

SUMMARY
While an essential role of HIV-1 integrase (IN) for integration of viral cDNA into human chromosome is established, studies with IN mutants and allosteric IN inhibitors (ALLINIs) have suggested that IN can also influence viral particle maturation. However, it has remained enigmatic as to how IN contributes to virion morphogenesis. Here we demonstrate that IN directly binds the viral RNA genome in virions. These interactions have specificity as IN exhibits distinct preference for select viral RNA structural elements. We show that IN substitutions that selectively impair its binding to viral RNA result in eccentric, non-infectious virions without affecting nucleocapsid-RNA interactions. Likewise, ALLINIs impair IN binding to viral RNA in virions of wild type but not escape mutant virus. These results reveal an unexpected biological role of IN binding to the viral RNA genome during virion morphogenesis and elucidate the mode of action of ALLINIs.
In brief
HIV-1 integrase binding to the viral RNA genome is necessary for formation of infectious viral particles and can be impaired by allosteric integrase inhibitors.
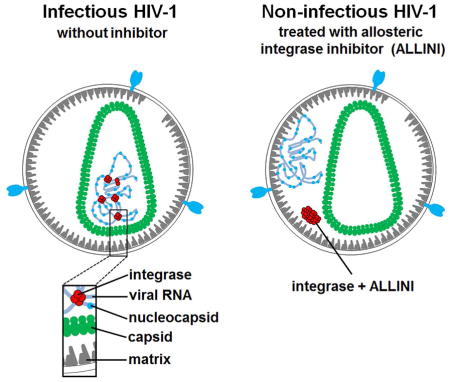
INTRODUCTION
HIV-1 integrase (IN) catalyzes the covalent insertion of the reverse transcribed viral genome into the host chromosome during the early steps of HIV-1 infection. A tetramer of IN binds both double stranded viral DNA ends to form the stable synaptic complex (SSC) and associates with cellular protein LEDGF/p75, which both enhances integration efficiency and preferentially guides HIV-1 integration to actively transcribed genes (reviewed in (Debyser et al., 2015; Engelman and Cherepanov, 2008)). The catalytic activity of IN has been exploited as a therapeutic target and three FDA approved inhibitors (Raltegravir, Elvitegravir and Dolutegravir) have witnessed widespread clinical success.
Mutational studies have unexpectedly revealed that various substitutions in the IN coding region can influence not only integration but also other viral replication steps (reviewed in (Engelman, 1999)). Therefore, IN mutations have been grouped into two general classes: substitutions that selectively impair integration have been assigned to class I, and IN mutants that adversely affect multiple steps of virus replication have been grouped as class II (reviewed in (Engelman, 1999)). Some class II IN mutations impair correct virus particle maturation during the late stage of replication. A hallmark of this phenotype is the mislocalization of the ribonucleoprotein complexes (RNPs) to an eccentric position between the empty capsid (CA) core and the particle membrane in mature virions, whereas normal virions contain RNPs within the CA core. For the past 20 years it has remained enigmatic as to how IN could contribute to proper viral particle maturation.
Allosteric IN inhibitors (ALLINIs, which are also known as LEDGINs, NCINIs or INLAIs) have recently emerged as a promising class of antiretroviral agents and select compounds are currently in clinical trials (Christ et al., 2010; Fader et al., 2014; Gupta et al., 2014; Kessl et al., 2012; Le Rouzic et al., 2013; van Bel et al., 2014). ALLINIs bind at the IN catalytic core domain (CCD) dimer interface and induce aberrant IN multimerization. Unexpectedly, ALLINIs potently impair the late stage of HIV-1 replication. In particular, these inhibitors selectively interfere with proper virus particle maturation and yield non-infectious particles with eccentrically positioned RNPs closely resembling those seen for certain class II IN mutants (Balakrishnan et al., 2013; Desimmie et al., 2013; Fontana et al., 2015; Jurado et al., 2013). However, it is unclear how ALLINIs, through the promotion of aberrant IN multimerization, adversely affect virion maturation and result in the mislocalization of the RNPs outside the CA core.
These observations prompted us to test the hypothesis that HIV-1 IN interacts with the viral RNA genome in virions and that these interactions are essential for correct viral particle morphogenesis. Further rationale for this notion was provided by an earlier study which had selected RNA oligonucleotides from a random pool of in vitro transcribed molecules of artificial sequences and showed that they can bind recombinant HIV-1 IN with high affinity (Allen et al., 1995). However, it remained unknown whether IN can interact with RNA in virions. To examine this possibility we have employed a crosslinking-immunoprecipitation sequencing (CLIP-seq) approach (Kutluay et al., 2014), which allows for the identification of RNA molecules bound by an RNA-binding protein of interest in physiologically relevant settings at near-nucleotide resolution. Our results have revealed that HIV-1 IN binds to the viral RNA genome in virions and that ALLINIs inhibit these interactions.
RESULTS
HIV-1 integrase binds the viral RNA genome in virions
To examine whether IN is bound to RNA in virions we carried out CLIP-seq experiments (Kutluay et al., 2014) with wild type (WT) HIV-1NL4-3. Incorporation of 4-thiouridine (4SU) in the nascent RNAs allowed us to crosslink interacting protein-RNA complexes. Subsequent immunoprecipitation with IN antibody followed by autoradiography revealed the IN-RNA adducts (Figure 1A) indicating that IN is directly bound to RNA in virions. The IN-RNA adducts were not seen in control experiments with HIV-1NL4-3 ΔIN, which yields eccentric, non-infectious virions (Bukovsky and Gottlinger, 1996; Engelman et al., 1995; Jurado et al., 2013). CLIP-seq analysis allowed us to identify crosslinking sites in RNA by the appearance of T to C mutations arising during reverse transcription. Figure 1B shows that ~95% of IN-bound RNAs were derived from viral RNAs, whereas a much smaller fraction (~5%) was of cellular origin. The higher levels of IN binding to viral versus cellular RNA seen in our CLIP-seq experiments is consistent with the enriched levels of viral (~80%) versus cellular (~20%) RNA present in virions as determined by RNA-seq (Kutluay et al., 2014). In parallel control experiments, only residual reads were seen with virions produced from HIV-1NL4-3 ΔIN (Figures 1C). Importantly, IN binding to the viral RNA genome was not uniform across the viral sequence but instead displayed evidence of selectivity with highly reproducible binding sites evident at numerous discrete positions across the genome (Figure S1).
Figure 1. CLIP-seq experiments showing HIV-1 IN binding to viral RNA in virions.

(A) A representative autoradiogram of IN-RNA crosslinked adducts from HIV-1NL4-3 and HIV-1NL4-3 ΔIN virions (upper panel). Corresponding western blot analysis of IN in immunoprecipitated fractions (lower panel). (B) Percentage of reads that map to cellular and viral RNA obtained from 2 independent CLIP-Seq analysis of IN-RNA adducts from WT virions. Error bars indicate standard errors. (C) CLIP-seq results showing read densities (frequency distribution of nucleotide occurrence) mapped to the viral genome obtained from HIV-1NL4-3 (red) and HIV-1NL4-3 ΔIN (blue) progeny virions. The read densities have been normalized with respect to total reads for WT HIV-1NL4-3. A collinear schematic diagram of the HIV-1 genome is shown above.
Comparison of binding sites along the viral genome in virions identified by CLIP-seq approaches for IN and previously reported NC (Kutluay et al., 2014) revealed distinct binding patterns for these two proteins (Figure S2A and B). For example, IN but not NC showed a strong preference for binding the trans-activation response (TAR) element (peak centered on nucleotide position 30) (Figure S2C). In contrast, IN did not bind the Psi packaging element (peak centered on nucleotide position 337), a preferred binding region for NC (Figure S2C). Additionally, differential binding by NC and IN was evident in most other regions of the viral RNA genome (Figure S2D, E and F). Both IN and NC interacted with the rev response element (RRE), but the local binding sites of these proteins within this RNA structural element exhibited both similarities and differences (Figure S2F). For example, both IN and NC bound to nucleotide positions 7516 and 7570 in RRE, but only IN exhibited preference for additional RRE sites (nucleotide positions 7364 and 7437). Overall, comparison of the 25 most frequent binding sites for IN and NC revealed only three overlapping sites. Taken together, the CLIP-seq results indicate largely distinct interactions for IN and NC with the viral RNA genome in virions.
IN preferentially binds to selected RNA elements in vitro
We next analyzed the binding of WT full-length recombinant IN to synthetic RNA molecules in vitro. First, we examined IN binding to an in vitro transcribed viral RNA segment, vRNA(1-850), using size exclusion chromatography (SEC) (Figure 2A), which had been used to monitor IN binding to synthetic viral DNA (~ 1 kbp) (Kessl et al., 2011). Unliganded IN is retained by the SEC column after centrifugation (Figure 2B, lane 3), whereas larger complexes including unbound nucleic acids and nucleic acids in the complex with IN are excluded from the column. Using this approach, we examined two reaction conditions. Duplicate IN-nucleic acid complexes were formed in the physiologically relevant (100 mM NaCl) ionic strength buffer. After complex formation, one set of samples was adjusted to higher ionic strength buffer (1 M NaCl). Both conditions were then subjected to SEC in their corresponding buffer (Figure 2B). Under physiologically relevant conditions IN was excluded from the column in the presence of viral DNA or viral RNA (Figure 2B, lanes 4 and 5) indicating that IN binds either nucleic acid. However, in samples subjected to higher ionic strength buffer prior to SEC, the SSC formed between IN and viral DNA remained intact, as expected (Figure 2B, lane 6) (Kessl et al., 2011; Kotova et al., 2010), whereas the preformed IN-viral RNA complex dissociated (Figure 2B, lane 7).
Figure 2. HIV-1 IN binding to synthetic viral RNA segments in vitro.

(A) Schematic for the SEC approach to test IN interactions with nucleic acids. (B) SDS-PAGE and immunoblotting with anti-IN antibodies of SEC fractions. Lane 1: molecular weight markers; Lane 2: 1/2 input IN; Lane 3: unliganded IN subjected to SEC; Lanes 4 and 5: IN+vDNA or IN+vRNA(1-850) complexes were assembled in the buffer containing 0.1 M NaCl and subjected to SEC in the same buffer. Lanes 6 and 7: IN+vDNA or IN+vRNA(1-850) complexes were formed in the buffer containing 0.1 M NaCl. Then NaCl concentration was increased to 1M followed by SEC. (C) AlphaScreen assay design to monitor direct binding between 6xHis-tagged WT IN and biotinylated RNA oligonucleotides (only IN dimer shown for simplicity). “A” and “D” indicate anti-His Acceptor and streptavidin coated Donor beads bound to 6xHis-tagged IN or biotin respectively. (D) Representative AlphaScreen binding curves for IN binding to vRNA(1-57)-TAR in the presence of 0.1 M or 1 M NaCl. The average values of three independent experiments and corresponding standard deviations are shown. (E and F) Summary of AlphaScreen-based assays for IN binding affinities for various RNA oligonucleotides with standard errors of three independent experiments shown.
To determine the relative binding affinity of recombinant WT IN for various viral RNA structural elements and cellular RNAs we have developed an AlphaScreen approach (Figure 2C). We first examined IN binding to several structurally distinct viral RNA elements of similar nucleotide length derived from the 5′-UTR. The AlphaScreen binding assay demonstrated a tight interaction between IN and vRNA(1-57)-TAR in physiologically relevant ionic strength buffer of 0.1 M NaCl (Figure 2D). In agreement with the SEC data, high ionic strength buffers (1 M NaCl) abolished IN-vRNA(1-57)-TAR binding (Figure 2D). Figure 2E shows that IN bound to vRNA(1-57)-TAR with significantly higher affinity (2.8 ± 0.4 nM) than vRNA(58-104)-polyadenylation signal (poly-A), vRNA(237-279)-dimerization initiation sequence (DIS), vRNA(278-305)-splice donor (SD), or vRNA(311-336)-Psi. Overall, these in vitro findings are consistent with the IN binding pattern seen in CLIP-seq experiments (Figures 1C, S1 and S2).
We used the vRNA(1-57)-TAR segment as an example to dissect whether IN exhibits preference for certain RNA nucleotide sequence and/or structural features (Figure S3). Since CLIP-seq data (Figures 1C, S2C and S3) revealed that nucleotide 30, located in the loop of TAR, was the primary 4SU nucleotide binding site (accounting for ~80% of T-to-C conversions within TAR) to IN (Figure S3A), our mutagenesis experiments targeted the loop and adjacently located 3-nt bulge of TAR (Figure S3B). Deletions of these structural elements reduced the IN binding affinity to vRNA(1-57)-TAR by ~14-fold while substitutions in the loop and 3-nt bulge did not significantly compromise IN binding to vRNA(1-57)-TAR (Figure S3C). Furthermore, the Tat derived L50 peptide (Athanassiou et al., 2007), which selectively engages both the loop and 3-nt bulge but not the double stranded stem of TAR, potently inhibited IN binding to vRNA(1-57)-TAR (Figure S3D). Collectively, these results indicate that IN displays a preference for select structural RNA elements of vRNA(1-57)-TAR including the adjacently located loop and 3-nt bulge, as opposed to a specific nucleotide sequence. Furthermore, these in vitro findings are in excellent agreement with the CLIP-seq results which identified the TAR loop as a major binding site for IN (Figures 1C, S1 and S2).
Next, we compared the relative binding affinities of IN to larger RNA segments of viral and cellular origin (Figure 2F). Due to the varying length of different RNAs used in these experiments, we normalized the binding affinities based on the overall nucleotide concentration in the reaction. IN bound with similar affinities to vRNA(1-850) and vRNA(7350-8200) containing TAR and RRE, respectively. However, IN exhibited ~8, 16 and 180-fold reduced affinities for human mRNA mix, yeast tRNA mix and human tRNALys, respectively (Figure 2F).
IN modulates RNA structure in vitro
To examine whether IN can affect RNA structure, we monitored the interaction of recombinant full-length WT IN with in vitro transcribed vRNA(1-850) using atomic force microscopy (AFM) (Figures 3 and S3). Figures 3A and S3E show that IN alone did not yield any detectable signal, whereas vRNA(1-850) molecules were uniformly dispersed (Figures 3B and S3F). The measured vRNA(1-850) volumes were compatible with predicted in silico calculations (Figure S3H). Strikingly, AFM images of IN-vRNA(1-850) complexes markedly differed in both shape and size compared to unliganded vRNA(1-850) (compare Figure 3C with B, and Figure S3G with F). Specifically, IN promoted the formation of large nucleoprotein complexes; IN did not coat the vRNA(1-850) but instead appeared to provide nucleation points to bridge between several vRNA(1-850) molecules (Figure 3C and D). Furthermore, measured volumes of bound IN (bright spots indicated by arrows in Figure 3D) corresponded to the sizes of dimers or tetramers (Figure S3I).
Figure 3. Biophysical analysis of IN-vRNA(1-850) complexes.

AFM images of SEC purified samples: IN alone (A), vRNA(1-850) alone (B) and IN+vRNA(1-850) (C and D). (D) The zoomed image of the box in (C) with arrows pointing to the bright spots corresponding to IN bound to vRNA(1-850). (E) The design of AlphaScreen-based RNA bridging assay (only IN dimer shown for simplicity). “A” and “D” indicate anti-DIG Acceptor and streptavidin coated Donor beads bound to either biotin or digoxin (DIG), respectively. (F) The results of the RNA bridging assay with 25 nM of total RNA incubated with indicated concentrations of IN and RT. The average values of three independent experiments and corresponding standard deviations are shown. (G) BS3 crosslinking to analyze the multimeric state of IN in the context of unliganded IN (lanes 2 and 3), IN+ribonucleotides mixture (rNTPs) (lanes 5 and 6), IN+vRNA(237-279)-DIS (lanes 8 and 9), and IN+vRNA(1-850) (lanes 11 and 12) in vitro compared to the multimeric states of IN in virions of HIV-1NL4-3 (lanes 14 and 15). Positions of monomeric (M) and dimeric (D) forms as well as higher-order aggregates are indicated.
To evaluate the stoichiometry of IN and vRNA(1-850) interactions, we added a molar excess of IN to vRNA(1-850) and subjected the complex to SEC to remove unbound protein (Figure 2B). Immunoblotting (Figure 2B) and AFM (Figures 3C and S3G) analysis confirmed that purified samples contained IN-vRNA(1-850) complexes and not unbound IN or vRNA(1-850). Examination of the purified complex in six independent experiments by immunoblotting for IN and UV spectroscopy (OD260 nm) for RNA indicated a ratio of ~4 IN per one vRNA(1-850) molecule (Figure S3J).
Because these data suggested that IN bridges separate RNA molecules, we developed an AlphaScreen-based RNA bridging assay (Figure 3E). This approach is complementary to the AFM experiments, which used longer vRNA(1-850) (Figure 3C), as the RNA bridging assay utilized short vRNA(1-57)-TAR oligonucleotides to avoid potential non-specific RNA coating induced complex aggregation. Furthermore, we varied the IN concentration from 0 nM to 1600 nM to delineate whether RNA bridging could be induced by soluble low-order oligomeric forms and/or higher order aggregates of the protein (Figure 3F). For example, in our reaction conditions containing 100 mM NaCl, 10 to 100 nM IN constitutes a range of soluble low-order oligomers such that monomeric IN predominates at the lower concentrations and dimeric and tetrameric forms prevail with increasing IN concentrations (Deprez et al., 2000). However, subsequent increased concentrations of IN, in the same physiologically relevant buffer causes the formation of higher order aggregates (Deprez et al., 2000). Figure 3F revealed a bell-shaped dose response curve where the AlphaScreen signal increased markedly with increasing IN concentrations up to 100 nM, corresponding to increasing dimeric and tetrameric forms of IN. In contrast, subsequent increases of IN concentrations, corresponding to increased higher order aggregates, yielded reduced AlphaScreen signal. Of note, parallel experiments with reverse transcriptase (RT) protein did not yield any detectable AlphaScreen signal, indicating that this RNA binding viral protein was unable to bridge two RNA molecules (Figure 3F). These results corroborate the AFM results (Figures 3C and D) and indicate that only soluble low-oligomeric forms (dimers and tetramers) of IN rather than higher order aggregates are capable of bridging RNA molecules.
The above findings suggested the possibility that RNA binding might stabilize functional low- order oligomeric forms of IN. The rationale for this idea is based on the fact that unliganded HIV-1 IN is poorly soluble, but LEDGF/p75 has been shown to stabilize a soluble IN tetramer both in vitro and in cells (Llano et al., 2006; McKee et al., 2008). While experiments with LEDGF/p75 knockout cells have demonstrated the key role of this chromatin bound protein during HIV-1 integration, LEDGF/p75 is fully dispensable during particle morphogenesis as virions produced in the presence or absence of LEDGF/p75 are equally infectious (Fadel et al., 2014). This raises the question as to how functional forms of IN are maintained in the virions following proteolytic cleavage of Gag-Pol. To test whether viral RNA binding can modulate recombinant IN multimerization we used bis(sulfosuccinimidyl)suberate (BS3) to crosslink interacting protein subunits. We assayed 200 nM IN in physiologically relevant 100 mM NaCl buffer, where unliganded protein forms both low-order oligomers (dimers and tetramers) as well as higher-order, functionally compromised aggregates (Figure 3G, lane 3). Addition of ribonucleotide mixture or vRNA(237-279)-DIS, which do not bind IN, before BS3 crosslinking had no impact on IN multimerization (lanes 6 and 9). However, the addition of vRNA(1-850) to IN under the same reaction conditions markedly reduced the formation of higher-order aggregates and instead stabilized low-order oligomers (compare lane 12 with lanes 3, 6 and 9). Importantly, the profile of BS3 crosslinked IN-vRNA(1-850) complexes in vitro closely resembles that in virions (compare lane 15 with lane 12).
Identification of HIV-1 IN residues interacting with vRNA
To map IN residues interacting with viral RNA we employed a mass spectrometry (MS)-based protein footprinting approach (Kvaratskhelia and Le Grice, 2008). The Lys specific small modifying reagent NHS-biotin was used to compare surface topologies of recombinant IN in the presence or absence of synthetic vRNA(1-57)-TAR (Figure 4). The Lys residues that are surface exposed in both unliganded IN and the IN-vRNA(1-57)-TAR complex are readily modified by NHS-biotin, whereas the residues that engage viral RNA are shielded from the modification in the nucleoprotein complex. We detected modifications of 12 Lys residues (K14, K159, K160, K186, K215, K219, K244, K258, K264, K266, K266, K273) in unliganded IN. Strikingly, only 3 adjacently located Lys residues (K264, K266 and K273) were protected in the presence of viral RNA (Figure 4).
Figure 4. Mapping IN residues binding to vRNA(1-57)-TAR using MS-based protein footprinting.

Representative segments of MS spectra showing that two IN peptides 263-269 (containing modified K264 and K266) and 270-284 (containing modified K273) are detected with unliganded IN (middle panels) but are significantly diminished in the IN-vRNA(1-57)-TAR complex (lower panels). In contrast, the peptide 216-224 (containing modified K219) persists with both unliganded IN and the IN-vRNA(1-57)-TAR complex. The unmodified peptides 200-211 and 245-262 serve as internal controls. Shown below is a graphical representation of HIV-1 IN protein with three domains and identified RNA binding residues indicated.
To verify the significance of identified IN residues for viral RNA binding we prepared two mutant recombinant INs containing either K264A/K266A or R269A/K273A substitutions. R269A was included in our mutagenesis studies due to its adjacent location with the identified Lys residues and a high degree of conservation amongst different HIV-1 variants. In vitro AlphaScreen assays revealed that both K264A/K266A and R269A/K273A INs failed to bind vRNA(1-57)-TAR (Figure 5A).
Figure 5. Biochemical characterization of IN mutants.

(A) Interactions of recombinant WT, K264A/K266A and R269A/K273A mutant INs with vRNA(1-57)-TAR measured by AlphaScreen. The average values of three independent experiments and corresponding standard deviations are shown. (B) Analytical SEC of WT, K264A/K266A and R269A/K273A mutant INs. Monomeric (M) and tetrameric (T) INs are indicated. (C) Affinity-pull-down assays showing binding of WT and mutant INs to LEDGF/p75. Lane 1: molecular weight markers; Lanes 2–5 loads of 6xHis-tagged WT IN, IN(K264A/K266A), IN(R269A/K273A) and tag-free LEDGF/p75; Lanes 6–9: affinity pull-down using Ni beads of LEDGF/p75 with 6xHis-tagged WT IN, IN(K264A/K266A) and IN(R269A/K273A). (D) Agarose gel analysis of concerted integration products. Lane 1: DNA markers (BIOLINE Quanti-Marker, 1 kb); Lane 2: linearized target DNA; Lanes 4–6: activities of WT, K264A/K266A and R269A/K273A mutant INs in the absence of LEDGF/p75; Lanes 7–9: activities of WT, K264A/K266A and R269A/K273A mutant INs in the presence of LEDGF/p75. Target and donor DNA substrates as well as single-site and concerted integration products are indicated.
Given the pleiotropic nature of many IN mutations (Engelman, 1999; Rihn et al., 2015), we also examined the effects of K264A/K266A and R269A/K273A substitutions on other functions of the recombinant protein. WT and R269A/K273A INs exhibited a very similar multimerization pattern with clearly separated tetrameric and monomeric peaks detected by SEC (Figure 5B). K264A/K266A IN also displayed tetrameric and monomeric peaks but had a slightly broader shoulder between the two peaks, possibly due to higher content of a dimeric form. WT, K264A/K266A and R269A/K273A INs similarly bound to LEDGF/p75 (Figure 5C). Figure 5D shows that IN R269A/K273A efficiently catalyzed LEDGF/p75-dependent concerted integration in vitro, whereas IN K264A/K266A was inactive. In summary, the K264A/K266A substitutions displayed pleiotropic effects, whereas R269A/K273A IN was selectively impaired for RNA binding without significantly affecting other known functions of the protein in vitro.
IN mutants impaired for viral RNA binding yield viral particles with eccentric RNPs
We next introduced IN K264A/K266A and R269A/K273A substitutions in HIV-1NL4-3 to examine their effects on HIV-1 replication and IN binding to RNA in virions. These amino acid changes did not detectably affect proteolytic processing of HIV-1 polyproteins (Figure S4A), genomic RNA packaging (Figure S4B) or particle release (Figure S4C). However, these particles were not infectious (Figure S4D) and failed to initiate reverse transcription in target cells (Figure S4E).
CLIP-seq experiments with virions isolated from producer cells transfected with HIV-1NL4-3 IN(K264A/K266A) or HIV-1NL4-3 IN(R269A/K273A) revealed that these substitutions greatly reduced IN binding to the viral RNA genome in virions (Figure 6A, B). Importantly, this finding was not due to reduced levels of IN protein in virions (Figure 6A). Moreover, and in sharp contrast, comparable levels of NC-RNA complexes were detected in virions of WT and IN-mutant viruses (Figure 6A).
Figure 6. Characterization of mutant viruses.

(A) Autoradiograms of IN-RNA and NC-RNA crosslinked adducts from WT HIV-1NL4-3, HIV-1NL4-3 IN(K264A/K266A) and HIV-1NL4-3 IN(R269A/K273A) virions (upper two panels). Corresponding western blot analysis of IN and NC in immunoprecipitated fractions (lower two panels). (B) CLIP-seq results for IN-viral RNA crosslinks from WT HIV-1NL4-3, HIV-1NL4-3 IN(K264A/K266A) and HIV-1NL4-3 IN(R269A/K273A) virions (only the first 1000 nucleotides shown). (C) Representative EM images of virions from WT HIV-1NL4-3, HIV-1NL4-3 IN(K264A/K266A) and HIV-1NL4-3 IN(R269A/K273A). (D) Sucrose density gradient separation of capsid cores from detergent-lysed virions of WT HIV-1NL4-3, HIV-1NL4-3 IN(K264A/K266A) and HIV-1NL4-3 IN(R269A/K273A). (E) Quantitation of triplicate experiments of HIV-1 CA-p24 signal intensity from (D) using ImageJ software. The error bars indicate standard deviations.
Further inspection of HIV-1NL4-3 IN(K264A/K266A) and HIV-1NL4-3 IN(R269A/K273A) particles with electron microscopy revealed eccentric cores with the characteristic class II phenotype where the electron dense material corresponding to RNPs was mislocalized outside of the translucent CA cores (Figure 6C). Sucrose density gradient fractionation experiments (Figures 6D and E) reproducibly corroborated these findings; WT CA cores containing the RNPs migrated to higher density fractions compared with CA cores isolated from HIV-1NL4-3 IN(K264A/K266A) or HIV-1NL4-3 IN(R269A/K273A) particles, which did not contain RNPs (Figure 6C) and were found in lower density fractions (Figure 6D and E).
IN-RNA interactions are critical for formation of infectious virions
During HIV-1 particle maturation IN is cleaved from the Gag-Pol precursor by the viral protease (PR). It was possible that IN-RNA interactions, and IN R269A/K273A substitutions, exert their effects early during assembly as part of the Gag-Pol precusor, or later after IN has been liberated. Moreover, it was possible that the effects of the mutations caused a dominant perturbation of virion assembly or a recessive defect as a consequence of the loss of IN-RNA binding. To distinguish between these possibilities, we complemented HIV-1NL4-3 IN(R269A/K273A) with a Vpr-IN fusion construct, containing either WT or the RNA binding defective IN R269A/K273A mutant (Figure S5). Immunoblotting analysis revealed that both WT and R269A/K273A mutant INs were effectively incorporated in virions and cleaved from their respective Vpr-IN fusion proteins by viral PR (compare lanes 2 and 3 with 1 in Figure S5A). Complementation of HIV-1NL4-3 IN(R269A/K273A) with Vpr-IN(R269A/K273A) failed to yield detectable levels of IN-RNA adducts, whereas similar levels of WT Vpr-IN supplied in trans to HIV-1NL4-3 IN(R269A/K273A) resulted in a marked increase of IN-RNA adducts in virions (Figure S5B). Correspondingly, transcomplementation with WT Vpr-IN but not with Vpr-IN (R269A/K273A) markedly enhanced infectivity of HIV-1NL4-3 IN(R269A/K273A) virions (Figure S5C).
In parallel experiments with WT HIV-1NL4-3 (Figure S6), overexpression of Vpr-IN (R269A/K273A) did not significantly alter the levels of IN-RNA adducts compared with WT HIV-1NL4-3 (compare lanes 1 and 2 in Figures S6A and B) indicating that IN R269A/K273A substitutions are recessive in nature. Incorporation of overexpressed levels of WT Vpr-IN into virions correspondingly increased levels of IN-RNA adducts (compare lanes 1 and 3 in Figures S6A and B). Infectivity assays (Figure S6C) have revealed that incorporation of WT Vpr-IN fusion protein into WT HIV-1NL4-3 virions resulted in modest (~40%) loss of infectivity. Importantly, a similar decrease in infectivity was observed for the complementation of WT HIV-1NL4-3 with mutant Vpr-IN (Figure S6C) suggesting that incorporation of overexpressed Vpr-IN fusion proteins into virions rather than IN R269A/K273A substitutions led to the observed reductions in infectivity. In addition, the similar levels of infectivity, that are detected in the presence of WT or mutant Vpr-IN (Figure S6C), indicates that RNA binding defective IN R269A/K273A substitutions did not have unintended, additional consequences on WT HIV-1NL4-3 particle maturation. Furthermore, these results suggest that the levels of IN-RNA adducts observed with WT HIV-1NL4-3 (lane 1 in Figure S6B) are sufficent for formation of the infectious virions as further increases of IN-RNA adducts by overexpression of WT Vpr-IN (lane 3 in Figure S6B) did not stimulate infectivity of WT HIV-1NL4-3 virions (Figure S6C). Collectively, because we have been able to specifically supply IN to the virion (rather than Gag-Pol) through the Vpr-IN fusion protein, we conclude that IN-RNA interactions are critical for formation of infectious virions.
ALLINIs impair IN-viral RNA interactions in virions
Since ALLINI treatment of HIV-1 infected producer cells have been shown to yield eccentric viral particles with mislocalized RNPs, which resemble those of certain IN class II mutants, we examined the effects of ALLINIs on IN-viral RNA interactions in virions. We first tested the best characterized archetypal IN inhibitor BI-B2 (also known as ALLINI-2) (Figure S7A) in conjunction with the WT virus and HIV-1NL4-3 IN(A128T), a mutation that confers significant resistance to this inhibitor (Figure 7A) (Feng et al., 2013; Fontana et al., 2015; Shkriabai et al., 2014). BI-B2 treatment of virus producer cells substantially inhibited the binding of WT IN to RNA in virions, while the treatment of HIV-1NL4-3 IN(A128T) with the inhibitor yielded IN-RNA crosslink levels that were comparable with untreated WT virus (Figures 7B and S7B). Further analysis of CLIP-seq data revealed that BI-B2 treatment of cells producing HIV-1NL4-3 blocked the binding of WT IN throughout the entire viral genome, while virions isolated from BI-B2 treated cells producing HIV-1NL4-3 IN(A128T) displayed the characteristic pattern of WT IN interaction with the viral RNA genome (Figure 7C).
Figure 7. Effects of BI-B2 on HIV-1 IN interactions with viral RNA in virions.

(A) Virions from WT HIV-1NL4-3 and the escape mutant HIV-1NL4-3 IN(A128T) were prepared in the presence and absence of 10 μM BI-B2 in virus producer cells and used to infect target cells to determine relative infectivity. The average values of three independent experiments and corresponding standard deviations are shown. (B) Autoradiograms of IN-RNA and NC-RNA crosslinks adducts from virions of WT HIV-1NL4-3 and HIV-1NL4-3 IN(A128T) prepared in the presence and absence of 10 μM BI-B2 (upper two panels). Corresponding western blot analysis of IN and NC in immunoprecipitated fractions (lower two panels). (C) CLIP-seq analysis of IN binding to viral RNA genome from virions of WT HIV-1NL4-3 (upper panel), WT HIV-1NL4-3 + BI-B2 (middle panels), and HIV-1NL4-3 IN(A128T) + BI-B2 (lower panel).
In addition, we have analyzed another potent ALLINI BI-D (Figure S7A), which unlike BI-B2 retains significant antiviral activity with respect to HIV-1NL4-3 IN(A128T) (Le Rouzic et al., 2013). In full agreement with the antiviral activity results, BI-D impaired IN binding to RNA in virions from both WT and IN A128T mutant viruses (Figures S7B). Importantly, neither BI-B2 nor BI-D had any detectable effects on NC binding to RNA in mature virions (Figures 7B and S7C).
DISCUSSION
The key findings of the present study are: i) HIV-1 IN binds to the viral RNA genome in virions and exhibits a marked preference for certain viral RNA elements. Importantly, CLIP-seq results show different but highly reproducible binding patterns for IN and NC (Figure S2) suggesting that these proteins can bind the viral RNA genome at the same time in the virion; ii) IN R269A/K273A substitutions compromise IN binding to the viral RNA genome without significantly affecting other known functions of this viral protein and result in non-infectious particles with eccentrically mislocalized RNPs; iii) transcomplementation of HIV-1NL4-3 IN(R269A/K273A) with WT IN but not the RNA binding defective IN R269A/K273A mutant markedly enhances IN-RNA binding and infectivity of the virions; iv) ALLINI treatments of virus producer cells infected with WT HIV-1, which result in non-infectious eccentric particles, are now shown to impair IN binding to the viral RNA genome in virions; v) Unlike IN-RNA interactions, NC-RNA complexes are not affected in the eccentric non-infectious virions of HIV-1NL4-3 IN(R269A/K273A) or ALLINI treated WT virus. These results strongly suggest an essential role for IN binding to the viral RNA genome during virion morphogenesis.
During particle maturation, viral PR cleaves Gag-Pol and Gag polyproteins into individual domains, triggering profound structural rearrangements within the virion and leading to the RNPs being localized into the conical CA core. The RNPs contain two copies of viral genome, some cellular RNAs, a number of viral proteins including NC, IN and RT as well as a subset of cellular proteins. While the role of NC in the condensation of RNPs has been widely accepted (Mori et al., 2015), many of the mechanistic details and molecular interactions required for correct viral particle morphogenesis are poorly understood. Our results show that when IN binding to viral RNA is impaired by IN R269A/K273A mutations (which do not affect any other known function of the protein) or ALLINI treatment, the resulting virions contain WT levels of NC-RNA complexes in eccentrically mislocalized RNPs. This finding demonstrates that the assembly of NC-RNA complexes, while essential, is not sufficient for correct virion morphogenesis and that IN binding to the viral RNA genome is also likely important for formation of infectious virions. We suggest that IN interactions with the viral RNA genome has the following dual roles: i) IN binding to viral RNA ensures the incorporation of this viral protein within the protective CA core and stabilizes low-oligomeric forms of IN, which is likely to be essential for its function during subsequent infection when it forms the catalytically active SSC with viral DNA in target cells; ii) In turn, IN contributes to the formation of the correct architecture and/or localization of RNPs within matured, infectious virions.
In vitro experiments with recombinant IN and NC have been instrumental to understand the mechanistic basis of their binding to model nucleic acids (reviewed in (Cherepanov et al., 2011; Mori et al., 2015)). In order to induce effective aggregation of the nucleoprotein complexes, NC is typically added at a ratio that would allow for NC to cover the entire nucleic acid with each molecule occupying ~7 nts segment. In contrast, IN does not coat viral RNA, even when added in significant excess, as evidenced by the stoichiometry of approximately four subunits of IN bound per one molecule of vRNA(1-850). Instead, IN appears to provide nucleation points by bridging separate RNA molecules (Figure 3).
Taken together, these in vitro results suggest the following complementary roles for NC and IN during particle maturation: ~2000 copies of NC could coat large segments of viral RNA and thus contribute to RNP condensation while ~200 copies of IN could markedly impact the architecture of RNPs, effectively compacting the two copies of viral genome by binding and bridging between several sites. Such interpretations are consistent with the CLIP-seq results that show distinct binding patterns for IN and NC to the viral RNA genome (Figure S2). The combined action of both NC and IN could be required for timely formation and correct localization of the RNPs within the CA core. Alternatively, IN could mediate the correct localization of RNPs in mature virions through potential bridging interactions between the viral RNA genome and CA cores. Further studies are warranted to better understand the complex interplay between multiple viral, and possibly cellular, components during particle maturation.
Surprisingly, our in vitro and in virion experiments show that IN, a double stranded DNA binding protein, displays marked preference for highly structured RNA elements. Such structural features in the viral RNA genome could be recognized by IN and account for the highly reproducible binding pattern seen in our CLIP-seq experiments (Figures 1 and S1). These multiple binding sites would enable IN multimers to effectively bridge different segments of the viral RNA genome and thus promote effective compaction of RNPs. In turn, the redundancy of structural elements along the viral genome allows for the virus to compensate and to not depend on a single binding event of IN to a particular viral RNA element. For example, deletions of either TAR or RRE do not have noticeable consequences on virus particle morphogenesis (Das et al., 2007; Kutluay et al., 2014). In addition, we note that highly structured RNA elements are also present in cellular mRNAs (Wachter, 2014) and our in vitro assays show that IN binds human mRNA mix with low nanomolar affinity (Figure 2F). Therefore, it is likely that during the production of functional HIV-1 based lentiviral vector particles containing the transgenes of cellular origin, IN could interact with both viral RNA and the RNA region corresponding to the transgene to allow for effective bridging between these RNA segments.
Our site-directed mutagenesis studies have revealed both similarities and differences between IN interactions with viral DNA and viral RNA. The IN K264A/K266A substitutions impaired its ability to bind both viral DNA and viral RNA, suggesting an overlapping interface for interaction with either nucleic acid. In contrast, IN R269A/K273A mutant was unable to bind viral RNA, yet exhibited effective LEDGF/p75-mediated concerted integration activity. Another important difference between IN binding to viral DNA versus viral RNA is seen when preformed IN-nucleic acid complexes are subjected to high ionic treatments (1 M NaCl). When IN forms the SSC with cognate viral DNA, it is resistant to high ionic treatments. The formation of such a stable complex is likely to be important during the early steps of HIV-1 replication. For example, stably bound IN tetramer could protect the viral DNA ends from cellular DNA modifying enzymes and also would allow the preintegration complex to retain its integrity while traversing through the nuclear pore and associating with LEDGF/p75 and chromatin for effective integration. In contrast, IN binding to viral RNA can be reversed by high ionic treatments suggesting that there is a dynamic interplay between IN and viral RNA, which could be important for allowing HIV-1 RT to effectively reverse transcribe the viral genome.
We and others have shown that the primary mechanism of action of ALLINIs is to induce aberrant IN multimerization in virions, which leads to the formation of eccentric cores with mislocalized RNPs (Balakrishnan et al., 2013; Desimmie et al., 2013; Fontana et al., 2015; Jurado et al., 2013; Le Rouzic et al., 2013; Sharma et al., 2014). However, it was not clear how altering the state of IN multimerization during the late stage of virus replication affected virion morphology. Here we demonstrate that representative ALLINIs, BI-B2 and BI-D, impair IN binding to the viral RNA genome in virions. All reported ALLINIs, including BI-B2 and BI-D, bind at the IN CCD dimer interface (Feng et al., 2013; Jurado et al., 2013). In addition, the CTD plays a critical role for ALLINI induced aberrant IN multimerization (Gupta et al., 2014; Shkriabai et al., 2014). In particular, our MS-based hydrogen-deuterium exchange studies have revealed that ALLINI binding to the CCD promotes multiple protein-protein contacts, which include the extreme C-terminal segment (aa 249-288) (Shkriabai et al., 2014). The identified viral RNA interacting residues (K264, K266, R269 and K273) are located within this protein segment and could be shielded in the ALLINI induced aberrant IN multimers and are hence not available to productively engage viral RNA (Figure S7D and E). These observations coupled with our findings that ALLINIs do not affect NC binding to RNA in virions (Figures 7 and S7C) suggest the importance of IN binding to viral RNA for proper virus particle morphogenesis.
In summary, our studies elucidate a previously undescribed non-catalytic role of IN in HIV-1 biology as it interacts with the viral RNA genome during particle morphogenesis. Furthermore, these findings significantly extend our understanding of the mechanism of action of ALLINIs, which are currently in clinical trials, and have uncovered a functionally essential interface between IN and the viral RNA genome that could serve as an attractive antiretroviral target.
Supplementary Material
(A and B) CLIP-seq results of two independent experiments. (C) The overlay of two independent CLIP-seq experiments shows the nearly perfect overlap between preferred IN binding sites on the viral genome.
CLIP-seq results for IN (A) and NC (B) binding to viral RNA in mature virions. In order to compare two different proteins (IN and NC), nucleotide reads are graphed as percentage of the total viral RNA genome as opposed to normalized read densities. (C–F) Zoomed in view for IN and NC binding sites to certain viral RNA regions.
Where V is the estimated volume, M is the molecular mass of the oligomer, Vp is the partial specific volume of the protein (~0.73–0.74 cm3/g), d is the hydration ratio (0.3–0.4), VW is the partial specific volume of water (1 cm3/g), and Na is the Avogadro’s number. The variations of the predicted volumes are due to varying Vp and d values (see above). (J) The stoichiometry for the HIV-1 IN binding to vRNA(1-850). IN+vRNA(1-850) complexes were assembled and purified using SEC (Figure 2A and B). The concentrations were measured by immunoblotting for IN and UV spectroscopy (OD260 nm) for vRNA(1-850). The average values of six independent experiments and corresponding standard deviations are shown.
(A) Gag processing by HIV-1 protease in the absence (lanes 2, 4 and 6) or presence (lanes 1, 3 and 4) of saquinavir (saq). A representative immunoblot probed with a CA antibody is shown. Unprocessed Gag (GAG-p55) and processed capsid (CA-p24) are indicated. Lane 7: molecular weight markers. (B) Analysis of the levels of packaged viral RNA in virions of WT HIV-1NL4-3, HIV-1NL4-3 IN(K264A/K266A) and HIV-1NL4-3 IN(R269A/K273A). (C) HIV-1 CA-p24 production levels for WT HIV-1NL4-3, HIV-1NL4-3 IN(K264A/K266A) and HIV-1NL4-3 IN(R269A/K273A) as determined by ELISA. (D) Infectivity of virions from producer cells infected with WT HIV-1NL4-3, HIV-1NL4-3 IN(K264A/K266A) and HIV-1NL4-3 IN(R269A/K273A). (E) Analysis of early reverse transcription (ERT) products by qPCR of subsequent infection with virions from WT HIV-1NL4-3, HIV-1NL4-3 IN(K264A/K266A) and HIV-1NL4-3 IN(R269A/K273A). The average values of three independent experiments and corresponding standard deviations are shown in B, C, D and E.
(A) Western blot analysis of IN and CA in virion lysates of HIV-1NL4-3 IN(R269A/K273A) + empty plasmid (lane 1), HIV-1NL4-3 IN(R269A/K273A)+Vpr-IN (R269A/K273A) (lane 2), and HIV-1NL4-3 IN(R269A/K273A)+WT Vpr-IN (lane 3). (B) Corresponding autoradiogram of IN-RNA crosslinked adducts. (C) Infectivity of virions from producer cells that were transfected with HIV-1NL4-3 IN(R269A/K273A) and additionally transcomplemented with either empty plasmid, Vpr-IN (R269A/K273A) or WT Vpr-IN. Infectivity is depicted as % infection normalized to WT control (WT HIV-1NL4-3 + WT Vpr-IN). The average values of three independent experiments and corresponding standard deviations are shown.
(A) Western blot analysis of IN and CA in virion lysates of WT HIV-1NL4-3 + empty plasmid (lane 1), WT HIV-1NL4-3 + Vpr-IN (R269A/K273A) (lane 2), and HIV-1NL4-3 + WT Vpr-IN (lane 3). (B) Corresponding autoradiogram of IN-RNA crosslinked adducts. (C) Infectivity of virions from producer cells that were transfected with HIV-1NL4-3 and additionally transcomplemented with either empty plasmid, Vpr-IN (R269A/K273A) or WT Vpr-IN for transcomplementation. Infectivity is depicted as % infection normalized to WT control (WT HIV-1NL4-3 + empty vector control). The average values of three independent experiments and corresponding standard deviations are shown.
(A) Chemical structures of BI-B2 and BI-D. (B and C) Virions from WT HIV-1NL4-3 and HIV-1NL4-3 IN(A128T) were prepared in the presence or absence of 10 μM BI-B2 or BI-D in virus producer cells. IN-RNA (B) and NC-RNA (C) complexes were immunoprecipitated using respective antibodies (Bouyac-Bertoia et al., 2001; Kutluay et al., 2014). Autoradiograms of immunoprecipitated IN-RNA and NC-RNA adducts are shown. Corresponding western blot analysis of IN and NC in immunoprecipiated material are shown below the autoradiograms. (D and E) A molecular model of the IN tetramer (Kessl et al., 2011) with viral RNA and ALLINI binding sites indicated. Individual full length IN subunits are colored yellow, magenta, cyan and green. The C-terminal domain (CTD) of each subunit is indicated. (D) IN residues interacting with viral RNA (K264, K266, R269 and K273) are depicted as black spheres. An IN tetramer could potentially promote bridging and compaction of the viral RNA genome with each CTD domain binding to separate sites on the viral RNA genome. (E) BI-B2 inhibitors (in red) bound at CCD-CCD dimer interfaces of the IN tetramer are shown. The protein segments shielded from the surface (shown in black) in the complex with BI-B2 significantly extend beyond the direct inhibitor binding sites (indicated by red arrows) and include CTD segments (aa 249-288) (Shkriabai et al., 2014). While our model only shows a single IN tetramer, BI-B2 binding promotes the formation of higher-order, aberrant IN multimers (not shown), where inhibitor induced protein-protein interactions shield the C-terminal segments of IN, including RNA binding residues K264, K266, R269 and K273, from the surface and could thus impede the ability of IN to bind viral RNA.
Table S1. RNA adapters and DNA primers for CLIP-seq experiments
Highlights.
HIV-1 integrase binds viral RNA in virions
Integrase exhibits distinct preference for select RNA structural elements
RNA binding defective mutants of integrase yield non-infectious virions
Allosteric integrase inhibitors impair integrase binding to viral RNA in virions
Acknowledgments
This work was supported by NIH grants R01AI062520, P50GM103368 and R01AI110310 (to M.K.), R01AI116258 and P50GM103297 (to S.B.K.), R01AI110310 (J.R.F.), R21AI110270 (to J.J.K), P50GM103297 and R01AI050111 (to P.D.B.).
Footnotes
AUTHOR CONTRIBUTIONS
J.J.K., S.B.K., P.D.B. and M.K. conceived and designed experiments. J.J.K., S.R., A.S., R.C.L., N.S. and N.B. performed biochemistry and virology experiments. S.B.K. and D.T. performed CLIP-seq experiments. J.R.F. provided BI-B2 and BI-D inhibitors. M.K. wrote the manuscript, with contributions from all other authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allen P, Worland S, Gold L. Isolation of high-affinity RNA ligands to HIV-1 integrase from a random pool. Virology. 1995;209:327–336. doi: 10.1006/viro.1995.1264. [DOI] [PubMed] [Google Scholar]
- Andersen ES, Contera SA, Knudsen B, Damgaard CK, Besenbacher F, Kjems J. Role of the transactivation response element in dimerization of HIV-1 RNA. The Journal of biological chemistry. 2004;279:22243–22249. doi: 10.1074/jbc.M314326200. [DOI] [PubMed] [Google Scholar]
- Athanassiou Z, Patora K, Dias RL, Moehle K, Robinson JA, Varani G. Structure-guided peptidomimetic design leads to nanomolar beta-hairpin inhibitors of the Tat-TAR interaction of bovine immunodeficiency virus. Biochemistry. 2007;46:741–751. doi: 10.1021/bi0619371. [DOI] [PubMed] [Google Scholar]
- Balakrishnan M, Yant SR, Tsai L, O’Sullivan C, Bam RA, Tsai A, Niedziela-Majka A, Stray KM, Sakowicz R, Cihlar T. Non-Catalytic Site HIV-1 Integrase Inhibitors Disrupt Core Maturation and Induce a Reverse Transcription Block in Target Cells. PloS one. 2013;8:e74163. doi: 10.1371/journal.pone.0074163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouyac-Bertoia M, Dvorin JD, Fouchier RA, Jenkins Y, Meyer BE, Wu LI, Emerman M, Malim MH. HIV-1 infection requires a functional integrase NLS. Molecular cell. 2001;7:1025–1035. doi: 10.1016/s1097-2765(01)00240-4. [DOI] [PubMed] [Google Scholar]
- Bukovsky A, Gottlinger H. Lack of integrase can markedly affect human immunodeficiency virus type 1 particle production in the presence of an active viral protease. Journal of virology. 1996;70:6820–6825. doi: 10.1128/jvi.70.10.6820-6825.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochemical pharmacology. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Cherepanov P. LEDGF/p75 interacts with divergent lentiviral integrases and modulates their enzymatic activity in vitro. Nucleic acids research. 2007;35:113–124. doi: 10.1093/nar/gkl885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherepanov P, Maertens GN, Hare S. Structural insights into the retroviral DNA integration apparatus. Current opinion in structural biology. 2011;21:249–256. doi: 10.1016/j.sbi.2010.12.005. [DOI] [PubMed] [Google Scholar]
- Christ F, Voet A, Marchand A, Nicolet S, Desimmie BA, Marchand D, Bardiot D, Van der Veken NJ, Van Remoortel B, Strelkov SV, et al. Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication. Nature chemical biology. 2010;6:442–448. doi: 10.1038/nchembio.370. [DOI] [PubMed] [Google Scholar]
- Corcoran DL, Georgiev S, Mukherjee N, Gottwein E, Skalsky RL, Keene JD, Ohler U. PARalyzer: definition of RNA binding sites from PAR-CLIP short-read sequence data. Genome biology. 2011;12:R79. doi: 10.1186/gb-2011-12-8-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das AT, Harwig A, Vrolijk MM, Berkhout B. The TAR hairpin of human immunodeficiency virus type 1 can be deleted when not required for Tat-mediated activation of transcription. Journal of virology. 2007;81:7742–7748. doi: 10.1128/JVI.00392-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debyser Z, Christ F, De Rijck J, Gijsbers R. Host factors for retroviral integration site selection. Trends in biochemical sciences. 2015;40:108–116. doi: 10.1016/j.tibs.2014.12.001. [DOI] [PubMed] [Google Scholar]
- Deprez E, Tauc P, Leh H, Mouscadet JF, Auclair C, Brochon JC. Oligomeric states of the HIV-1 integrase as measured by time-resolved fluorescence anisotropy. Biochemistry. 2000;39:9275–9284. doi: 10.1021/bi000397j. [DOI] [PubMed] [Google Scholar]
- Desimmie BA, Schrijvers R, Demeulemeester J, Borrenberghs D, Weydert C, Thys W, Vets S, Van Remoortel B, Hofkens J, De Rijck J, et al. LEDGINs inhibit late stage HIV-1 replication by modulating integrase multimerization in the virions. Retrovirology. 2013;10:57. doi: 10.1186/1742-4690-10-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman A. In vivo analysis of retroviral integrase structure and function. Advances in virus research. 1999;52:411–426. doi: 10.1016/s0065-3527(08)60309-7. [DOI] [PubMed] [Google Scholar]
- Engelman A, Cherepanov P. The lentiviral integrase binding protein LEDGF/p75 and HIV-1 replication. PLoS pathogens. 2008;4:e1000046. doi: 10.1371/journal.ppat.1000046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman A, Englund G, Orenstein JM, Martin MA, Craigie R. Multiple effects of mutations in human immunodeficiency virus type 1 integrase on viral replication. Journal of virology. 1995;69:2729–2736. doi: 10.1128/jvi.69.5.2729-2736.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadel HJ, Morrison JH, Saenz DT, Fuchs JR, Kvaratskhelia M, Ekker SC, Poeschla EM. TALEN knockout of the PSIP1 gene in human cells: analyses of HIV-1 replication and allosteric integrase inhibitor mechanism. Journal of virology. 2014;88:9704–9717. doi: 10.1128/JVI.01397-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fader LD, Malenfant E, Parisien M, Carson R, Bilodeau F, Landry S, Pesant M, Brochu C, Morin S, Chabot C, et al. Discovery of BI 224436, a Noncatalytic Site Integrase Inhibitor (NCINI) of HIV-1. ACS medicinal chemistry letters. 2014;5:422–427. doi: 10.1021/ml500002n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Sharma A, Slaughter A, Jena N, Koh Y, Shkriabai N, Larue RC, Patel PA, Mitsuya H, Kessl JJ, et al. The A128T resistance mutation reveals aberrant protein multimerization as the primary mechanism of action of allosteric HIV-1 integrase inhibitors. The Journal of biological chemistry. 2013;288:15813–15820. doi: 10.1074/jbc.M112.443390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana J, Jurado KA, Cheng N, Ly NL, Fuchs JR, Gorelick RJ, Engelman AN, Steven AC. Distribution and Redistribution of HIV-1 Nucleocapsid Protein in Immature, Mature, and Integrase-Inhibited Virions: a Role for Integrase in Maturation. Journal of virology. 2015;89:9765–9780. doi: 10.1128/JVI.01522-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta K, Brady T, Dyer BM, Malani N, Hwang Y, Male F, Nolte RT, Wang L, Velthuisen E, Jeffrey J, et al. Allosteric Inhibition of Human Immunodeficiency Virus Integrase: LATE BLOCK DURING VIRAL REPLICATION AND ABNORMAL MULTIMERIZATION INVOLVING SPECIFIC PROTEIN DOMAINS. The Journal of biological chemistry. 2014;289:20477–20488. doi: 10.1074/jbc.M114.551119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurado KA, Wang H, Slaughter A, Feng L, Kessl JJ, Koh Y, Wang W, Ballandras-Colas A, Patel PA, Fuchs JR, et al. Allosteric integrase inhibitor potency is determined through the inhibition of HIV-1 particle maturation. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:8690–8695. doi: 10.1073/pnas.1300703110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessl JJ, Jena N, Koh Y, Taskent-Sezgin H, Slaughter A, Feng L, de Silva S, Wu L, Le Grice SF, Engelman A, et al. A multimode, cooperative mechanism of action of allosteric HIV-1 integrase inhibitors. The Journal of biological chemistry. 2012;287:16801–16811. doi: 10.1074/jbc.M112.354373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessl JJ, Li M, Ignatov M, Shkriabai N, Eidahl JO, Feng L, Musier-Forsyth K, Craigie R, Kvaratskhelia M. FRET analysis reveals distinct conformations of IN tetramers in the presence of viral DNA or LEDGF/p75. Nucleic acids research. 2011;39:9009–9022. doi: 10.1093/nar/gkr581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotova S, Li M, Dimitriadis EK, Craigie R. Nucleoprotein intermediates in HIV-1 DNA integration visualized by atomic force microscopy. Journal of molecular biology. 2010;399:491–500. doi: 10.1016/j.jmb.2010.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkosky J, BouHamdan M, Geist A, Pomerantz RJ. A novel Vpr peptide interactor fused to integrase (IN) restores integration activity to IN-defective HIV-1 virions. Virology. 1999;255:77–85. doi: 10.1006/viro.1998.9544. [DOI] [PubMed] [Google Scholar]
- Kutluay SB, Zang T, Blanco-Melo D, Powell C, Jannain D, Errando M, Bieniasz PD. Global changes in the RNA binding specificity of HIV-1 gag regulate virion genesis. Cell. 2014;159:1096–1109. doi: 10.1016/j.cell.2014.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvaratskhelia M, Le Grice SF. Structural analysis of protein-RNA interactions with mass spectrometry. Methods Mol Biol. 2008;488:213–219. doi: 10.1007/978-1-60327-475-3_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Rouzic E, Bonnard D, Chasset S, Bruneau JM, Chevreuil F, Le Strat F, Nguyen J, Beauvoir R, Amadori C, Brias J, et al. Dual inhibition of HIV-1 replication by integrase-LEDGF allosteric inhibitors is predominant at the post-integration stage. Retrovirology. 2013;10:144. doi: 10.1186/1742-4690-10-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Llano M, Saenz DT, Meehan A, Wongthida P, Peretz M, Walker WH, Teo W, Poeschla EM. An essential role for LEDGF/p75 in HIV integration. Science. 2006;314:461–464. doi: 10.1126/science.1132319. [DOI] [PubMed] [Google Scholar]
- McKee CJ, Kessl JJ, Shkriabai N, Dar MJ, Engelman A, Kvaratskhelia M. Dynamic modulation of HIV-1 integrase structure and function by cellular lens epithelium-derived growth factor (LEDGF) protein. The Journal of biological chemistry. 2008;283:31802–31812. doi: 10.1074/jbc.M805843200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori M, Kovalenko L, Lyonnais S, Antaki D, Torbett BE, Botta M, Mirambeau G, Mely Y. Nucleocapsid Protein: A Desirable Target for Future Therapies Against HIV-1. Current topics in microbiology and immunology. 2015;389:53–92. doi: 10.1007/82_2015_433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munk C, Brandt SM, Lucero G, Landau NR. A dominant block to HIV-1 replication at reverse transcription in simian cells. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:13843–13848. doi: 10.1073/pnas.212400099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nocera TM, Chen J, Murray CB, Agarwal G. Magnetic anisotropy considerations in magnetic force microscopy studies of single superparamagnetic nanoparticles. Nanotechnology. 2012;23:495704. doi: 10.1088/0957-4484/23/49/495704. [DOI] [PubMed] [Google Scholar]
- Rihn SJ, Hughes J, Wilson SJ, Bieniasz PD. Uneven genetic robustness of HIV-1 integrase. Journal of virology. 2015;89:552–567. doi: 10.1128/JVI.02451-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Slaughter A, Jena N, Feng L, Kessl JJ, Fadel HJ, Malani N, Male F, Wu L, Poeschla E, et al. A New Class of Multimerization Selective Inhibitors of HIV-1 Integrase. PLoS pathogens. 2014;10:e1004171. doi: 10.1371/journal.ppat.1004171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shkriabai N, Dharmarajan V, Slaughter A, Kessl JJ, Larue RC, Feng L, Fuchs JR, Griffin PR, Kvaratskhelia M. A critical role of the C-terminal segment for allosteric inhibitor-induced aberrant multimerization of HIV-1 integrase. The Journal of biological chemistry. 2014;289:26430–26440. doi: 10.1074/jbc.M114.589572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaughter A, Jurado KA, Deng N, Feng L, Kessl JJ, Shkriabai N, Larue RC, Fadel HJ, Patel PA, Jena N, et al. The mechanism of H171T resistance reveals the importance of Ndelta-protonated His171 for the binding of allosteric inhibitor BI-D to HIV-1 integrase. Retrovirology. 2014;11:100. doi: 10.1186/s12977-014-0100-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuruyama T, Nakai T, Ohmori R, Ozeki M, Tamaki K, Yoshikawa K. Dialysis purification of integrase-DNA complexes provides high-resolution atomic force microscopy images: dimeric recombinant HIV-1 integrase binding and specific looping on DNA. PloS one. 2013;8:e53572. doi: 10.1371/journal.pone.0053572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bel N, van der Velden Y, Bonnard D, Le Rouzic E, Das AT, Benarous R, Berkhout B. The allosteric HIV-1 integrase inhibitor BI-D affects virion maturation but does not influence packaging of a functional RNA genome. PloS one. 2014;9:e103552. doi: 10.1371/journal.pone.0103552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachter A. Gene regulation by structured mRNA elements. Trends in genetics : TIG. 2014;30:172–181. doi: 10.1016/j.tig.2014.03.001. [DOI] [PubMed] [Google Scholar]
- Wang H, Jurado KA, Wu X, Shun MC, Li X, Ferris AL, Smith SJ, Patel PA, Fuchs JR, Cherepanov P, et al. HRP2 determines the efficiency and specificity of HIV-1 integration in LEDGF/p75 knockout cells but does not contribute to the antiviral activity of a potent LEDGF/p75-binding site integrase inhibitor. Nucleic acids research. 2012;40:11518–11530. doi: 10.1093/nar/gks913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Chen X. Carbon nanotubes: a promising standard for quantitative evaluation of AFM tip apex geometry. Ultramicroscopy. 2007;107:293–298. doi: 10.1016/j.ultramic.2006.08.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A and B) CLIP-seq results of two independent experiments. (C) The overlay of two independent CLIP-seq experiments shows the nearly perfect overlap between preferred IN binding sites on the viral genome.
CLIP-seq results for IN (A) and NC (B) binding to viral RNA in mature virions. In order to compare two different proteins (IN and NC), nucleotide reads are graphed as percentage of the total viral RNA genome as opposed to normalized read densities. (C–F) Zoomed in view for IN and NC binding sites to certain viral RNA regions.
Where V is the estimated volume, M is the molecular mass of the oligomer, Vp is the partial specific volume of the protein (~0.73–0.74 cm3/g), d is the hydration ratio (0.3–0.4), VW is the partial specific volume of water (1 cm3/g), and Na is the Avogadro’s number. The variations of the predicted volumes are due to varying Vp and d values (see above). (J) The stoichiometry for the HIV-1 IN binding to vRNA(1-850). IN+vRNA(1-850) complexes were assembled and purified using SEC (Figure 2A and B). The concentrations were measured by immunoblotting for IN and UV spectroscopy (OD260 nm) for vRNA(1-850). The average values of six independent experiments and corresponding standard deviations are shown.
(A) Gag processing by HIV-1 protease in the absence (lanes 2, 4 and 6) or presence (lanes 1, 3 and 4) of saquinavir (saq). A representative immunoblot probed with a CA antibody is shown. Unprocessed Gag (GAG-p55) and processed capsid (CA-p24) are indicated. Lane 7: molecular weight markers. (B) Analysis of the levels of packaged viral RNA in virions of WT HIV-1NL4-3, HIV-1NL4-3 IN(K264A/K266A) and HIV-1NL4-3 IN(R269A/K273A). (C) HIV-1 CA-p24 production levels for WT HIV-1NL4-3, HIV-1NL4-3 IN(K264A/K266A) and HIV-1NL4-3 IN(R269A/K273A) as determined by ELISA. (D) Infectivity of virions from producer cells infected with WT HIV-1NL4-3, HIV-1NL4-3 IN(K264A/K266A) and HIV-1NL4-3 IN(R269A/K273A). (E) Analysis of early reverse transcription (ERT) products by qPCR of subsequent infection with virions from WT HIV-1NL4-3, HIV-1NL4-3 IN(K264A/K266A) and HIV-1NL4-3 IN(R269A/K273A). The average values of three independent experiments and corresponding standard deviations are shown in B, C, D and E.
(A) Western blot analysis of IN and CA in virion lysates of HIV-1NL4-3 IN(R269A/K273A) + empty plasmid (lane 1), HIV-1NL4-3 IN(R269A/K273A)+Vpr-IN (R269A/K273A) (lane 2), and HIV-1NL4-3 IN(R269A/K273A)+WT Vpr-IN (lane 3). (B) Corresponding autoradiogram of IN-RNA crosslinked adducts. (C) Infectivity of virions from producer cells that were transfected with HIV-1NL4-3 IN(R269A/K273A) and additionally transcomplemented with either empty plasmid, Vpr-IN (R269A/K273A) or WT Vpr-IN. Infectivity is depicted as % infection normalized to WT control (WT HIV-1NL4-3 + WT Vpr-IN). The average values of three independent experiments and corresponding standard deviations are shown.
(A) Western blot analysis of IN and CA in virion lysates of WT HIV-1NL4-3 + empty plasmid (lane 1), WT HIV-1NL4-3 + Vpr-IN (R269A/K273A) (lane 2), and HIV-1NL4-3 + WT Vpr-IN (lane 3). (B) Corresponding autoradiogram of IN-RNA crosslinked adducts. (C) Infectivity of virions from producer cells that were transfected with HIV-1NL4-3 and additionally transcomplemented with either empty plasmid, Vpr-IN (R269A/K273A) or WT Vpr-IN for transcomplementation. Infectivity is depicted as % infection normalized to WT control (WT HIV-1NL4-3 + empty vector control). The average values of three independent experiments and corresponding standard deviations are shown.
(A) Chemical structures of BI-B2 and BI-D. (B and C) Virions from WT HIV-1NL4-3 and HIV-1NL4-3 IN(A128T) were prepared in the presence or absence of 10 μM BI-B2 or BI-D in virus producer cells. IN-RNA (B) and NC-RNA (C) complexes were immunoprecipitated using respective antibodies (Bouyac-Bertoia et al., 2001; Kutluay et al., 2014). Autoradiograms of immunoprecipitated IN-RNA and NC-RNA adducts are shown. Corresponding western blot analysis of IN and NC in immunoprecipiated material are shown below the autoradiograms. (D and E) A molecular model of the IN tetramer (Kessl et al., 2011) with viral RNA and ALLINI binding sites indicated. Individual full length IN subunits are colored yellow, magenta, cyan and green. The C-terminal domain (CTD) of each subunit is indicated. (D) IN residues interacting with viral RNA (K264, K266, R269 and K273) are depicted as black spheres. An IN tetramer could potentially promote bridging and compaction of the viral RNA genome with each CTD domain binding to separate sites on the viral RNA genome. (E) BI-B2 inhibitors (in red) bound at CCD-CCD dimer interfaces of the IN tetramer are shown. The protein segments shielded from the surface (shown in black) in the complex with BI-B2 significantly extend beyond the direct inhibitor binding sites (indicated by red arrows) and include CTD segments (aa 249-288) (Shkriabai et al., 2014). While our model only shows a single IN tetramer, BI-B2 binding promotes the formation of higher-order, aberrant IN multimers (not shown), where inhibitor induced protein-protein interactions shield the C-terminal segments of IN, including RNA binding residues K264, K266, R269 and K273, from the surface and could thus impede the ability of IN to bind viral RNA.
Table S1. RNA adapters and DNA primers for CLIP-seq experiments