

Abstract
The anti-arthritic gold-containing drug Auranofin is lethal to the protozoan intestinal parasite Entamoeba histolytica, the causative agent of human amebiasis, in both culture and animal models of the disease. A putative mechanism of Auranofin action proposes that monovalent gold, Au(I), released from the drug, can bind to the redox-active dithiol group of thioredoxin reductase (TrxR). Au(I) binding in the active site is expected to prevent electron transfer to the downstream substrate thioredoxin (Trx), thus interfering with redox homeostasis in the parasite. To clarify the molecular mechanism of Auranofin action in more detail, we determined a series of atomic resolution x-ray structures for E. histolytica thioredoxin (EhTrx) and thioredoxin reductase (EhTrxR), the latter with and without Auranofin. Only the disulfide-bonded form of the active site dithiol (Cys140-Cys143) was invariably observed in crystals of EhTrxR in spite of the addition of reductants in various crystallization trials, and no gold was found associated with these cysteines. Non-catalytic Cys286 was identified as the only site of modification, but further mutagenesis studies using the C286Q mutant demonstrated that this site was not responsible for inhibition of EhTrxR by Auranofin. Interestingly, we obtained both of the catalytically-relevant conformations of this bacterial-like, low molecular weight TrxR in crystals without requiring an engineered disulfide linkage between Cys mutants of TrxR and Trx (as was originally done with E. coli TrxR and Trx). We note that the –CXXC– catalytic motif, even if reduced, would likely not provide space sufficient to bind Au(I) by both cysteines of the dithiol group.
Keywords: human amebiasis, Entamoeba histolytica, Auranofin, thioredoxin reductase, thioredoxin, x-ray structure
INTRODUCTION
Entamoeba histolytica is a microaerobic intestinal parasite causing human amebiasis (Choudhuri and Rangan, 2012), the fourth leading cause of death and the third leading cause of morbidity from parasitic diseases worldwide, predominantly affecting areas with poor water quality and inadequate provision for sewage (Debnath et al., 2012a). High-throughput screening of a collection of US Food and Drug Administration (FDA)-approved drugs against E. histolytica singled out the oral anti-inflammatory drug Auranofin (Kean et al., 1997) as a potent E. histolytica inhibitor in culture (EC50 of 0.5 μM). Auranofin, an Au(I) complex of tetraacetyl-thio-glucopyranoside thiolate stabilized by triethyl phosphine (Fig. 1), was 10-fold more potent than metronidazole (Debnath et al., 2012b), the drug currently in widespread use for amebiasis treatment in humans (Freeman et al., 1997). Based on in vivo efficacy in animal models of amebic colitis and liver abscess, Auranofin has been granted Orphan Drug Status from the US FDA. Transcriptional profiling, thioredoxin reductase (TrxR) assays, and redox Western blot of thioredoxin (Trx) recovered from the organism suggest that Auranofin targets Trx metabolism in E. histolytica, enhancing the sensitivity of trophozoites to reactive oxygen-mediated killing (Debnath et al., 2012b). A recently-published study also indicates the utility of using auranofin to treat Giardia lamblia (Tejman-Yarden et al., 2013).
Figure 1.
Chemical structure of Auranofin.
The thioredoxin system is a major contributor toward thiol homeostasis in anaerobic protozoa, including pathogenic E. histolytica and Giardia, both of which lack glutathione and contain cysteine as their major low-molecular mass thiol (Brown et al., 1993; Fahey et al., 1984). Two major classes of TrxR have evolved, distinguished by molecular weight (Mr), architecture of protein domains, lack or presence of a cysteine- or selenocysteine-containing second redox site, and finally, structure and location of the disulfide/dithiol motif (Williams et al., 2000). E. histolytica TrxR (EhTrxR) (Arias et al., 2007) groups with bacterial (Lennon et al., 1999; Waksman et al., 1994) and low-eukaryotic (Brown et al., 1996) enzymes which are distinguished by having a smaller Mr due to entirely missing the interface domain, by the absence of the second redox site, and by the C(X)2C structure of the disulfide/dithiol motif associated with the NADPH domain, in which redox-active cysteines are spaced by two instead of four residues as they are in the high Mr TrxR C(X)4C motif associated with the FAD domain (Williams et al., 2000). TrxR transfers reducing equivalents from NADPH via FAD and an internal redox-active disulfide center to the Trx substrate by disulfide/dithiol interchange between the two proteins (Veine et al., 1998). In low Mr TrxR, this occurs by means of a large-scale conformational change which requires the NADPH-domain to rotate at least once during the catalytic cycle relative to the FAD domain. During catalysis, TrxR adopts a conformation compatible either with internal reduction of the redox-active disulfide bond by FADH2 or with delivery of electrons to Trx (Lennon et al., 1999; Waksman et al., 1994). The switch between two conformations allows acceptance of reducing equivalents from NADPH and internal reduction of the disulfide bond from the same face (the re face) of the flavin isoalloxazine ring. In contrast, domain orientation in high Mr TrxR permits receiving reducing equivalents from NADPH at the re face of the isoalloxazine ring, and then passing it to a disulfide positioned at the opposite si face, then out to the redox center in the C-terminal tail (Williams et al., 2000). Without need for major conformational changes, small substrates (glutathione, trypanothione) can directly access the active site dithiol, while the larger Trx substrate may use the second redox center, a selenosulfide or disulfide, as mediator to communicate with the active site dithiol (Williams et al., 2000).
Reagents that commonly inhibit sulfhydryl-dependent reactions inhibit enzymatic properties of TrxRs. Various metal-containing compounds have been shown to inhibit TrxRs (reviewed in (Cai et al., 2012)), presumably via metal binding to redox-active cysteine pairs. Similarly, a putative mechanism of Auranofin action hinges on release of a monovalent gold, Au(I), that subsequently binds to a protein target (Omata et al., 2006). Auranofin was reported to inhibit human TrxR with IC50 of 20 nM (Gromer et al., 1998), which is much lower than reported for EhTrxR (IC50 of 0.4 μM) (Debnath et al., 2012b). The higher inhibitory effect of metal-containing compounds toward high Mr TrxRs is attributed to a second selenosulfide redox site which is thought to facilitate metal release (Angelucci et al., 2009). While the precise mechanism and molecular target(s) of Auranofin’s anti-inflammatory activity have not been established, in a number of human parasites Auranofin is believed to inhibit antioxidant pathways maintaining the intracellular redox environment by targeting thiol redox enzymes such as thioredoxin reductase, thioredoxin-glutathione reductase (TGR) or trypanothione reductase (Angelucci et al., 2009; Caroli et al., 2012; Ilari et al., 2012; Prast-Nielsen et al., 2011; Sannella et al., 2008). In addition to E. histolytica and Giardia, unicellular human parasites in which Auranofin induces oxidative stress include Trypanosoma cruzi (da Silva et al., 2015), Leishmania infantum (Ilari et al., 2012), Leishmania donovani (Sharlow et al., 2014) and Plasmodium falciparum (Caroli et al., 2012), as well as parasitic flatworms Schistosoma mansoni (Angelucci et al., 2009), Schistosoma japonicum (Song et al., 2012) and Taenia crassiceps (Martinez-Gonzalez et al., 2015). Also, Auranofin exerts broad-spectrum bactericidal activities by targeting thiol-redox homeostasis (Harbut et al., 2015).
Following exposure to Auranofin, x-ray structure analysis of thioredoxin-glutathione reductase from Schistosoma mansoni (Angelucci et al., 2010; Angelucci et al., 2009) as well as the related trypanothione reductase from Leishmania infantum (Ilari et al., 2012) shows Au(I) binding to cysteine thiol groups in the catalytic C(X)4C motif, albeit with low site occupancy; any Au(I) at the C-terminal selenol/thiol center of TGR could not be identified because that region of the protein is disordered in the crystal structures. In addition to interacting with cysteine thiol groups, Au(I) was found trapped in a hydrophobic pocket of S. mansoni TGR (Angelucci et al., 2009). Trypanothione reductase of L. infantum (Ilari et al., 2012) revealed, albeit at low resolution, evidence for Auranofin’s tetra-acetyl-thio-glucopyranoside moiety binding to the trypanothione binding site, suggesting interference with the substrate as another possible mechanism of inhibition. Dual inhibition of inflammatory and redox pathways by Auranofin makes it a promising candidate for new applications as an anti-cancer agent (Li et al., 2015; Topkas et al., 2015; You and Park, 2015) and in treating of parasitic infections in humans (Debnath et al., 2012a; Madeira et al., 2012; Tejman-Yarden et al., 2013).
To further explore the molecular mechanism of Auranofin action against E. histolytica, we have structurally characterized EhTrx and EhTrxR, the latter both with and without Auranofin. No Au(I) binding to the redox active dithiol was observed. Instead, non-catalytic Cys286 of EhTrxR was identified as a single low affinity Au(I)-binding site. Furthermore, the CXXC catalytic motif in EhTrxR crystals (1) invariably favored an oxidized disulfide state and (2) did not provide enough space to bind Au(I) by the catalytic dithiol group. These results challenge the prevailing hypothesis of the mechanism of Auranofin action against thioredoxin reductases.
MATERIALS AND METHODS
Thioredoxin reductase cloning, mutagenesis, expression and purification
For expression of wild type EhTrxR, the coding region for expressing EhTrxR PCR-amplified from an earlier vector (Debnath et al., 2012b) was cloned into the expression vector pTHCm, a derivative of Invitrogen’s vector pTrcHisA with the ampicillin resistance exchanged for chloramphenicol resistance (Nelson et al., 2008b). This plasmid was used to express a non-tagged, native form of EhTrxR under the control of a strong trc promoter. The C140S, C286Q and double C140SC286Q mutants were generated using the QuikChange II procedure (Stratagene) and the primersGGCAAAATGGAGTTAGTGCTAGCGCCATTTGTGATGGGGCTG and CATGTGGGGATGTACAGGATAGAGTATATAGACAAGC (mutated bases in bold), respectively, paired with their complementary primers. The wild type and mutant proteins were expressed in E. coli strain B834, using Studier’s ZYM-5052 auto-induction media at 37 °C. Harvested cells were resuspended in 50 mM potassium phosphate, pH 7.0, 2.5 mM EDTA with the addition of 0.5 mM 4-(2-Aminoethyl) benzenesulfonyl fluoride and Complete Protease Inhibitor cocktail (Roche). Cells were disrupted using an Avestin EmulsiFlex-C5 homogenizer, and nucleic acids were removed from the crude extract by addition of 2% (w/v) streptomycin sulfate. The cleared lysate was loaded onto a Q-Sepharose HP (GE Healthcare Life Sciences) column and eluted using a 0 to 1 M NaCl gradient. Fractions containing EhTrxR (wild type or C286Q) were pooled, dialyzed extensively against 12.5 mM potassium phosphate, pH 7.0, 0.5 mM EDTA and loaded onto a Blue Sepharose 6 Fast Flow column (GE Life Sciences) to remove contaminating proteins. The flow-through was loaded onto a 2′5′ ADP Sepharose 4B column (GE Life Sciences) and eluted with a gradient of 0 to 1 M NaCl. Pure protein was concentrated and buffer-exchanged into 25 mM potassium phosphate pH 7.0, 1 mM EDTA before aliquoting and freezing at −80 °C.
Thioredoxin expression and purification
Expression vectors for E. histolytica thioredoxin (EhTrx), wild type and C34S mutant, described elsewhere (Leitsch et al., 2007; Schlosser et al., 2013), were kindly provided by the Michael Duchene laboratory (Institute of Specific Prophylaxis and Tropical Medicine Center for Pathophysiology, Infectiology and Immunology, Vienna, Austria). Wild-type and mutant proteins were expressed in BL21DE3 with induction by 0.5 mM isopropyl-β-D-thiogalactopyranoside (IPTG) overnight at 25 °C. All purification steps were carried out at 4 °C. Cells were resuspended in 50 mM Tris-Cl, pH 8.0, and broken with the Emulsiflex homogenizer, and nucleic acids removed, as described above. The extract was loaded to a cobalt-NTA column, and washed with 50 mM sodium phosphate pH 8.0, 0.3 M NaCl and 25 mM imidazole. Protein was eluted by increasing the imidazole concentration to 0.5 M. Fractions containing the thioredoxin were pooled, and protein precipitated by adding ammonium sulfate to 75% saturation. The pellet was dissolved in a small volume of 20 mM Tris-Cl pH 8.0, 1 mM EDTA and 100 mM NaCl, and loaded onto a 2.6 × 100 cm Sephadex G-50 gel filtration column. Fractions containing pure thioredoxin were pooled, concentrated and frozen at −80 °C. E. coli Trx1 was expressed from E. coli strain CHW170 (Veine et al., 1998), which was a gift from Dr. Charles H. Williams at the University of Michigan, and was purified as described previously (Nelson et al., 2008a).
Crystallization, data collection and structure determination
Prior to crystallization, EhTrxR, wild type and mutants, was diluted to 10 mg/ml by mixing with 10 mM Tris-HCl, pH 7.5, alone or supplemented with 0.6–5.0 mM Auranofin or 0.6 mM AuCN, 0.6 mM NADP+ or 0.6 mM NADPH, or 3.0 mM TCEP when indicated. Crystallization conditions in each case were determined using commercial high-throughput screening kits available in deep-well format (Hampton Research), a nanoliter drop-setting Mosquito robot (TTP LabTech) operating with 96-well plates, and a hanging drop crystallization protocol. Crystals of two different morphologies were generated for EhTrxR from distinct crystallization conditions and further optimized in 24-well plates for diffraction data collection (Table 1). Plate-shaped crystals diffracting in the P21 space group formed in the absence of NADPH or NADP+ under pH <8.3, whereas rod-shaped crystals of the holoenzyme diffracting in the P212121 space group grew from pH 8.5–9.0.
Table 1.
EhTrxR x-ray data collection and refinement statistics
| Protein | EhTrxR | EhTrxR | EhTrxR | EhTrxR | EhTrxRC140S,C2 86Q | EhTrxR |
|---|---|---|---|---|---|---|
|
| ||||||
| Ligand | NADPH | NADPH/AuCN | NADPH/Auranofin | NADP+ | NADP+ | Auranofin |
|
| ||||||
| PDB ID | 4A5L | 4A65 | 4CBQ | 4CCQ | 4UP3 | 4CCR |
| Data collection | ||||||
| Space group | P212121 | P212121 | P212121 | P212121 | P212121 | P21 |
| Cell dimensions | ||||||
| a, b, c (Å) | 65.8, 91.9, 103.8 | 65.5, 91.4, 101.3 | 65.6, 91.6, 103.7 | 65.7, 92.2, 103.5 | 65.8, 92.0, 102.9 | 83.8, 91.0, 90.2 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 105.8, 90 |
| Molecules in AU | 2 | 2 | 2 | 2 | 2 | 4 |
| Wavelength | 1.11587 | 1.11587 | 1.11587 | 1.11587 | 1.11587 | 1.11587 |
| Resolution (Å) | 1.66 | 1.7 | 1.94 | 1.50 | 1.44 | 2.28 |
| Rsym or Rmerge (%) | 4.4 (38.3)1 | 5.0 (44.0) | 7.3 (69.6) | 5.2 (43.1) | 4.0 (54.0) | 8.4 (76.3) |
| I/σI | 12.7 (1.6) | 13.2 (2.0) | 9.2 (1.6) | 11.4 (1.7) | 15.0 (1.3) | 8.9 (1.5) |
| Completeness (%) | 80.6 (32.8) | 76.6 (28.8) | 99.1 (97.1) | 95.7 (77.8) | 81.2 (36.4) | 99.7 (99.9) |
| Redundancy | 3.5 (2.2) | 4.3 (3.3) | 3.9 (3.8) | 3.8 (1.9) | 3.7 (2.2) | 3.6 (3.7) |
| Crystallization conditions | 20% PEG 4000 0.1 M ammonium sulfate 0.1 M Bicine, pH 9.0 0.01 M MgCl2 0.6 mM NADPH |
22% PEG 4000 0.1 M ammonium sulfate 0.1 M Bicine, pH 9.0 1 mM AuCN 0.6 mM NADPH |
25% PEG 3350 0.2 M lithium sulfate 0.1 M Tris HCl, pH 8.9 3 mM Auranofin 0.6 mM NADPH |
20% PEG 3350 0.2 M lithium acetate 0.6 mM NADP+ |
20% PEG 3350 0.2 M lithium acetate 0.6 mM NADP+ |
20% PEG 3350 0.2 M ammonium tartrate, pH 7.0 3 mM Auranofin |
| Refinement | ||||||
| No. reflections | 56952 | 48807 | 44071 | 91863 | 87129 | 56315 |
| Rwork/Rfree (%) | 16.9/22.1 | 16.7/23.0 | 16.8/21.8 | 17.2/20.7 | 12.6/18.1 | 20.3/26.5 |
| No. atoms | ||||||
| Protein | 4786 | 4853 | 4766 | 4928 | 4791 | 8395 |
| FAD | 106 | 106 | 106 | 106 | 106 | 212 |
| NADPH/NADP+ | 632 | 63 | 87 | 88 | 144 | N/A |
| Au(I) | N/A | 2 | 2 | N/A | N/A | 4 |
| Solvent | 610 | 713 | 417 | 864 | 767 | 237 |
| Mean B value | 24.7 | 25.2 | 34.9 | 20.0 | 24.8 | 43.7 |
| B-factors | ||||||
| Protein | 23.5 | 23.9 | 34.8 | 17.7 | 23.9 | 44.4 |
| FAD | 20.1 | 20.2 | 31.7 | 17.5 | 22.9 | 36.6 |
| NADPH/NADP+ | 31.1 | 37.2 | 46.4 | 26.2 | 32.8 | N/A |
| Au(I) | N/A | 36.6 | 47.4 | N/A | N/A | 88.9 |
| Solvent | 35.8 | 35.4 | 43.7 | 32.5 | 36.8 | 40.2 |
| R.m.s deviations | ||||||
| Bond lengths (Å) | 0.026 | 0.022 | 0.017 | 0.026 | 0.019 | 0.013 |
| Bond angles (°) | 2.111 | 1.971 | 1.940 | 2.312 | 2.032 | 1. 637 |
Values in parentheses are for highest-resolution shell
number of the NADPH/NADP+ atoms used in refinement varied depending on quality of electron density map
The EhTrx crystals were obtained for the C34S mutant at 10 mg/ml with an asymmetric unit constituted by a covalent dimer cross-linked via an intermolecular disulfide bond between the active site residues Cys31. Prior to data collection, all crystals were cryo-protected by plunging them into a drop of reservoir solution supplemented with 20–25% glycerol or ethylene glycol, then flash frozen in liquid nitrogen.
Diffraction data were collected at 100–110 K at beamline 8.3.1, Advanced Light Source, Lawrence Berkeley National Laboratory, USA. Data indexing, integration, and scaling were conducted using MOSFLM (Leslie, 1992) and the programs implemented in the ELVES software suite (Holton and Alber, 2004). The crystal structure was initially determined by molecular replacement using as a search model isolated FAD- and NADPH-binding domains of Saccharomyces cerevisiae TrxR (PDB ID 3ITJ) having 61% sequence identity to EhTrxR. The EhTrxR model was built using the BUCCANEER (1994 Collaborative Computational Project; Cowtan, 2006) and COOT (Emsley and Cowtan, 2004) programs. Refinement was performed by using REFMAC5 software (1994 Collaborative Computational Project; Murshudov et al., 1997). The final coordinates were used as the molecular replacement model for the entire series of EhTrxR structures determined in this work. Data collection and refinement statistics are shown in Table 1. EhTrx structure was determined using as the molecular replacement model thioredoxin from parasitic trematode Fasciola hepatica (PDB ID 2VIM) (Line et al., 2008) having 45% sequence identity. Data collection and refinement statistics are shown in Table 2.
Table 2.
EhTrx x-ray data collection and refinement statistics
| Protein | EhTrx C34S |
|---|---|
|
| |
| PDB ID | 4CW9 |
| Data collection | |
| Space group | P212121 |
| Cell dimensions | |
| a, b, c (Å) | 35.5, 48.0, 117.4 |
| α, β, γ (°) | 90, 90, 90 |
| Molecules in AU | 2 |
| Wavelength | 1.11587 |
| Resolution (Å) | 1.84 |
| Rsym or Rmerge (%) | 4.2 (36.1)1 |
| I/σI | 14.1 (1.9) |
| Completeness (%) | 82.3 (40.7) |
| Redundancy | 3.5 (2.4) |
| Crystallization conditions | 25% PEG 3350 0.2 M NaCl 0.1 M Bis-Tris, pH 5.5 |
| Refinement | |
| No. reflections | 14028 |
| Rwork/Rfree (%) | 19.0/26.9 |
| No. atoms | |
| Protein | 1643 |
| Solvent | 99 |
| Mean B value | 35.3 |
| B-factors | |
| Protein | 36.0 |
| Solvent | 39.1 |
| R.m.s deviations | |
| Bond lengths (Å) | 0.017 |
| Bond angles (°) | 1.787 |
Values in parentheses are for highest-resolution shell
Electrospray ionization mass spectrometry
Fifty micrograms of purified protein was desalted by buffer exchange against 0.5% formic acid in water using Nanosep 10K Omega ultrafiltration devices (Pall Corporation). The retentate was recovered in a volume of 100 μl, mixed with equal volume of acetonitrile, and infused at 10 μl/min into a QSTAR XL mass spectrometer (AB Sciex). Multi-charge time-of-flight spectra were acquired in 700–1500 amu range. Zero-charge spectra were obtained by Bayesian reconstruction in 20–40 kDa range.
Assays to monitor inhibition of EhTrxR with auranofin
The thionitrobenzoate (TNB)-coupled assay for TrxR activity was modified from Mulrooney (Mulrooney, 1997). Following a pre-incubation of EhTrxR (45 nM) with auranofin (predissolved in ethanol), 20 μM E. coli Trx1 and 200 μM 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) for 3 min at 25 °C in assay buffer (50 mM Tris-HCl, pH 7.5, with 100 mM NaCl), reaction mixtures (600 μl) were supplemented with 100 μM NADPH, then monitored continuously at 412 nm to observe TNB release (ε412 for TNB = 13,600 M−1 cm−1, and two TNB molecules are released per turnover of EhTrxR). In assays where the E.coli Trx1 was replaced with the E. histolytica TrxA8 (EhTrx), the thioredoxin concentration was decreased to 2 μM.
Inhibition of EhTrxR with auranofin during turnover with high levels of DTNB (4 mM) in the absence of Trx employed the same conditions as above, except for the inclusion of a higher level of EhTrxR, at 0.5 μM. Data from the region between 80 and 100 s were used as the final linear rates for replotting vs. auranofin concentrations.
RESULTS
Overall structure of EhTrxR
Crystal structures of multiple forms of EhTrxR holoenzyme (including both pyridine nucleotide and FAD) were determined, all to resolutions < 2 Å (Table 1). Lower resolution, 2.28 Å, was achieved for EhTrxR with only FAD bound. At the time of x-ray data collection, it is likely that all NADPH was oxidized to NADP+. As expected, EhTrxR is a dimer with the interface formed mainly by the FAD domains of the monomers (Fig. 2A, 2B). Three major conformations were observed for wild type EhTrxR without resorting to protein engineering, two of them in the presence and one in the absence of pyridine nucleotide (NADPH or NADP+) during crystallization. In the absence of added NADPH, the redox-active disulfide bond was invariably positioned over the re-face of the flavin isoalloxazine ring permitting internal transfer of reducing equivalents from reduced FAD to the active-site disulfide (PDB ID 4CCR; Fig. 2A, 2C). Due to high domain mobility in the absence of the pyridine nucleotide cofactor, the NADPH-binding domain in chain D of the 4CCR structure could not be localized and, thus, was omitted from the atomic coordinates. In the presence of NADP(H), two alternative NADPH-domain orientations were observed in EhTrxR dimer. In one monomer, the nicotinamide moiety was positioned over the same flavin face with the Cys140-Cys143 disulfide bond now exposed to enable interactions with Trx. (Fig. 2B, 2D). Consistent with earlier studies on Escherichia coli TrxR (Lennon et al., 1999; Lennon et al., 2000; Waksman et al., 1994), a switch between the two conformations involves a ~ 66° rotation of the NADPH-domain relative to the FAD-domain largely along the β-sheet linker connecting both domains.
Figure 2. The EhTrxR x-ray structure.

A–B, EhTrxR dimer in conformations consistent with FAD (magenta) oxidation and intramolecular Cys140-Cys143 disulfide bond (cyan) reduction (A) and FAD reduction with NADPH (yellow) and thioredoxin binding (B). Monomers are represented by green and blue ribbons. Rotation between protein domains is achieved by rewrapping the two antiparallel β-strands which work as a domain swivel. C–D, A fragment of 2Fo-Fc electron density map (blue mesh) with 1.0 σ cutoff delineates positions of the disulfide loop (C) and bound NADP+ (D) relative to FAD. Electron density for NADP+ is progressively less defined toward the nicotinamide moiety in all structures resolved in this work. PDB ID of the corresponding structures are shown in parentheses.
An alternative orientation of the NADPH-domain observed in EhTrxR is incompatible with the electron transfer. This domain disposition is also distinct from that observed in other structurally characterized low Mr TrxRs sharing highest sequence identity with EhTrxR: S. cerevisiae (61% sequence identity to EhTrxR) (Oliveira et al., 2010), Arabidopsis thaliana (61%) (Dai et al., 1996) and barley (59%) (Kirkensgaard et al., 2009), followed by the bacterial species, Mycobacterium tuberculosis (44%) (Akif et al., 2005), E. coli (43%) (Lennon et al., 1999; Lennon et al., 2000; Waksman et al., 1994), Brucella melitensis (43%) and Deinococcus radiodurans (42%) (Obiero et al., 2010), to name a few. The disposition of the NADPH- and FAD-domains varies significantly in these structures, maybe a result of crystal packing interference with the “ball-and-socket” motion enabling the switch between two functional conformations.
A notable feature of the lower eukaryotic TrxR including EhTrxR is a 5-amino acid insert in the FAD domain that is missing from bacterial sequences. This insert forms a loop serving as a cap over the FAD adenine moiety. As judged based on the E. coli TrxR/Trx complex, this structural addition should affect Trx binding in a manner consistent with species-specific TrxR reactivity toward cognate Trx documented in the literature (Discola et al., 2009; Oliveira et al., 2010; Zhang et al., 2009).
In contrast to E. coli TrxR (Lennon et al., 2000; Waksman et al., 1994), the data described above indicate that the presence of the NADP(H) cofactor in EhTrxR was sufficient to stabilize a conformation compatible with Trx binding and electron transfer even in the absence of Trx. The E. coli TrxR-Trx complex was trapped in similar conformation by intermolecular cross-linking via a disulfide bond connecting two cysteines, one belonging to TrxR and the other to the Trx active site. A second cysteine in each site was substituted by serine (Lennon et al., 2000). Further, an NADP+ analog, 3-aminopyridine adenine dinucleotide phosphate, was used in co-crystallization to further stabilize the E. coli TrxR-Trx complex (Lennon et al., 2000). Consistent with the dithiol exposure in the absence of the Trx substrate, EhTrxR is known to reduce the small-molecule substrates methylene blue, quinones, ferricyanide and S-nitrosothiols (Arias et al., 2012).
Redox status of the Cys140-Cys143 redox center
According to the hypothetical mechanism of Auranofin action, the redox active disulfide bond in TrxR should be reduced to dithiol in order for Au(I) to bind. To validate that this condition was fulfilled, the redox status of the Cys140-Cys143 was monitored prior to mixing with Auranofin by electrospray ionization mass spectrometry (ESI-MS). Upon treatment with NADPH, the Mr of EhTrxR increased by 2 Da (Fig. 3A), suggesting that one disulfide bond had been reduced to dithiol, potentially allowing Au(I) to bind. However, regardless of whether NADPH or NADP+ was used in the crystallization, the Cys140-Cys143 pair was always oxidized to a disulfide bond in the crystals. We speculate that Au(I) binding, if any, must have been reversed during the course of crystallization due to dithiol oxidation back to a disulfide bond. Structurally, the 14-atom C(X)2C loop favors disulfide bond formation (Fig. 3B). In our studies, mutated S140XXC143 motif tends to preserve helical conformation (PDB ID 4UP3), albeit with temperature factors notably above average, particularly in chain B, where electron density for the motif is barely defined suggesting high degree of flexibility and lack of structure in the reduced state. The conformation of the disulfide bonded ring in TrxR proteins is virtually identical to that of thioredoxin despite the differences in the structural context of the C(X)2C motif between two proteins (Waksman et al., 1994).
Figure 3. Reduced and oxidized EhTrxR.

A, ESI-MS analysis of reduced (red) and oxidized (blue) EhTrxR. 2 Da experimental mass difference between NADPH-plus and NADPH-minus samples corresponds to reduction of one disulfide bond in the NADPH-treated sample. B, 2Fo-Fc electron density map (blue mesh) contoured at 1.0 σ shows the oxidized redox-active sulfhydryl groups forming a 4-residue disulfide loop.
Binding of Au(I) to non-conserved Cys286
In all of the crystals of EhTrxR grown in the presence of Auranofin, instead of binding the Cys140-Cys143 site, Au(I) was bound to the non-conserved Cys286 (Fig. 4A). Also, Cys286 was the only site modified when AuCN was used in co-crystallization instead of Auranofin (Fig. 4B). Tris(2-carboxyethyl)phosphine (TCEP), if used as an alternative to NADPH to reduce EhTrxR prior to crystallization, prevented Au(I) binding to Cys286 (Fig. 4C, D). Being a phosphine, TCEP binds gold, albeit with relatively low affinity (Dauksaite et al., 2007), thus competing with the protein thiol groups. Attenuated affinity of Cys286 for gold is suggested by two alternative conformations observed in the crystals, with only one being exposed for interactions with Au(I) (Fig. 4C–E). Analysis of temperature factors indicated that the occupancy of Au(I) is partial, ranging from 0.3 to 0.5. The propensity of Au(I) to bind protein thiol groups is not particularly surprising as it is routinely utilized in x-ray crystallography to introduce heavy atom sites. The precedent of Auranofin causing modification of cysteine residues other than the catalytic redox pair has also been previously reported (Angelucci et al., 2009; Ilari et al., 2012).
Figure 4. Cys286 gold-binding site in EhTrxR.

A–B, Gold atom originated from Auranofin (A) or AuCN (B) bound to Cys286. C–E, Free Cys286 adopts alternative conformations, only one of which is accessible for interactions with Au(I). Cys282 points away from Cys286 in all the structures resolved in this work. Fragments of 2Fo-Fc electron density map (blue mesh) contoured at 1.0 σ show gold binding site under different crystallization conditions. Au(I) is shown as a golden sphere, protein is drawn as sticks with heteroatoms colored according to the elements: oxygen in red, nitrogen in blue, and sulfur in yellow.
While Cys140-Cys143 functionality is strictly conserved across the TrxR family (Fig. 5A), Cys286 is not. The only other species where cysteine is present at a position equivalent to residue 286 in E. histolytica are Giardia species (Fig. 5B), both those sensitive and those resistant to metronidazole therapy (Tejman-Yarden et al., 2013). In other eukaryotic organisms, glutamine normally occupies this spot. In prokaryotes, the equivalent position is variable. The EhTrxRC286Q mutant generated by site-directed mutagenesis preserved the catalytic activity of the wild type and was equally susceptible to Auranofin inhibition, indicating that inhibition of EhTrxR is not through Au(I) binding to Cys286 (Fig. 6). Another pair of the semi-conserved cysteine residues, C301(X)5C307, is found at the C-terminus (Fig. 5B); both cysteines are missing from the Giardia sequences. Their sulfhydryl groups are not in suitable positions to form a disulfide bond nor to coordinate a monovalent metal ion. No redox function has been thus far associated with this pair.
Figure 5. Sequence alignments.

TrxRs share a number of conserved residues (highlighted in red) in the redox-active site (A) and the C-terminus (B). The catalytic pair Cys140-Cys143 is strictly conserved across the low Mr TrxR family, while Cys286, the site of Au(I) binding, is not. UniProt database (http://www.uniprot.org/) accession numbers are provided for each protein sequence used in alignment: Candida tropicalis (C5MIU4), Candida albicans (Q5AG89), Pichia angusta (E7R0D2), Entamoeba histolytica (C4LW95), Dictyostelium purpureum (F0ZN16), Giardia intestinalis (Giardia lamblia) (E2RU27), Trichomonas vaginalis (Q8IEV3), Escherichia coli (P0A9P4), Mycobacterium tuberculosis (P52214), Treponema pallidum (O83790). Residue numbering corresponds to EhTrxR sequence.
Figure 6. Inhibition of EhTrxR by Auranofin.
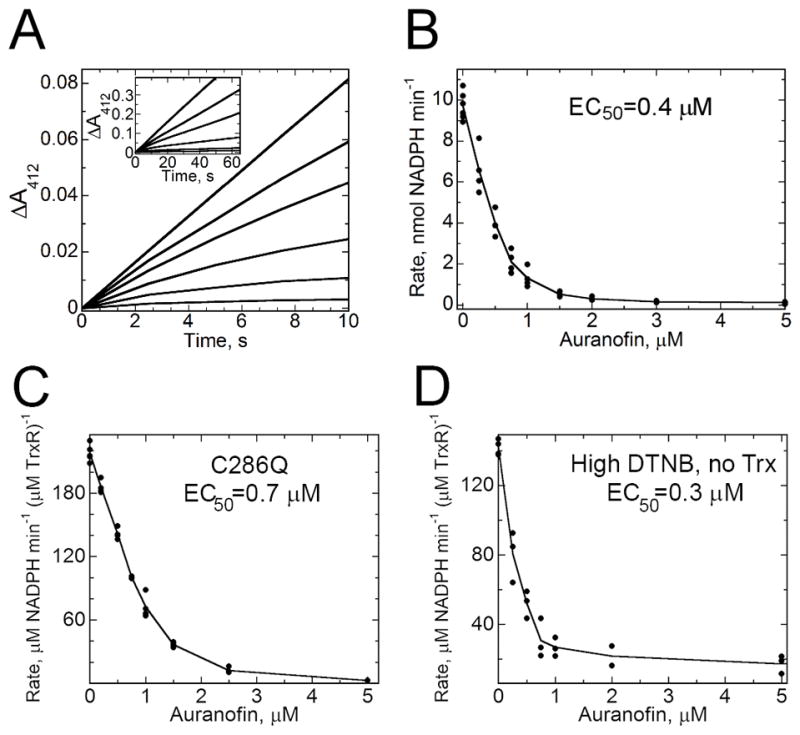
A, Non-linear increase in the absorbance of the reaction product, TNB, over the first 50 s of turnover in the presence of auranofin. EhTrxR (45 nM) was preincubated with auranofin, 2 μM EhTrx1 and 200 μM DTNB for 3 min at 25 °C in assay buffer (50 mM Tris-HCl, pH 7.5, with 100 mM NaCl), supplemented with 100 μM NADPH, then monitored continuously at 412 nm. Auranofin was included at concentrations of 0, 0.25, 0.5, 1, 2 and 5 μM, in the order of decreasing slopes. With no Auranofin added, a linear increase in 412 nm absorbance is observed for over several minutes. B, Concentration dependence of the linear rates of reaction after 50 s at each Auranofin concentration yields an EC50 value for Auranofin of 0.4 μM. C, Concentration dependence of inhibition as in B, but using C286Q TrxR. D, Concentration dependence of the linear rates of reaction over 80–100 s after addition of 4 mM DTNB (in the absence of Trx) to a pre-incubated mixture of EhTrxR (45 μM), 100 μM NADPH and auranofin at various concentrations.
In crystallization studies using a double mutant of EhTrxR lacking both C286 and C140 (C140S, C286Q), no auranofin or gold was observed bound to the protein, indicating that C143 cannot itself bind Au(I) and/or this protein does not actively release gold from its bound form in auranofin (PDB ID 4UP3).
EhTrx structure
The authentic substrate of EhTrxR, EhTrx, characterized elsewhere as TrxA8 (Leitsch et al., 2007; Schlosser et al., 2013), in this work was used in inhibition assays with EhTrxR and characterized structurally. Although both wild type and the C34S mutant of EhTrx were subjected to crystallization, only EhTrxC34S crystals were obtained and a crystal structure of the mutant to a resolution of 1.84 Å was determined (Table 2). Formation of the covalent dimer via intrasubunit Cys31-Cys31′ bond possibly forced by the crystal lattice may explain the higher crystallization propensity of the mutant compared to the wild type. The EhTrx structure has a typical Trx scaffold consisting of a central 5-stranded β-sheet surrounded by four α-helices (Fig. 7A). The conserved active site dithiol motif C31XXC34 is located at the N-terminus of α-helix 2. Replacement of one cysteine residue with a serine in C34S prevents formation of the intramolecular disulfide bond thus structurally imitating the reduced state of the motif. Unlike studies both by NMR (Jeng et al., 1994; Qin et al., 1994) and x-ray crystallography (Line et al., 2008) reporting very similar structures for oxidized and reduced Trx proteins, the EhTrxC34S mutant exhibits two distinct conformations (in chain A and chain B constituting an asymmetric unit of 4CW9) in the region harboring the dithiol group (present as an intersubunit disulfide, as mentioned above) (Fig. 7B).
Figure 7. Flexibility of the dithiol containing region in EhTrx.

A, In the crystal structure (PDB ID 4CW9), the EhTrxC34S mutant (shown in ribbon) is cross-linked via formation of the disulfide bond (in stick representation) formed between the C31 thiol groups. B, In the EhTrxC34S mutant, N-terminus of the α-helix 2 accommodating catalytic dithiol motif adopts α-helical structure in chain B which unwinds in chain A. In B, both chains are shown in the same orientation derived from superimposition of their structures.
Turnover with thioredoxin or DTNB is required for TrxR inhibition by Auranofin
Failure of Au(I) to stably bind to the Cys140-Cys143 dithiol group even when AuCN was added to the reduced (NADPH or DTT treated) EhTrxR indicates that interaction of Auranofin with EhTrxR alone may not be sufficient to cause inhibition. Consistent with this suggestion is the observation that turnover with Trx (utilizing E. coli Trx1 or EhTrx proteins) is required to initiate TrxR inactivation (Fig. 6). When EhTrxR pre-incubated with auranofin is mixed with NADPH, Trx and DTNB, turnover of the whole system is monitored by Trx-dependent TNB release at 412 nm. A slow onset of inhibition is observed (over ~50 s) as a curvature in the rate of TNB formation which then becomes linear with time after 50 s (Debnath et al., 2012b). Inclusion of NADPH in the pre-incubation mixture did not change this slow onset of inhibition under these conditions, unlike in the assays conducted previously with S. mansoni TGR (Angelucci et al., 2009). Even extension of the pre-incubation period to overnight did not affect the onset of inhibition. However, if a large excess of DTNB (4 mM) was included in the assay in the absence of Trx, it was able to act as a direct substrate of TrxR and exhibited a slow onset (non-linear progress curves as described above) as with Trx (Fig. 6D). Future studies will be conducted to address the nature of the inhibited EhTrxR complex(es) for this system.
DISCUSSION
A previously proposed mechanism of EhTrxR inhibition by Auranofin suggests that monovalent gold binds to the redox-active C(X)2C motif. The structures obtained of EhTrxR with bound gold instead showed its association with Cys286, remote from the active site. Follow-up experiments with a mutant lacking this cysteine (C286Q) demonstrated that this is a “decoy” target of gold rather than the site through which auranofin inhibition occurs. By x-ray structure analysis we were unable to demonstrate Au(I) binding to the 4-residue disulfide loop under any conditions explored in this study. It is also notable that the active site dithiol was only ever detected in its disulfide form in our structures even when high concentrations (up to 5 mM) of the reducing agents, NADPH, 2-mercaptoethanol, TCEP or DTT, alone or in combinations, were used to maintain a reducing environment throughout the course of crystallization. This either interfered with the crystal growth or still resulted in the oxidized disulfide in the crystal structure. We speculate that the short spacer of the C(X)2C motif favors re-oxidation of dithiol back to a disulfide bond rather than formation of the stable linear two-coordinate Au(I) complex that is typical for monovalent metal ions. An explanation for this phenomenon lies in the fundamentals of inorganic chemistry, where the primary determinant of metal ion binding is dictated by the metal coordination geometry – coordination number, metal-ligand bond length and ligand-metal-ligand dihedral angles (Pennella and Giedroc, 2005). In the perfect geometry of the chelate structure, monovalent metal binding achieves an extraordinary zeptomolar (10−21 M) affinity, as demonstrated by the E. coli copper, silver and gold sensor, CueR, which possesses the C(X)7C metal binding motif (Changela et al., 2003). In CueR, the coordinate bonds between Au(I) and two sulfur atoms exhibit bond distances of 2.32 and 2.39 Å with an essentially linear bond angle of 176° (Changela et al., 2003) (Fig. 8A). The shortening of the spacer between two cysteines should eventually compromise coordination geometry and reduce metal binding affinity. Consistent with this model, Au(I) binding was observed, albeit with partial occupancy, between the sulfhydryl groups of the C(X)4C motif of thioredoxin-glutatione reductase from S. mansoni (Angelucci et al., 2009), and in trypanothione reductase from L. infantum (Ilari et al., 2012) (Fig. 8B). With the spacer shortened to two amino acids in the C(X)2C motif of EhTrxR, the room between the thiol groups becomes insufficient to accommodate Au(I), and the 14-atom disulfide bonded ring is favored by bond geometry (Fig. 8C).
Figure 8. Dithiol C(X)nC motif.

A, A perfect geometry of the zeptomolar (10−21 M) chelate gold complex in CueR (Changela et al., 2003). B, C(X)4C motif of the high Mr thioredoxin-glutathione reductase (TGR) of S. mansoni (Angelucci et al., 2009) provides space to accommodate Au(I) but is too short to establish a two-coordinate linear configuration. Being in alternative conformations, one of the two cysteine residues separated by one α-helix turn, barely participates in gold binding. C, Spacing between the sulfhydryl groups in the C(X)2C motif of EhTrxR is insufficient to accommodate Au(I) and favors oxidation to disulfide instead.
Similarly to E. coli TrxR, Cys143 is expected to form a disulfide bond with cysteine in the Trx active site (Lennon et al., 2000) leaving Cys140 and a second cysteine of the Trx active site in the form of free thiols. Hypothetically, based on the involvement of an event of catalytic turnover in EhTrxR inhibition by Auranofin evidenced by our studies, the TrxR-reduced thioredoxin may be positioned more favorably to provide a second coordination bond to Au(I). High conformational plasticity of the loops harboring dithiol groups demonstrated by the crystal structures of both EhTrxR and EhTrx allow us to speculate that once EhTrx is reduced, the N-terminal helical turn unwinds exposing the cysteine thiol group (particularly Cys31) for interactions with electron acceptors or, possibly, metal coordination. However, we were unable to capture such a complex crystallographically.
Alternatively, a transient binding of Au(I) to the catalytic dithiol group, although undetectable by x-ray crystallography, may affect large-scale domain motion indispensable for coupling of the oxidative and reductive halves of the reaction in low Mr TrxR by forcing TrxR into a catalytically non-productive irreversible conformation. Multiple orientations of the NADPH-domain observed in the TrxR crystal structures, including EhTrxR, point to such a possibility.
Conclusions
Thus, the molecular mechanism of Auranofin action remains enigmatic and may differ between low and high Mr TrxRs with different catalytic motif structures and different needs for large-scale conformational changes. Given that substrate turnover is a prerequisite of the EhTrxR inhibition with Auranofin, an intermediate of this reaction may be the ultimate target. Alternatively, Auranofin may affect large-scale domain motion possibly via accumulation of an EhTrxR conformer with impaired mechanism of oxidation-reduction coupling mediated by the large-scale conformational changes. Finally, the last resort to stick to the prevailing hypothesis of the mechanism of Auranofin action is to speculate that even transient gold binding to the catalytic motif that is not detected by x-ray crystallography may slow TrxR enough to severely interfere with its physiological function.
Acknowledgments
We thank Prof. Michael Duchene for kindly providing E. histolytica thioredoxin expression vector, Mr. Potter Wickware for proof reading the manuscript, the staff members of beamline 8.3.1, James Holton, George Meigs and Jane Tanamachi, at the Advanced Light Source at Lawrence Berkeley National Laboratory, for assistance with data collection. This work was supported by National Institutes of Health grants RO1 GM050389 (to L.B.P.) and UO1 AI077882 (to S.L.R. and J.H.M.). A.D. was supported by the Bill and Melinda Gates Foundation. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. Molecular graphics images were produced in part using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41 RR001081).
ABBREVIATIONS
- Trx
thioredoxin
- TrxR
thioredoxin reductase
- TGR
thioredoxin-glutathione reductase
- DTNB
5,5′-dithiobis(2-nitrobenzoic acid)
- TNB
thionitrobenzoate
Footnotes
Accession Codes
The atomic coordinates and structure factors (EhTrxR PDB ID codes 4A5L, 4A65, 4CBQ, 4CCQ, 4CCR and 4UP3; EhTrx PDB ID code: 4CW9) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/)
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Collaborative Computational Project, Number 4. Acta Crysallogr D. 50:760–763. [Google Scholar]
- 2.Akif M, Suhre K, Verma C, Mande SC. Conformational flexibility of Mycobacterium tuberculosis thioredoxin reductase: crystal structure and normal-mode analysis. Acta Crystallogr D Biol Crystallogr. 2005;61:1603–1611. doi: 10.1107/S0907444905030519. [DOI] [PubMed] [Google Scholar]
- 3.Angelucci F, Dimastrogiovanni D, Boumis G, Brunori M, Miele AE, Saccoccia F, Bellelli A. Mapping the catalytic cycle of Schistosoma mansoni thioredoxin glutathione reductase by x-ray crystallography. The Journal of biological chemistry. 2010;285:32557–32567. doi: 10.1074/jbc.M110.141960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Angelucci F, Sayed AA, Williams DL, Boumis G, Brunori M, Dimastrogiovanni D, Miele AE, Pauly F, Bellelli A. Inhibition of Schistosoma mansoni thioredoxin-glutathione reductase by auranofin: structural and kinetic aspects. The Journal of biological chemistry. 2009;284:28977–28985. doi: 10.1074/jbc.M109.020701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arias DG, Gutierrez CE, Iglesias AA, Guerrero SA. Thioredoxin-linked metabolism in Entamoeba histolytica. Free Radic Biol Med. 2007;42:1496–1505. doi: 10.1016/j.freeradbiomed.2007.02.012. [DOI] [PubMed] [Google Scholar]
- 6.Arias DG, Regner EL, Iglesias AA, Guerrero SA. Entamoeba histolytica thioredoxin reductase: molecular and functional characterization of its atypical properties. Biochim Biophys Acta. 2012;1820:1859–1866. doi: 10.1016/j.bbagen.2012.08.020. [DOI] [PubMed] [Google Scholar]
- 7.Brown DM, Upcroft JA, Upcroft P. Cysteine is the major low-molecular weight thiol in Giardia duodenalis. Mol Biochem Parasitol. 1993;61:155–158. doi: 10.1016/0166-6851(93)90169-x. [DOI] [PubMed] [Google Scholar]
- 8.Brown DM, Upcroft JA, Upcroft P. A thioredoxin reductase-class of disulphide reductase in the protozoan parasite Giardia duodenalis. Mol Biochem Parasitol. 1996;83:211–220. doi: 10.1016/s0166-6851(96)02776-4. [DOI] [PubMed] [Google Scholar]
- 9.Cai W, Zhang L, Song Y, Wang B, Zhang B, Cui X, Hu G, Liu Y, Wu J, Fang J. Small molecule inhibitors of mammalian thioredoxin reductase. Free Radic Biol Med. 2012;52:257–265. doi: 10.1016/j.freeradbiomed.2011.10.447. [DOI] [PubMed] [Google Scholar]
- 10.Caroli A, Simeoni S, Lepore R, Tramontano A, Via A. Investigation of a potential mechanism for the inhibition of SmTGR by Auranofin and its implications for Plasmodium falciparum inhibition. Biochem Biophys Res Commun. 2012;417:576–581. doi: 10.1016/j.bbrc.2011.12.009. [DOI] [PubMed] [Google Scholar]
- 11.Changela A, Chen K, Xue Y, Holschen J, Outten CE, O’Halloran TV, Mondragón A. Molecular basis of metal-ion selectivity and zeptomolar sensitivity by CueR. Science. 2003;301:1383–1387. doi: 10.1126/science.1085950. [DOI] [PubMed] [Google Scholar]
- 12.Choudhuri G, Rangan M. Amebic infection in humans. Indian J Gastroenterol. 2012 doi: 10.1007/s12664-012-0192-2. [DOI] [PubMed] [Google Scholar]
- 13.Cowtan K. The Buccaneer software for automated model building. 1 Tracing protein chains. Acta Crystallogr D Biol Crystallogr. 2006;62:1002–1011. doi: 10.1107/S0907444906022116. [DOI] [PubMed] [Google Scholar]
- 14.da Silva MT, Silva-Jardim I, Portapilla GB, de Lima GM, Costa FC, Anibal FF, Thiemann OH. In vivo and in vitro auranofin activity against Trypanosoma cruzi: Possible new uses for an old drug. Exp Parasitol. 2015 doi: 10.1016/j.exppara.2015.05.012. [DOI] [PubMed] [Google Scholar]
- 15.Dai S, Saarinen M, Ramaswamy S, Meyer Y, Jacquot JP, Eklund H. Crystal structure of Arabidopsis thaliana NADPH dependent thioredoxin reductase at 2.5 A resolution. J Mol Biol. 1996;264:1044–1057. doi: 10.1006/jmbi.1996.0695. [DOI] [PubMed] [Google Scholar]
- 16.Dauksaite V, Lorentzen M, Besenbacher F, Kjems J. Antibody-based protein detection using piezoresistive cantilever arrays. Nanotechnology. 2007;18:125503. [Google Scholar]
- 17.Debnath A, Ndao M, Reed SL. Reprofiled drug targets ancient protozoans: Drug discovery for parasitic diarrheal diseases. Gut microbes. 2012a;4 doi: 10.4161/gmic.22596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Debnath A, Parsonage D, Andrade RM, He C, Cobo ER, Hirata K, Chen S, Garcia-Rivera G, Orozco E, Martinez MB, Gunatilleke SS, Barrios AM, Arkin MR, Poole LB, McKerrow JH, Reed SL. A high-throughput drug screen for Entamoeba histolytica identifies a new lead and target. Nature medicine. 2012b;18:956–960. doi: 10.1038/nm.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Discola KF, de Oliveira MA, Rosa Cussiol JR, Monteiro G, Barcena JA, Porras P, Padilla CA, Guimaraes BG, Netto LE. Structural aspects of the distinct biochemical properties of glutaredoxin 1 and glutaredoxin 2 from Saccharomyces cerevisiae. J Mol Biol. 2009;385:889–901. doi: 10.1016/j.jmb.2008.10.055. [DOI] [PubMed] [Google Scholar]
- 20.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 21.Fahey RC, Newton GL, Arrick B, Overdank-Bogart T, Aley SB. Entamoeba histolytica: a eukaryote without glutathione metabolism. Science. 1984;224:70–72. doi: 10.1126/science.6322306. [DOI] [PubMed] [Google Scholar]
- 22.Freeman CD, Klutman NE, Lamp KC. Metronidazole. A therapeutic review and update. Drugs. 1997;54:679–708. doi: 10.2165/00003495-199754050-00003. [DOI] [PubMed] [Google Scholar]
- 23.Gromer S, Arscott LD, Williams CH, Jr, Schirmer RH, Becker K. Human placenta thioredoxin reductase. Isolation of the selenoenzyme, steady state kinetics, and inhibition by therapeutic gold compounds. The Journal of biological chemistry. 1998;273:20096–20101. doi: 10.1074/jbc.273.32.20096. [DOI] [PubMed] [Google Scholar]
- 24.Harbut MB, Vilcheze C, Luo X, Hensler ME, Guo H, Yang B, Chatterjee AK, Nizet V, Jacobs WR, Jr, Schultz PG, Wang F. Auranofin exerts broad-spectrum bactericidal activities by targeting thiol-redox homeostasis. Proc Natl Acad Sci U S A. 2015;112:4453–4458. doi: 10.1073/pnas.1504022112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holton J, Alber T. Automated protein crystal structure determination using ELVES. Proc Natl Acad Sci U S A. 2004;101:1537–1542. doi: 10.1073/pnas.0306241101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ilari A, Baiocco P, Messori L, Fiorillo A, Boffi A, Gramiccia M, Di Muccio T, Colotti G. A gold-containing drug against parasitic polyamine metabolism: the X-ray structure of trypanothione reductase from Leishmania infantum in complex with auranofin reveals a dual mechanism of enzyme inhibition. Amino acids. 2012;42:803–811. doi: 10.1007/s00726-011-0997-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jeng MF, Campbell AP, Begley T, Holmgren A, Case DA, Wright PE, Dyson HJ. High-resolution solution structures of oxidized and reduced Escherichia coli thioredoxin. Structure. 1994;2:853–868. doi: 10.1016/s0969-2126(94)00086-7. [DOI] [PubMed] [Google Scholar]
- 28.Kean WF, Hart L, Buchanan WW. Auranofin. Br J Rheumatol. 1997;36:560–572. doi: 10.1093/rheumatology/36.5.560. [DOI] [PubMed] [Google Scholar]
- 29.Kirkensgaard KG, Hagglund P, Finnie C, Svensson B, Henriksen A. Structure of Hordeum vulgare NADPH-dependent thioredoxin reductase 2. Unwinding the reaction mechanism. Acta Crystallogr D Biol Crystallogr. 2009;65:932–941. doi: 10.1107/S0907444909021817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leitsch D, Kolarich D, Wilson IB, Altmann F, Duchene M. Nitroimidazole action in Entamoeba histolytica: a central role for thioredoxin reductase. PLoS biology. 2007;5:e211. doi: 10.1371/journal.pbio.0050211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lennon BW, Williams CH, Jr, Ludwig ML. Crystal structure of reduced thioredoxin reductase from Escherichia coli: structural flexibility in the isoalloxazine ring of the flavin adenine dinucleotide cofactor. Protein Sci. 1999;8:2366–2379. doi: 10.1110/ps.8.11.2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lennon BW, Williams CH, Jr, Ludwig ML. Twists in catalysis: alternating conformations of Escherichia coli thioredoxin reductase. Science. 2000;289:1190–1194. doi: 10.1126/science.289.5482.1190. [DOI] [PubMed] [Google Scholar]
- 33.Leslie AGW. Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4 ESF-EAMCB Newslett. Protein Crystallogr. 1992;26 [Google Scholar]
- 34.Li H, Hu J, Wu S, Wang L, Cao X, Zhang X, Dai B, Cao M, Shao R, Zhang R, Majidi M, Ji L, Heymach JV, Wang M, Pan S, Minna J, Mehran RJ, Swisher SG, Roth JA, Fang B. Auranofin-mediated inhibition of PI3K/AKT/mTOR axis and anticancer activity in non-small cell lung cancer cells. Oncotarget. 2015 doi: 10.18632/oncotarget.6516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Line K, Isupov MN, Garcia-Rodriguez E, Maggioli G, Parra F, Littlechild JA. The Fasciola hepatica thioredoxin: High resolution structure reveals two oxidation states. Molecular and Biochemical Parasitology. 2008;161:44–48. doi: 10.1016/j.molbiopara.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 36.Madeira JM, Gibson DL, Kean WF, Klegeris A. The biological activity of auranofin: implications for novel treatment of diseases. Inflammopharmacology. 2012;20:297–306. doi: 10.1007/s10787-012-0149-1. [DOI] [PubMed] [Google Scholar]
- 37.Martinez-Gonzalez JJ, Guevara-Flores A, Rendon JL, del Arenal IP. Auranofin-induced oxidative stress causes redistribution of the glutathione pool in Taenia crassiceps cysticerci. Mol Biochem Parasitol. 2015;201:16–25. doi: 10.1016/j.molbiopara.2015.05.001. [DOI] [PubMed] [Google Scholar]
- 38.Mulrooney SB. Application of a single-plasmid vector for mutagenesis and high-level expression of thioredoxin reductase and its use to examine flavin cofactor incorporation. Protein Expr Purif. 1997;9:372–378. doi: 10.1006/prep.1996.0698. [DOI] [PubMed] [Google Scholar]
- 39.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 40.Nelson KJ, Day AE, Zeng BB, King SB, Poole LB. Isotope-coded, iodoacetamide-based reagent to determine individual cysteine pK(a) values by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Anal Biochem. 2008a;375:187–195. doi: 10.1016/j.ab.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nelson KJ, Parsonage D, Hall A, Karplus PA, Poole LB. Cysteine pK(a) values for the bacterial peroxiredoxin AhpC. Biochemistry. 2008b;47:12860–12868. doi: 10.1021/bi801718d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Obiero J, Pittet V, Bonderoff SA, Sanders DA. Thioredoxin system from Deinococcus radiodurans. J Bacteriol. 2010;192:494–501. doi: 10.1128/JB.01046-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oliveira MA, Discola KF, Alves SV, Medrano FJ, Guimaraes BG, Netto LE. Insights into the specificity of thioredoxin reductase-thioredoxin interactions. A structural and functional investigation of the yeast thioredoxin system. Biochemistry. 2010;49:3317–3326. doi: 10.1021/bi901962p. [DOI] [PubMed] [Google Scholar]
- 44.Omata Y, Folan M, Shaw M, Messer RL, Lockwood PE, Hobbs D, Bouillaguet S, Sano H, Lewis JB, Wataha JC. Sublethal concentrations of diverse gold compounds inhibit mammalian cytosolic thioredoxin reductase (TrxR1) Toxicol In Vitro. 2006;20:882–890. doi: 10.1016/j.tiv.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 45.Pennella MA, Giedroc DP. Structural determinants of metal selectivity in prokaryotic metalresponsive transcriptional regulators. Biometals : an international journal on the role of metal ions in biology, biochemistry, and medicine. 2005;18:413–428. doi: 10.1007/s10534-005-3716-8. [DOI] [PubMed] [Google Scholar]
- 46.Prast-Nielsen S, Huang HH, Williams DL. Thioredoxin glutathione reductase: its role in redox biology and potential as a target for drugs against neglected diseases. Biochim Biophys Acta. 2011;1810:1262–1271. doi: 10.1016/j.bbagen.2011.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qin J, Clore GM, Gronenborn AM. The high-resolution three-dimensional solution structures of the oxidized and reduced states of human thioredoxin. Structure. 1994;2:503–522. doi: 10.1016/s0969-2126(00)00051-4. [DOI] [PubMed] [Google Scholar]
- 48.Reeves SA, Parsonage D, Nelson KJ, Poole LB. Kinetic and thermodynamic features reveal that Escherichia coli BCP is an unusually versatile peroxiredoxin. Biochemistry. 2011;50:8970–8981. doi: 10.1021/bi200935d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sannella AR, Casini A, Gabbiani C, Messori L, Bilia AR, Vincieri FF, Majori G, Severini C. New uses for old drugs. Auranofin, a clinically established antiarthritic metallodrug, exhibits potent antimalarial effects in vitro: Mechanistic and pharmacological implications. FEBS letters. 2008;582:844–847. doi: 10.1016/j.febslet.2008.02.028. [DOI] [PubMed] [Google Scholar]
- 50.Schlosser S, Leitsch D, Duchene M. Entamoeba histolytica: identification of thioredoxintargeted proteins and analysis of serine acetyltransferase-1 as a prototype example. Biochem J. 2013;451:277–288. doi: 10.1042/BJ20121798. [DOI] [PubMed] [Google Scholar]
- 51.Sharlow ER, Leimgruber S, Murray S, Lira A, Sciotti RJ, Hickman M, Hudson T, Leed S, Caridha D, Barrios AM, Close D, Grogl M, Lazo JS. Auranofin is an apoptosis-simulating agent with in vitro and in vivo anti-leishmanial activity. ACS Chem Biol. 2014;9:663–672. doi: 10.1021/cb400800q. [DOI] [PubMed] [Google Scholar]
- 52.Song L, Li J, Xie S, Qian C, Wang J, Zhang W, Yin X, Hua Z, Yu C. Thioredoxin glutathione reductase as a novel drug target: evidence from Schistosoma japonicum. PloS one. 2012;7:e31456. doi: 10.1371/journal.pone.0031456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tejman-Yarden N, Miyamoto Y, Leitsch D, Santini J, Debnath A, Gut J, McKerrow JH, Reed SL, Eckmann L. A reprofiled drug, auranofin, is effective against metronidazole-resistant Giardia lamblia. Antimicrob Agents Chemother. 2013;57:2029–2035. doi: 10.1128/AAC.01675-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Topkas E, Cai N, Cumming A, Hazar-Rethinam M, Gannon OM, Burgess M, Saunders NA, Endo-Munoz L. Auranofin is a potent suppressor of osteosarcoma metastasis. Oncotarget. 2015 doi: 10.18632/oncotarget.5704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Veine DM, Mulrooney SB, Wang PF, Williams CH., Jr Formation and properties of mixed disulfides between thioredoxin reductase from Escherichia coli and thioredoxin: evidence that cysteine-138 functions to initiate dithiol-disulfide interchange and to accept the reducing equivalent from reduced flavin. Protein Sci. 1998;7:1441–1450. doi: 10.1002/pro.5560070621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Waksman G, Krishna TS, Williams CH, Jr, Kuriyan J. Crystal structure of Escherichia coli thioredoxin reductase refined at 2 A resolution. Implications for a large conformational change during catalysis. J Mol Biol. 1994;236:800–816. [PubMed] [Google Scholar]
- 57.Williams CH, Arscott LD, Muller S, Lennon BW, Ludwig ML, Wang PF, Veine DM, Becker K, Schirmer RH. Thioredoxin reductase two modes of catalysis have evolved. Eur J Biochem. 2000;267:6110–6117. doi: 10.1046/j.1432-1327.2000.01702.x. [DOI] [PubMed] [Google Scholar]
- 58.You BR, Park WH. Auranofin induces mesothelioma cell death through oxidative stress and GSH depletion. Oncol Rep. 2015 doi: 10.3892/or.2015.4382. [DOI] [PubMed] [Google Scholar]
- 59.Zhang Z, Bao R, Zhang Y, Yu J, Zhou CZ, Chen Y. Crystal structure of Saccharomyces cerevisiae cytoplasmic thioredoxin reductase Trr1 reveals the structural basis for species-specific recognition of thioredoxin. Biochim Biophys Acta. 2009;1794:124–128. doi: 10.1016/j.bbapap.2008.09.011. [DOI] [PubMed] [Google Scholar]