

Abstract
Numerous clinical trials are utilizing mesenchymal stem cells (MSC) to treat critical limb ischemia, primarily for their ability to secrete signals that promote revascularization. These cells have demonstrated clinical safety, but their efficacy has been limited, possibly because these paracrine signals are secreted at subtherapeutic levels. In these studies the combination of cell and gene therapy was evaluated by engineering MSC with a lentivirus to overexpress vascular endothelial growth factor (VEGF). To achieve clinical compliance, the number of viral insertions was limited to 1–2 copies/cell and a constitutive promoter with demonstrated clinical safety was used. MSC/VEGF showed statistically significant increases in blood flow restoration as compared with sham controls, and more consistent improvements as compared with nontransduced MSC. Safety of MSC/VEGF was assessed in terms of genomic stability, rule-out tumorigenicity, and absence of edema or hemangiomas in vivo. In terms of retention, injected MSC/VEGF showed a steady decline over time, with a very small fraction of MSC/VEGF remaining for up to 4.5 months. Additional safety studies completed include absence of replication competent lentivirus, sterility tests, and absence of VSV-G viral envelope coding plasmid. These preclinical studies are directed toward a planned phase 1 clinical trial to treat critical limb ischemia.
Introduction
Critical limb ischemia (CLI) is the most severe manifestation of peripheral arterial disease. This atherosclerotic disease affects up to 15% of patients over age 65, with increasing prevalence expected as the incident of diabetes in an aging population increases.1,2 Due to the systemic nature of this disease, which precludes many patients from surgical options because of occluded or diffusely diseased distal arteries, patients are left with nonviable limbs that require amputation. Within this population of no-option patients, only 56% are expected to live with both limbs within 1 year of diagnosis.3 The need for alternative methods of revascularization for these patients is dire.
Previous CLI clinical trials have tested the administration of angiogenic factors as recombinant proteins or as gene therapies.4 Despite the short half-life and poor retention of most recombinant protein formulations, nephrotoxicity and other side effects associated with systemic growth factor administration have been observed in several of these trials.4 Gene therapy represents a more promising alternative, where expression of specific angiogenic growth factors is induced in the ischemic tissue to achieve a more sustained local delivery than what the recombinant protein could provide.
A number of studies have evaluated vascular endothelial growth factor (VEGF) as an angiogenic gene therapy candidate, using both plasmid and adenovirus.5 The trials that had used intramuscular administration of VEGF-A165 as naked DNA transfer and adenoviral-mediated gene transfer had demonstrated increases in collateral blood vessel formation, although transient edema formation was observed.6–8 The RAVE study, which utilized an adenovirus coding for VEGF-A121, demonstrated safety after 1 year of follow up, but unfortunately concluded with no differences in outcome between Ad/VEGF-A121 and placebo treated patients.9 Though clinical efficacy has been disappointing, there has been little evidence of clinically significant toxicity.
Another potential alternative for treatment of ischemic diseases is cell therapy. Multiple trials have tested the administration of mesenchymal stem cells (MSC) for treatment of CLI (reviewed in ref. 10). These cells are ideal for this application due to their ease of isolation and expansion, low immunogenicity in allogeneic settings,11 ability to secrete paracrine factors that stimulate regeneration, and demonstrated safety.12 Hundreds of clinical studies provide extensive safety and provisional efficacy data for nonmatched allogeneic bone marrow-derived MSC administration in patients through FDA-approved clinical trials.11 Furthermore, MSC show tropism toward sites of hypoxia13 and are stimulated to express angiogenic factors in hypoxic environments (reviewed in ref. 14), which make them particularly advantageous for application in ischemic disease. While these cells have shown great promise in animal models, the limited success in human clinical trials demonstrates the need for development of more potent treatment strategies or cell formulations. By combining cell and gene therapy, MSC that are genetically modified to over express VEGF could be an optimal approach, emphasizing all aspects of MSC that make them ideal for treatment of CLI.15 In the current studies, a clinically compliant MSC/VEGF product was developed and tested for efficacy and potential safety risks associated with a combined stem cell and gene therapy product.
Results
Number of viral insertions and VEGF secretion
Generation of MSC/VEGF was done using increasing multiplicities of infection (MOI) under current good manufacturing practices conditions. Bone marrow derived MSC were transduced using MOI 1, 5, or 20 (i.e., 0.4, 2, or 8 µl virus/105 cells) and compared with nontransduced MSC. The number of viral insertions per cell for each MOI was determined by polymerase chain reaction (PCR), as shown in Figure 1a. Here. MOI 1 leads to an average of 0.5 viral insertions per cell. In order to determine whether any single cell has a very high number of viral insertions (increasing the risk of insertional mutagenesis) we addressed the range of insertions by flow cytometry using a lentiviral vector with eGFP in place of VEGF (Supplementary Figure S1a). The eGFP intensity was within one order of magnitude, suggesting only a minimal variation of gene insertions within the transduced population.
Figure 1.

Quantification of transgene insertions and VEGF secretion. MSC were transduced with the VEGF lentivirus at the indicated MOI, media was changed, and supernatants were collected 24 hours later and cells counted. (a) Total DNA was extracted and analyzed by quantitative polymerase chain reaction (qPCR) to determine the number of viral copies per cell. (b) Supernatants were tested for secreted VEGF by enzyme linked immunosorbant assay (ELISA) (n = 3, each measured in triplicate). MSC, mesenchymal stem cells; VEGF, vascular endothelial growth factor; MOI, multiplicities of infection.
The amount of VEGF synthesized and secreted by MSC/VEGF in vitro was determined by enzyme linked immunosorbant assay (ELISA). At MOI 1, the protocol ideal for clinical manufacturing, 1,000 MSC/VEGF secreted ~70 pg/ml VEGF per day into the culture supernatant (Figure 1b). Depending on basal VEGF levels detected in unmodified MSC, overexpression of VEGF can result in an almost 10-fold increase in VEGF secretion. To ensure that VEGF levels are not excessive in the MSC microenvironment, we compared secreted VEGF to cell-associated and extracellular matrix-bound VEGF in culture. As shown in Supplementary Figure S1b, >97% of the total VEGF was detected as soluble molecules in the culture medium, <2% was inside or attached to the cells, and <1% was bound to the extracellular matrix. Thus, the majority of the VEGF produced by MSC/VEGF in vitro is secreted and should be available in ischemic tissue following administration.
Autocrine effects of overexpressing VEGF in MSC
We have previously shown that, in contrast to transduction with other growth factors, such as FGF-2, PDGF, and TGF-b1, overexpression of VEGF does not alter the proliferation, morphology, or differentiation potential of MSC.15 These results were also confirmed in the current studies, using clinically compliant products and protocols. MSC/VEGF and nontransduced MSC had very similar abilities to undergo osteogenic and adipogenic differentiation (not shown).
In order to address whether the number of viral insertions could confer a proliferative advantage to transduced MSC, proliferation of MSC transduced with MOI 1, 5, and 20 was compared with nontransduced MSC. Supplementary Figure S2a shows that transduction at MOI 1 had a minimal effect on growth of MSC, while transduction with MOI of 5 and 20 showed progressively more inhibition of proliferation. These results suggest that transduction does not lead to outgrowth of highly proliferative cells in vitro. In contrast, we observed that a high viral load inhibits cell growth. Further studies related to the issue of cell proliferation in the context of potential tumorigenesis are described below.
Angiogenic activity of MSC/VEGF
To confirm functionality of MSC/VEGF in vitro, a wound/scratch assay was used to assess the ability of these cells to induce migration of human umbilical cord vein endothelial cells (HUVEC). It has been established that VEGF activates Erk1/2 and Akt signaling pathways16,17 in endothelial cells and that the specific VEGF receptor inhibitor Axitinib18 can effectively block VEGF signaling. Figure 2a,b show that both MSC and MSC/VEGF conditioned culture medium contain factors that activate migration of HUVEC, indicating that the protein product of the gene construct is biologically active, and that the over-expression of VEGF in MSC/VEGF induces more migration of HUVEC than unmodified MSC. Of note, the inhibition exerted by Axitinib suggests that the increased migration of HUVEC is directly due to higher VEGF levels and not to other potential changes in the secretome of MSC. Axitinib showed a minor reduction in migration of control MSC, consistent with inhibition of the low levels of VEGF that are secreted by the unmodified cells.
Figure 2.

MSC/VEGF promotes migration of endothelial cells in vitro. Human umbilical cord vein endothelial cells (HUVEC) were exposed in a wound/scratch assay to conditioned media from MSC (CM-MSC), that were either non-transduced (no virus) or transduced to overexpress VEGF using an MOI = 1. Each condition was also treated with either DMSO (vehicle only, control) or the VEGF receptor inhibitor Axitinib (100nmol/l). Cell migration (i.e.,wound closure) was measured 12 hours after started the assay. (a) Representative images, 12 hours after started the assay. (b) Quantification, where average of six independent experiments performed in triplicate are shown (n = 6). *P < 0.05; **P < 0.005. MSC, mesenchymal stem cells; VEGF, vascular endothelial growth factor; MOI, multiplicities of infection.
To determine the angiogenic activity of MSC/VEGF in vivo, an immune deficient mouse model of hind limb ischemia (HLI) was used as previously described.15,19,20 Figure 3a shows representative Laser Doppler Perfusion Imaging (LDPI) images 10 weeks after surgery and treatment, and as demonstrated in Figure 3b, starting at 6 weeks, animals treated with MSC/VEGF recovered more blood flow in the ischemic limb as compared with control (vehicle only) mice. In contrast, blood flow of mice treated with nontransduced MSC was inconsistent and did not show a significant improvement as compared with control-treated animals. As an initial dose-finding study, this experiment was then repeated to include an arm with a low dose of MSC/VEGF (2.5 × 104 cells). In this new experiment, increased efficacy of MSC/VEGF (5 × 105 cells) over controls was confirmed, while both nontransduced MSC (5 × 105 cells) and MSC/VEGF (low dose) showed an intermediate effect, not significantly superior to controls (Normosol) (Figure 3c).
Figure 3.

Revascularization potential of MSC/VEGF. (a) Representative laser Doppler images of blood flow in NSG mice following hind limb ischemia surgery and intramuscular injection of Normosol (control), MSC, or MSC/VEGF. Degree of blood flow is indicated from red (strongest flow) to blue (weakest flow). Changes in blood flow were measured as a ratio of the treated limb to the untreated limb in the same animal over time. (b) Laser Doppler measurements in NSG mice over 11 weeks. Cell dose (MSC and MSC/VEGF) was 5 × 105 cells/mouse. (c) Laser Doppler measurements in NSG mice over 5 weeks. Cell dose for MSC and MSC/VEGF (high) was 5 × 105 cells/mouse, while MSC/VEGF (low) was 2.5 × 104 cells/mouse. For both experiments b and c, n = 12 mice/group. Statistical analysis was performed comparing MSC/VEGF (high) to Normosol, where *P < 0.05. MSC, mesenchymal stem cells; VEGF, vascular endothelial growth factor; NSG, NOD/SCIDIL2RY-/-.
We then compared MSC/VEGF-treated NOD/SCID β2M null (B2M) mice to controls using histological methods. Eight weeks after HLI-induction/cell administration we observed a significant increase in perfused blood vessels (i.e., FITC-Dextran label) and a trend toward a higher density of murine CD31 endothelial cells (Figure 4). These results suggest that MSC/VEGF may contribute by stabilizing blood vessels (favoring greater perfusion), while not necessarily promoting an increase in number of endothelial cells. Also in this mouse strain, MSC/VEGF showed a significant increase in blood flow restoration as compared with controls (see Supplementary Figure S3). Altogether, our efficacy studies demonstrate a superior angiogenic potential of MSC/VEGF as compared with sham controls and nontransduced MSC in vitro and in vivo. Consequently, we then focused on demonstrating the safety of MSC/VEGF.
Figure 4.

MSC/VEGF increases the density of perfused blood vessels. MSC/VEGF (5 × 105 cells/mouse) or vehicle (Normosol) was injected into NOD/SCID β2M null (B2M) mice. After 56 days, mice were perfused with FITC-Dextran under anesthesia and sacrificed for histological analysis. (a) Representative images of histological stainings. (b) Average quantification of mCD31 and FITC-Dextran label from five animals per condition and four random sections per animal. *P < 0.05 as calculated using a nonpaired Student’s t-test. MSC, mesenchymal stem cells; VEGF, vascular endothelial growth factor; NOD/SCID, nonobese diabetic/severe combined immune deficient; FITC, fluorescein isothiocyanate.
Genetic stability and rule-out tumorigenicity of MSC/VEGF
The use of lentiviral vectors raises the possibility that replication competent virus could emerge from the cell product and infect other cells when the product is used as a therapeutic agent. To ensure that replication competent lentivirus is not being generated after transduction of MSC with the pCCLc-MNDU3-VEGF-WPRE vector, a p24 assay on culture media from cells exposed to media collected from cells 8 passages after transduction was done. This method can detect replication competent lentivirus in concentrations as low as 3.9 pg/ml. No replication competent lentivirus particles were generated post-transduction with the VEGF vector (not shown).
Several studies were then conducted to examine the genetic stability of MSC/VEGF. Karyotypic analysis of MSC/VEGF was performed to determine whether transduction of MSC with pCCLc-MNDU3-VEGF-WPRE induces chromosomal instability in the cells. The MSC/VEGF product was manufactured using standard manufacturing protocols except that the MOI was varied to increase the number of vector integrants per cell to increase the possibility that vector would cause a karyotypic change. After expanding cells for an additional passage after transduction, no chromosomal abnormalities were detected at any of the tested MOI (up to 20 times higher than the intended use; see Supplementary Figure S2b).
Integration stability was assessed to determine whether transduction with the vector would lead to stable DNA integration into the chromosomes of the MSC, or if this insertion could suffer genetic rearrangements. Stability of the vector was assessed using PCR of genomic DNA from MSC transduced using standard manufacturing conditions at MOI 1 and 10. Cells were grown for one additional passage and then genomic DNA was isolated from these MSC/VEGF for PCR analysis. Results are shown in Figure 5. Genomic DNA from nontransduced MSC did not amplify the vector PCR products. In contrast, PCR bands corresponding to pCCLc-MNDU3-VEGF-WPRE were detected in genomic DNA from MSC/VEGF and these were identical to PCR bands amplified from the vector plasmid used in lentiviral vector preparations, indicating that no deletions or insertions of vector DNA occurred following transduction in either MSC/VEGF transduced with MOI 1 or 10.
Figure 5.
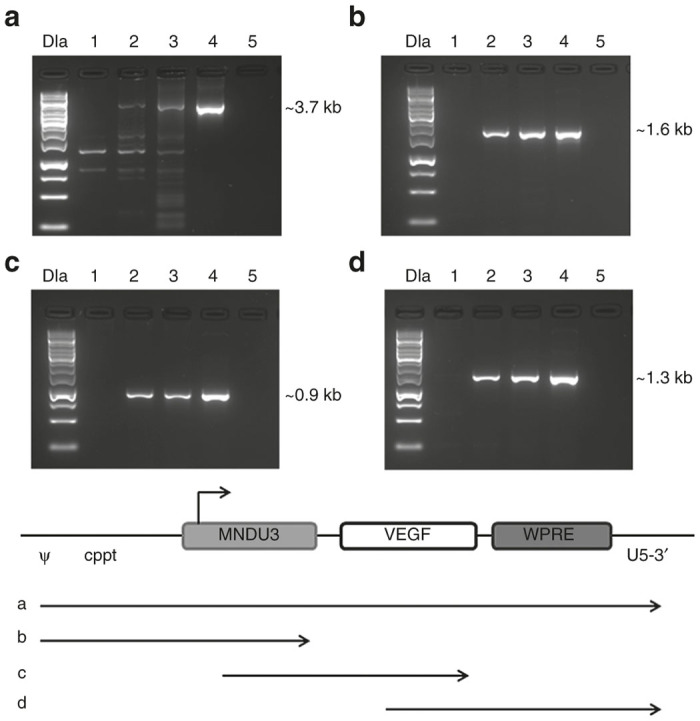
Stability of the clinical vector in transduced cells. Total genomic DNA was extracted from MSC/VEGF transduced at multiplicities of infection (MOI) 1 or 10, or nontransduced MSC, and analyzed by polymerase chain reaction (PCR) with primers specific for the vector segments shown. (a) ψ (forward) and U5-3′ (reverse), (b) ψ (forward) and MNDU3 (reverse), (c) MNDU3 (forward) and VEGF (reverse), and (d) VEGF (forward) and U5-3′ (reverse). Lanes were assigned as follows: One kilobase DNA ladder (DLa), nontransduced MSC (negative control, lane 1), MSC/VEGF MOI 1 (lane 2), MSC-VEFG MOI 10 (lane 3), VEGF vector plasmid (positive control, lane 4), no template control (lane 5). A schematic of the PCR products is below the panels. Ψ, psi packaging sequence; VEGF, vascular endothelial growth factor; WPRE, Woodchuck hepatitis virus post-transcriptional regulatory element; kb, kilobase.
The potential for tumorigenicity of MSC/VEGF in vivo was addressed. Mice were injected with MSC/VEGF transduced using MOI of 1, 10, and 20, or nontransduced MSC, then analyzed for tumorigenicity either 2 or 4 months after cell administration. No tumors arose in mice injected with nontransduced MSC or MSC/VEGF regardless of transduction MOI, while 13 out of 14 mice injected with our positive controls (Reh, human induced pluripotent stem cells or human embryonic stem cells) developed large tumors within 1 month (Figure 6). One mouse treated with human embryonic stem cells that did not develop a palpable tumor exhibited additional pathologies as a result of the human embryonic stem cells injection, which were not seen in MSC or MSC/VEGF treated animals (not shown). Of the 46 animals treated with nontransduced MSC or MSC/VEGF and analyzed by the UC Davis Pathology Department, no tumor formation was observed.
Figure 6.

Rule-out tumorigenicity assay. NSG mice were injected subcutaneously with 106 cells suspended in matrigel in the flank. Positive control mice were injected with either Reh cells, human embryonic stem cells (hESC), or human induced pluripotent stem cells (hiPSC) and sent to pathology for analysis when tumors were ≤1.5 cm in diameter. At that time, mice were photographed to document tumor formation. Additional mice were similarly injected with 106 nontransduced MSC or MSC/VEGF transduced with MOI 1, 10, or 20. No evidence of tumor formation was found in any of the 32 mice treated with MSC or MSC/VEGF, while 13 out of 14 mice injected with positive control cells grew tumors within one month after cell injection. (a) Representative images. (b) Percentage of mice showing tumors one month after cell injection. MSC, mesenchymal stem cells; VEGF, vascular endothelial growth factor; NSG, NOD/SCIDIL2RY-/-.
In vivo edema and hemangioma formation after MSC/VEGF administration
Though the majority of previous clinical trials using either MSC or VEGF protein administration in CLI have demonstrated overall safety, concerns of edema formation and vascular tumor (hemangioma) formation are not unfounded.21 In this studies, two separate experiments to address these issues. First, edema formation was addressed by measuring potential swelling of the foot and ankle of healthy mice injected with either MSC or MSC/VEGF transduced at MOI 1. There were no significant differences in ankle or foot diameters in any of the treatment groups (see Supplementary Figure S4a,b). This study was repeated in the HLI mouse model, using MSC transduced with the VEGF vector at MOI 1 and 10. In this experiment, the diameter of the ankle was measured using the laser Doppler photographs of each animal over time (see Supplementary Figure S4c). Again, no differences were found among the treatment groups. Lack of swelling in response to MSC/VEGF suggests that the level of secreted VEGF is not sufficient to cause edema, despite being sufficient to induce tissue reperfusion in the HLI model (as demonstrated in Figures 3 and 4).
To address the potential for hemangioma formation after MSC/VEGF administration, NSG mice were injected intramuscularly in each hind limb with Normosol, nontransduced MSC or MSC/VEGF transduced at MOI 1. After 7, 14, and 30 days, animals were sacrificed and tissues sent to the pathology laboratory at UC Davis for histological analysis. No evidence of hemangioma, other vascular tumors, tissue disruption, or edema was found in these animals (not shown).
Retention of MSC/VEGF in vivo
Finally, the duration of in vivo persistence of cells after intramuscular injection was determined. For this, MSC/VEGF was transduced to express the luciferase gene in order to enable tracking by bioluminescent imaging following in vivo administration. As shown in Figure 7a,b, the number of cells decreased ~10-fold per week until day 28, at which time the signal to background noise become too high to accurately quantify cells using this methodology. A second method was then employed, using PCR to detect human and mouse DNA. In vitro, as little as 1 pg of human DNA was detected in 200 ng of mouse DNA (not shown). Using this sensitive PCR-based detection method, human DNA was detected in 36 out of 37 mice at 2 months after cell administration. After 4.5 months, human DNA was detected in seven out of seven mice examined, but no human DNA could be detected in any animals tested 6 months after injection (Figure 7c). These results suggest that retention of MSC declines during the first weeks after administration, but a small subset of cells remains for several months.
Figure 7.

Retention of MSC/VEGF. Cells were cotransduced to express luciferase and VEGF, then injected into the hamstring muscles of healthy NSG mice. (a) bioluminescence in a representative animal over 28 days after cell injection. (b) Cell survival as correlated with luciferase signal intensity was measured by luminescence every 7 days (n = 6). (c) Polymerase chain reaction (PCR) detection of human DNA in mouse muscles up to 6 months after injection. MSC, mesenchymal stem cells; VEGF, vascular endothelial growth factor; NSG, NOD/SCIDIL2RY-/-.
Discussion
Currently, no-option patients with CLI require amputation of the ischemic limb, signifying a serious unmet clinical need. Although phase 1 and 2 clinical trials of angiogenic growth factor gene therapy showed potential clinical benefit in treatment of CLI, phase 3 studies in this indication did not achieve significant efficacy, potentially due to suboptimal mechanisms of delivery of therapeutic growth factors.22 Similarly, local administration of MSC into patients with CLI have demonstrated safety, but only limited efficacy.10 Our approach combines cell and gene therapy strategies by utilizing allogeneic MSC that are genetically modified to overexpress VEGF for sustained local delivery at the sites of ischemia. The preclinical studies presented here are aimed at demonstrating safety and efficacy of MSC/VEGF. Investigational new drug -enabling studies will further evaluate safety of the planned intramuscular dosage in human patients. These investigational new drug-enabling studies include toxicology, tissue distribution and persistence, and rule-out tumorigenicity studies performed with a larger number of animals per group than those presented in the current studies.
The rationale for using a lentiviral vector to deliver the VEGF transgene in MSC is supported by numerous gene therapy trials with demonstrated safety.23 Similarly, the constitutive promoter MNDU3 was selected based on over ten years of follow-up data in patients treated with a viral vector using this promoter with demonstrated safety.24 Additionally, the WPRE enhancer maximizes VEGF expression, while limiting the viral load. Controlling for the number of insertions in MSC is essential to minimize risks associated with insertional mutagenesis. Here we demonstrate that the target viral load (MOI = 1) for MSC/VEGF is safe and efficacious, and confirmed low variation within the transduced population.24 Of note, our studies did not address the specific insertion sites of the vector, because under standard culture conditions MSC do not undergo clonal expansion, hence the genomic insertion sites are as many as cells transduced. Our studies on MSC/VEGF proliferation, karyotype, and potential for tumorigenicity support the notion that no subset of cells gained a high proliferative advantage even when using very high amounts of lentivirus.
Our results indicate that the VEGF vector stably transduces MSC, leading to at least a 10-fold increase in VEGF secretion. Interestingly, the number of viral integrations per cell was proportional to the viral load, while secreted VEGF did not show this proportional increase, presumably limited by the capacity of cells to produce and secrete the transgene. This further supports the strategy to transduce MSC with a low viral load, as increased viral load not only increases risks associated with a combined cell and gene therapy, but also negatively affects the proliferation of the cells without the benefit of substantially increased VEGF secretion. Finally, excessive VEGF levels could cause edemas and hemangiomas, therefore, it is important to limit the amount of secreted VEGF.21 We confirmed the that even at microenvironmental level (i.e., in cells or associated to extracellular matrix) the VEGF production is within acceptable levels, which was further validated by the absence of edema or hemangiomas in mice. We had previously show that VEGF expression does not exert an autocrine effect in MSC,15 consistent with the finding that MSC do not express VEGF receptors.25,26
After confirming the biological activity of the MSC/VEGF transgene product in vitro, we addressed the revascularization potential of the cells in an immune compromised mouse model of HLI. As previously shown in our proof-of concept studies,15 we found that MSC/VEGF treated mice exhibited faster and more sustained blood flow restoration in the ischemic limb compared with mice treated with vehicle alone. Nontransduced MSC only had a mild, nonsignificant effect, with blood flow levels typically ranging in between control and MSC/VEGF levels.
It is known that choice of mouse model can have profoundly varying effects on revascularization potential in HLI studies.27–30 Most HLI models imply the use of immune competent mouse strains. In fact, it is know that the immune system plays an essential role in vascular remodeling.30–32 We utilized immune deficient mouse strains, NSG and B2M, to allow survival and function of xeno-transplanted human MSC. Previous works have demonstrated substantial increases in revascularization of ischemic mouse limbs within 28 days of surgery, while our model does not show more than 50% blood flow perfusion, regardless of treatment, until 45 days after surgery.19,27–30 The deficits in immune function of this strain may also be attributed to the overall low recovery of blood flow, regardless of treatment. Of note, our model offers the advantage of establishing an acutely induced but chronic development of ischemia, mimicking the human condition. Still, our studies were conducted on healthy, young adult mice, while the target clinical population is largely elderly and/or diabetic, among many other diverse complications. Revascularization after HLI surgery in diabetic mice has been shown to be impaired,33 and age and chronic stress cause dysfunction in collateral vessel formation in mouse models of angiogenesis.34–36 These are important factors that may influence the efficacy of MSC/VEGF in a clinical setting.
Our studies addressing MSC/VEGF retention after administration demonstrate that, while increased revascularization is seen in MSC/VEGF treated mice (mostly after 4–5 weeks), most of the cells clear within 4 weeks. Only a small subset of cells, which possibly engraft as pericytes in the developing vasculature,37,38 persist for up for several months. We therefore, propose that MSC/VEGF cells initiate angiogenesis that is then carried on by endogenous mechanisms. In conclusion, our work regarding efficacy and safety of MSC/VEGF strongly supports their application for treatment of no-option patients with CLI. In addition, our work encourages further exploration of other new therapeutics combining cell and gene therapy, where MSC (with an outstanding safety prolife) can be used as delivery platforms for therapeutic transgenes.25,26
Materials and Methods
MSC isolation and culture
Isolation and expansion of human bone marrow derived MSC was conducted under current good manufacturing practices conditions. Bone marrow aspirates were purchased commercially (All Cells, Emeryville, CA). Whole bone marrow was plated in flasks with MSC media consisting of Minimum Essential Medium Alpha (MEMα) (Hyclone Thermo Scientific, Waltham, MA) and 10% Premium Select Fetal Bovine Serum (Atlanta Biologicals, Flowery Branch, GA), supplemented with 1% L-Glutamine (Hyclone) and 1% Penicillin-streptomycin (Hyclone). After 2 days, nonadherent cells were discarded by washing twice with phosphate-buffered saline, and fresh MSC media added to the adherent cells. Cells were incubated at 37°C in 5% CO2, and 20% O2, while media changed every 2–3 days. MSC at passage 3–6 were used for experimentation. Cells that were used for in vivo studies were incubated at 1% O2 in dedicated hypoxic incubators for 48 hours immediately prior to administration.
Lentiviral vectors and MSC transduction
A third generation lentiviral vector based on the CCLc-x backbone39 was used to generate a vector of the general form pCCLc-MNDU3-X-WPRE, where X is the site of insertion for full length cDNA of human VEGF-A165, GFP, or Luciferase. Transgene expression is driven by the constitutive MNDU3 promoter and the enhancer element, WPRE, acting in cis. Lentivirus was produced in HEK-293 T-cells, as previously described,40 and tittered after vector harvest. Transductions were performed when MSC reached ~70% confluence, using 20 µg/ml protamine sulfate. The volume of lentivirus to be used was determined from titer results.
Vector integration copy number
MSC were transduced with increasing amounts of the VEGF-coding lentivirus. After several days in culture, cells were lifted by trypsin treatment (Hyclone) and total DNA was isolated using the Quick-gDNA MiniPrep kit (Zymo Research, Irvine, CA) following manufacturer’s instructions. Genomic DNA was quantified using NanoDrop (Thermo Fisher Scientific, Grand Island, NY). To determine viral copy number per cell, we used real-time PCR. 25 ng of gDNA were mixed with SYBRGreen MasterMix (Life Technologies, Grand Island, NY) and primers targeting either WPRE (specific for the viral construct), VEGF-A (present in both, the construct and human cells) and GAPDH (loading control). The primers were WPRE-fwd: 5′’-TTACGCTATGTGGATACGCTG-3′, WPRE-rev: 5′-TCATAAAGAGACAGCAACCAGG-3′, VEGFA-fwd: 5′-TCTTCAAGCCATCCTGTGTG-3′, VEGFA-rev: 5′-CTGCATGGTG ATGTTGGACT-3′ GAPDH-fwd: 5′-ACAGTCAGCCGCATCTTC-3′ and GAPDH-rev: 5′-CTCCGACCTTCACCTTCC-3′. Two standard curves were generated using serial dilutions with the plasmid pCCLc-MNDU3-VEGF-WPRE; one for WPRE and one for VEGF-A. Using linear regression and considering that each cell carries two copies of VEGF-A gene, Ct values of nontransduced MSC were translated to copy numbers in order to determine the number of cells examined. GAPDH measurements were used to confirm homogeneity of gDNA amount loaded per sample. WPRE measurements and standard curve were used to determine viral copy number.
VEGF detection
Cells were plated at 10,000 cells per cm2 in six-well plates. MSC/VEGF were transduced at the indicated MOI, and cultured for an additional 2 days. Then, medium was changed to standard culture medium. After 24 hours, supernatant was collected and cells were lifted with trypsin (Hyclone) and counted using Trypan blue exclusion dye (Life Technologies) and a hemocytometer. VEGF levels in the supernatant were determined by enzyme linked immunosorbant assay (R&D Systems, Minneapolis, MN) and normalized to cell number for each sample.
Endothelial cells migration assay
MSC were plated at 10,000 cells per cm2 in 12-well plates, and transduced with the VEGF lentivirus as described above. After 2 days, medium was changed to standard culture medium, and left for additional 2 days. Supernatants were then collected and tested for effects on migration of HUVEC. HUVEC were isolated as previously described15 and cultured in endothelial growth media 2 (Lonza, Basel, Switzerland), with media changes for every 3 days. For the migration assay, HUVEC were plated in 24 well plates at 1.5 × 105 cells per well with inserts from CytoSelect 24-well wound healing assay (Cell Biolabs, San Diego, CA), and left over night. Then, inserts were removed to create a 0.5 mm diameter gap in the HUVEC monolayer, and media changed to the conditioned media from transduced or nontransduced MSC with or without Axitinib (100 nmol/l), a selective inhibitor of VEGF receptors. Conditions without Axitinib received an equivalent volume of dimethyl sulfoxide (DMSO, solvent). Each well was photographed using an inverted phase contrast microscope at time 0 and 12 hours after the addition of conditioned MSC media. Changes in the open area in the photographs were quantified using TScratch Software (version 1.0. Available at: http://www.cse-lab.ethz.ch/).41
Vector stability in transduced cells
PCR was performed using genomic DNA from MSC and MSC/VEGF to determine whether genomic rearrangements or deletions occurred in vector transduced cells. The Quick gDNA MiniPrep kit (Zymo Research) was used for total DNA extraction from non-transduced MSC and MSC/VEGF that were transduced with MOI 1 and 10. PCR was then performed using FideliTaq polymerase and MasterMix (Affymetrix, Santa Clara, CA), and primers corresponding to the following vector specific transgenes: ψ-fwd: 5′-ACCTGAAAGCGAAAGGGAAAC-3′, U5-3′-rev: 5′-CTGCTAGAGATTTTCCACACTGAC-3′, MNDU3-fwd: 5′-CGCCCTCAGCAGTT TCTAG-3′, MNDU3-rev: 5′-CTATCTATGGCTCGTACTCTATA-3′, VEGF primers are described above. PCR products were then visualized on an agarose gel against a 1 kilobase DNA ladder (Life Technologies).
Hind limb ischemia model
Prior to surgery, hair in the surgical area was removed by nair. NOD/SCID-IL2Rγ-/- (NSG) or NOD/SCID β2M null (B2M) mice were anesthetized with inhaled isoflurane, and skin cleaned with betadine and wiped with an alcohol pad. Unilateral hind limb artery ligation was performed, as previously described.16 In brief, a 1 cm segment of the right femoral artery and all major collateral vessels are ligated using 5-0 monofilament suture (Moore Medical, Farmington, CT) to induce complete HLI. Mice were injected with two 20 µl injections of 5 × 105 non-transduced MSC, MSC-VEGF, or Normosol (vehicle) (Hospira, San Jose, CA) in the hamstrings of the surgical limb 24 hours after surgery. Blood flow was measured by Laser Doppler Perfusion Imaging (Moor Instruments, Devon, UK) before surgery, the day of cell injection, and weekly thereafter for 10 weeks. Animals were anesthetized and placed on a heating pad for 10 minutes prior to imaging. Blood flow, expressed as flux over the area of each foot, was calculated as the flux ratio of the surgical limb to the non-surgical limb of each animal. All surgical procedures were conducted in compliance with UC Davis IACUC policies on rodent survival surgery. NSG mice were bred at the UC Davis Institute for Regenerative Cures and B2M mice were acquired from The Jackson Laboratory and bred at the UC Davis Institute for Regenerative Cures.
Histological analysis
The day after HLI surgery, described above, MSC/VEGF (5 × 105 cells) or Normosol solution were injected intramuscularly into the ischemic limb of B2M mice. Blood flow was measured weekly by Laser Doppler Perfusion Imaging, as described above. Mice were perfused with 1 mg/100 ml FITC-Dextran (Sigma-Aldrich, St. Louis, MO) via tail vein to label all blood flow in the animal, and euthanized 10 minutes after the injection, at the indicated time point. All muscles of the ischemic limb isolated by dissection, preserved in Optimal Temperature Compound, and stored at −80°. Tissues were then sectioned at 20 µm thickness and placed onto glass slides. Tissue samples were fixed using an acetone fixation protocol, washed with phosphate-buffered saline, blocked with 1% bovine serum albumin, then immunostained with rat anti-mouse CD31 (BioLegend, San Diego, CA) at a 1:50 dilution, overnight. A secondary antirat IgG antibody was applied at 1:500 dilution and left for 1 hour incubation. All samples were imaged using a BioRevo Keyence BZ-9000 fluorescence microscope (Keyence, Itasca, IL). Data analysis was conducted using NIS Elements BR Object Count Software version 4.0 (Nikon, Tokyo, Japan).
Rule-out tumorigenicity assay
To address whether transduction with the VEGF vector can induce oncogenic transformation in MSC, cells were transduced with lentivirus at MOI 1, 10, and 20 and cultured for five passages. Then, cells were harvested and 106 MSC/VEGF or nontransduced MSC were injected with Matrigel (Corning, Corning, NY) as vehicle into the flank of NSG mice. An aggressive leukemia cell line, (Reh), embryonic stem cells (ESC, H9), and induced pluripotent stem cells generated in our laboratory, were injected as positive controls, using similar methods. Following UC Davis IACUC guidelines, positive control mice were sent to the UC Davis Pathology Laboratory for analysis of tumor formation once tumors were ≤ 1.5 cm in diameter, within one month of injection. MSC and MSC/VEGF injected mice were sent for pathological analysis without palpable tumors at 2 and 4 months after injection.
Edema and hemangioma assays
To determine whether in vivo administration of MSC/VEGF could lead to edema formation, NSG mice were injected intramuscularly in each leg with 2, 20 µl injections of 5 × 105 MSG/VEGF, MSC/GFP (a control cell population transduced with the pCCLc-MNDU3-GFP-WPRE vector), or Normosol (vehicle). Using calipers, ankle or foot diameter measurements were taken every 3 days after cell injection, up to day 12. Additionally, Laser Doppler Perfusion Imaging photographs from mice that received HLI surgery and treatment with MSC/VEGF at MOI 1 or 10, nontransduced MSC, or Normosol 24 hours after surgery were used to measure ankle diameters once per week over 28 days.
To address potential hemangioma formation after MSC/VEGF administration, MSC or MSC/VEGF transduced at MOI 1 were injected intramuscularly in 20 µl Normosol in NSG mice. Animals were sent to the UC Davis Pathology Laboratory for assessment of hemangioma formation at the injection site 7, 14, and 30 days postinjection.
Retention studies
To generate Luciferase-expressing MSC, a third generation lentiviral vector with the general form pCCLc-MNDU3-Luciferase-PGK-eGFP-WPRE was used, following the transduction protocol outlined above. Immune compromised NSG mice were anesthetized with inhaled isoflurane and then injected with 20 µl of Normosol containing the luciferase-expressing MSC into the hamstring muscles. Anesthetized animals were injected intraperitoneally with 100 ml of 20 mg/ml D-Luciferin Firefly (Life Technologies) 10 minutes prior to imaging via IVIS Spectrum (Perkin Elmer, Waltham, MA). Mice were imaged the day of cell injection (day 0) and then weekly thereafter for 28 days.
To determine the potential presence of human cells after intramuscular injection by PCR, the hamstring muscles were carefully isolated from injected mice. Tissues were subjected to proteinase K (Zymo) digestion (20 mg/ml), and DNA was isolated using Genomic DNA-Tissue MidiPrep (Zymo Research, Irvine, CA) according to the manufacturer’s instructions. The presence/absence of human cells was evaluated using human-specific primers and probe, detecting the human endogenous retrovirus gene ERV3. Samples underwent a preamplification step of 15 cycles, using 150 ng of gDNA, 12.5 μl of 2X TaqMan Universal MasterMix (Life Technologies), 50 μmol/l of each of the hERV3 primers, and water to a 25 μl final total volume. The conditions of preamplification were: 2 minutes at 50°C, 10 minutes at 95°C, followed by 15 cycles of 15 seconds at 95°C, and 1minute at 60°C. 3 μl of the preamplification reaction product were assayed in a second PCR reaction containing 10 μl of 2X TaqMan Universal MasterMix, 50 μmol/l of each of the hERV3 primers, 50 μmol/l of probe and water to a 20 μl final volume. The reactions were performed using a StepOne Plus PCR instrument under the same conditions as described above, but for 40 cycles. As a loading control, we used mouse GAPDH. The primers used for those experiments were: hERV3-fwd: 5′-CATGGGAAGCAAGGGAACTAATG-3′, hERV3-rev: 5′-CCCAGCGAGCAATACAG AATTT-3′, and 5′-6-fluorescein (FAM)-containing probe 5′-/56-FAM/TCTTCCCTCGAACCT GCACCATCAAGTCA/36-TAMTSp/-3′. mGAPDH-fwd: 5′-ACCACGAGAAATATGACAA CTCA-3′, mGAPDH-rev: 5′-CCCACTGCCTACAT ACCATGAGC-3′, and FAM-containing probe 5′-/56-FAM/ TCAGCAATGCAT CCTGCACCACCAACT/36-TAMTSp/-3′.
Data representation and statistical analysis
All studies have been conducted in accordance with UC Davis policy; under an active Biological Use Authorization and active IACUC protocol for animal studies. All experiments were performed at least independent times with three different donors, except where otherwise noted. The specific number of replicates for each experiment is indicated in the figure legends as n. All results are represented as mean ± SEM. Statistical differences were calculated using a Student’s t-test between the two specified conditions and indicated time points, where P < 0.05 was considered significantly different and denoted by *;** signifies P < 0.005.
Acknowledgments
This project was funded by Disease Team Grant DR2A-05423 for CLI from the California Institute for Regenerative Medicine (CIRM), CIRM Early Translational Grant TR2-01787 (N.A.J.) and NIH Transformative Grant 1R01GM099688 (JAN). S.K. is supported by a fellow ship from the Dickensons and P.Z. is the 2015 Wing Fat Fellow. A.H. and N.L.M are scholars of the Bridges to Stem cell Research Programs #TB1-01190 and #TB1-01184 from CIRM to promote undergraduate training in stem cell biology and regenrerative medicine. All authors declare no conflicts of interest regarding this publication or any information related to it.
References
- Minar, E (2009). Critical limb ischaemia. Hamostaseologie 29: 102–109. [PubMed] [Google Scholar]
- Mahoney, EM, Wang, K, Keo, HH, Duval, S, Smolderen, KG, Cohen, DJ et al. Reduction of Atherothrombosis for Continued Health (REACH) Registry Investigators. (2010). Vascular hospitalization rates and costs in patients with peripheral artery disease in the United States. Circ Cardiovasc Qual Outcomes 3: 642–651. [DOI] [PubMed] [Google Scholar]
- Selvin, E, Wattanakit, K, Steffes, MW, Coresh, J and Sharrett, AR (2006). HbA1c and peripheral arterial disease in diabetes: the Atherosclerosis Risk in Communities study. Diabetes Care 29: 877–882. [DOI] [PubMed] [Google Scholar]
- Grochot-Przeczek, A, Dulak, J and Jozkowicz, A (2013). Therapeutic angiogenesis for revascularization in peripheral artery disease. Gene 525: 220–228. [DOI] [PubMed] [Google Scholar]
- Birk, DM, Barbato, J, Mureebe, L and Chaer, RA (2008). Current insights on the biology and clinical aspects of VEGF regulation. Vasc Endovascular Surg 42: 517–530. [DOI] [PubMed] [Google Scholar]
- Shyu, KG, Chang, H, Wang, BW and Kuan, P (2003). Intramuscular vascular endothelial growth factor gene therapy in patients with chronic critical leg ischemia. Am J Med 114: 85–92. [DOI] [PubMed] [Google Scholar]
- Isner, JM (1998). Arterial gene transfer of naked DNA for therapeutic angiogenesis: early clinical results. Adv Drug Deliv Rev 30: 185–197. [DOI] [PubMed] [Google Scholar]
- Mäkinen, K, Manninen, H, Hedman, M, Matsi, P, Mussalo, H, Alhava, E et al. (2002). Increased vascularity detected by digital subtraction angiography after VEGF gene transfer to human lower limb artery: a randomized, placebo-controlled, double-blinded phase II study. Mol Ther 6: 127–133. [DOI] [PubMed] [Google Scholar]
- Rajagopalan, S, Mohler, ER 3rd, Lederman, RJ, Mendelsohn, FO, Saucedo, JF, Goldman, CK et al. (2003). Regional angiogenesis with vascular endothelial growth factor in peripheral arterial disease: a phase II randomized, double-blind, controlled study of adenoviral delivery of vascular endothelial growth factor 121 in patients with disabling intermittent claudication. Circulation 108: 1933–1938. [DOI] [PubMed] [Google Scholar]
- Liew, A and O’Brien, T (2012). Therapeutic potential for mesenchymal stem cell transplantation in critical limb ischemia. Stem Cell Res Ther 3: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ankrum, JA, Ong, JF and Karp, JM (2014). Mesenchymal stem cells: immune evasive, not immune privileged. Nat Biotechnol 32: 252–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendicino, M, Bailey, AM, Wonnacott, K, Puri, RK and Bauer, SR (2014). MSC-based product characterization for clinical trials: an FDA perspective. Cell Stem Cell 14: 141–145. [DOI] [PubMed] [Google Scholar]
- Gruenloh, W, Kambal, A, Sondergaard, C, McGee, J, Nacey, C, Kalomoiris, S et al. (2011). Characterization and in vivo testing of mesenchymal stem cells derived from human embryonic stem cells. Tissue Eng Part A 17: 1517–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, CC, Yew, TL, Yang, DC, Huang, WH and Hung, SC (2012). Benefits of hypoxic culture on bone marrow multipotent stromal cells. Am J Blood Res 2: 148–159. [PMC free article] [PubMed] [Google Scholar]
- Fierro, FA, Kalomoiris, S, Sondergaard, CS and Nolta, JA (2011). Effects on proliferation and differentiation of multipotent bone marrow stromal cells engineered to express growth factors for combined cell and gene therapy. Stem Cells 29: 1727–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber, HP, McMurtrey, A, Kowalski, J, Yan, M, Keyt, BA, Dixit, V et al. (1998). Vasular endothelian growth factor regulates endothelial cell survival through the phosphatidylinositol 3’-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J Biol Chem 273: 30336–30343. [DOI] [PubMed] [Google Scholar]
- Olsson, AK, Dimberg, A, Kreuger, J and Claesson-Welsh, L (2006). VEGF receptor signalling- in control of vascular function. Nat Rev Mol Call Biol 7: 359–371. [DOI] [PubMed] [Google Scholar]
- Kelly, RJ and Rixe, O (2009). Axitinib --a selective inhibitor of the vascular endothelial growth factor (VEGF) receptor. Targeted Oncol 4: 297–305. [DOI] [PubMed] [Google Scholar]
- Capoccia, BJ, Robson, DL, Levac, KD, Maxwell, DJ, Hohm, SA, Neelamkavil, MJ et al. (2009). Revascularization of ischemic limbs after transplantation of human bone marrow cells with high aldehyde dehydrogenase activity. Blood 113: 5340–5351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosová, I, Dao, M, Capoccia, B, Link, D and Nolta, JA (2008). Hypoxic preconditioning results in increased motility and improved therapeutic potential of human mesenchymal stem cells. Stem Cells 26: 2173–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa, CR, Banfi, A, Glazer, NL, Thurston, G, Springer, ML, Kraft, PE et al. (2004). Microenvironmental VEGF concentration, not total dose, determines a threshold between normal and aberrant angiogenesis. J Clin Invest 113: 516–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mughal, NA, Russell, DA, Ponnambalam, S and Homer-Vanniasinkam, S (2012). Gene therapy in the treatment of peripheral arterial disease. Br J Surg 99: 6–15. [DOI] [PubMed] [Google Scholar]
- Naldini, L (2015). Gene therapy returns to centre stage. Nature 526: 351–360. [DOI] [PubMed] [Google Scholar]
- Candotti, F, Shaw, KL, Muul, L, Carbonaro, D, Sokolic, R, Choi, C et al. (2012). Gene therapy for adenosine deaminase-deficient severe combined immune deficiency: clinical comparison of retroviral vectors and treatment plans. Blood 120: 3635–3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball, SG, Shuttleworth, CA and Kielty, CM (2007). Vascular endothelial growth factor can signal through platelet-derived growth factor receptors. J Cell Biol 177: 489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delorme, B, Ringe, J, Gallay, N, Le Vern, Y, Kerboeuf, D, Jorgensen, C et al. (2008). Specific plasma membrane protein phenotype of culture-amplified and native human bone marrow mesenchymal stem cells. Blood 111: 2631–2635. [DOI] [PubMed] [Google Scholar]
- Helisch, A. et al. (2006). Impact of mouse strain differences in innate hindlimb collateral vasculature. Arteriosclerosis, thrombosis, and vascular biology 26: 520–526. [DOI] [PubMed] [Google Scholar]
- Chalothorn, D, Clayton, JA, Zhang, H, Pomp, D and Faber, JE (2007). Collateral density, remodeling, and VEGF-A expression differ widely between mouse strains. Physiol Genomics 30: 179–191. [DOI] [PubMed] [Google Scholar]
- Greenberg, JI, Suliman, A, Barillas, S and Angle, N (2008). Chapter 7. Mouse models of ischemic angiogenesis and ischemia-reperfusion injury. Methods Enzymol 444: 159–174. [DOI] [PubMed] [Google Scholar]
- Shireman, PK and Quinones, MP (2005). Differential necrosis despite similar perfusion in mouse strains after ischemia. J Surg Res 129: 242–250. [DOI] [PubMed] [Google Scholar]
- Heil, M and Schaper, W (2004). Influence of mechanical, cellular, and molecular factors on collateral artery growth (arteriogenesis). Circ Res 95: 449–458. [DOI] [PubMed] [Google Scholar]
- Ishikawa, F, Yasukawa, M, Lyons, B, Yoshida, S, Miyamoto, T, Yoshimoto, G et al. (2005). Development of functional human blood and immune systems in NOD/SCID/IL2 receptor {gamma} chain(null) mice. Blood 106: 1565–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landázuri, N, Joseph, G, Guldberg, RE and Taylor, WR (2012). Growth and regression of vasculature in healthy and diabetic mice after hindlimb ischemia. Am J Physiol Regul Integr Comp Physiol 303: R48–R56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassance-Soares, RM, Sood, S, Chakraborty, N, Jhamnani, S, Aghili, N, Nashin, H et al. (2014). Chronic stress impairs collateral blood flow recovery in aged mice. J Cardiovasc Transl Res 7: 749–755. [DOI] [PubMed] [Google Scholar]
- Wang, J, Peng, X, Lassance-Soares, RM, Najafi, AH, Alderman, LO, Sood, S et al. (2011). Aging-induced collateral dysfunction: impaired responsiveness of collaterals and susceptibility to apoptosis via dysfunctional eNOS signaling. J Cardiovasc Transl Res 4: 779–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber, JE, Zhang, H, Lassance-Soares, RM, Prabhakar, P, Najafi, AH, Burnett, MS et al. (2011). Aging causes collateral rarefaction and increased severity of ischemic injury in multiple tissues. Arterioscler Thromb Vasc Biol 31: 1748–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Khaldi, A, Al-Sabti, H, Galipeau, J and Lachapelle, K (2003). Therapeutic angiogenesis using autologous bone marrow stromal cells: improved blood flow in a chronic limb ischemia model. Ann Thorac Surg 75: 204–209. [DOI] [PubMed] [Google Scholar]
- Huang, WH, Chen, HL, Huang, PH, Yew, TL, Lin, MW, Lin, SJ et al. (2014). Hypoxic mesenchymal stem cells engraft and ameliorate limb ischaemia in allogeneic recipients. Cardiovasc Res 101: 266–276. [DOI] [PubMed] [Google Scholar]
- Dull, T, Zufferey, R, Kelly, M, Mandel, RJ, Nguyen, M, Trono, D et al. (1998). A third-generation lentivirus vector with a conditional packaging system. J Virol 72: 8463–8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalomoiris, S, Lawson, J, Chen, RX, Bauer, G, Nolta, JA and Anderson, JS (2012). CD25 preselective anti-HIV vectors for improved HIV gene therapy. Hum Gene Ther Methods 23: 366–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebäck, T, Schulz, MM, Koumoutsakos, P and Detmar, M (2009). TScratch: a novel and simple software tool for automated analysis of monolayer wound healing assays. Biotechniques 46: 265–274. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.