

Abstract
System noise was analyzed in 77 Affymetrix 6.0 samples from a previous clinical study of copy number variation (CNV). Twenty-three samples were classified as eligible for CNV detection, 29 samples as ineligible and 25 were classified as being of intermediate quality. New software (“noise-free-cnv”) was developed to visualize the data and reduce system noise. Fresh DNA preparations were more likely to yield eligible samples (p < 0.001). Eligible samples had higher rates of successfully genotyped SNPs (p < 0.001) and lower variance of signal intensities (p < 0.001), yielded fewer CNV findings after Birdview analysis (p < 0.001), and showed a tendency to yield fewer PennCNV calls (p = 0.053). The noise-free-cnv software visualized trend patterns of noise in the signal intensities across the ordered SNPs, including a wave pattern of noise, being co-linear with the banding pattern of metaphase chromosomes, as well as system deviations of individual probe sets (per-SNP noise). Wave noise and per-SNP noise occurred independently and could be separately removed from the samples. We recommend a two-step procedure of CNV validation, including noise reduction and visual inspection of all CNV calls, prior to molecular validation of a selected number of putative CNVs.
Keywords: copy number variation (CNV), variance, wave noise, per-SNP noise, noise-free-cnv software, noise reduction, validation of CNV findings
1. Introduction
Genomic copy number variation (CNV) was associated with a variety of clinical phenotypes [1,2,3,4,5,6]. Hence, the study of CNV is of diagnostic importance. CNV identification from high-density SNP-microarrays may be unreliable, particularly in noisy data [7,8,9]. Therefore, extensive validation of CNV findings is needed. Since CNV detection software may identify hundreds of putative CNVs in each sample and since validation of CNV findings by qPCR, or by other molecular methods, is laborious, we searched for simple strategies to evaluate large numbers of CNV findings.
Rigorous studies revealed that several components of system error occur in copy number data [10,11,12,13]. Here we focus on two major types of noise and present the noise-free-cnv software package for the visualization of copy number data and for the reduction of noise. This software enables large-scale inspection of CNV findings (produced by PennCNV [14], Birdview [15,16], or other specialized software packages). For illustration, we used 77 microarrays from a previous study of patients with cervical artery dissection from Switzerland and Southern Germany (age: 42.5 ± 9.8 years; 31 (40.3%) women) [17]. DNA was isolated from peripheral blood samples (no DNA from lymphoblastoid cell lines was used). DNA extraction, array hybridization, and array scanning were performed according to the manufacturer’s instructions [17]. The LRR and BAF values were obtained from the CEL files with the Affymetrix Power Tools software (APT). The quantile normalization was done in APT. The LRR and BAF can be then imported to PennCNV, to other CNV detections software packages (QuantiSNP, MAD), or to noise-free-cnv.
The Affymetrix 6.0 microarrays used for CNV detection contain a total of 906,600 single nucleotide polymorphisms (SNPs) and 946,000 non-polymorphic copy number probes (CNPs) covering all human chromosomes. In the present article, the notion of SNP is used for all analyzed probe sets (SNPs as well as CNPs).
2. Noise Components
Figure 1 shows two samples (visualized by noise-free-cnv), displaying signal intensity (LRR—upper panel) and B-allele frequency (BAF—lower panel) of all SNPs ordered along the chromosomes. The Log R Ratio (LRR) is a normalized measure of the total signal intensity for two alleles of the SNP. The B-Allele Frequency (BAF) is a normalized measure of the allelic intensity ratio of two alleles [18]. Signal intensities in sample ID 2355 show larger variance than in ID 1022. Moreover, a prominent pattern of waves is apparent in sample ID 2355. In many samples, we observed similar wave patterns. The noise-free-cnv software identified waves using a Gaussian filter with a large standard deviation, for instance comprising 1,000 SNPs. This filter “blurs” the values as shown in Figure 2(G,H). We called the resulting wave data the wave component of the LRR values. The variance of the blurred LRR values is a measure for the prominence of waves, the wave variance.
Figure 1.

Signal strength (LRR) and B-allele frequency (BAF) of samples from two male patients (ID 2355 and ID 1022). SNPs were visualized in increasing position along the chromosomes. LRR values of patient ID 2355 have larger variance and show pronounced wave noise.
Figure 2.

Wave noise. Ideograms of pro-metaphase (A) and metaphase (B) chromosome 7 were compared with signal intensities of SNPs of chromosome 7 of two patients (C,D) and with a human prometaphase (E) and metaphase (F) chromosome 7. Signal intensities shown in C and D were smoothed (noise-free-cnv software, function “blur” across 1,000 probe sets) to visualize genomic waves (G,H).
This wave pattern was compared with the banding pattern of metaphase chromosomes (Figure 2). Human metaphase chromosomes were stained with the Giemsa-trypsine procedure, which induces a banding pattern. AT-rich regions are more frequent in Giemsa-dark bands than in Giemsa-light bands [19,20]. In our study samples, Giemsa-dark bands corresponded to genomic regions with reduced probe set signals. This pattern of noise was described by others as “genomic waves” or “CG-waves” [10,11,12,13]. The co-linearity of genomic waves with Giemsa bands illustrates that genomic waves follow a similar pattern in all samples.
After subtraction of the wave component, the resulting LRR values follow an approximately normal distribution around zero. We called the resulting values per-SNP component and their variance the per-SNP variance. The decomposition of system noise in wave component and per-SNP component is shown for one sample in Figure 3. Wave variance and per-SNP variance components were calculated for all samples in Table A1.
Figure 3.

Noise components. LRR values of a noisy sample (A), split up in wave component (B) and per-SNP component (C). All SNPs of chromosomes 1–3 were shown (chromosomes indicated on top of panel A).
The system deviations of individual SNP signal intensities are strongly correlated across samples (Figure 4). To quantify the correlation of the noise (variance) components between different samples, we computed two additional data series: for each SNP the median through all 77 per-SNP components was computed and saved as the per-SNP profile. For the wave profile the same procedure was applied to the wave components. We then computed, for each sample, the correlation between the wave profile and the (individual) wave component as well as the correlation between the per-SNP profile and the (individual) per-SNP component. Details of the algorithm are described in Appendix. The high correlations found in our 77 samples confirmed that wave noise and per-SNP noise are system noise, i.e., follow highly non-random patterns. On average, the correlation was 0.843 for the wave component and 0.568 for the per-SNP component.
Figure 4.

per-SNP system noise. Signal intensities in genomic region 2: 189766706–189891527 shown for four patients (ID 1020; ID 1022; ID1026; ID 1028). The lower panel shows the per-SNP median profile (median signal intensities) of all samples (n = 77). Arrows and arrowheads indicate SNPs with LRR values far above and below the mean.
3. Factors Associated with Quality of Copy Number Data
The resolution of a classical chromosome study depends on the quality of the chromosomes and is expressed as the total number of visible cytogenetic bands (400 bands: low to moderate quality; 850 bands: excellent quality). According to our knowledge, no comparable quality metric for molecular karyotyping exists. Quality control in most copy number studies consists of rejecting samples with outlier numbers of CNV findings. A quality metric for the resolution of a CNV study (relating the size of a CNV and the likelihood of its detection) has not yet been defined.
In the current study we propose a preliminary quality metric based on the median number of SNPs per chromosome with copy number state (CN) ≠ 2 (numbers/chromosome for all cases are shown in Table A1). Copy Number state of each SNP was determined by the Affymetrix Power Tools software package (APT). SNPs located in common CNVs were excluded from this analysis. To identify SNPs located in common CNVs, we analyzed 403 control samples without visible waves and with highest genotype call rates selected from a large German population (PopGen [21]), as described before [17]. The median number of SNPs with CN ≠ 2 per chromosome was considered as a preliminary quality metric. The quality of a sample was related to the chromosomal background of SNPs with abnormal copy number (Figure 5). We defined deliberate quality categories: samples were classified as eligible, if the median number of SNPs per chromosome with CN ≠ 2 was zero, those with >100 SNPs with CN ≠ 2 were classified as ineligible.
Figure 5.

Quality of copy number samples. Number of SNPs with CN ≠ 2 per chromosome were scored. Sample ID 715 is eligible for CNV studies (most chromosomes without SNPs with CN ≠ 2). Accumulation of aberrant SNPs in chromosome 7 and 18 indicates presence of rare CNVs. Sample ID 50 is of intermediate quality. Sample ID 062 was classified as ineligible for CNV studies (>100 SNPs with CN ≠ 2 in most chromosomes).
Samples were classified according to the defined quality categories in Table 1. The use of freshly prepared DNA (compared to DNA samples that were used since years and had been thawed and frozen repeatedly) was a significant determinant of eligible samples (p < 0.001). Samples with high call rate (rate of successfully genotyped SNPs) were more likely to be suitable for copy number studies than those with lower call rates (p < 0.001). Low levels of wave variance as well as per-SNP variance were associated with eligibility for CNV analysis (p < 0.001). Eligibility for CNV studies was not significantly associated with the median number of calls by PennCNV (p = 0.053). However, eligible samples had between 63 and 165 calls, while the range of calls was much broader in ineligible samples. Birdview yielded significantly more calls in ineligible samples (p < 0.001). The proportion of putative false positive Birdview calls increased with decreasing confidence rates: The number of CNV findings with confidence below 2.5 was most strongly elevated.
Table 1.
Characteristics of 77 analyzed samples, classified according to eligibility for copy number variation (CNV) analysis. Numbers indicate mean values and range (lowest–highest value). Mean values were compared between groups with the Chi-2 test or the Kruskal-Wallis test.
| Ineligible | Intermediate | Eligible | Chi-2/ kruskal-wallis |
|
|---|---|---|---|---|
| (n = 29) | (n = 25) | (n = 23) | p | |
| Fresh DNA preparation | 0 (0.0 %) | 6 (20.7 %) | 14 (60.9 %) | <0.001 |
| Genotyping call rate | 94.7 [80.9–97.3] | 96.6 [94.8–98.3] | 97.7 [96.6–98.5] | <0.001 |
| Autosomal variance | 0.2291 [0.115–0.706] | 0.1343 [0.068–0.208] | 0.0870 [0.062–0.114] | <0.001 |
| wave noise | 0.0109 [0.002–0.058] | 0.0034 [0.001–0.017] | 0.0015 [0.001–0.013] | <0.001 |
| per–SNP noise | 0.2259 [0.082–0.696] | 0.1281 [0.067–0.204] | 0.0811 [0.060–0.164] | <0.001 |
| PennCNV, No. of calls | 238 [14–1821) | 103 [34–1024] | 98 [63–165] | 0.053 |
| PennCNV, % of deletions | 18.6 [1.3–81.3] | 27.4 [0.7–65.9] | 40.0 [10.3–54.8] | 0.164 |
| Birdview No. of calls | 527 [163–8,203] | 225 [154–1,339] | 208 [163–348] | <0.001 |
| Birdview (cf > 10) | 15 [2–717] | 12 [5–33] | 14 [4–20] | 0.048 |
| Birdview (cf = 10) | 89 [76–145] | 92 [74–105] | 94 [77–102] | 0.209 |
| Birdview (cf 2.5–10) | 93 [14–3344] | 19 [10–361] | 21 [11–45] | <0.001 |
| Birdview (cf < 2.5) | 370 [52–5665] | 106 [35–857] | 85 [42–194] | <0.001 |
Figure 6 summarizes salient aspects of system noise in SNP microarrays. Figure 6(A) plots for each sample the variances of wave component and per-SNP component. Wave variance and per-SNP variance seem to occur independently from each other: the observed correlation between both noise components (r = 0.124) was not significant (p = 0.401). Figure 6(B) illustrates the relation between sample eligibility and noise components in the eligible (n = 23) and ineligible (n = 29) cases. Eligible samples (i.e., those that are supposed to be excellent for copy number studies) have low levels of per-SNP variance. Samples with high wave variance are inappropriate for copy number studies.
Figure 6.

Wave variance and per-SNP variance. (A) Noise components in all 77 samples and (B) in samples of low (O) and high (●) quality (samples of intermediate quality were not included in (B)).
4. Noise Reduction in Copy Number Samples
The noise-free-cnv software package permits the visualization of samples, the isolation of noise components and the subtraction of isolated noise components. The next two examples (Figure 7 and Figure 8) illustrate noise reduction by comparing a test sample with a reference sample. We finally demonstrate the use of the noise-free-cnv-filter algorithm for the evaluation of CNVs.
Figure 7.

Signal intensities (y-axis: LRR values) of all SNPs from chromosome 18q up to chromosome 22. (A) Patient ID 1091; (B) reference sample ID 2355. After subtraction of the samples, a deletion in chromosome 20 became apparent (arrow).
Figure 8.

Sample with mosaic large deletion in chromosome 5q. (A,B) LRR- and BAF-values of SNPs of chromosomes 5 and 6 of patient. (C) LRR values of reference sample. (D) Signal intensities after subtraction of reference sample. Arrows indicate region with reduced LRR values. (E) LRR values after application of noise-free-cnv blur over 2,000 SNPs. (Bottom panel) Chromosome analysis of cultured peripheral blood lymphocytes from patient (courtesy of Johannes W.G. Janssen, Department of Human Genetics, University of Heidelberg). Arrow points to 5q-minus chromosome.
Figure 7 shows a deletion in chromosome 20 of patient ID 1091, which was detected by PennCNV and Birdview analysis. Due to strong waves, reduced signal intensities in the region of the putative deletion are not easily seen. Visual inspection of the LRR values of chromosome 20 after subtraction of a reference sample (A–B) suggested the presence of a true deletion in this patient.
Figure 8 illustrates the analysis of a mosaic deletion. Although sample ID D62 was classified as ineligible for CNV studies, analysis of SNPs with CN ≠ 2 per chromosome revealed significant clustering on chromosome 5 (Table A1; Figure 5). Neither PennCNV nor Birdsuite identified a large CNV on chromosome 5. After noise reduction, LRR and BAF values were suggestive for the presence of a mosaic deletion [22,23,24] (Figure 8(B,D)). To confirm the diagnosis of a mosaic deletion, a conventional chromosome analysis was performed: Some rare 5q chromosomes were observed amongst a majority of normal chromosome sets. Interestingly, it was recently demonstrated that the identification of mosaic abnormalities by microarray analysis is unreliable [25].
We developed the noise-free-cnv-filter algorithm for optimized noise reduction (Appendix). In the samples of our study population, noise-free-cnv-filter analysis resulted in an average reduction of the wave variance by 74.2%, of per-SNP variance by 35.3% and of the overall variance by 38.1%. Noise-reduction according to this algorithm supports the evaluation of CNV findings, in particular when the putative CNVs are small (Figure 9).
Figure 9.

Validation of CNV findings. Left panels show crude LRR values, left panels show LRR values after noise-free-cnv-filter analysis. Samples were renamed with suffix “nf” after noise-free-cnv-filter analysis. Bars indicate putative CNV findings.
In patient ID 715, both Birdview and PennCNV identified a deletion on chromosome 18 (green bar in Figure 9). Noise-free-cnv-filter analysis of the sample (ID 715 nf) suggested that the deletion was true. Subsequent molecular analysis confirmed the finding: the joining segment of the deletion was identified by a case-specific PCR and the breakpoints of the deletion were identified by DNA sequencing following standard procedures [17,26]. Two putative duplications in patients ID 412 were evaluated after noise-free-cnv-filter analysis. We considered the duplication in chromosome 1 (region 222 Mb) as spurious (red bar), but the duplication in chromosome 9 as probably true. As a consequence, this putative duplication is a candidate for further validation by molecular methods.
5. Conclusions—Proposal of a Two-Step Procedure for the Validation of CNV Findings
Our analysis had the following key findings: (1) Copy number samples may be noisy, which interferes—above a certain level of noise—with reliable identification of CNVs; (2) Eligible copy number samples were more likely when fresh DNA was used for microarray hybridization; (3) wave component and per-SNP component of noise are independent; (4) noise-free-cnv software enables noise reduction by subtracting wave and per-SNP noise components from samples; and (5) noise-free-cnv software supports the quality control of copy number data and the validation of copy number findings.
The current noise-free-cnv version was developed for the analysis of SNP microarray samples and was not designed for noise reduction in array based comparative genomic hybridization samples. The present study highlighted the value of noise reduction for large scale CNV validation (after software-assisted CNV detection). However, the value of noise reduction before software-assisted CNV detection is to be analyzed in future studies.
Based on our analysis of noise in real-life copy number samples we suggested a two-step procedure of CNV validation. As a first step of preliminary CNV validation we proposed large-scale inspection of CNV findings after noise reduction, to select putative candidate CNVs and reject false positive findings. In a second stage, this selection of putative CNV calls is analyzed further by independent molecular methods for final validation [17,26].
Acknowledgments
This work was supported by a grant from the Swiss Heart Foundation.
Appendix: Comments to the Noise-Free-CNV Software
A1. Noise-Free-CNV
The noise-free-cnv program package was specifically developed to analyze copy number variation in SNP-microarray samples and to manipulate the data in order to reduce noise. It was written in C++ and released as free software under the GNU General Public License version 3. Installer packages are available for Debian-based Linux systems and Windows. For the computation of the Fast Fourier Transform, we used the FFTW library [27]. Noise-free-cnv is compatible with the file format used by PennCNV [14].
The central program of the noise-free-cnv package is noise-free-cnv-gtk, a visual editor for interactive visualization and manipulation of SNP microarray data. Besides functioning as a browser for direct inspection and verification of CNV findings, it allows the user to perform many operations on the data. These include the Gaussian filters and variance computation referred to in the article. For further information, see the project homepage http://noise-free-cnv.sourceforge.net. A second program, noise-free-cnv-filter, implements a specific algorithm for system noise reduction, as described below. It is usable as a command line program to be easily applied to a batch of samples.
A2. The Noise-Free-CNV-Filter Algorithm
The noise reduction algorithm noise-free-cnv-filter consists of two main steps. In the first step, a genomic wave profile and a per-SNP noise profile are deduced from a batch of samples. In the second step, these profiles are used to modify the individual samples.
A2.1. System Noise Assessment
For each individual sample:
-
(1)
The non-autosomal data is removed and the Log R Ratio values are normalized towards an average value of zero.
-
(2)
The wave component is computed by applying a Gaussian filter with a standard deviation of 1,000 SNPs to the Log R Ratio sequence
-
(3)
The wave component is subtracted from the Log R Ratio values to calculate the per-SNP component.
Subsequently, the batch-specific wave is computed by regarding each SNP throughout the wave components of all samples and taking the median value. The same is done for the per-SNP profile utilizing the per-SNP components.
A2.2. System Noise Removal
In the second step, we use the median profiles to adjust the original samples.
For each individual:
-
(1)
The covariance of the wave component and the batch-specific wave profile is divided by the variance of the wave profile.
-
(2)The result is used as a scaling factor for the wave profile, the scaled profile is then subtracted from the wave component
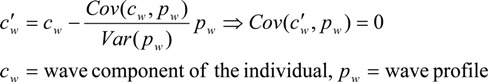
The same procedure is repeated on the per-SNP components.
-
(3)
Finally, the corrected components are added together and yield the corrected Log R Ratio values.
A3. Program Usage
Noise-free-cnv-filter was implemented as a command-line program. In the most simple case, it receives the file names of several SNP microarray samples in the PennCNV file format (due to the nature of the algorithm, application on a single sample is pointless). It then computes the profiles (saved as “wave_profile” and “per-snp_profile”) and the cleaned versions of all provided samples, which it saves as “<original filename>.nf”. As additional options, noise-free-cnv-filter allows the use of pre-computed profile sequences and the inclusion of the sex chromosomes into the analysis. As an example, noise-free-cnv-filter—verbose individuals/* applies the algorithm to all files in the directory individuals, discards the sex chromosomes and outputs detailed information about the progress and statistical information about the samples. For further help, type: noise-free-cnv-filter—help.
Table A1.
Eligibility of samples.
| ID | call rate | var. | wave var. | per_SNP var. | Chr1 | Chr2 | Chr3 | Chr4 | Chr5 | Chr6 | Chr7 | Chr8 | Chr9 | Chr10 | Chr11 | Chr12 | Chr13 | Chr14 | Chr15 | Chr16 | Chr17 | Chr18 | Chr19 | Chr20 | Chr21 | Chr22 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3 | 98.33 | 0.068 | 0.001 | 0.067 | 2 | 0 | 0 | 2 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 11 | 0 | 1 | 0 | 0 | 2 | 2 | 0 | 0 | 0 | 102 |
| 15 | 96.02 | 0.144 | 0.001 | 0.142 | 10 | 4 | 38 | 8 | 11 | 2 | 0 | 50 | 16 | 11 | 8 | 150 | 13 | 0 | 78 | 22 | 11 | 21 | 19 | 0 | 4 | 0 |
| 36 | 96.00 | 0.243 | 0.004 | 0.238 | 506 | 845 | 678 | 751 | 1,376 | 503 | 977 | 974 | 722 | 385 | 379 | 639 | 593 | 232 | 541 | 752 | 356 | 397 | 364 | 225 | 262 | 203 |
| 38 | 95.76 | 0.183 | 0.001 | 0.181 | 179 | 80 | 102 | 48 | 140 | 80 | 91 | 268 | 33 | 84 | 122 | 44 | 27 | 144 | 20 | 47 | 114 | 41 | 155 | 0 | 40 | 20 |
| 48 | 96.16 | 0.174 | 0.007 | 0.167 | 0 | 39 | 60 | 141 | 23 | 42 | 90 | 69 | 56 | 5 | 10 | 59 | 110 | 18 | 32 | 361 | 29 | 32 | 0 | 94 | 15 | 55 |
| 49 | 94.81 | 0.174 | 0.007 | 0.167 | 203 | 31 | 35 | 111 | 40 | 28 | 33 | 22 | 14 | 6 | 22 | 9 | 8 | 15 | 41 | 0 | 0 | 23 | 12 | 0 | 7 | 0 |
| 50 | 97.14 | 0.103 | 0.002 | 0.101 | 46 | 11 | 0 | 14 | 22 | 242 | 2 | 0 | 8 | 0 | 0 | 65 | 0 | 0 | 11 | 2 | 3 | 0 | 45 | 0 | 2 | 2 |
| 62 | 97.92 | 0.090 | 0.003 | 0.087 | 0 | 14 | 0 | 14 | 3 | 19 | 15 | 11 | 2 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 6 | 16 | 0 | 0 | 0 | 2 |
| 71 | 93.92 | 0.202 | 0.002 | 0.200 | 667 | 490 | 326 | 78 | 149 | 40 | 269 | 252 | 215 | 116 | 45 | 231 | 96 | 163 | 142 | 266 | 134 | 148 | 248 | 38 | 97 | 50 |
| 76 | 93.71 | 0.229 | 0.002 | 0.226 | 511 | 251 | 200 | 65 | 229 | 59 | 336 | 467 | 352 | 422 | 457 | 233 | 85 | 185 | 170 | 252 | 285 | 161 | 462 | 112 | 167 | 49 |
| 97 | 89.52 | 0.291 | 0.008 | 0.282 | 3,123 | 5,467 | 11,613 | 3,795 | 6,729 | 4,870 | 5,374 | 4,898 | 4,455 | 4,169 | 5,721 | 6,492 | 3,486 | 3,020 | 3,709 | 4,031 | 4,120 | 3,177 | 3,581 | 2,466 | 1,775 | 1,281 |
| 101 | 97.85 | 0.077 | 0.002 | 0.075 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 9 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 |
| 111 | 96.51 | 0.175 | 0.003 | 0.172 | 70 | 76 | 14 | 49 | 287 | 187 | 29 | 76 | 64 | 266 | 105 | 47 | 61 | 17 | 15 | 20 | 0 | 74 | 10 | 136 | 6 | 21 |
| 112 | 94.70 | 0.147 | 0.011 | 0.134 | 2,378 | 2,988 | 2,743 | 2,514 | 2,823 | 4,455 | 2,774 | 3,014 | 2,837 | 2,707 | 4,227 | 3,054 | 2,315 | 1,595 | 1,905 | 1,087 | 2,085 | 2,044 | 1,562 | 650 | 580 | 602 |
| 129 | 96.61 | 0.139 | 0.003 | 0.135 | 7 | 31 | 51 | 9 | 19 | 35 | 3 | 94 | 30 | 77 | 23 | 2 | 3 | 10 | 0 | 96 | 0 | 2 | 74 | 0 | 22 | 0 |
| 131 | 96.45 | 0.199 | 0.002 | 0.196 | 319 | 229 | 139 | 314 | 108 | 125 | 248 | 304 | 172 | 363 | 279 | 68 | 96 | 93 | 118 | 212 | 182 | 71 | 83 | 28 | 13 | 28 |
| 141 | 96.44 | 0.121 | 0.017 | 0.101 | 266 | 121 | 174 | 173 | 147 | 64 | 195 | 121 | 258 | 24 | 43 | 291 | 45 | 36 | 59 | 202 | 105 | 96 | 208 | 90 | 86 | 72 |
| 144 | 94.72 | 0.176 | 0.005 | 0.170 | 251 | 746 | 130 | 464 | 250 | 328 | 253 | 410 | 390 | 139 | 799 | 55 | 553 | 63 | 193 | 178 | 52 | 286 | 195 | 9 | 15 | 45 |
| 168 | 94.36 | 0.315 | 0.036 | 0.275 | 5,205 | 5,382 | 5,272 | 4,479 | 4,337 | 5,737 | 6,682 | 4,504 | 3,966 | 2,627 | 2,901 | 5,294 | 2,492 | 3,065 | 2,487 | 3,126 | 2,559 | 2,658 | 2,179 | 1,405 | 961 | 948 |
| 182 | 95.02 | 0.316 | 0.002 | 0.313 | 1,189 | 1,988 | 2,051 | 1,096 | 2,301 | 2,991 | 1,814 | 2,365 | 1,408 | 687 | 1,144 | 1,314 | 839 | 654 | 689 | 815 | 569 | 1,953 | 427 | 517 | 456 | 377 |
| 188 | 90.12 | 0.474 | 0.011 | 0.461 | 14,534 | 15,554 | 28,322 | 10,245 | 10,904 | 16,212 | 12,471 | 14,300 | 6,642 | 6,499 | 8,681 | 9,241 | 5,595 | 6,784 | 7,503 | 5,150 | 6,048 | 7,028 | 3,978 | 2,417 | 3,536 | 2,274 |
| 189 | 97.32 | 0.097 | 0.012 | 0.084 | 120 | 34 | 155 | 12 | 35 | 41 | 74 | 69 | 0 | 29 | 47 | 34 | 16 | 6 | 1 | 0 | 0 | 0 | 30 | 0 | 35 | 2 |
| 193 | 97.34 | 0.093 | 0.004 | 0.088 | 53 | 3 | 0 | 3 | 0 | 0 | 0 | 5 | 71 | 6 | 2 | 0 | 47 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 412 | 97.51 | 0.092 | 0.006 | 0.086 | 0 | 0 | 62 | 2 | 0 | 0 | 20 | 0 | 2 | 14 | 0 | 10 | 22 | 0 | 0 | 0 | 2 | 2 | 0 | 0 | 0 | 4 |
| 415 | 98.23 | 0.103 | 0.001 | 0.102 | 5 | 11 | 0 | 5 | 3 | 10 | 35 | 0 | 2 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 9 | 0 | 0 | 0 | 2 |
| 421 | 96.73 | 0.165 | 0.055 | 0.102 | 0 | 7 | 0 | 0 | 0 | 5 | 28 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 4 | 0 | 0 | 0 | 0 |
| 422 | 96.10 | 0.074 | 0.002 | 0.073 | 37,996 | 36,654 | 32,343 | 29,935 | 29,248 | 29,062 | 23,198 | 24,951 | 21,457 | 22,172 | 22,247 | 21,574 | 15,560 | 14,542 | 14,525 | 12,574 | 10,444 | 14,095 | 8,616 | 10,294 | 7,218 | 4,456 |
| 430 | 96.39 | 0.160 | 0.019 | 0.138 | 10,614 | 10,096 | 9,196 | 16,278 | 15,383 | 7,959 | 7,758 | 9,465 | 6,757 | 6,295 | 6,291 | 6,559 | 4,955 | 3,487 | 2,491 | 3,459 | 3,074 | 5,404 | 1,525 | 2,399 | 3,569 | 1,187 |
| 438 | 89.76 | 0.463 | 0.057 | 0.399 | 45,981 | 56,162 | 42,403 | 45,750 | 55,976 | 37,901 | 44,212 | 34,069 | 30,387 | 26,842 | 26,461 | 38,930 | 23,963 | 20,800 | 18,258 | 19,160 | 16,624 | 18,367 | 9,637 | 13,485 | 8,790 | 5,479 |
| 442 | 97.82 | 0.084 | 0.008 | 0.076 | 486 | 382 | 0 | 0 | 14 | 0 | 9 | 57 | 2 | 0 | 33 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 67 | 0 | 4 | 3 |
| 451 | 95.96 | 0.205 | 0.004 | 0.200 | 109 | 154 | 49 | 363 | 75 | 361 | 107 | 213 | 124 | 100 | 85 | 150 | 124 | 20 | 63 | 4 | 60 | 116 | 50 | 59 | 0 | 19 |
| 461 | 80.86 | 0.706 | 0.007 | 0.696 | 64,822 | 74,302 | 41,655 | 64,993 | 56,987 | 58,825 | 49,369 | 55,433 | 45,357 | 47,372 | 35,753 | 49,377 | 30,870 | 27,158 | 25,448 | 29,590 | 14,637 | 22,231 | 15,372 | 19,289 | 12,431 | 8,971 |
| 613 | 97.64 | 0.090 | 0.001 | 0.088 | 2 | 6 | 3 | 7 | 4 | 0 | 49 | 15 | 0 | 0 | 22 | 2 | 10 | 2 | 0 | 0 | 0 | 0 | 7 | 0 | 2 | 0 |
| 647 | 95.49 | 0.157 | 0.002 | 0.154 | 15 | 1 | 30 | 166 | 0 | 47 | 9 | 0 | 6 | 45 | 41 | 58 | 0 | 5 | 28 | 41 | 24 | 0 | 14 | 4 | 4 | 11 |
| 653 | 98.22 | 0.079 | 0.004 | 0.074 | 12 | 2 | 14 | 1,618 | 26 | 7 | 12 | 7 | 0 | 9 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 4 | 4 |
| 665 | 97.32 | 0.123 | 0.037 | 0.082 | 5,071 | 8,140 | 6,289 | 5,520 | 6,417 | 6,816 | 5,890 | 6,894 | 4,238 | 5,326 | 4,503 | 5,246 | 2,641 | 2,696 | 2,301 | 1,766 | 1,445 | 4,211 | 1,699 | 1,399 | 2,448 | 932 |
| 670 | 96.74 | 0.152 | 0.043 | 0.105 | 3,160 | 4,486 | 3,895 | 3,913 | 3,812 | 3,038 | 3,309 | 3,676 | 2,359 | 2,112 | 3,327 | 2,791 | 1,591 | 1,595 | 1,142 | 969 | 1,028 | 2,043 | 543 | 1,213 | 1,034 | 286 |
| 675 | 97.67 | 0.084 | 0.009 | 0.074 | 0 | 0 | 3 | 0 | 4 | 1 | 3 | 37 | 0 | 0 | 58 | 4 | 0 | 0 | 0 | 11 | 0 | 0 | 48 | 0 | 0 | 0 |
| 676 | 98.15 | 0.078 | 0.001 | 0.077 | 0 | 0 | 0 | 0 | 45 | 2 | 18 | 0 | 0 | 0 | 14 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 677 | 95.58 | 0.208 | 0.003 | 0.204 | 25 | 149 | 161 | 54 | 77 | 57 | 267 | 157 | 38 | 210 | 63 | 13 | 52 | 64 | 34 | 44 | 57 | 137 | 121 | 16 | 56 | 75 |
| 693 | 95.72 | 0.133 | 0.005 | 0.128 | 4 | 73 | 18 | 6 | 31 | 12 | 15 | 27 | 7 | 0 | 10 | 8 | 73 | 14 | 10 | 0 | 5 | 3 | 20 | 2 | 0 | 12 |
| 715 | 97.44 | 0.095 | 0.006 | 0.089 | 3 | 5 | 0 | 0 | 4 | 0 | 471 | 0 | 2 | 0 | 0 | 0 | 0 | 4 | 0 | 4 | 0 | 24 | 0 | 4 | 0 | 0 |
| 717 | 96.60 | 0.134 | 0.001 | 0.132 | 0 | 2 | 22 | 21 | 29 | 41 | 0 | 17 | 0 | 6 | 57 | 10 | 0 | 2 | 0 | 15 | 0 | 0 | 34 | 0 | 0 | 0 |
| 729 | 94.75 | 0.189 | 0.001 | 0.187 | 115 | 23 | 115 | 184 | 49 | 55 | 84 | 91 | 42 | 45 | 101 | 111 | 16 | 57 | 103 | 80 | 57 | 61 | 192 | 0 | 22 | 0 |
| 733 | 95.84 | 0.198 | 0.009 | 0.188 | 23 | 28 | 84 | 218 | 218 | 0 | 306 | 180 | 40 | 34 | 59 | 11 | 32 | 3 | 33 | 62 | 108 | 6 | 8 | 38 | 46 | 0 |
| 735 | 95.28 | 0.173 | 0.003 | 0.169 | 94 | 21 | 111 | 290 | 81 | 41 | 116 | 14 | 220 | 79 | 0 | 87 | 47 | 31 | 34 | 43 | 16 | 13 | 181 | 0 | 0 | 22 |
| 742 | 96.83 | 0.114 | 0.013 | 0.099 | 0 | 0 | 3 | 0 | 2 | 0 | 1 | 0 | 4 | 0 | 2 | 0 | 0 | 15 | 8 | 2 | 18 | 0 | 137 | 0 | 0 | 0 |
| 744 | 97.26 | 0.108 | 0.017 | 0.089 | 0 | 135 | 126 | 332 | 73 | 148 | 263 | 0 | 135 | 31 | 52 | 150 | 180 | 34 | 15 | 40 | 0 | 166 | 114 | 0 | 94 | 0 |
| 746 | 95.72 | 0.253 | 0.004 | 0.248 | 283 | 895 | 350 | 378 | 287 | 388 | 227 | 723 | 594 | 712 | 229 | 559 | 323 | 206 | 98 | 478 | 398 | 689 | 341 | 73 | 184 | 173 |
| 750 | 97.15 | 0.114 | 0.015 | 0.097 | 2,389 | 642 | 1,750 | 1,501 | 1,370 | 1,792 | 707 | 1,440 | 997 | 615 | 674 | 750 | 559 | 225 | 544 | 63 | 187 | 814 | 458 | 177 | 184 | 0 |
| 752 | 97.80 | 0.103 | 0.004 | 0.099 | 88 | 121 | 119 | 155 | 196 | 83 | 118 | 187 | 93 | 206 | 66 | 147 | 27 | 35 | 50 | 41 | 32 | 70 | 15 | 36 | 27 | 25 |
| 796 | 94.23 | 0.247 | 0.002 | 0.244 | 2,165 | 482 | 568 | 1,127 | 539 | 263 | 360 | 591 | 440 | 697 | 908 | 806 | 106 | 630 | 256 | 498 | 262 | 565 | 127 | 275 | 67 | 48 |
| 1020 | 98.21 | 0.075 | 0.001 | 0.074 | 2 | 0 | 0 | 0 | 2 | 1 | 2 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 8 | 0 | 16 | 0 |
| 1022 | 97.77 | 0.082 | 0.001 | 0.081 | 6 | 16 | 2 | 30 | 2 | 2 | 2 | 0 | 12 | 0 | 11 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| 1026 | 98.34 | 0.066 | 0.001 | 0.064 | 1 | 0 | 0 | 0 | 5 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 11 | 9 | 0 | 0 | 0 | 3 | 0 | 0 | 0 |
| 1028 | 97.49 | 0.089 | 0.001 | 0.088 | 9 | 0 | 0 | 0 | 40 | 49 | 0 | 3 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 1029 | 98.54 | 0.062 | 0.001 | 0.060 | 3 | 64 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 2 | 50 | 0 | 25 | 0 | 2 | 0 | 0 | 0 | 0 |
| 1033 | 97.58 | 0.087 | 0.001 | 0.085 | 0 | 0 | 83 | 0 | 0 | 0 | 2 | 0 | 70 | 4 | 4 | 0 | 0 | 0 | 8 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 1034 | 97.50 | 0.092 | 0.010 | 0.081 | 0 | 0 | 0 | 2 | 0 | 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 1037 | 98.26 | 0.068 | 0.001 | 0.067 | 8 | 5 | 2 | 50 | 2 | 6 | 2 | 12 | 0 | 2 | 1 | 0 | 2 | 0 | 0 | 3 | 54 | 13 | 2 | 2 | 0 | 0 |
| 1040 | 96.56 | 0.114 | 0.001 | 0.113 | 9 | 0 | 17 | 53 | 3 | 1 | 4 | 0 | 0 | 0 | 0 | 0 | 17 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| 1041 | 97.16 | 0.094 | 0.001 | 0.093 | 0 | 0 | 0 | 4 | 2 | 2 | 0 | 0 | 0 | 0 | 13 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 1042 | 97.46 | 0.087 | 0.003 | 0.084 | 15 | 14 | 19 | 20 | 11 | 28 | 10 | 19 | 4 | 2 | 2 | 6 | 8 | 4 | 18 | 4 | 2 | 24 | 2 | 0 | 2 | 0 |
| 1056 | 97.11 | 0.098 | 0.003 | 0.095 | 2 | 0 | 2 | 2 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 15 | 0 | 4 | 0 | 0 | 35 | 0 | 10 | 0 | 0 | 18 |
| 1063 | 98.31 | 0.075 | 0.002 | 0.072 | 0 | 2 | 169 | 10 | 0 | 0 | 29 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 13 | 0 | 0 | 0 | 2 | 0 | 0 | 0 |
| 1065 | 98.23 | 0.068 | 0.003 | 0.065 | 0 | 4 | 2 | 7 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 8 | 0 | 0 | 0 | 0 | 58 | 4 | 0 | 0 | 0 |
| 1088 | 97.64 | 0.092 | 0.004 | 0.088 | 53 | 23 | 61 | 45 | 8 | 90 | 26 | 76 | 32 | 44 | 50 | 71 | 34 | 6 | 15 | 54 | 0 | 22 | 13 | 4 | 9 | 39 |
| 1091 | 96.69 | 0.138 | 0.029 | 0.105 | 7,631 | 6,080 | 7,146 | 6,512 | 7,299 | 4,006 | 4,952 | 6,666 | 4,169 | 2,579 | 3,990 | 4,012 | 2,388 | 2,194 | 1,415 | 2,613 | 1,030 | 2,828 | 646 | 3,697 | 1,862 | 451 |
| 1147 | 97.96 | 0.079 | 0.002 | 0.077 | 5 | 76 | 4 | 8 | 0 | 0 | 4 | 4 | 16 | 0 | 2 | 3 | 2 | 0 | 2 | 2 | 0 | 7 | 190 | 0 | 0 | 0 |
| 1151 | 97.90 | 0.087 | 0.001 | 0.086 | 2 | 4 | 0 | 0 | 3 | 0 | 0 | 8 | 0 | 4 | 9 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 11 | 2 |
| 2110 | 93.16 | 0.343 | 0.010 | 0.332 | 671 | 3,307 | 3,614 | 1,414 | 3,548 | 3,035 | 2,775 | 2,070 | 1,581 | 2,718 | 2,153 | 2,054 | 1,573 | 1,426 | 995 | 1,173 | 1,584 | 1,358 | 662 | 285 | 971 | 581 |
| 2134 | 95.73 | 0.134 | 0.008 | 0.125 | 144 | 52 | 81 | 51 | 51 | 100 | 28 | 88 | 70 | 0 | 35 | 77 | 3 | 55 | 7 | 16 | 56 | 7 | 41 | 2 | 18 | 1 |
| 2144 | 97.12 | 0.093 | 0.004 | 0.089 | 2 | 5 | 8 | 1 | 4 | 0 | 4 | 0 | 2 | 0 | 14 | 4 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 2 | 0 | 0 |
| 2240 | 94.48 | 0.299 | 0.004 | 0.294 | 488 | 1,656 | 1,019 | 1,734 | 2,180 | 1,605 | 754 | 1,750 | 1,179 | 531 | 916 | 1,728 | 916 | 751 | 643 | 921 | 501 | 427 | 713 | 356 | 339 | 374 |
| 2355 | 94.50 | 0.258 | 0.026 | 0.229 | 1,870 | 1,799 | 1,422 | 741 | 1,491 | 2,829 | 1,663 | 1,353 | 829 | 1,500 | 1,360 | 1,714 | 654 | 677 | 1,262 | 767 | 1,420 | 1,061 | 842 | 826 | 464 | 452 |
| 2406 | 94.78 | 0.195 | 0.011 | 0.183 | 67 | 125 | 136 | 122 | 201 | 80 | 142 | 109 | 62 | 61 | 5 | 51 | 70 | 68 | 34 | 7 | 60 | 83 | 122 | 53 | 43 | 0 |
| D_062 | 94.17 | 0.322 | 0.003 | 0.318 | 772 | 838 | 547 | 536 | 4,419 | 436 | 711 | 496 | 239 | 308 | 219 | 582 | 319 | 215 | 222 | 81 | 62 | 540 | 101 | 86 | 0 | 94 |
For each sample, genotype call rate, variance, wave variance and per-SNP variance were calculated. The remaining columns show for each chromosome (chromosome number indicated) the number of probe sets with CN ≠ 2. This analysis included only probe sets that had normal copy number (CN = 2) in 403 samples from a population based German study (for details and references see [17]). A non-random distribution of probe sets with CN ≠ 2 is highly suggestive for the existence of a rare CNV (for instance ID 1147 or ID653, in contrast to ID 1042 or ID 2034). Even in samples with high variance, non-random distribution can be detected (chromosome 5 of ID D_062, chromosomes 1 and 2 in ID 442).
Table A2.
Analysis of noise components in samples.
| ID | variance | wave variance | per-SNP variance | wave correlation | per-SNP correlation | wave subtraction factor | per-SNP subtraction factor |
|---|---|---|---|---|---|---|---|
| 3 | 0.068 | 0.001 | 0.067 | 0.804 | 0.676 | 0.409 | 0.800 |
| 15 | 0.144 | 0.001 | 0.142 | 0.567 | 0.564 | 0.393 | 0.972 |
| 36 | 0.243 | 0.004 | 0.238 | 0.877 | 0.388 | 0.997 | 0.865 |
| 38 | 0.183 | 0.001 | 0.181 | 0.456 | 0.502 | 0.314 | 0.977 |
| 48 | 0.174 | 0.007 | 0.167 | 0.939 | 0.508 | 1.405 | 0.949 |
| 49 | 0.174 | 0.007 | 0.167 | 0.959 | 0.527 | 1.419 | 0.985 |
| 50 | 0.103 | 0.002 | 0.101 | 0.881 | 0.536 | 0.626 | 0.779 |
| 62 | 0.090 | 0.003 | 0.087 | 0.929 | 0.609 | 0.886 | 0.820 |
| 71 | 0.202 | 0.002 | 0.200 | 0.365 | 0.589 | 0.274 | 1.203 |
| 76 | 0.229 | 0.002 | 0.226 | 0.580 | 0.529 | 0.511 | 1.151 |
| 97 | 0.291 | 0.008 | 0.282 | 0.875 | 0.360 | 1.427 | 0.873 |
| 101 | 0.077 | 0.002 | 0.075 | 0.906 | 0.727 | 0.717 | 0.910 |
| 111 | 0.175 | 0.003 | 0.172 | 0.914 | 0.482 | 0.903 | 0.914 |
| 112 | 0.147 | 0.011 | 0.134 | 0.864 | 0.578 | 1.671 | 0.968 |
| 129 | 0.139 | 0.003 | 0.135 | 0.898 | 0.620 | 0.964 | 1.043 |
| 131 | 0.199 | 0.002 | 0.196 | 0.875 | 0.417 | 0.790 | 0.844 |
| 141 | 0.121 | 0.017 | 0.101 | 0.949 | 0.703 | 2.297 | 1.024 |
| 144 | 0.176 | 0.005 | 0.170 | 0.881 | 0.591 | 1.141 | 1.115 |
| 168 | 0.315 | 0.036 | 0.275 | 0.936 | 0.406 | 3.234 | 0.975 |
| 182 | 0.316 | 0.002 | 0.313 | 0.809 | 0.386 | 0.704 | 0.990 |
| 189 | 0.097 | 0.012 | 0.084 | 0.930 | 0.668 | 1.874 | 0.883 |
| 193 | 0.093 | 0.004 | 0.088 | 0.960 | 0.704 | 1.174 | 0.956 |
| 412 | 0.092 | 0.006 | 0.086 | 0.950 | 0.691 | 1.304 | 0.925 |
| 421 | 0.103 | 0.001 | 0.102 | 0.898 | 0.680 | 0.598 | 0.991 |
| 422 | 0.165 | 0.055 | 0.102 | 0.873 | 0.523 | 3.763 | 0.763 |
| 425 | 0.074 | 0.002 | 0.073 | 0.908 | 0.572 | 0.653 | 0.705 |
| 430 | 0.160 | 0.019 | 0.138 | 0.896 | 0.457 | 2.265 | 0.778 |
| 438 | 0.463 | 0.057 | 0.399 | 0.913 | 0.354 | 4.006 | 1.021 |
| 442 | 0.084 | 0.008 | 0.076 | 0.942 | 0.663 | 1.496 | 0.835 |
| 451 | 0.205 | 0.004 | 0.200 | 0.927 | 0.453 | 1.111 | 0.927 |
| 461 | 0.706 | 0.007 | 0.696 | −0.310 | 0.228 | −0.460 | 0.868 |
| 613 | 0.090 | 0.001 | 0.088 | 0.850 | 0.565 | 0.589 | 0.767 |
| 647 | 0.157 | 0.002 | 0.154 | 0.766 | 0.589 | 0.671 | 1.059 |
| 653 | 0.079 | 0.004 | 0.074 | 0.938 | 0.569 | 1.141 | 0.707 |
| 665 | 0.123 | 0.037 | 0.082 | 0.896 | 0.555 | 3.159 | 0.726 |
| 670 | 0.152 | 0.043 | 0.105 | 0.906 | 0.565 | 3.422 | 0.837 |
| 675 | 0.084 | 0.009 | 0.074 | 0.951 | 0.672 | 1.647 | 0.837 |
| 676 | 0.078 | 0.001 | 0.077 | 0.646 | 0.635 | 0.313 | 0.807 |
| 677 | 0.208 | 0.003 | 0.204 | 0.901 | 0.465 | 0.870 | 0.960 |
| 693 | 0.133 | 0.005 | 0.128 | 0.953 | 0.581 | 1.179 | 0.952 |
| 715 | 0.095 | 0.006 | 0.089 | 0.950 | 0.667 | 1.308 | 0.911 |
| 717 | 0.134 | 0.001 | 0.132 | 0.522 | 0.650 | 0.351 | 1.081 |
| 729 | 0.189 | 0.001 | 0.187 | 0.606 | 0.611 | 0.411 | 1.209 |
| 733 | 0.198 | 0.009 | 0.188 | 0.947 | 0.488 | 1.628 | 0.967 |
| 735 | 0.173 | 0.003 | 0.169 | 0.901 | 0.609 | 0.917 | 1.144 |
| 742 | 0.114 | 0.013 | 0.099 | 0.956 | 0.649 | 2.017 | 0.936 |
| 744 | 0.108 | 0.017 | 0.089 | 0.921 | 0.570 | 2.186 | 0.779 |
| 746 | 0.253 | 0.004 | 0.248 | 0.906 | 0.414 | 1.033 | 0.943 |
| 750 | 0.114 | 0.015 | 0.097 | 0.940 | 0.612 | 2.137 | 0.874 |
| 752 | 0.103 | 0.004 | 0.099 | 0.937 | 0.471 | 1.033 | 0.679 |
| 796 | 0.247 | 0.002 | 0.244 | 0.015 | 0.527 | 0.012 | 1.192 |
| 1020 | 0.075 | 0.001 | 0.074 | 0.742 | 0.614 | 0.348 | 0.767 |
| 1022 | 0.082 | 0.001 | 0.081 | 0.909 | 0.644 | 0.640 | 0.836 |
| 1026 | 0.066 | 0.001 | 0.064 | 0.861 | 0.664 | 0.557 | 0.770 |
| 1028 | 0.089 | 0.001 | 0.088 | 0.742 | 0.643 | 0.380 | 0.871 |
| 1029 | 0.062 | 0.001 | 0.060 | 0.912 | 0.661 | 0.572 | 0.742 |
| 1033 | 0.087 | 0.001 | 0.085 | 0.820 | 0.703 | 0.518 | 0.940 |
| 1034 | 0.092 | 0.010 | 0.081 | 0.963 | 0.709 | 1.732 | 0.924 |
| 1037 | 0.068 | 0.001 | 0.067 | 0.782 | 0.633 | 0.390 | 0.748 |
| 1040 | 0.114 | 0.001 | 0.113 | 0.639 | 0.701 | 0.355 | 1.077 |
| 1041 | 0.094 | 0.001 | 0.093 | 0.850 | 0.672 | 0.546 | 0.937 |
| 1042 | 0.087 | 0.003 | 0.084 | 0.947 | 0.695 | 0.884 | 0.921 |
| 1056 | 0.098 | 0.003 | 0.095 | 0.941 | 0.686 | 0.893 | 0.966 |
| 1063 | 0.075 | 0.002 | 0.072 | 0.924 | 0.571 | 0.832 | 0.701 |
| 1065 | 0.068 | 0.003 | 0.065 | 0.959 | 0.657 | 0.904 | 0.764 |
| 1088 | 0.092 | 0.004 | 0.088 | 0.918 | 0.497 | 1.046 | 0.675 |
| 1091 | 0.138 | 0.029 | 0.105 | 0.912 | 0.537 | 2.854 | 0.797 |
| 1147 | 0.079 | 0.002 | 0.077 | 0.944 | 0.717 | 0.795 | 0.909 |
| 1151 | 0.087 | 0.001 | 0.086 | 0.688 | 0.572 | 0.311 | 0.769 |
| 2110 | 0.343 | 0.010 | 0.332 | 0.948 | 0.408 | 1.715 | 1.075 |
| 2134 | 0.134 | 0.008 | 0.125 | 0.936 | 0.551 | 1.575 | 0.893 |
| 2144 | 0.093 | 0.004 | 0.089 | 0.953 | 0.609 | 1.046 | 0.832 |
| 2240 | 0.299 | 0.004 | 0.294 | 0.909 | 0.433 | 1.060 | 1.075 |
| 2355 | 0.258 | 0.026 | 0.229 | 0.960 | 0.530 | 2.826 | 1.161 |
| 2406 | 0.195 | 0.011 | 0.183 | 0.940 | 0.570 | 1.833 | 1.113 |
| 188c | 0.474 | 0.011 | 0.461 | 0.899 | 0.314 | 1.715 | 0.975 |
| D62 | 0.322 | 0.003 | 0.318 | 0.819 | 0.340 | 0.761 | 0.878 |
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Girirajan S., Campbell C.D., Eichler E.E. Human copy number variation and complex genetic disease. Annu. Rev. Genet. 2011;45:203–226. doi: 10.1146/annurev-genet-102209-163544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang F., Gu W., Hurles M.E., Lupski J.R. Copy number variation in human health, disease, and evolution. Annu. Rev. Genomics Hum. Genet. 2009;10:451–481. doi: 10.1146/annurev.genom.9.081307.164217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fakhro K.A., Choi M., Ware S.M., Belmont J.W., Towbin J.A., Lifton R.P., Khokha M.K., Brueckner M. Rare copy number variations in congenital heart disease patients identify unique genes in left-right patterning. Proc. Natl. Acad. Sci. USA. 2011;108:2915–2920. doi: 10.1073/pnas.1019645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Priebe L., Degenhardt F., Strohmaier J., Breuer R., Herms S., Witt S.H., Hoffmann P., Kulbida R., Mattheisen M., Moebus S., et al. Copy number variants in german patients with schizophrenia. PLoS One. 2013;8:e64035. doi: 10.1371/journal.pone.0064035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vandeweyer G., Kooy R.F. Detection and interpretation of genomic structural variation in health and disease. Expert. Rev. Mol. Diagn. 2013;13:61–82. doi: 10.1586/erm.12.119. [DOI] [PubMed] [Google Scholar]
- 6.Southard A.E., Edelmann L.J., Gelb B.D. Role of copynumber variants in structural birth defects. Pediatrics. 2012;129:755–763. doi: 10.1542/peds.2011-2337. [DOI] [PubMed] [Google Scholar]
- 7.Zhang D., Qian Y., Akula N., Alliey-Rodriguez N., Tang J., The Bipolar Genome Study. Gershon E.S., Liu C. Accuracy of CNV detection from GWAS data. PLoS One. 2011;6:e14511. doi: 10.1371/journal.pone.0014511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dellinger A.E., Saw S.M., Goh L.K., Seielstad M., Young T.L., Li Y.J. Comparative analyses of seven algorithms for copy number variant identification from single nucleotide polymorphism arrays. Nucleic Acids Res. 2010;38:e105. doi: 10.1093/nar/gkq040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zheng X., Shaffer J.R., McHugh C.P., Laurie C.C., Feenstra B., Melbye M., Murray J.C., Marazita M.L., Feingold E. Using family data as a verification standard to evaluate copy number variation calling strategies for genetic association studies. Genet. Epidemiol. 2012;36:253–262. doi: 10.1002/gepi.21618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marioni J.C., Thorne N.P., Valsesia A., Fitzgerald T., Redon R., Fiegler H., Andrews T.D., Stranger B.E., Lynch A.G., Dermitzakis E.T., et al. Breaking the waves: Improved detection of copy number variation from microarray-based comparative genomic hybridization. Genome Biol. 2007;8:R228. doi: 10.1186/gb-2007-8-10-r228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Diskin S.J., Li M., Hou C., Yang S., Glessner J., Hakonarson H., Bucan M., Maris J.M., Wang K. Adjustment of genomic waves in signal intensities from whole-genome SNP genotyping platforms. Nucleic Acids Res. 2008;36:e126. doi: 10.1093/nar/gkn556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van de Wiel M.A., Brosens R., Eilers P.H., Kumps C., Meijer G.A., Menten B., Sistermans E., Speleman F., Timmerman M.E., Ylstra B. Smoothing waves in array CGH tumor profiles. Bioinformatics. 2009;25:1099–1104. doi: 10.1093/bioinformatics/btp132. [DOI] [PubMed] [Google Scholar]
- 13.Lee Y.H., Ronemus M., Kendall J., Lakshmi B., Leotta A., Levy D., Esposito D., Grubor V., Ye K., Wigler M., et al. Reducing system noise in copynumber data using principal components of self-self hybridizations. Proc. Natl. Acad. Sci. USA. 2012;109:E103–E110. doi: 10.1073/pnas.1106233109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang K., Li M., Hadley D., Liu R., Glessner J., Grant S.F., Hakonarson H., Bucan M. PennCNV: An integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17:1665–1674. doi: 10.1101/gr.6861907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Korn J.M., Kuruvilla F.G., McCarroll S.A., Wysoker A., Nemesh J., Cawley S., Hubbell E., Veitch J., Collins P.J., Darvishi K., et al. Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat. Genet. 2008;40:1253–1260. doi: 10.1038/ng.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McCarroll S.A., Kuruvilla F.G., Korn J.M., Cawley S., Nemesh J., Wysoker A., Shapero M.H., de Bakker P.I., Maller J.B., Kirby A., et al. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat. Genet. 2008;40:1166–1174. doi: 10.1038/ng.238. [DOI] [PubMed] [Google Scholar]
- 17.Grond-Ginsbach C., Chen B., Pjontek R., Wiest T., Burwinkel B., Tchatchou S., Krawczak M., Schreiber S., Brandt T., Kloss M., et al. Copy number variation in patients with cervical artery dissection. Eur. J. Hum. Genet. 2012;20:1295–1299. doi: 10.1038/ejhg.2012.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang K., Bucan M. Copy number variation detection via high-density SNP genotyping. Cold Spring Harb. Protoc. 2008;2008 doi: 10.1101/pdb.top46. [DOI] [PubMed] [Google Scholar]
- 19.Niimura Y., Gojobori T. In silico chromosome staining: Reconstruction of Giemsa bands from the whole human genome sequence. Proc. Natl. Acad. Sci. USA. 2002;99:797–802. doi: 10.1073/pnas.022437999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Costantini M.L., Clay O., Federico C., Saccone S., Auletta F., Bernardi G. Human chromosomal bands: Nested structure, high-definition map and molecular basis. Chromosoma. 2007;116:29–40. doi: 10.1007/s00412-006-0078-0. [DOI] [PubMed] [Google Scholar]
- 21.Krawczak M., Nikolaus S., von Eberstein H., Croucher P.J., El Mokhtari N.E., Schreiber S. PopGen: Population-based recruitment of patients and controls for the analysis of complex genotype-phenotype relationships. Community Genet. 2006;9:55–61. doi: 10.1159/000090694. [DOI] [PubMed] [Google Scholar]
- 22.Piotrowski A., Bruder C.E., Andersson R., Diaz de Ståhl T., Menzel U., Sandgren J., Poplawski A., von Tell D., Crasto C., Bogdan A., et al. Somatic mosaicism for copy number variation in differentiated human tissues. Hum. Mutat. 2008;29:1118–1124. doi: 10.1002/humu.20815. [DOI] [PubMed] [Google Scholar]
- 23.Jasmine F., Rahaman R., Dodsworth C., Roy S., Paul R., Raza M., Paul-Brutus R., Kamal M., Ahsan H., Kibriya M.G. A genome-wide study of cytogenetic changes in colorectal cancer using SNP microarrays: Opportunities for future personalized treatment. PLoS One. 2012;7:e31968. doi: 10.1371/journal.pone.0031968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laurie C.C., Laurie C.A., Rice K., Doheny K.F., Zelnick L.R., McHugh C.P., Ling H., Hetrick K.N., Pugh E.W., Amos C., et al. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat. Genet. 2012;44:642–650. doi: 10.1038/ng.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bi W., Borgan C., Pursley A.N., Hixson P., Shaw C.A., Bacino C.A., Lalani S.R., Patel A., Stankiewicz P., Lupski J.R., et al. Comparison of chromosome analysis and chromosomal microarray analysis: What is the value of chromosome analysis in today’s genomic array era? Genet. Med. 2013;15:450–457. doi: 10.1038/gim.2012.152. [DOI] [PubMed] [Google Scholar]
- 26.Vissers L.E., Bhatt S.S., Janssen I.M., Xia Z., Lalani S.R., Pfundt R., Derwinska K., de Vries B.B., Gilissen C., Hoischen A., et al. Rare pathogenic microdeletions and tandem duplications are microhomology-mediated and stimulated by local genomic architecture. Hum. Mol. Genet. 2009;18:3579–3593. doi: 10.1093/hmg/ddp306. [DOI] [PubMed] [Google Scholar]
- 27.Frigo M., Johnson S.G. The design and implementation of FFTW3. Proc. IEEE. 2005;93:216–231. doi: 10.1109/JPROC.2004.840301. [DOI] [Google Scholar]