1 signaling contribute to the synergistic neurotoxic effects of opioids and Tat [20].
Based on these findings we sought to investigate the mechanism(s) by which HIV-1 Tat protein mediated the disruption of CX3CL1/CX3CR1 axis, which in turn, could underlie exaggerated microglial activation and CNS pathology. The data presented herein suggests that HIV-1 Tat can downregulate CX3CR1 expression in primary rat microglial cells and BV2 cells at both the mRNA and protein levels. The mechanism by which Tat downregulated CX3CR1 in microglia involved the NF-κB-YY1 regulatory pathway. Importantly, CX3CR1 down-regulation induced by Tat decreased the responsiveness of microglia cells to CX3CL1.
Methods
Reagents and Plasmids
HIV Tat (1–72, derived from HIV-1 clade B) and mutant Tat Δ31–61 were obtained from UK College of Medicine, Lexington, KY. Recombinant Rat CX3CL1 was purchased from R&D Systems (Minneapolis, MN, USA). Iκκ-2 inhibitor SC514 was ordered from Sigma Chemicals (St. Louis, MO, USA). The YY1 expression plasmid and short hairpin (sh) RNA plasmid were gifts from Y. Shi (Harvard University) and used as described previoulsy [21,22]. The pCMV-p65 and pIκBα-mut plasmid were gifts from X.M. Cheng (Creighton University).
Cell cultures
Primary cultures of rat mixed glia were prepared from the cerebral cortices of neonatal Sprague-Dawley rats. Briefly, cerebral cortices from neonatal rats were dissected and freed of meninges. Cells were dissociated by mild trypsinization followed by triturating and passing through a nylon mesh. Cells (2×107) were plated in 75 cm2 culture flasks in 20mL culture medium consisted of DMEM supplemented with 10% fetal bovine serum, 2mM L-glutamine, Penicillin (100U/ml)/Streptomycin (100μg/ml) (Invitrogen, Carlsbad, CA, USA). Cultures were kept at 37°C in a humidified incubator gassed with 5% CO2 and air. On 12th to 14th day in vitro, confluent cultures of mixed glia were shaken for 3hr at 180 rpm in a rotary shaker to separate microglia. Detached microglia were plated and cultured for additional 24hr. Human fetal microglia cultures were prepared from fetal brain tissue-derived microglia-astrocytes mixed cultures as previously described [23]. Enriched microglia were plated and cultured in the presence of M-CSF (0.25ng/ml, macrophage colony-stimulating factor; PeproTech, Rocky Hill, NJ) for additional 7d prior to experimentation. BV2, widely used murine microglia cell lines, were grown in Dulbecco’s Modified Eagle’s Medium (DMEM, Invitrogen, Carlsbad, CA, USA) supplemented with 10 % fetal bovine serum, Penicillin/Streptomycin at 37°C in 5% CO2.
Reverse transcription and real-time RT-PCR
Total RNA was extracted with Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Total mRNA (1 μg) was reverse transcribed using cDNA RT Kits (Thermo, Rockford, IL, USA) according to the manufacturer’s manual. Real-time PCR was performed using SYBR Green Master Mix (SABiosciences, Frederick, MD, USA) on an Applied Biosystems 7500 Fast Real-Time PCR System (Foster City, CA, USA). The cycle threshold (Ct) values were analyzed by using the comparative Ct (ΔΔCt) method. The amount of target was obtained by normalizing to the endogenous reference (GAPDH) and shown as the relative to the control (non-treated cells). The following oligonucleotides were used as primers: human CX3CR1 (forward, 5′- GACGGTTGCATTTAGCCATT -3′; reverse, 5′- TGCTCAGAACACTTCCATGC -3′), mouse CX3CR1 (forward, 5′- TGAGTGACTGGCACTTCCTG -3′; reverse, 5′- CGAGGACCACCAACAGATTT -3′), rat CX3CR1 (forward, 5′- TCACCATGCCTACCTCCTTC -3′; reverse, 5′- ACCAGACCGAACGTGAAGAC -3′), mouse YY1 (forward, 5′- ACCTGGCATTGACCTCTCAG -3′; reverse, 5′- TTCTCATGGCCGAGTTATCC -3′), GAPDH (forward, 5′- TGCACCACCAACTGCTTAGC -3′; reverse, 5′- GGCATGGACTGTGGTCATGAG -3′). The primers for TNF-α, IL-1β, LI-6, CXCL10 were purchased from SABiosciences.
Western blotting
Treated cells were lysed using the Mammalian Cell Lysis kit (Sigma, St Louis, MO, USA). Equal amounts of the proteins were electrophoresed in a sodium dodecyl sulfate-polyacrylamide gel (12%) under reducing conditions followed by transfer to PVDF membranes. The blots were blocked with 5% non-fat dry milk in phosphate buffered saline. The western blots were then probed with antibodies recognizing the NF-κB P65, Histone, T-ERK, p-ERK, T-JNK, p-JNK, T-P38, p-P38 (1:200; Cell Signaling Technology, Danvers, MA, USA), YY1 (1:200, Santa-Cruz Biotechnology, Santa Cruz, CA, USA), CX3CR1 (1:200, Abcam, Cambridge, MA, USA) and β-actin (1:4000; Sigma). The secondary antibodies were alkaline phosphatase conjugated to goat anti mouse/rabbit IgG (1:5000). Signals were detected by chemiluminescence and imaged on the FLA-5100 (Fujifilm, Valhalla, NY, USA) digital image scanner; Quantification was performed by densitometry using Image J software (NIH).
Immunochemistry
For immunocytochemistry cells were plated on coverslips. After treatment, cells were fixed with 4% paraformaldehyde for 15 min at room temperature followed by permeabilization with 0.3% Triton X-100 in PBS. Cells were then incubated with a blocking buffer containing 10% NGS in PBS for 1h at room temperature followed by incubation of cells with rabbit anti-CX3CR1 (1:100; Abcam, Cambridge, MA, USA) overnight at 4°C. The secondary AlexaFluor 488 goat anti-rabbit IgG was then added at a 1:500 dilution for 2h to detect the expression of CX3CR1. Cells were washed 3 times and mounted with ProLong® Gold antifade reagent with DAPI onto slides (Invitrogen, Carlsbad, CA, USA).
Transfection
Plasmid transfection was performed using Lipofectamine reagent as suggested by the manufacturer (Invitrogen, Carlsbad, CA, USA). Briefly, BV2 cells were seeded in 24-well plate at 3×105 cells/well for 24 hrs, following which the medium was changed and replaced with serum free OPTI-MEM (Gibco). For transfection, 0.8μg DNA vector in 100μl of serum-free medium was mixed with 2μl lipofectamine 2000, incubated at room temperature for 20 min and then added to the cell culture. Following gentle shaking and spinning at 1200g for 5 min, transfected BV2 cells were cultured at 37°C for 24h.
Chromatin immunoprecipicocaineion (ChIP) Assay
ChIP assay was performed according to the manufacturer’s instructions (Millipore, Temecula, CA, USA). The purified DNA was subjected to PCR to identify the promoter region containing consensus YY1 binding site (C/G)(G/T/A)CCATNTTN. The sequence of the primers used to identify the CX3CR1 promoter bound by transcription factor YY1 was as follows: sense: 5′- GCTGGGCTTCTGGTTTAATG -3′, anti-sense:5′- ATGCCTGTCTGCTCTCTGGT -3′.
Measurement of calcium transients
The changes in Ca2+ flux were monitored using Fluo-4/AM (Invitrogen, Eugene, OR, USA) according to manufacturers’ instructions. Following treatment, the rat primary microglia cultured on Lab-Tek® Chamber Slides (Thermo, Rockford, IL, USA) were rinsed thrice with the Bath solution (140mM NaCl, 5mM KCl, 1mM CaCl2, 0.5mM MgCl2, 10mM glucose, 5.5mM HEPES, pH 7.4), and incubated in Bath solution containing 5μM Fluo-4/AM at 37°C for 20 min. Cells were then rinsed twice with the Bath solution, followed by addition of 100ng/ml CX3CL1 to the Bath solution and scanned every 10 seconds using confocal microscopy (X400) (510 Meta; Zeiss). Fluorescence was quantified at excitation and emission wavelengths of 488 and 515 nm. All analyses for Ca2+ transients were processed at a single-cell level. Intracellular calcium concentration was expressed as the fluorescence intensity.
Microglial migration
Migration of microglia in vitro was determined using Boyden Chambers (Millipore) as previously described [24,25]. Rat primary microglia was pretreated with Tat (200ng/ml) for 24hrs, collected and washed with PBS and fluorescently labeled with 10μM cell tracker green (Molecular Probes, Eugene, OR) for 10min at 37°C. Labeled cells (2×105 cells) were added to the upper compartment of transwell inserts in serum-free medium. CX3CL1 was placed in the lower chamber. The transwell plates were incubated for 6h at 37°C followed by quantification of microglial migration by measuring the number of migrated cells following detachment of cells from the insert using a Synergy Mx fluorescence plate reader (BioTek Instruments, Winooski, VT, USA).
Statistical analysis
Data were expressed as mean ± standard deviation (SD) from at least three separate experiments. Significance of differences between groups was determined by Student’s t test. Results were judged statistically significant if p<0.05 by analysis of variance.
Result
Tat-induced suppression of microglial CX3CR1 mRNA
To investigate the effect of Tat on the expression of CX3CR1, primary cultured rat and human microglial cells and BV2 cells were treated with Tat (200 ng/ml) followed by extraction of total RNA. In both the cell types treatment with Tat significantly decreased CX3CR1 mRNA in a time-dependent manner as demonstrated by real-time PCR analysis (Fig. 1A). As expected, mutant Tat Δ31–61 or heated Tat failed to reduce expression of CX3CR1 (Fig. 1B). Additionally, Tat exposure resulted induction of microglial activation-related genes including TNF-α, MCP-1 and CXCL10 (Fig. 1C). These results demonstrated that suppression of CX3CR1 occurred in parallel to Tat-mediated induction of proinflammatory responses.
Fig. 1. Tat-induced suppression of microglial CX3CR1 mRNA.
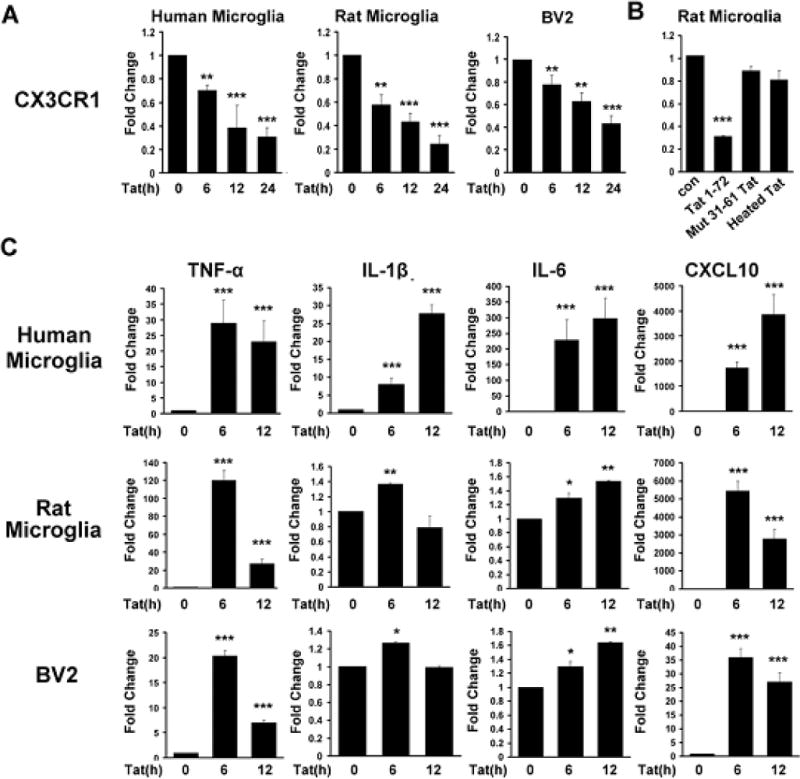
Microglia were stimulated with Tat (1–72, 200ng/ml) mutant Tat (Δ31–61) or heated Tat for 24hrs for the indicated time periods and analyzed for the expression of mRNA for CX3CR1 (A–B) and pro-inflammatory cytokines (C) by real-time RT-PCR and normalized to mRNA levels of GAPDH. All the data are indicated as mean±SD of triplicate independent experiments. *p<0.05, **p<0.01, ***p<0.001 vs control group.
Tat-induced suppression of microglial CX3CR1 protein
Based on the mRNA results we next sought to determine the effect of Tat on the expression of CX3CR1 protein in primary cultured microglial cells and BV2 cells. The results demonstrated that Tat treatment markedly reduced CX3CR1 expression as shown by both flow cytometry (Fig. 2A) and western blotting (Fig. 2B). These findings were further confirmed by immunostaining as evidenced by the fact that Tat treatment resulted in decreased expression of CX3CR1 in primary human, rat microglial cells as well as BV2 cells (Fig. 2C). Taken together, these data suggested that Tat treatment suppressed the expression of CX3CR1 protein in microglia.
Fig. 2. Tat-induced down regulation of microglial CX3CR1 protein.
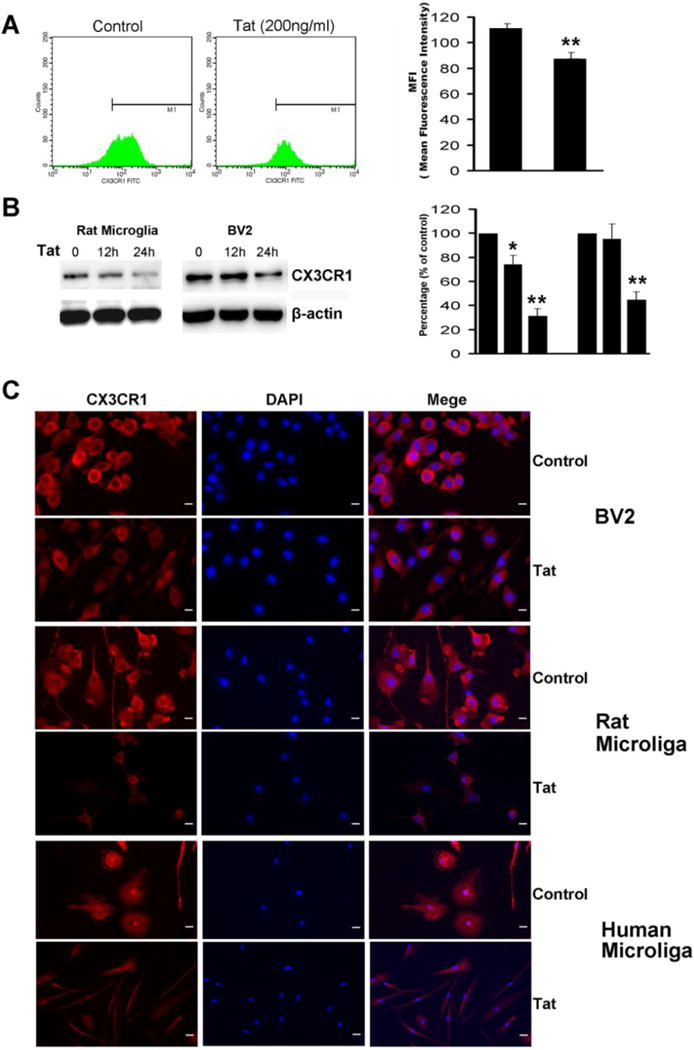
A) Microglial cells were treated with Tat (1–72, 200ng/ml) for the indicated times. Surface expression of CX3CR1 in rat microglia was analyzed by flow cytometry. B) Cell extracts were prepared from treated cultures and analyzed by immunoblotting for expression of CX3CR1 protein. C) Microglial cells treated with Tat were stained using antibodies specific for CX3CR1 (red). Nuclei were stained with DAPI (Blue). Representative images from three independent experiments are presented. Scale bars: 20μm. All the data are presented as mean±SD of three individual experiments.*p<0.05, **p<0.01 vs control group.
NF-κB negatively regulates CX3CR1 expression in microglial cells
It is well recognized that NF-κB signaling plays critical role in the activation of microglia following HIV infection [6,9,26]. Next, we sought to dissect the role of NF-κB in down regulation of CX3CR1 expression as well as concomitant microglial activation. As shown in Figure 3A, treatment of primary rat microglia with Tat protein resulted in increased nuclear translocation of NF-κB. Next, to validate the role of NF-κB we employed both a pharmacological (using a specific inhibitor of inhibitory Kappa B kinase (IKK) SC514 [15μM, IC50 = 14.5μM] as well as a genetic approach [pCMV-p65 and dominant-negative vector (pIκBα-mut)]. Real time RT-PCR results demonstrated that inhibition of NF-κB signaling reversed Tat-induced down-regulation of CX3CR1 expression, and that transfection of p65 resulted in increased inhibition of CX3CR1 mRNA level in BV2 cells (Fig. 3B). Following transfection of BV2 cells with either the pCMV-p65 or pIκBα-mut, CX3CR1 protein expression mirrored the effects observed at the mRNA level (Fig. 3C and D). These findings thus suggested the role of NF-κB activation in Tat-induced suppression of CX3CR1 expression. Intriguingly and as expected, transfection of BV-2 cells with the pIκbα-mut vector also resulted in abrogation of Tat-mediated activation of microglia (data not shown).
Fig. 3. NF-κB negatively regulates CX3CR1 expression in microglial cells.
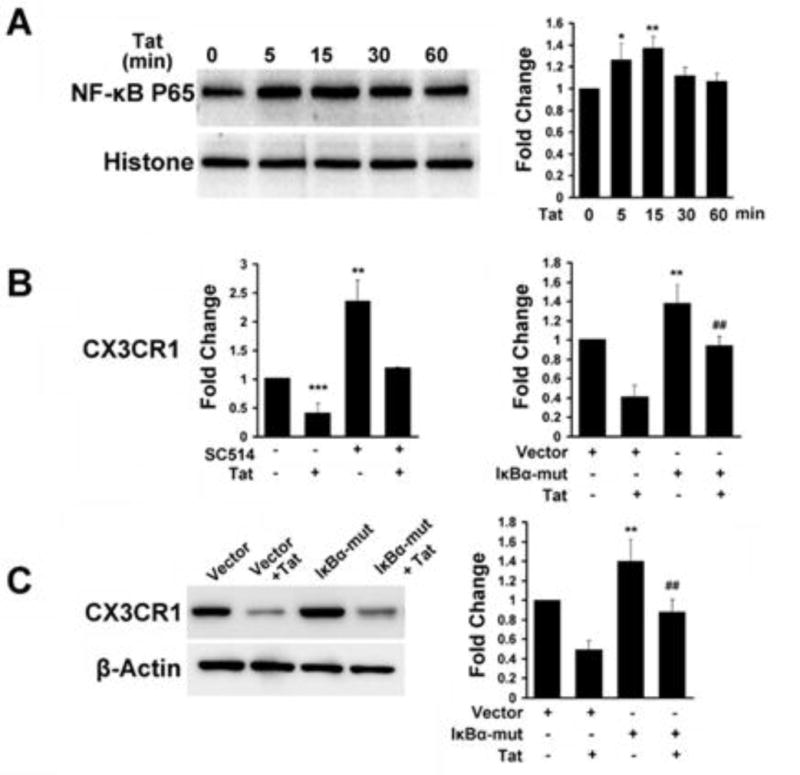
A) Primary rat microglia was treated with Tat (1–72, 200ng/ml) for the indicated times, and cell extracts were prepared and analyzed by immunoblotting for NF-κB p65 in the nuclear fractions. B) Cells were pretreated with SC514, followed by treatment of primary rat microglia with HIV Tat for 12h. BV2 cells were also treated with Tat for 12h following transfection with either pCMV-p65 or pIκBα-mut plasmids. CX3CR1 mRNA levels were then measured by real-time RT-PCR. C) Following transfection of cells with the IκBα-mut plasmid, BV2 cells were treated with Tat (for 24h, and assessed for CX3CR1 protein levels by western blot. All the data are presented as mean±SD of three individual experiments. *p<0.05, **p<0.01, ***p<0.001 vs vector group. #p<0.05, ##p<0.01 vs IκBα-mut alone.
NF-κB-YY1 Negatively Regulates CX3CR1 in Microglial cells
It has been previously documented that NF-κB activation regulates the transcriptional repressor YY1, which in turn, inhibits transcription of myofibrillar genes and miR-29 expression in skeletal myogenesis [22,27]. These findings prompted us to investigate whether the repressor YY1 also played a role in Tat-mediated down regulation of CX3CR1. As predicted from TFSEARCH (http://www.cbrc.jp/research/db/TFSEARCH.html), there is a putative YY1 binding site (C/G) (G/T/A)CCATNTTN) ~10 kB upstream of CX3CR1 promoter in rats, mice and humans. In order to examine whether YY1 physically bound to the CX3CR1 promoter, CHIP assays were performed. Intriguingly, treatment of BV2 cells with Tat resulted in the binding of YY1 with the CX3CR1 promoter, thereby suggesting that YY1 binds to a putative regulatory element of the CX3CR1 promoter (Fig. 4A). To further investigate the regulation of CX3CR1 by YY1, BV2 cells were transfected with YY1 overexpression plasmid. As expected, using quantitative real-time RT-PCR, CX3CR1 levels decreased almost 0.68-fold in the presence of YY1 (Fig. 4B). In contrast, transfection of cells with shRNA plasmid that knocked down the expression of YY1 resulted in significant enhancement of expression of CX3CR1 (Fig. 4B). These findings implicated the role of YY1 as a repressor of CX3CR1. These findings were also confirmed at the protein level as demonstrated in Fig. 4C.
Fig. 4. NF-κB-YY1 negatively regulates CX3CR1 expression in microglial cells.
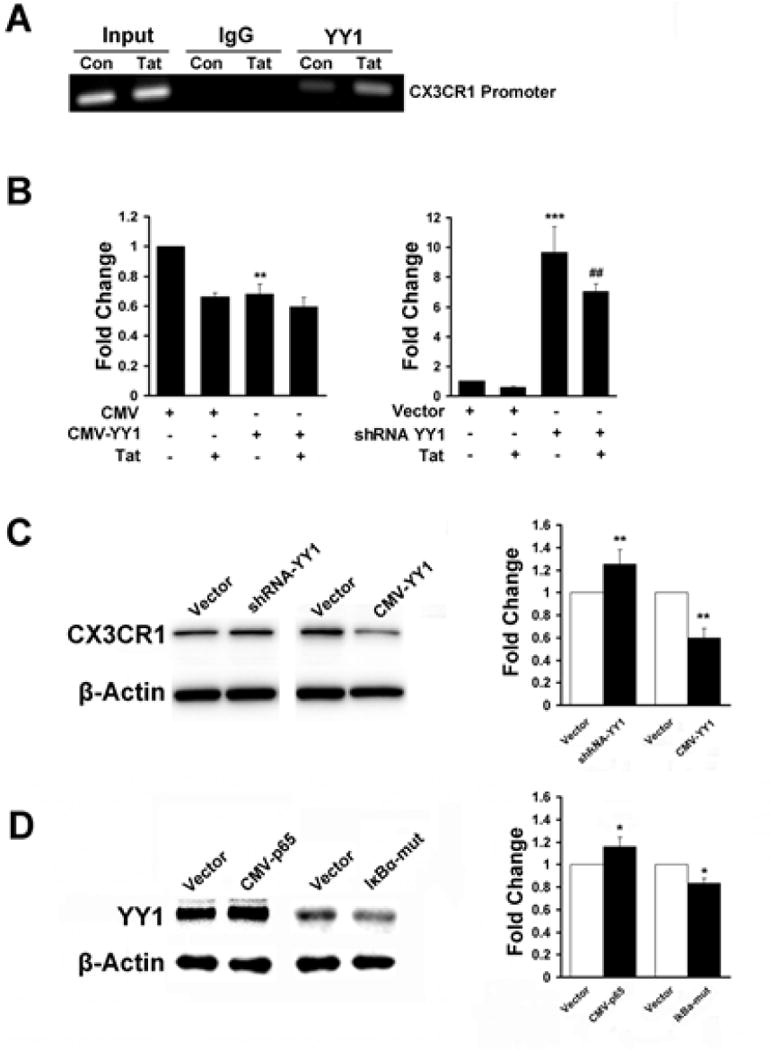
A) CHIP assays with either the YY1 or control IgG was performed on chromatin derived from either control or Tat treated BV2 cells. B) BV2 cells were transfected with either pCMV-YY1 or the shRNA-YY1 plasmid followed by treatment with Tat for 12h, and assessed for CX3CR1 mRNA levels by real-time RT-PCR. C) Following transfection of BV2 cells with either the pCMV-YY1 or shRNA-YY1 plasmid, cells were treated with Tat for 24h, and assessed for CX3CR1 protein levels by western blotting. D) BV2 cells were transfected with either the pCMV-p65 or pIκBα-mut plasmid and assessed for YY1 expression by western blotting. All the data are presented as mean±SD of three individual experiments.*p<0.05, **p<0.01, ***p<0.001 vs control group. ##p<0.01 vs shRNA-YY1 alone.
In order to examine the relationship between NF-κB and YY1, BV2 cells were transfected with either the pCMV-p65 or the pIκBα-mut plasmid and assessed for expression of YY1. As shown in Fig. 4D, overexpressing p65 resulted in increased expression of YY1 and in contrast, transfection of cells with the pIκBα-mut plasmid inhibited expression of the repressor. These data thus suggested that activation of the NF-κB-YY1 pathway mediated suppression of CX3CR1 expression.
Tat-induced down-regulation of CX3CR1 dampens CX3CL1-induced calcium transients
A common feature of chemokine receptors is their capacity to mobilize calcium following interaction with their specific ligand(s). Previous studies have shown that CX3CL1 stimulates a rapid increase in [Ca2+]i transients in microglial cells in the presence of its functional receptor [10,28]. To investigate the effect of CX3CR1 down regulation on calcium influx, we treated microglial cells with Tat for 24h and assessed the changes in [Ca2+]i transients. As shown in Fig. 5, pretreatment of cells with Tat resulted in dampening of the CX3CL1 (100ng/ml)-mediated calcium response, suggesting thereby that Tat-induced down-regulation of CX3CR1 expression contributed to the decreased [Ca2+]i response of cells to CX3CL1.
Fig. 5. Tat-induced down-regulation of CX3CR1 dampens CX3CL1-induced calcium mobilization.
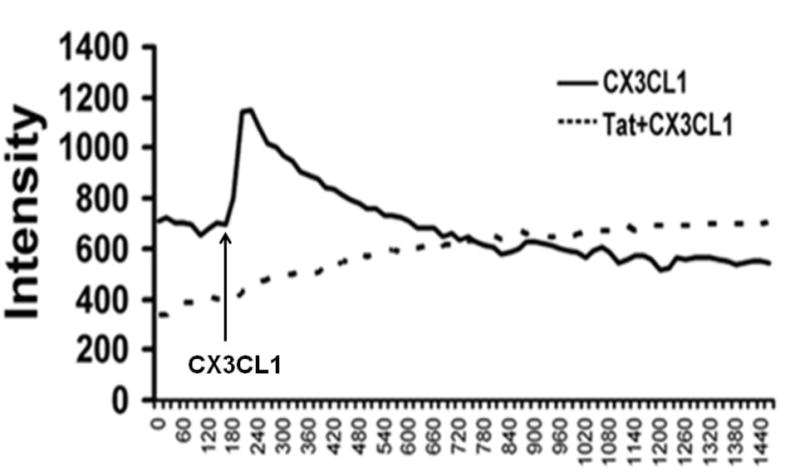
Response curve of the peak amplitude of the [Ca2+] response to CX3CL1 (100ng/ml) in non-treated and Tat (1–72, 200ng/ml) treated for 24h. Rat primary microglia was incubated with Fura-4AM followed by examination of intracellular [Ca2+] influx using confocal microscope.
Tat-induced down-regulation of CX3CR1 dampens CX3CL1-mediated activation of MAPK
CX3CL1 is reportedly involved in the activation of various intracellular signaling pathways, including ERK1/2, JNK, and p38 MAPKs, and PI3K [28–32]. Having determined that Tat significantly down regulated the expression of CX3CR1 we sought to examine whether Tat treatment that leads to reduced expression of CX3CR1 would also result in abrogation of CX3CL1 mediated activation of MAPK and PI3K. Pretreatment of microglia with Tat for 24h followed by treatment with CX3CL1 (100ng/ml) resulted in inhibition of CX3CL1-mediated activation of ERK and JNK MAPKs compared to cells not treated with Tat (Fig. 6). These results thus demonstrate that microglial cells in the presence of Tat exhibited reduced ability to respond to CX3CL1.
Fig. 6. Tat-induced down-regulation of CX3CR1 dampens CX3CL1-induced activation of MAPK.
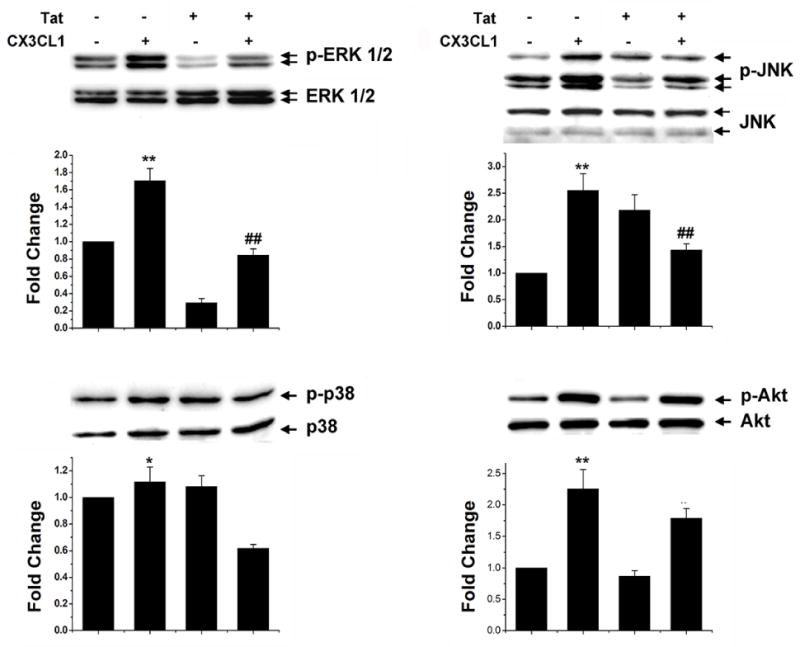
Rat primary microglia was pretreated with HIV or left untreated for 24h, followed by exposure of cells to CX3CL1 (100ng/ml) for 30min. Whole cell lysates of the treated cells were then analyzed for activation of MAPKs and PI3K using western blot. Representative blots are shown from three individual experiments.
Tat-induced down-regulation of CX3CR1 dampens CX3CL1-mediated migration of microglia
The CX3CL1/CX3CR1 axis plays a key role in microglia migration [28,33]. We thus sought to investigate the effects of CX3CL1 on microglial movement in cells pretreated with Tat, using the Boyden Chamber. Our findings demonstrated that as expected, in the presence of varying concentrations of recombinant soluble CX3CL1 microglia exhibited stronger migratory activity with maximal migration at 300ng/ml CX3CL1 (Fig. 7A). In cells pretreated with Tat however, there was a significant attenuation of migration in response to CX3CL1 (Fig. 7B), suggesting thereby that Tat-induced down regulation of CX3CR1 contributes to the reduced migration of microglia.
Fig. 7. Tat-mediated down-regulation of CX3CR1 dampens CX3CL1-induced migration of microglia.
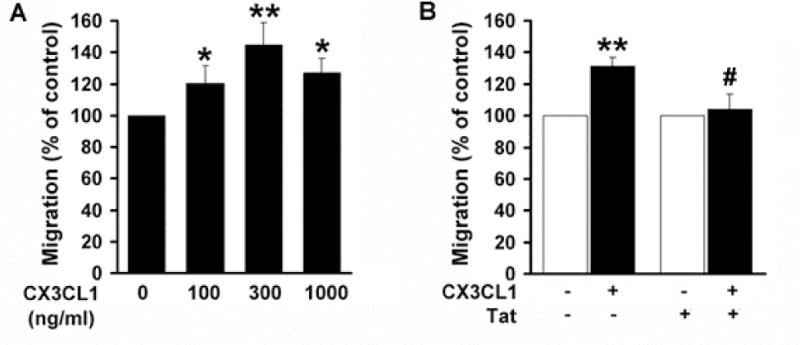
A) Concentration-dependent chemotaxis of microglia in response to CX3CL1 was measured using Boyden chambers. B) Pretreatment of rat primary microglia with HIV Tat for 24h resulted in reduced responsiveness of cells to CX3CL1. * p<0.05, **p<0.01 vs control group; (#p<0.01 vs CX3CL1-treated group. All the data are presented as mean±SD of three individual experiments.
Discussion
A key aspect of brain function is the bidirectional interplay and cross talk between the neurons and the neighboring glial cells. This has largely been possible due to the ligand-receptor axis that facilitates communication between various cell types. Growing evidence demonstrates that the regulation of chemokine receptor expression during cell activation or deactivation is an important inducible immune response, often elicited during normal and/or pathologic conditions. For example, Human Herpesvirus 8 (HHV-8) infection alters the chemokine receptor expression in dendritic cells (DCs) and chemokine-induced migration [34]; Platelet-derived CXC chemokine ligand 4 (CXCL4) induces down-regulation of CC chemokine receptors (CCR) 1, −2, and −5, resulting in dramatic impairment of monocyte chemotactic migration towards their cognate CC chemokine ligands (CCL) for these receptors [35]; Primary EBV infection of tonsillar B cells leads to a reduced cell surface expression of CCR7 and CXCR5, as well as to altered expression of several chemokine receptors and chemokines [36].
In the CNS, the chemokine CX3CL1, is constitutively expressed in neuronal cells, while its G protein coupled receptor, CX3CR1, is primarily expressed on the resident brain microglia and on peripheral immune cells including macrophages [10]. A well-organized reciprocal communication between neurons and microglia is essential for maintaining homeostasis in the CNS during both physiological and inflammatory conditions. The CX3CL1/CX3CR1 axis has evolved as a communication link between neurons and microglial cells. Intriguingly, while on one hand CX3CL1 is known to prevent excess microglial activation in the absence of injury, on the other hand, it is capable of promoting microglial activation in the presence of a toxic inflammatory stimuli. CX3CL1 is also unique as its chemokine domain is tethered to the cell membrane by a long, highly glycosylated stem attached to a transmembrane and the intracellular domain [13]. It can be cleaved by metalloproteinases such as ADAM 10 [37]. Thus both soluble and insoluble forms of CX3CL1 exist and while both forms are known to mediate chemotaxis, membrane bound form is the one that captures the microglia and dampens glial activation.
Previous reports have demonstrated that microbial agents and/or inflammatory mediators can mediate down-regulation of CX3CR1 [16,19,38,39]. In fact, in three different clinically relevant in vivo models including systemic inflammation (LPS), MPTP model of Parkinson’s disease and a model of ALS, CX3CR1 deficiency has been shown to play a key role in microglial neurotoxicity [12].
The current study was undertaken to examine the potential effect of HIV Tat on disruption of CX3CL1/CX3CR1 signaling and to investigate the molecular mechanism(s) involved in this process. Our findings demonstrated that Tat exposure of microglial cells significantly down-regulated the expression of CX3CR1 both at the mRNA and protein levels. Furthermore, Tat-induced down-regulation of CX3CR1 expression was accompanied with concomitant activation of microglia as evidenced by up-regulated expression of pro-inflammatory mediators such as TNF-α, IL-1β, IL-6 and CXCL10. Interestingly pretreatment of cells with Tat followed by exposure to CX3CL1 also resulted in decreased activation of both ERK and JNK mitogen-activated protein kinases (MAPK). Since CX3CR1 ligation is known to trigger phosphatidylinositol-3 kinase (PI3K) dependent Ca2+ influx, with downstream activation of MAPK, it is not surprising that Tat-mediated downregulation of CX3CR1 dampened this effect. It should be mentioned that Tat is known to also activate inflammatory mediators and MAPK. A possible explanation therefore for downregulation of MAPK in Tat-pretreated cells in response to CX3CL1 could be attributed to the timing of the effect. It is often the early and acute effects of Tat that lead to time-dependent early activation of MAPK [40]. In our study the cells were pretreated with Tat for 24 hrs followed by treatment with CX3CL1 for 30min and then assessed for activation of MAPK. It is likely that the effects observed in our system could be attributed to dampened effects of reduced CX3CR1 expression. Our findings also implicated the role of the NF-κB-YY1 pathway Tat-mediated down regulation of CX3CR1. Functional implications of these findings were demonstrated in Tat-treated microglia, which exhibited dampened response to: CX3CL1-induced calcium mobilization, MAPKs activation and migration.
Over the last few years, there have been escalating reports implying that the absence of CX3CR1 is associated with a worse outcome possibly due to the lack of CX3CL1-mediated capture of microglia resulting in activation and neuroinflammation [12,41–43]. Conflicting reports have also suggested that loss of signaling via CX3CR1 can either be protective [44–46] or have no effect at all [47,48]. These disparate observations indicate complexity and disease-specific regulation of the neuron-glia cross-talk via the CX3CL1/CX3CR1 axis. Interestingly, in the acute models of CNS injury (transient and permanent brain ischemia) absence of CX3CR1 has been shown to reduce ischemic damage and inflammation [44,49,50]. While the role of microglial activation, generation of inflammatory mediators and phagocytosis has been well studied in the context of CX3CR1 deficiency, it remains less clear as to how CX3CR1 deficiency impacts the microglia in the context of HIV protein Tat. Viral Tat is secreted by HIV-1-infected cells and is taken up by neighboring uninfected cells [5]. This protein has also been shown to activate microglia [51,52], resulting in both loss of supportive function for the neurons [53] while also inflicting neuronal injury via the release of inflammatory mediators.
Given the down-regulation of CX3CR1 expression in cells exposed to Tat protein, we explored the mechanism(s) underlying this process. Previous studies have shown that CX3CR1 expression can be regulated via activation of the PI3K/Akt [16] and TGF-β1 pathways [32]. However, an important mechanism involved in chemokine receptor regulation is that at the messenger RNA (mRNA) level, via both transcriptional regulation and increased stability of the corresponding mRNA. Our findings demonstrate that both the transcription factors NF-κB and YY1 negatively regulate CX3CR1 expression in microglia. Gain and loss-of-function studies demonstrate that Tat protein activates the NF-κB (p65) pathway with a concomitant increase in YY1 expression as summarized in Figure 8. Interestingly, it has been previously shown that NF-κB-mediated regulation of YY1 inhibits both gene and mRNA expression [22,27]. Intriguingly, our findings identified a regulatory element containing a YY1 binding site located ~10 kb upstream from the CX3CR1 promoter and demonstrated the binding of the repressor to the CX3CR1 promoter, thus revealing a novel mechanism of the involvement of the NF-κB-YY1 pathway in Tat-induced down-regulation of CX3CR1 in microglia.
Fig. 8. Schematic illustration demonstrating the role of NF-κB/repressor YY1 in Tat-mediated the decreased expression of CX3CR1.
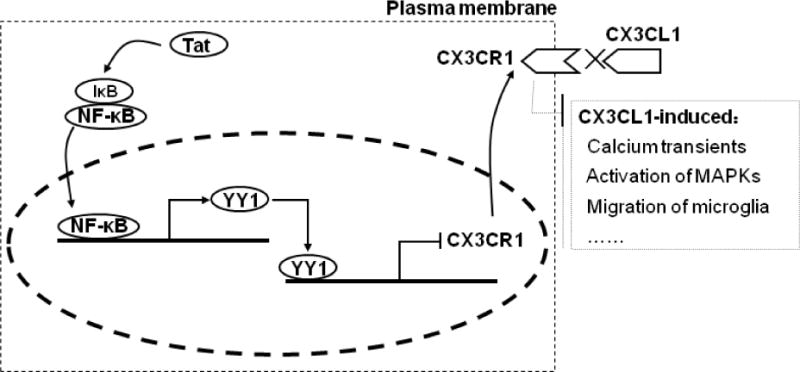
Tat treatment resulted in activation of NF-κB/YY1 with subsequent decreased expression of CX3CR1. Tat protein suppresses CX3CR1 expression in microglia via NF-κB-YY1, which attenuates CX3CL1-induced functional response of microglia such as calcium transients, activation of MAPK s and migration of microglia.
In the CNS, activation of CX3CR1 results in an increase in intracellular calcium levels [10,28], the phosphorylation of the MAPKs and PI3K in microglia [28,29,33]. Our findings showed that Tat exposure resulted in dampening of the CX3CL1-induced calcium mobilization and MAPK activation in microglia, suggesting that Tat exposure disrupts the CX3CL1/CX3CR1 communication axis via down-regulation expression of CX3CR1. Our findings are thus in agreement with previous reports that have demonstrated that microbial agents and/or inflammatory mediators such as Tat can mediate down-regulation of microglial CX3CR1 [16,19,38,39] leading ultimately to microglial neurotoxicity [12]. Clearly, additional studies are warranted to further define the role of CX3CL1/CX3CR1 signaling in HAND pathogenesis.
Acknowledgments
This work was supported by grants MH068212-08, DA027729, DA033150, DA036157, DA035203 from the National Institutes of Health in addition to Nebraska Tobacco Settlement Biomedical Research.
Footnotes
Disclosure
The authors declare no competing financial interests.
References
- 1.Antinori A, Arendt G, Becker JT, Brew BJ, Byrd DA, et al. Updated research nosology for HIV-associated neurocognitive disorders. Neurology. 2007;69:1789–1799. doi: 10.1212/01.WNL.0000287431.88658.8b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yadav A, Collman RG. CNS inflammation and macrophage/microglial biology associated with HIV-1 infection. J Neuroimmune Pharmacol. 2009;4:430–447. doi: 10.1007/s11481-009-9174-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bansal AK, Mactutus CF, Nath A, Maragos W, Hauser KF, et al. Neurotoxicity of HIV-1 proteins gp120 and Tat in the rat striatum. Brain Res. 2000;879:42–49. doi: 10.1016/s0006-8993(00)02725-6. [DOI] [PubMed] [Google Scholar]
- 4.Hudson L, Liu J, Nath A, Jones M, Raghavan R, et al. Detection of the human immunodeficiency virus regulatory protein tat in CNS tissues. J Neurovirol. 2000;6:145–155. doi: 10.3109/13550280009013158. [DOI] [PubMed] [Google Scholar]
- 5.Chang HC, Samaniego F, Nair BC, Buonaguro L, Ensoli B. HIV-1 Tat protein exits from cells via a leaderless secretory pathway and binds to extracellular matrix-associated heparan sulfate proteoglycans through its basic region. AIDS. 1997;11:1421–1431. doi: 10.1097/00002030-199712000-00006. [DOI] [PubMed] [Google Scholar]
- 6.Bruce-Keller AJ, Barger SW, Moss NI, Pham JT, Keller JN, et al. Pro-inflammatory and pro-oxidant properties of the HIV protein Tat in a microglial cell line: attenuation by 17 beta-estradiol. J Neurochem. 2001;78:1315–1324. doi: 10.1046/j.1471-4159.2001.00511.x. [DOI] [PubMed] [Google Scholar]
- 7.Polazzi E, Levi G, Minghetti L. Human immunodeficiency virus type 1 Tat protein stimulates inducible nitric oxide synthase expression and nitric oxide production in microglial cultures. J Neuropathol Exp Neurol. 1999;58:825–831. doi: 10.1097/00005072-199908000-00005. [DOI] [PubMed] [Google Scholar]
- 8.Turchan-Cholewo J, Dimayuga FO, Gupta S, Keller JN, Knapp PE, et al. Morphine and HIV-Tat increase microglial-free radical production and oxidative stress: possible role in cytokine regulation. J Neurochem. 2009;108:202–215. doi: 10.1111/j.1471-4159.2008.05756.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Minghetti L, Visentin S, Patrizio M, Franchini L, Ajmone-Cat MA, et al. Multiple actions of the human immunodeficiency virus type-1 Tat protein on microglial cell functions. Neurochem Res. 2004;29:965–978. doi: 10.1023/b:nere.0000021241.90133.89. [DOI] [PubMed] [Google Scholar]
- 10.Harrison JK, Jiang Y, Chen S, Xia Y, Maciejewski D, et al. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc Natl Acad Sci U S A. 1998;95:10896–10901. doi: 10.1073/pnas.95.18.10896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Limatola C, Lauro C, Catalano M, Ciotti MT, Bertollini C, et al. Chemokine CX3CL1 protects rat hippocampal neurons against glutamate-mediated excitotoxicity. J Neuroimmunol. 2005;166:19–28. doi: 10.1016/j.jneuroim.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 12.Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9:917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- 13.Bazan JF, Bacon KB, Hardiman G, Wang W, Soo K, et al. A new class of membrane-bound chemokine with a CX3C motif. Nature. 1997;385:640–644. doi: 10.1038/385640a0. [DOI] [PubMed] [Google Scholar]
- 14.Sechler JM, Barlic J, Grivel JC, Murphy PM. IL-15 alters expression and function of the chemokine receptor CX3CR1 in human NK cells. Cell Immunol. 2004;230:99–108. doi: 10.1016/j.cellimm.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 15.Barlic J, Sechler JM, Murphy PM. IL-15 and IL-2 oppositely regulate expression of the chemokine receptor CX3CR1. Blood. 2003;102:3494–3503. doi: 10.1182/blood-2003-03-0946. [DOI] [PubMed] [Google Scholar]
- 16.Saitoh Y, Koizumi K, Sakurai H, Minami T, Saiki I. RANKL-induced down-regulation of CX3CR1 via PI3K/Akt signaling pathway suppresses Fractalkine/CX3CL1-induced cellular responses in RAW264.7 cells. Biochem Biophys Res Commun. 2007;364:417–422. doi: 10.1016/j.bbrc.2007.09.137. [DOI] [PubMed] [Google Scholar]
- 17.Corona AW, Huang Y, O’Connor JC, Dantzer R, Kelley KW, et al. Fractalkine receptor (CX3CR1) deficiency sensitizes mice to the behavioral changes induced by lipopolysaccharide. J Neuroinflammation. 2010;7:93. doi: 10.1186/1742-2094-7-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boddeke EW, Meigel I, Frentzel S, Biber K, Renn LQ, et al. Functional expression of the fractalkine (CX3C) receptor and its regulation by lipopolysaccharide in rat microglia. Eur J Pharmacol. 1999;374:309–313. doi: 10.1016/s0014-2999(99)00307-6. [DOI] [PubMed] [Google Scholar]
- 19.Wynne AM, Henry CJ, Huang Y, Cleland A, Godbout JP. Protracted downregulation of CX3CR1 on microglia of aged mice after lipopolysaccharide challenge. Brain Behav Immun. 2010;24:1190–1201. doi: 10.1016/j.bbi.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suzuki M, El-Hage N, Zou S, Hahn YK, Sorrell ME, et al. Fractalkine/CX3CL1 protects striatal neurons from synergistic morphine and HIV-1 Tat-induced dendritic losses and death. Mol Neurodegener. 2011;6:78. doi: 10.1186/1750-1326-6-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu S, Hu YC, Liu H, Shi Y. Loss of YY1 impacts the heterochromatic state and meiotic double-strand breaks during mouse spermatogenesis. Mol Cell Biol. 2009;29:6245–6256. doi: 10.1128/MCB.00679-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang H, Garzon R, Sun H, Ladner KJ, Singh R, et al. NF-kappaB-YY1-miR-29 regulatory circuitry in skeletal myogenesis and rhabdomyosarcoma. Cancer Cell. 2008;14:369–381. doi: 10.1016/j.ccr.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chao CC, Gekker G, Hu S, Peterson PK. Human microglial cell defense against Toxoplasma gondii. The role of cytokines. J Immunol. 1994;152:1246–1252. [PubMed] [Google Scholar]
- 24.Miller AM, Stella N. Microglial cell migration stimulated by ATP and C5a involve distinct molecular mechanisms: quantification of migration by a novel near-infrared method. Glia. 2009;57:875–883. doi: 10.1002/glia.20813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ryu JK, Cho T, Choi HB, Wang YT, McLarnon JG. Microglial VEGF receptor response is an integral chemotactic component in Alzheimer’s disease pathology. J Neurosci. 2009;29:3–13. doi: 10.1523/JNEUROSCI.2888-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Swingler S, Morris A, Easton A. Tumour necrosis factor alpha and interleukin-1 beta induce specific subunits of NFKB to bind the HIV-1 enhancer: characterisation of transcription factors controlling human immunodeficiency virus type 1 gene expression in neural cells. Biochem Biophys Res Commun. 1994;203:623–630. doi: 10.1006/bbrc.1994.2228. [DOI] [PubMed] [Google Scholar]
- 27.Wang H, Hertlein E, Bakkar N, Sun H, Acharyya S, et al. NF-kappaB regulation of YY1 inhibits skeletal myogenesis through transcriptional silencing of myofibrillar genes. Mol Cell Biol. 2007;27:4374–4387. doi: 10.1128/MCB.02020-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maciejewski-Lenoir D, Chen S, Feng L, Maki R, Bacon KB. Characterization of fractalkine in rat brain cells: migratory and activation signals for CX3CR-1-expressing microglia. J Immunol. 1999;163:1628–1635. [PubMed] [Google Scholar]
- 29.Meucci O, Fatatis A, Simen AA, Miller RJ. Expression of CX3CR1 chemokine receptors on neurons and their role in neuronal survival. Proc Natl Acad Sci U S A. 2000;97:8075–8080. doi: 10.1073/pnas.090017497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cambien B, Pomeranz M, Schmid-Antomarchi H, Millet MA, Breittmayer V, et al. Signal transduction pathways involved in soluble fractalkine-induced monocytic cell adhesion. Blood. 2001;97:2031–2037. doi: 10.1182/blood.v97.7.2031. [DOI] [PubMed] [Google Scholar]
- 31.Noda M, Doi Y, Liang J, Kawanokuchi J, Sonobe Y, et al. Fractalkine attenuates excito-neurotoxicity via microglial clearance of damaged neurons and antioxidant enzyme heme oxygenase-1 expression. J Biol Chem. 2011;286:2308–2319. doi: 10.1074/jbc.M110.169839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen S, Luo D, Streit WJ, Harrison JK. TGF-beta1 upregulates CX3CR1 expression and inhibits fractalkine-stimulated signaling in rat microglia. J Neuroimmunol. 2002;133:46–55. doi: 10.1016/s0165-5728(02)00354-5. [DOI] [PubMed] [Google Scholar]
- 33.Zhang M, Xu G, Liu W, Ni Y, Zhou W. Role of fractalkine/CX3CR1 interaction in light-induced photoreceptor degeneration through regulating retinal microglial activation and migration. PLoS One. 2012;7:e35446. doi: 10.1371/journal.pone.0035446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cirone M, Conte V, Farina A, Valia S, Trivedi P, et al. HHV-8 reduces dendritic cell migration through down-regulation of cell-surface CCR6 and CCR7 and cytoskeleton reorganization. Virol J. 2012;9:92. doi: 10.1186/1743-422X-9-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwartzkopff F, Petersen F, Grimm TA, Brandt E. CXC chemokine ligand 4 (CXCL4) down-regulates CC chemokine receptor expression on human monocytes. Innate Immun. 2012;18:124–139. doi: 10.1177/1753425910388833. [DOI] [PubMed] [Google Scholar]
- 36.Ehlin-Henriksson B, Liang W, Cagigi A, Mowafi F, Klein G, et al. Changes in chemokines and chemokine receptor expression on tonsillar B cells upon Epstein-Barr virus infection. Immunology. 2009;127:549–557. doi: 10.1111/j.1365-2567.2008.03029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hundhausen C, Misztela D, Berkhout TA, Broadway N, Saftig P, et al. The disintegrin-like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1-mediated cell-cell adhesion. Blood. 2003;102:1186–1195. doi: 10.1182/blood-2002-12-3775. [DOI] [PubMed] [Google Scholar]
- 38.Ramos MV, Fernandez GC, Brando RJ, Panek CA, Bentancor LV, et al. Interleukin-10 and interferon-gamma modulate surface expression of fractalkine-receptor (CX(3)CR1) via PI3K in monocytes. Immunology. 2010;129:600–609. doi: 10.1111/j.1365-2567.2009.03181.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pachot A, Cazalis MA, Venet F, Turrel F, Faudot C, et al. Decreased expression of the fractalkine receptor CX3CR1 on circulating monocytes as new feature of sepsis-induced immunosuppression. J Immunol. 2008;180:6421–6429. doi: 10.4049/jimmunol.180.9.6421. [DOI] [PubMed] [Google Scholar]
- 40.Bethel-Brown C, Yao H, Callen S, Lee YH, Dash PK, et al. HIV-1 Tat-mediated induction of platelet-derived growth factor in astrocytes: role of early growth response gene 1. J Immunol. 2011;186:4119–4129. doi: 10.4049/jimmunol.1002235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pabon MM, Bachstetter AD, Hudson CE, Gemma C, Bickford PC. CX3CL1 reduces neurotoxicity and microglial activation in a rat model of Parkinson’s disease. J Neuroinflammation. 2011;8:9. doi: 10.1186/1742-2094-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cho SH, Sun B, Zhou Y, Kauppinen TM, Halabisky B, et al. CX3CR1 protein signaling modulates microglial activation and protects against plaque-independent cognitive deficits in a mouse model of Alzheimer disease. J Biol Chem. 2011;286:32713–32722. doi: 10.1074/jbc.M111.254268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Briones TL, Woods J, Wadowska M. Chronic neuroinflammation and cognitive impairment following transient global cerebral ischemia: role of fractalkine/CX3CR1 signaling. J Neuroinflammation. 2014;11:13. doi: 10.1186/1742-2094-11-13. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 44.Fumagalli S, Perego C, Ortolano F, De Simoni MG. CX3CR1 deficiency induces an early protective inflammatory environment in ischemic mice. Glia. 2013;61:827–842. doi: 10.1002/glia.22474. [DOI] [PubMed] [Google Scholar]
- 45.Cipriani R, Villa P, Chece G, Lauro C, Paladini A, et al. CX3CL1 is neuroprotective in permanent focal cerebral ischemia in rodents. J Neurosci. 2011;31:16327–16335. doi: 10.1523/JNEUROSCI.3611-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tang Z, Gan Y, Liu Q, Yin JX, Shi J, et al. CX3CR1 deficiency suppresses activation and neurotoxicity of microglia/macrophage in experimental ischemic stroke. J Neuroinflammation. 2014;11:26. doi: 10.1186/1742-2094-11-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, et al. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol. 2000;20:4106–4114. doi: 10.1128/mcb.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- 49.Denes A, Ferenczi S, Halasz J, Kornyei Z, Kovacs KJ. Role of CX3CR1 (fractalkine receptor) in brain damage and inflammation induced by focal cerebral ischemia in mouse. J Cereb Blood Flow Metab. 2008;28:1707–1721. doi: 10.1038/jcbfm.2008.64. [DOI] [PubMed] [Google Scholar]
- 50.Donnelly DJ, Longbrake EE, Shawler TM, Kigerl KA, Lai W, et al. Deficient CX3CR1 signaling promotes recovery after mouse spinal cord injury by limiting the recruitment and activation of Ly6Clo/iNOS+ macrophages. J Neurosci. 2011;31:9910–9922. doi: 10.1523/JNEUROSCI.2114-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McManus CM, Weidenheim K, Woodman SE, Nunez J, Hesselgesser J, et al. Chemokine and chemokine-receptor expression in human glial elements: induction by the HIV protein, Tat, and chemokine autoregulation. Am J Pathol. 2000;156:1441–1453. doi: 10.1016/S0002-9440(10)65013-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.D’Aversa TG, Yu KO, Berman JW. Expression of chemokines by human fetal microglia after treatment with the human immunodeficiency virus type 1 protein Tat. J Neurovirol. 2004;10:86–97. doi: 10.1080/13550280490279807. [DOI] [PubMed] [Google Scholar]
- 53.Li W, Li G, Steiner J, Nath A. Role of Tat protein in HIV neuropathogenesis. Neurotox Res. 2009;16:205–220. doi: 10.1007/s12640-009-9047-8. [DOI] [PubMed] [Google Scholar]