

Abstract
A series of 2-substituted 6-hydroxy-1,2,4-triazine-3,5(2H,4H)-dione derivatives were synthesized as inhibitors of d-amino acid oxidase (DAAO). Many compounds in this series were found to be potent DAAO inhibitors, with IC50 values in the double-digit nanomolar range. The 6-hydroxy-1,2,4-triazine-3,5(2H,4H)-dione pharmacophore appears metabolically resistant to O-glucuronidation unlike other structurally related DAAO inhibitors. Among them, 6-hydroxy-2-(naphthalen-1-ylmethyl)-1,2,4-triazine-3,5(2H,4H)-dione 11h was found to be selective over a number of targets and orally available in mice. Furthermore, oral coadministration of d-serine with 11h enhanced the plasma levels of d-serine in mice compared to the oral administration of d-serine alone, demonstrating its ability to serve as a pharmacoenhancer of d-serine.
Introduction
d-Serine, a full agonist at the glycine modulatory site of the N-methyl-d-aspartate (NMDA) receptor, has been actively explored as a potential therapeutic agent for the treatment of schizophrenia.1 Several clinical studies with oral d-serine have shown promising results for the treatment of not only positive symptoms but also negative symptoms which have not responded well to existing drugs.1 Further clinical development of d-serine, however, could be hampered by the high doses of d-serine (60–120 mg/kg) required for the optimal efficacy2 since d-serine was reported to be nephrotoxic in rats at high doses.3d-Amino acid oxidase (DAAO, EC 1.4.3.3) is a flavoenzyme that catalyzes the oxidation of d-amino acids including d-serine to the corresponding α-keto acids. In mammals, DAAO is present in kidneys, liver, and brain. Because the highest DAAO activity is found in the kidneys,4 a substantial amount of orally administered d-serine is metabolized in the kidneys, contributing to its rapid clearance. Indeed, pharmacokinetics studies of d-serine in mice lacking DAAO unmasked the predominant role of DAAO in the plasma clearance of d-serine.5 Furthermore, d-serine-induced nephrotoxicity is most likely due to the production of hydrogen peroxide as a byproduct of DAAO-mediated oxidation because it does not occur in mutant rats lacking DAAO activity.6 These findings collectively suggest that inhibition of DAAO would exert dual beneficial effects on d-serine therapy: (i) enhancement of d-serine bioavailability and (ii) attenuation of d-serine induced nephrotoxicity.
In the past decade, concurrent with the functional studies of DAAO,7 substantial strides have been made in identifying potent and selective DAAO inhibitors of broad structural diversity (Figure 1).8,9 Carboxylate-based DAAO inhibitors 1(10) and 2(11) presumably originated from a prototype DAAO inhibitor, benzoic acid, have evolved into a new class of compounds containing carboxylate bioisosteres such as 3(12) and 4.13 Crystal structure of 4 bound to DAAO revealed that the α-hydroxycarbonyl moiety of 4 retains the key interactions seen between the DAAO active site and carboxylate-based inhibitors.13 Subsequently, a variety of other heterocyclic frameworks bearing an α-hydroxycarbonyl moiety were explored in the pursuit of new scaffolds for potent DAAO inhibitors including 5(14) and 6.15 Structural insights gained from DAAO in complex with imino-DOPA16 led to the discovery of the latest DAAO inhibitors possessing an additional substituent that extends to the secondary binding site adjacent to the active site of DAAO. For example, our group exploited this secondary binding site using kojic acid derivatives such as compound 7 substituted at its 2-methyl group.17 Similarly, Hondo et al. reported potent DAAO inhibitors 8a–b based on a 4-hydroxypyridazin-3(2H)-one scaffold with a phenethyl group extending to the secondary binding site.18 The secondary binding site was also exploited by carboxylate-based DAAO inhibitors such as 9(19) and 10.20
Figure 1.

Representative inhibitors of DAAO.
Recently, the pharmacokinetics profile of several representative DAAO inhibitors has been systematically compared in mice and rats.21 Although most of the DAAO inhibitors were reported to show negligible distribution to the brain, the poor CNS permeability does not pose a major concern for our objective of increasing exposure to orally administered d-serine by inhibition of peripheral DAAO. To our surprise, however, carboxylate-based DAAO inhibitors exhibited superior oral bioavailability relative to those containing a carboxylate bioisostere. Subsequent investigation revealed that most of these carboxylate bioisosteres are subject to glucuronidation in liver microsomes, presumably contributing to their poor oral bioavailability.22
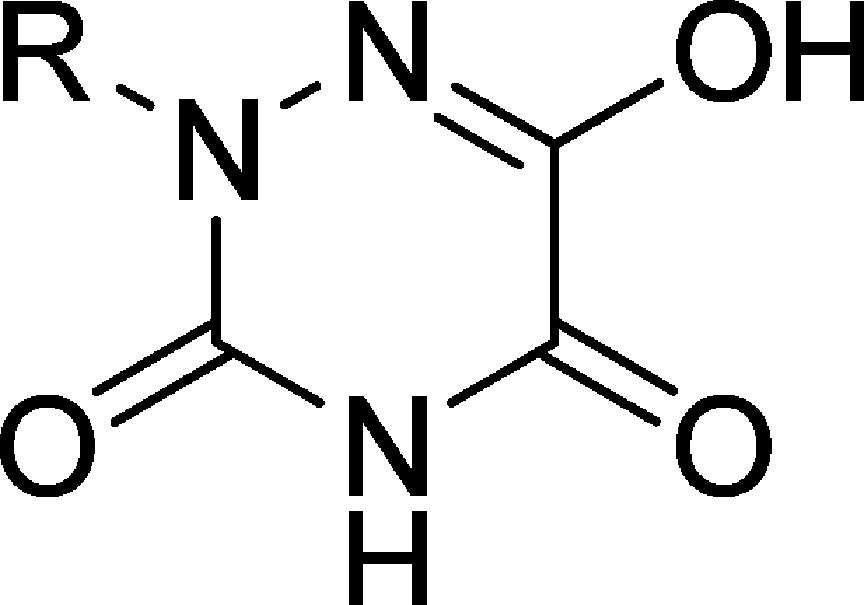
As a part of our continuous effort to identify new orally available DAAO inhibitors, we have searched for a pharmacophore providing not only potent DAAO inhibition but also considerable resistance to glucuronidation. During the course of screening a library of FDA-approved small molecule drugs, ceftriaxone (Figure 2) was identified as a moderate DAAO inhibitor with an IC50 value of 10 μM. Although ceftriaxone does not offer a particularly attractive molecular template for developing SAR studies, the 3-mercapto-2-methyl-1,2-dihydro-1,2,4-triazine-5,6-dione portion of ceftriaxone has caught our attention as its tautomer, 6-hydroxy-3-mercapto-2-methyl-1,2,4-triazin-5(2H)-one, has a structural feature common to other DAAO inhibitors. Interestingly, 6-hydroxy-2-methyl-1,2,4-triazine-3,5(2H,4H)-dione 11a, one of the degradation products of ceftriaxone in aqueous solution,23 inhibited DAAO with an IC50 value of 2.8 μM, prompting us to further examine the potential of 2-substituted 6-hydroxy-1,2,4-triazine-3,5(2H,4H)-dione derivatives as new DAAO inhibitors. Here we present a new class of DAAO inhibitors based on the 6-hydroxy-1,2,4-triazine-3,5(2H,4H)-dione scaffold. With appropriate substitutions at the 2-position, some 6-hydroxy-1,2,4-triazine-3,5(2H,4H)-dione derivatives exhibited good potency as well as improved metabolic stability. One of these derivatives was also tested for its oral bioavailability as well as effects on d-serine plasma levels following oral coadministration with d-serine in mice.
Figure 2.

Ceftriaxone and compound 11a.
Chemistry
The majority of 2-substituted 6-hydroxy-1,2,4-triazine-3,5(2H,4H)-dione derivatives were synthesized using 6-bromo-1,2,4-triazine-3,5(2H,4H)-dione 12(24) as a starting material (Scheme 1). Alkylation of the 2-position of 12 with alkyl halides mediated by bis(trimethylsilyl)acetamide (BSA)25 gave 2-substituted derivatives 13a–z. Subsequently, the 6-bromo group of 13a–z was replaced by the benzyloxy group by treating with benzyl alcohol in the presence of potassium carbonate to give 14a–z. The final products 11a–z were obtained by removing the benzyl group of 14a–z by either catalytic hydrogenation or boron tribromide. Anisole derivatives 11u–w are further converted into the corresponding phenolic derivatives 15u–w by boron tribromide.
Scheme 1. Synthesis of 11a–z and 15u–w.

Reagents and conditions: (a) RI or RBr, BSA, acetonitrile, 82 °C; (b) K2CO3, BnOH, 150 °C; (c) H2, Pd/C, MeOH, rt; (d) BBr3, dichloromethane, rt.
Some derivatives were synthesized using 6-bromo-4-[(phenylmethoxy)methyl]-1,2,4-triazine-3,5(2H,4H)-dione 16(26) as a starting material (Scheme 2). Mitsunobu reaction of 16 and alcohols with the aid of DIAD and PPh3 provided the 2-substituted derivatives 17a–f. Reaction with benzyl alcohol gave the benzyloxy protected intermediates 18a–f, which were subsequently converted into the desired products 19a–f by either catalytic hydrogenation or boron tribromide. Synthesis of ketone derivative 22 began with base-mediated coupling of 2-bromoacetophenone to 16. The product 20 was converted into 21 by treating with benzyl alcohol. The removal of benzyl and BOM groups from 21 by boron tribromide afforded 22. Synthetic intermediate 21 was also transformed to the hydroxyl derivative 23. Synthesis of 2-phenyl derivative 26 involves Chan–Lam coupling of 16 to phenylboronic acid to give 24. Subsequent benzyloxylation and deprotection by BBr3 provided 26.
Scheme 2. Synthesis of 19a–f, 22, 23, and 26.

Reagents and conditions: (a) ROH, Ph3P, DIAD, THF, 66 °C; (b) BnOH, NaH, DMF, 0 °C; (c) H2, Pd/C, MeOH, rt; (d) BBr3, dichloromethane, rt; (e) BrCH2COPh, NaH, DMF, rt; (f) (i) H2, [Rh(COD)Cl]2, Et3N, MeOH-EtOAc, rt, (ii) BBr3, dichloromethane, rt; (g) phenylboronic acid, pyridine, copper(II) acetate, dichloromethane, rt.
As shown in Scheme 3, we synthesized two structurally close analogues of 11e, where its carboxylate bioisostere moiety is slightly modified. 3-Thioxo derivative 30 was prepared using (2-phenylethyl)hydrazine 27 as a starting material. Reaction of 27 with ammonium thiocyanate provided hydrazinecarbothioamide 28,27 which was subsequently treated with methyl chlorooxoacetate to give 29.28 Cyclization of 29 in the presence of DBU afforded the desired product 30.28 Uracil derivative 32 was prepared by BSA-mediated alkylation of 5-hydroxypyrimidine-2,4(1H,3H)-dione 31 with phenethyl iodide.
Scheme 3. Synthesis of 30 and 32.

Reagents and conditions: (a) NH4SCN, EtOH, 78 °C; (b) methyl chlorooxoacetate, THF, rt; (c) DBU, THF, 50 °C; (d) PhCH2CH2I, BSA, acetonitrile, 82 °C.
Results and Discussion
The inhibitory potency of the synthesized compounds were determined using recombinant human DAAO as previously reported.12 In vitro DAAO inhibitory data are summarized in Tables 1 and 2. As shown in Table 1, 6-hydroxy-1,2,4-triazine-3,5(2H,4H)-dione derivatives containing a variety of substituents at the 2-position displayed varying degrees of inhibitory potency against DAAO. All compounds but 11c and 26 were found to be potent DAAO inhibitors, with IC50 values in the low-micromolar to nanomolar range. Within the compounds containing an aromatic ring, phenethyl derivative 11e and naphthalen-1-ylmethyl derivative 11h exhibited the most potent inhibitory activity, with IC50 values of 70 and 50 nM, respectively. Incorporation of a heterocyclic ring (compounds 19a–d) did not result in improved potency. We conducted inhibitory kinetic studies of compound 11h at various concentrations of d-serine to determine its mode of inhibition. As shown in Figure 3, a double reciprocal plot of DAAO activity versus the d-serine concentrations produced a pattern indicative of competitive inhibition with a Ki value of 60 nM. Furthermore, escalation of FAD concentration from 10 to 100 μM did not affect IC50 value for 11h, providing additional supporting evidence that compound 11h competes with d-serine for the substrate-binding site distinct from the FAD-binding site of DAAO.
Table 1. Inhibition of Human DAAO by 6-Hydroxy-1,2,4-triazine-3,5(2H,4H)-dione Derivatives.

Values are mean ± SD of at least four experiments.
Table 2. Inhibition of Human DAAO by Analogues of 11e.
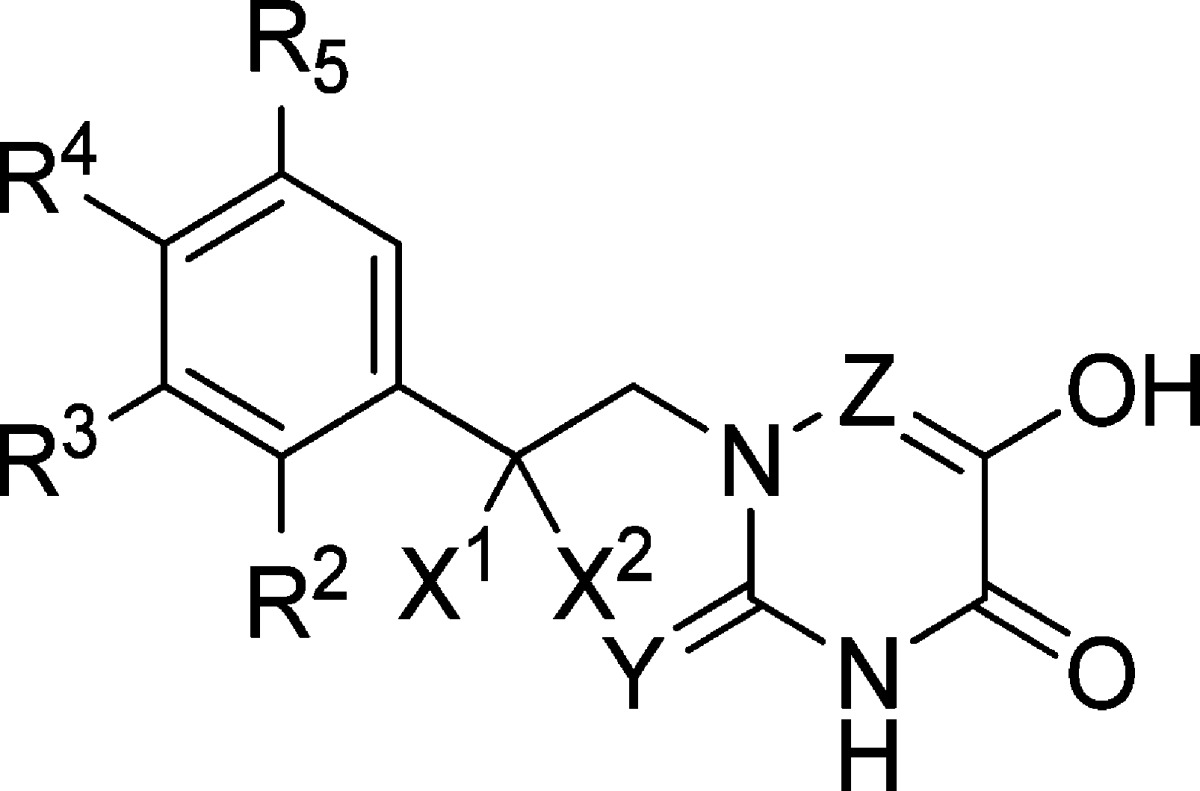
| compd | R2 | R3 | R4 | X1 | X2 | Y | Z | IC50 (μM)a |
|---|---|---|---|---|---|---|---|---|
| 11e | H | H | H | H | H | O | N | 0.07 ± 0.01 |
| 11j | Cl | H | H | H | H | O | N | 0.10 ± 0.05 |
| 11k | H | Cl | H | H | H | O | N | 0.06 ± 0.02 |
| 11l | H | H | Cl | H | H | O | N | 0.04 ± 0.01 |
| 11m | F | H | H | H | H | O | N | 0.08 ± 0.03 |
| 11n | H | F | H | H | H | O | N | 0.06 ± 0.02 |
| 11o | H | H | F | H | H | O | N | 0.05 ± 0.02 |
| 11p | CH3 | H | H | H | H | O | N | 0.08 ± 0.03 |
| 11q | H | CH3 | H | H | H | O | N | 0.07 ± 0.02 |
| 11r | H | H | CH3 | H | H | O | N | 0.09 ± 0.02 |
| 11s | H | CF3 | H | H | H | O | N | 0.10 ± 0.02 |
| 11t | H | H | CF3 | H | H | O | N | 0.08 ± 0.04 |
| 11u | OCH3 | H | H | H | H | O | N | 0.27 ± 0.07 |
| 11v | H | OCH3 | H | H | H | O | N | 0.12 ± 0.03 |
| 11w | H | H | OCH3 | H | H | O | N | 0.12 ± 0.03 |
| 15u | OH | H | H | H | H | O | N | 0.09 ± 0.04 |
| 15v | H | OH | H | H | H | O | N | 0.16 ± 0.05 |
| 15w | H | H | OH | H | H | O | N | 0.11 ± 0.03 |
| 11x | H | OPh | H | H | H | O | N | 0.24 ± 0.05 |
| 11y | H | H | OPh | H | H | O | N | 0.08 ± 0.02 |
| 11z | H | H | Ph | H | H | O | N | 0.05 ± 0.02 |
| 19e | H | H | H | CH3 | H | O | N | 3.3 ± 0.6 |
| 19f | H | H | H | F | F | O | N | 0.40 |
| 22 | H | H | H | O | O | O | N | 0.85 ± 0.13 |
| 23 | H | H | H | OH | H | O | N | 0.47 ± 0.23 |
| 30 | H | H | H | H | H | S | N | 0.05 ± 0.01 |
| 32 | H | H | H | H | H | O | CH | 0.08 ± 0.01 |
Values are mean ± SD of at least four experiments except for compound 19f (n = 2).
Figure 3.

Double-reciprocal plot of the oxidation of d-serine by human DAAO in the presence of compound 11h. The straight lines represent the least-squares fit of the data obtained by plotting the reciprocal of the initial rate of change in the absorbance at 411 nm versus the reciprocal of the d-serine concentrations (mM).
The potent inhibitory activity of 11e prompted us to conduct more focused SAR studies using 11e as a molecular template (Table 2). A variety of substitutions were examined at the terminal phenyl group of 11e (compounds 11j–z and 15u–w). Overall, there was no significant change in inhibitory potency, with IC50 values ranging from 40 to 270 nM. Analogues containing a large substituent such as compounds 11x–z retained IC50 values in the nanomolar range. Para substitutions were particularly well tolerated, as demonstrated by phenoxy substituted derivative 11y and phenyl substituted 11z with IC50 values of 80 and 50 nM, respectively. These substituents are presumably oriented toward the entrance of the DAAO active site, possessing a high degree of steric tolerance. In contrast, a notable decrease in potency was seen in compound 19e containing a branched methyl group at the linker connecting the phenyl and the 6-hydroxy-1,2,4-triazine-3,5(2H,4H)-dione moieties. Other modifications at the linker (compounds 19f, 22, and 23) were not as disruptive as the methyl group of 19e although these compounds still showed somewhat lower potency compared to 11e. Compounds 30 and 32 possessing slightly modified carboxylate bioisosteres retained potent DAAO inhibitory activity with IC50 values 50 and 80 nM, respectively, nearly identical to that of 11e.
As illustrated in Figure 4, proposed binding mode of 11e (white) to the active site of DAAO is superposed onto the crystal structure of DAAO in complex with 8a (cyan).18 In this model, the α-hydroxycarbonyl moiety forms key hydrogen bonds with Tyr228 and Arg283. It is conceivable that the 4-position nitrogen group of 11e forms a hydrogen bond with the backbone carbonyl of Gly313 in a similar manner as the 2-nitrogen atom of 4-hydroxypyridazin-3(2H)-one moiety in 8a. This explains the relatively high inhibitory potency of DAAO inhibitors with a 6-hydroxy-1,2,4-triazine-3,5(2H,4H)-dione scaffold compared to the kojic acid based compounds17 represented by 7, which lacks a nitrogen atom in the core ring. In contrast, the nitrogen atom at the 1-position of 11e does not seem to be involved in the interaction with the active site of DAAO. This is consistent with the similar inhibitory potency of the uracil derivative 32, in which the corresponding position is replaced by a carbon atom. 3-Thioxo derivative 30 was also as potent as 11e, presumably due to the preservation of all of the key interactions involved in binding of 11e to DAAO. The secondary binding pocket occupied by the 2-phenylethyl group of 11e appears quite spacious and capable of accommodating larger substituents owing to the movement of Tyr224 away from the active site. This is in a good agreement with the high inhibitory potency retained by compounds with bulky substituents such as compounds 11x–z.
Figure 4.
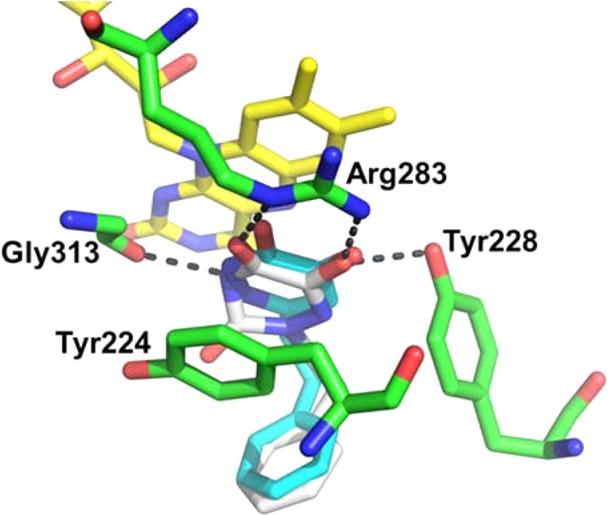
Proposed binding mode of 11e (white) to the active site of DAAO (3W4K). Key residues and FAD are shown in green and yellow, respectively. Hydrogen-bonding interactions between 11h and the key residues are shown as gray dashed lines. Compound 8a (cyan) of 3W4K is also shown for comparison.
Table 3 summarizes in vitro metabolic stability of selected compounds in mouse liver microsomes. As mentioned earlier, many of the DAAO inhibitors containing carboxylate bioisosteres are known to be subject to a varying degree of glucuronidation in liver microsomes.22 For example, a kojic acid based DAAO inhibitor 7 is extensively glucuronidated in mouse liver microsomes in the presence of UDPGA, while compound 8b with the 4-hydroxypyridazin-3(2H)-one scaffold shows some resistance to glucuronidation. 6-Hydroxy-1,2,4-triazine-3,5(2H,4H)-dione derivatives 11e and 11h were found to be completely resistant to glucuronidation in mouse liver microsomes. 3-Thioxo derivative 30 was also metabolically stable against glucuronidation. In sharp contrast, uracil-based DAAO inhibitor 32 was fully glucuronidated within 60 min of incubation. This trend appears consistent with our previous findings from other DAAO inhibitors, showing that increased topological polar surface area (tPSA) of the carboxylate bioisostere moiety contributes to higher resistance to glucuronidation.22 Compounds 11e and 11h were also stable in mouse liver microsomes in the presence of NADPH, a cofactor for CYP-dependent oxidation.
Table 3. Metabolic Stability of DAAO Inhibitors in Mouse Liver Microsomes.
| % remaining
after 60 min incubation |
|||
|---|---|---|---|
| compd | tPSA (Å2)a | w/UDPGA | w/NADPH |
| 7 | 47 | 5 | NDb |
| 8b | 62 | 59 | 92 |
| 11e | 82 | ≥99 | ≥99 |
| 11h | 82 | ≥99 | ≥99 |
| 30 | 97 | ≥99 | 87 |
| 32 | 70 | ≤1 | NDb |
Topological surface area of the carboxylate bioisostere moiety.
Not determined.
Further pharmacological characterization was conducted with compound 11h. No evidence of mutagenicity was observed in the Ames test using Salmonella typhimurium strain (TA100) in the presence or absence of metabolic activation by S9 mixture. Compound 11h was also tested in a panel of relevant in vitro assays (Table 4) including hERG channels, various sites of the NMDA receptors, and another class of flavoenzymes, monoamine oxidases A and B (MAO-A and MAO-B). Compound 11h showed no significant activity in any of these assays at the highest tested concentrations.
Table 4. In Vitro Pharmacological Characterization of 11h.
| assay | results |
|---|---|
| hERG channel | no inhibition at concentrations up to 25 μM |
| NMDA receptor (agonist) | ≤5% inhibition at 10 μM |
| NMDA receptor (glycine) | 22% inhibition at 10 μM |
| NMDA receptor (phencyclidine) | ≤5% inhibition at 10 μM |
| NMDA receptor (MK801) | ≤5% inhibition at 10 μM |
| glycine receptor (strychnine sensitive) | ≤5% inhibition at 10 μM |
| MAO-A | ≤5% inhibition at 10 μM |
| MAO-B | ≤5% inhibition at 10 μM |
In vivo pharmacokinetic studies of 11h were carried out in CD1 mice by intravenous and oral administration (30 mg/kg). The plasma pharmacokinetic parameters are summarized in Table 5, along with those for 8b for direct comparison. The plasma clearance of 11h in mice was 10.2 mL/min/kg with a terminal half-life of 0.9 h following intravenous administration. Oral absorption of 11h was rapid, and the plasma peak concentration was generally observed within the first hour of oral administration. As predicted from the metabolic stability data (Table 3), the oral bioavailability of 11h (F = 79%) is significantly higher than that of 8b (F = 31%) despite the increased polarity. The improvement in oral bioavailability of 11h over 8b clearly illustrates the significant impact of glucuronidation on the pharmacokinetics of DAAO inhibitors. Following oral administration, compound 11h showed negligible brain penetration with a brain to plasma ratio of 0.01 in mice. Thus, compound 11h is unlikely to increase the brain levels of endogenous d-serine by inhibiting DAAO expressed in the brain. Given the favorable plasma exposure, however, compound 11h should be capable of inhibiting peripheral DAAO and minimize the metabolism of orally taken d-serine.
Table 5. Mouse Pharmacokinetics of 8b and 11h (30 mg/kg).
| route | parameter | 8ba | 11h |
|---|---|---|---|
| iv | CL (mL/min/kg) | 38.7 | 10.2 |
| Vd (L/kg) | 0.5 | 0.8 | |
| T1/2 (h) | 0.2 | 0.9 | |
| AUClast (μg·h/mL) | 12.7b | 16.3 | |
| po | Tmax (h) | 0.25 | 0.08 |
| Cmax (μg/mL) | 6.6 | 13.8 | |
| AUClast (μg·h/mL) | 3.9 | 12.9 | |
| brain to plasma ratio | NDc | 0.01 | |
| po/iv | F (%) | 31 | 79 |
Intravenous administration of 8b was conducted at 10 mg/kg.
The value is dose-normalized to 30 mg/kg.
Not determined.
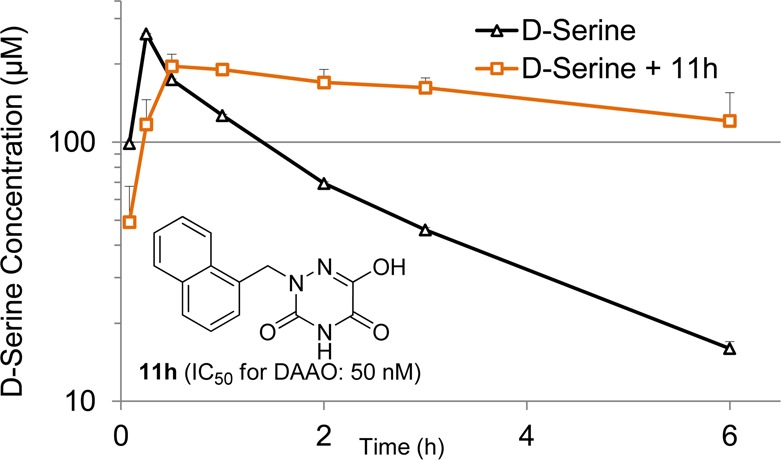
To determine the effects of 11h on d-serine plasma levels, mice (n = 3 per time point) were dosed with 11h (30 mg/kg, po) along with d-serine (30 mg/kg, po) simultaneously. As shown in Figure 5, compound 11h showed no ability to enhance plasma Cmax of d-serine. However, its plasma clearance was substantially reduced for a sustained period of time, resulting in the significant increase in d-serine AUC0–6h (96.9 μg*h/mL) compared to that of d-serine alone treatment (43.3 μg·h/mL). The curve of d-serine concentration versus time in mice coadministered with 11h is nearly identical to that of DAAO knockout mice treated with oral d-serine.5 The lack of enhancement in d-serine plasma Cmax in mice by coadministration with 11h is in a good agreement with the negligible levels of DAAO found in mouse liver as opposed to kidneys,29 where DAAO is abundant and most likely contributes to the plasma clearance of d-serine. Thus, the primary site of action for 11h in mice appears DAAO in kidneys, which explains its significant effects on plasma clearance of d-serine but not on plasma Cmax value. Given the previously reported cross-species variation in IC50 values between human and rat forms of DAAO,13 caution needs to be taken in establishing the PK/PD relationship in mice as we have not measured inhibitory potency of 11h in mouse DAAO. It is worth noting, however, that 4-hydroxypyridazin-3(2H)-one derivatives represented by 8b showed little difference in inhibitory potency between human and mouse DAAO.18 Given their structural similarity to the 6-hydroxy-1,2,4-triazine-3,5(2H,4H)-dione derivatives, we anticipate comparative inhibitory potencies of 11h against human and mouse forms of DAAO.
Figure 5.

Effects of compound 11h (30 mg/kg, po) on plasma pharmacokinetics of oral d-serine (30 mg/kg) in mice.
Conclusions
In the past decade, substantial efforts have been made by multiple groups to develop DAAO inhibitors of potential therapeutic value. These efforts have not only provided structurally diverse DAAO inhibitors but also contributed to the cumulative knowledge in this area, including the use of carboxylate bioisosteres, exploitation of the secondary binding pocket, and the strategy to minimize glucuronidation. We took full advantage of the prior knowledge in guiding our efforts to develop a new class of DAAO inhibitors based on the 6-hydroxy-1,2,4-triazine-3,5(2H,4H)-dione pharmacophore. The inhibitory potency of these compounds was dependent on the variation of the substituents at the 2-position. Many compounds containing an arylalkyl group at this position showed potent inhibitory activity against DAAO. Among these inhibitors, compound 11h exhibited good metabolic stability in liver microsomes and selectivity over other relevant targets. Compound 11h was also found to be orally available and enhance plasma levels of coadministered d-serine by reducing its clearance. The ability of compound 11h to sustain high plasma levels of d-serine should be particularly attractive for the treatment of chronic psychiatric disorders such as schizophrenia.
Experimental Section
General
All solvents were reagent grade or HPLC grade. Unless otherwise noted, all materials were obtained from commercial suppliers and used without further purification. All reactions were performed under nitrogen. Melting points were obtained on a Mel-Temp apparatus and are uncorrected. 1H NMR spectra were recorded at 400 MHz. 13C NMR spectra were recorded at 100 MHz. The HPLC solvent system consisted of distilled water and acetonitrile, both containing 0.1% formic acid. Preparative HPLC purification was performed on an Agilent 1200 series HPLC system equipped with an Agilent G1315D DAD detector using a Phenomenex Luna 5 μm C18 (2) column (21.2 mm × 250 mm, 5 μm). Analytical HPLC was performed on an Agilent 1200 series HPLC system equipped with an Agilent G1315D DAD detector (detection at 220 nm) and an Agilent 6120 quadrupole MS detector. Unless otherwise specified, the analytical HPLC conditions involve a gradient of 20% acetonitrile/80% water for 0.5 min followed by an increase to 85% acetonitrile/15% water over 4 min and continuation of 85% acetonitrile/15% water for 3.5 min with a Luna C18 column (2.1 mm × 50 mm, 3.5 μm) at a flow rate of 0.75 mL/min. All final compounds tested were confirmed to be of ≥95% purity by the HPLC methods described above. The model of compound 11h bound to human DAAO (Figure 4) was generated using AutoDock Vina.30
6-Bromo-2-methyl-1,2,4-triazine-3,5(2H,4H)-dione (13a)
To a solution of 5-bromo-6-azauracil 12 (0.30 g, 1.6 mmol) in acetonitrile (5 mL) was added N,O-bis(trimethylsilyl)acetamide (4.0 mmol, 1.0 mL). The mixture was heated at 82 °C for 3 h, after which methyl iodide (0.12 mL, 1.9 mmol) was added. After 24 h, more methyl iodide (0.5 equiv) was added and the mixture was stirred for additional 24 h. The reaction mixture was concentrated, and the resulting residue was dissolved in dichloromethane, washed with water and brine, dried over Na2SO4, and concentrated to give 0.11 g of 13a as a black solid (33% crude yield). This material was used in the next step without further purification. 1H NMR (DMSO-d6) δ 3.44 (s, 3H), 12.50 (s, 1H).
6-(Benzyloxy)-2-methyl-1,2,4-triazine-3,5(2H,4H)-dione (14a)
A mixture of 13a (0.10 g, 0.49 mmol) and K2CO3 (0.14 g, 1.0 mmol) in benzyl alcohol (1.0 mL) was heated overnight at 150 °C. The reaction mixture was partitioned between aqueous 10% KHSO4 and EtOAc. The organic layer was dried over Na2SO4 and concentrated. The residual material was purified using a Biotage Isolera One flash purification system with a silica gel flash cartridge (EtOAc/hexanes) to give 0.070 g of 14a as a white solid (61% yield). 1H NMR (DMSO-d6) δ 3.36 (s, 3H), 5.12 (s, 2H), 7.37–7.46 (m, 5H), 12.16 (s, 1H).
6-Hydroxy-2-methyl-1,2,4-triazine-3,5(2H,4H)-dione (11a)
To a solution of 14a (0.060 g, 0.26 mmol) in methanol (5 mL) was added one spatula tip of 10% Pd/C. The mixture was shaken under hydrogen (30 psi) for 1 h. The catalyst was removed by filtration, and the filtrate was concentrated to give 0.035 g of 11a as a beige powder (94% yield); mp 259 °C. 1H NMR (DMSO-d6) δ 3.27 (s, 3H), 11.11 (s, 1H), 12.03 (s, 1H). LCMS (5% acetonitrile/95% water for 0.5 min followed by an increase to 40% acetonitrile/60% water over 4 min and continuation of 40% acetonitrile/60% water for 3.5 min): retention time 5.05 min, m/z 144 [M + H]+.
6-Bromo-2-isopentyl-1,2,4-triazine-3,5(2H,4H)-dione (13b)
Compound 13b was prepared as described for the preparation of 13a, except 1-iodo-3-methylbutane (1.6 equiv) was used in place of methyl iodide. The crude product was purified by trituration with cold diethyl ether; yellow solid (27% yield). 1H NMR (DMSO-d6) δ 0.89 ((d, J = 6.3 Hz, 6H), 1.50 (m, 2H), 1.60 (m, 1H), 3.82 (t, J = 7.3 Hz, 2H), 12.48 (s, 1H).
6-(Benzyloxy)-2-isopentyl-1,2,4-triazine-3,5(2H,4H)-dione (14b)
Compound 14b was prepared from 13b as described for the preparation of 14a; clear oil (36% yield). 1H NMR (DMSO-d6) δ 0.87 (d, J = 6.3 Hz, 6H), 1.49 (m, 2H), 1.54 (m, 1H), 3.73 (t, J = 7.1 Hz, 2H), 5.14 (s, 2H), 7.36–7.45 (m, 5H), 12.14 (s, 1H).
6-Hydroxy-2-isopentyl-1,2,4-triazine-3,5(2H,4H)-dione (11b)
Compound 11b was prepared from 14b as described for the preparation of 11a; gray solid (quantitative yield); mp 171 °C. 1H NMR (DMSO-d6) δ 0.88 (d, J = 6.3 Hz, 6H), 1.47 (m, 2H), 1.54 (m, 1H), 3.66 (t, J = 7.3 Hz, 2H), 11.65 (s, 1H), 11.94 (s, 1H). LCMS: retention time 1.04 min, m/z 200 [M + H]+.
6-Bromo-2-(3,3-dimethylbutyl)-1,2,4-triazine-3,5(2H,4H)-dione (13c)
Compound 13c was prepared as described for the preparation of 13a, except 1-iodo-3,3-dimethylbutane (3.3 equiv) was used in place of methyl iodide; clear oil (19% yield). 1H NMR (DMSO-d6) δ 0.92 (s, 9H), 1.53 (m, 2H), 3.82 (m, 2H), 12.50 (s, 1H).
6-(Benzyloxy)-2-(3,3-dimethylbutyl)-1,2,4-triazine-3,5(2H,4H)-dione (14c)
Compound 14c was prepared from 13c as described for the preparation of 14a; clear oil (80% yield). 1H NMR (DMSO-d6) δ 0.90 (s, 9H), 1.48 (m, 2H), 3.72 (m, 2H), 5.15 (s, 2H), 7.35–7.45 (m, 5H), 12.15 (s, 1H).
2-(3,3-Dimethylbutyl)-6-hydroxy-1,2,4-triazine-3,5(2H,4H)-dione (11c)
Compound 11c was prepared from 14c as described for the preparation of 11a, except the material was purified by trituration with EtOAc/hexanes; white solid (77% yield); mp 219 °C. 1H NMR (DMSO-d6) δ 0.91 (m, 9H), 1.50 (m, 2H), 3.67 (m, 2H), 11.64 (s, 1H), 12.02 (s, 1H). LCMS: retention time 2.45 min, m/z 214 [M + H]+.
2-Benzyl-6-bromo-1,2,4-triazine-3,5(2H,4H)-dione (13d)
Compound 13d was prepared as described for the preparation of 13a, except benzyl bromide (1.2 equiv) was used in place of methyl iodide. The crude product was purified by trituration with cold diethyl ether; light-tan solid (71% yield). 1H NMR (DMSO-d6) δ 5.02 (s, 2H), 7.29–7.38 (m, 5H), 12.59 (s, 1H).
2-Benzyl-6-(benzyloxy)-1,2,4-triazine-3,5(2H,4H)-dione (14d)
Compound 14d was prepared from 13d as described for the preparation of 14a, except the product was purified by trituration with EtOAc/hexanes; white solid (70% yield). 1H NMR (DMSO-d6): δ 4.91 (s, 2H), 5.11 (s, 2H), 7.29–7.40 (m, 10H), 12.24 (s, 1H).
2-Benzyl-6-hydroxy-1,2,4-triazine-3,5(2H,4H)-dione (11d)
Compound 11d was prepared from 14d as described for the preparation of 11a, except the hydrogenation was performed overnight under atmospheric pressure of hydrogen; light-pink powder (81% yield); mp 242 °C. 1H NMR (DMSO-d6) δ 4.85 (s, 2H), 7.26–7.36 (m, 5H), 11.72 (bs, 1H), 11.94 (bs, 2H). LCMS (5% acetonitrile/95% water for 0.5 min followed by an increase to 40% acetonitrile/60% water over 4 min and continuation of 40% acetonitrile/60% water for 3.5 min): retention time 2.84 min, m/z 220 [M + H]+.
6-Bromo-2-phenethyl-1,2,4-triazine-3,5(2H,4H)-dione (13e)
Compound 13e was prepared as described for the preparation of 13a, except phenethyl iodide (2.5 equiv) was used in place of methyl iodide. The crude product was purified by trituration with cold diethyl ether; tan solid (64% yield). 1H NMR (DMSO-d6): δ 2.94 (t, J = 7.5 Hz, 2H), 4.04 (t, J = 7.6 Hz, 2H), 7.22 (m, 3H), 7.28 (m, 2H), 12.53 (s, 1H).
6-(Benzyloxy)-2-phenethyl-1,2,4-triazine-3,5(2H,4H)-dione (14e)
Compound 14e was prepared from 13e as described for the preparation of 14a, except the product was purified by trituration with EtOAc/hexanes; beige solid (74% yield). 1H NMR (DMSO-d6): δ 2.91 (t, J = 7.2 Hz, 2H), 5.05 (s, 2H), 3.96 (t, J = 7.2 Hz, 2H), 7.15 (m, 2H), 7.20 (m, 1H), 7.26–7.31 (m, 2H), 7.36–7.43 (m, 5H), 12.13 (s, 1H).
6-Hydroxy-2-phenethyl-1,2,4-triazine-3,5(2H,4H)-dione (11e)
Compound 11e was prepared from 14e as described for the preparation of 11a, except a mixture of methanol and acetic acid (3:1) was used as a solvent and that the hydrogenation was performed at 50 psi for 1.5 h; light-pink powder (60% yield); mp 200 °C. 1H NMR (DMSO-d6) δ 2.90 (t, J = 7.8 Hz, 2H), 3.87 (t, J = 7.6 Hz, 2H), 7.20 (m, 3H), 7.29 (m, 2H), 11.71 (s, 1H), 12.03 (s, 1H). LCMS: retention time 1.50 min, m/z 234 [M + H]+.
6-Bromo-2-(3-phenylpropyl)-1,2,4-triazine-3,5(2H,4H)-dione (13f)
Compound 13f was prepared as described for the preparation of 13a, except (3-iodopropyl)benzene (1.6 equiv) was used in place of methyl iodide; white solid (43% yield). 1H NMR (DMSO-d6): δ 1.94 (m, 2H), 2.63 (t, J = 7.7 Hz, 2H), 3.83 (t, J = 6.9 Hz, 2H), 7.15–7.22 (m, 3H), 7.25–7.29 (m, 2H), 12.45 (s, 1H).
6-(Benzyloxy)-2-(3-phenylpropyl)-1,2,4-triazine-3,5(2H,4H)-dione (14f)
Compound 14f was prepared from 13f as described for the preparation of 14a; clear oil (85% yield oil). 1H NMR (DMSO-d6): δ 1.93 (m, 2H), 2.60 (t, J = 7.7 Hz, 2H), 3.76 (t, J = 6.8 Hz, 2H), 5.13 (s, 2H), 7.18 (m, 3H), 7.24 (m, 2H), 7.35–7.46 (m, 5H), 12.09 (s, 1H).
6-Hydroxy-2-(3-phenylpropyl)-1,2,4-triazine-3,5(2H,4H)-dione (11f)
Compound 11f was prepared from 14f as described for the preparation of 11a, except a mixture of methanol and acetic acid (3:1) was used as a solvent; light-pink solid (85% yield); mp 153 °C. 1H NMR (DMSO-d6) δ 1.89 (m, 2H), 2.60 (t, J = 7.6 Hz, 2H), 3.67 (t, J = 6.9 Hz, 2H), 7.20 (m, 3H), 7.25 (m, 2H), 11.65 (bs, 1H), 11.93 (bs, 1H). LCMS: retention time 1.87 min, m/z 248 [M + H]+.
6-Bromo-2-(naphthalen-2-ylmethyl)-1,2,4-triazine-3,5(2H,4H)-dione (13g)
Compound 13g was prepared as described for the preparation of 13a, except 2-(bromomethyl)naphthalene (1.2 equiv) was used in place of methyl iodide. The reaction was heated overnight. The crude product was purified by trituration with cold diethyl ether; tan solid (56% yield). 1H NMR (DMSO-d6) δ 5.20 (s, 2H), 7.46 (dd, J = 1.5, 8.3 Hz, 1H), 7.51 (m, 2H), 7.22 (m, 1H), 7.86–7.92 (m, 4H), 12.62 (s, 1H).
6-(Benzyloxy)-2-(naphthalen-2-ylmethyl)-1,2,4-triazine-3,5(2H,4H)-dione (14g)
Compound 14g was prepared from 13g as described for the preparation of 14a; white solid foam (76% yield). 1H NMR (DMSO-d6) δ 5.09 (s, 2H), 5.12 (s, 2H), 7.26–7.31 (m, 3H), 7.36 (m, 2H), 7.45 (dd, J = 1.8, 7.6 Hz, 1H), 7.50–7.53 (m, 2H), 7.85–7.92 (m, 4H), 12.27 (s, 1H).
6-Hydroxy-2-(naphthalen-2-ylmethyl)-1,2,4-triazine-3,5(2H,4H)-dione (11g)
To a solution of compound 14g (0.26 g, 0.72 mmol) in dichloromethane (7.0 mL) was slowly added a 1.0 M solution of BBr3 (1.4 mL, 1.4 mmol) at room temperature. The reaction was stirred for 1 h, after which 2 additional equiv of BBr3 were added. Stirring continued at rt for an additional hour, and the reaction was quenched by the addition of water. The compound was extracted with EtOAc. The organic layer was washed with brine, dried over Na2SO4, and concentrated. The residual material was purified by preparative HPLC (20% acetonitrile/80% water for 5 min followed by an increase to 70% acetonitrile/30% water over 35 min and an increase to 100% acetonitrile over 10 min; flow rate 25 mL/min) to give 0.057 g of 11g as a white fluffy solid (29% yield); mp 264 °C. 1H NMR (DMSO-d6) δ 5.03 (s, 2H), 7.44 (dd, J = 1.3, 8.3 Hz, 1H), 7.49 (m, 2H), 7.80 (s, 1H), 7.88 (m, 3H), 12.07 (bs, 2H). LCMS (20% acetonitrile/80% water for 0.25 min followed by an increase to 85% acetonitrile/15% water over 1.5 min and continuation of 85% acetonitrile/15% water for 2.25 min; flow rate 1.25 mL/min): retention time 1.45 min, m/z 270 [M + H]+.
6-Bromo-2-(naphthalen-1-ylmethyl)-1,2,4-triazine-3,5(2H,4H)-dione (13h)
Compound 13h was prepared as described for the preparation of 13a, except 1-(bromomethyl)-naphthalene (1.2 equiv) was used in place of methyl iodide. The crude product was purified by trituration with cold diethyl ether; orange solid (65% yield). 1H NMR (DMSO-d6) δ 5.49 (s, 2H), 7.49 (m, 2H), 7.59 (m, 2H), 7.91 (m, 1H), 7.98 (m, 1H), 8.14 (d, J = 8.1 Hz, 1H), 12.64 (br s, 1H).
6-(Benzyloxy)-2-(naphthalen-1-ylmethyl)-1,2,4-triazine-3,5(2H,4H)-dione (14h)
Compound 14h was prepared from 13h as described for the preparation of 14a, except the product was purified by silica gel column chromatography (30% EtOAc in hexanes); white solid (40% yield). 1H NMR (DMSO-d6) δ 5.04 (s, 2H), 5.39 (s, 2H), 7.31 (s, 5H), 7.49 (d, J = 4.8 Hz, 2H), 7.57 (m, 2H), 7.91 (t, J = 4.8 Hz, 1H), 7.98 (m, 1H), 8.22 (d, J = 7.6 Hz, 1H), 12.29 (s, 1H).
6-Hydroxy-2-(naphthalen-1-ylmethyl)-1,2,4-triazine-3,5(2H,4H)-dione (11h)
Compound 11h was prepared from 14h as described for the preparation of 11a, with the exception that a mixture of methanol/ethyl acetate/acetic acid (1:1:0.1) was used as a solvent. The hydrogenation was performed at 20 psi for 3 h; white powder (90% yield); mp 260 °C (dec). 1H NMR (DMSO-d6) δ 5.33 (s, 2H), 7.41 (d, J = 7.1 Hz, 1H), 7.48 (t, J = 7.6 Hz, 1H), 7.57 (m, 2H), 7.88 (d, J = 8.1 Hz, 1H), 7.96 (m, 1H), 8.16 (d, J = 7.5 Hz, 1H), 11.68 (s, 1H), 12.20 (s, 1H). LCMS (20% acetonitrile/80% water for 0.25 min followed by an increase to 85% acetonitrile/15% water over 1.5 min and continuation of 85% acetonitrile/15% water for 2.25 min; flow rate 1.25 mL/min): retention time 1.43 min, m/z 270 [M + H]+. The corresponding sodium salt was used for all in vivo studies involving compound 11h. The salt was prepared by dissolving 11h in 1.0 equiv of a 0.103 N volumetric solution of NaOH, followed by the lyophilization.
6-Bromo-2-(2-(naphthalen-1-yl)ethyl)-1,2,4-triazine-3,5(2H,4H)-dione (13i)
Compound 13i was prepared as described for the preparation of 13a, except 1-(2-iodoethyl)naphthalene (2.0 equiv) was used in place of methyl iodide. The crude product was purified by trituration with cold diethyl ether; tan solid. (46% yield). 1H NMR (DMSO-d6) δ 3.41 (t, J = 7.5 Hz, 2H), 4.13 (t, J = 7.6 Hz, 2H), 7.43 (m, 2H), 7.55 (m, 2H), 7.81 (d, J = 8.3 Hz, 1H), 7.93 (d, J = 7.8 Hz, 1H), 8.13 (d, J = 8.3 Hz, 1H), 12.56 (s, 1H).
6-(Benzyloxy)-2-(2-(naphthalen-1-yl)ethyl)-1,2,4-triazine-3,5(2H,4H)-dione (14i)
Compound 14i was prepared from 13i as described for the preparation of 14a, except the product was purified by trituration with EtOAc/hexanes; yellow solid (56% yield). 1H NMR (DMSO-d6) δ 3.38 (t, J = 6.8 Hz, 2H), 4.06 (t, J = 7.1 Hz, 2H), 4.91 (s, 2H), 7.30 (m, 1H), 7.37–7.44 (m, 6H), 7.54 (m, 2H), 7.80 (d, J = 8.1 Hz, 1H), 7.92 (d, J = 6.6 Hz, 1H), 8.09 (d, J = 7.8 Hz, 1H) 12.15 (s, 1H).
6-Hydroxy-2-(2-(naphthalen-1-yl)ethyl)-1,2,4-triazine-3,5(2H,4H)-dione (11i)
Compound 11i was prepared from 14i as described for the preparation of 11a, except a mixture of methanol and acetic acid (2:1) was used as a solvent; light-pink powder (90% yield); mp 300 °C (dec). 1H NMR (DMSO-d6) δ 3.37 (t, J = 8.1 Hz, 2H), 3.93 (bs, 2H), 7.38 (bs, 1H), 7.44 (t, J = 7.5 Hz, 1H), 7.53 (t, J = 7.8 Hz, 1H), 7.59 (t, J = 7.2 Hz, 1H), 7.80 (d, J = 8.1 Hz, 1H), 7.92 (d, J = 7.8 Hz, 1H), 8.20 (bs, 1H), 11.95 (s, 1H), 12.28 (d, 1H). LCMS: retention time 2.99 min, m/z 284 [M + H]+.
6-Bromo-2-(2-chlorophenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (13j)
Compound 13j was prepared as described for the preparation of 13a, except 3-chloro-4-(2-iodoethyl)benzene (2.0 equiv) was used in place of methyl iodide. The crude product was purified by trituration with cold diethyl ether; yellow solid (28% yield). 1H NMR (DMSO-d6) δ 3.07 (t, J = 7.1 Hz, 2H), 4.07 (t, J = 7.1 Hz, 2H), 7.26–7.29 (m, 2H), 7.34 (m, 1H), 7.41 (m, 1H), 12.54 (s, 1H).
6-(Benzyloxy)-2-(2-chlorophenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (14j)
Compound 14j was prepared from 13j as described for the preparation of 14a; white solid (85% yield). 1H NMR (DMSO-d6) δ 3.05 (t, J = 6.7 Hz, 2H), 4.00 (t, J = 6.7 Hz, 2H), 4.92 (s, 2H), 7.22–7.27 (m, 4H), 7.34–7.43 (m, 5H), 12.15 (s, 1H).
2-(2-Chlorophenethyl)-6-hydroxy-1,2,4-triazine-3,5(2H,4H)-dione (11j)
Compound 11j was prepared from 14j as described for the preparation of 11g, with the exception that 2 equiv of BBr3 were used and that the crude product was purified by trituration with EtOAc/hexanes; white powder (39% yield); mp 211 °C. 1H NMR (DMSO-d6) δ 3.04 (t, J = 7.1 Hz, 2H), 3.91 (t, J = 7.1 Hz, 2H), 7.24–7.29 (m, 3H), 7.41 (m, 1H), 11.66 (s, 1H), 12.04 (s, 1H). LCMS: retention time 2.90 min, m/z 268 [M + H]+.
6-Bromo-2-(3-chlorophenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (13k)
Compound 13k was prepared as described for the preparation of 13a, except 3-chloro-4-(2-iodoethyl)benzene (2.0 equiv) was used in place of methyl iodide. The crude product was purified by trituration with cold diethyl ether; tan solid (73% yield). 1H NMR (DMSO-d6) δ 2.95 (t, J = 7.2 Hz, 2H), 4.06 (t, J = 7.3 Hz, 2H), 7.18 (m, 1H), 7.27–7.34 (m, 3H), 12.53 (s, 1H).
6-(Benzyloxy)-2-(3-chlorophenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (14k)
Compound 14k was prepared from 13k as described for the preparation of 14a, except the product was purified by trituration with EtOAc/hexanes; beige powder (70% yield). 1H NMR (DMSO-d6) δ 2.93 (t, J = 6.9 Hz, 2H), 3.98 (t, J = 6.9 Hz, 2H), 5.05 (s, 2H), 7.09 (m, 1H), 7.25–7.31 (m, 3H), 7.36–7.43 (m, 5H), 12.13 (s, 1H).
2-(3-Chlorophenethyl)-6-hydroxy-1,2,4-triazine-3,5(2H,4H)-dione (11k)
Compound 11k was prepared from 14k as described for the preparation of 11g, with the exception that 2 equiv of BBr3 were added at rt and that the reaction was stirred at rt for 1.5 h. The compound was purified by trituration with EtOAc/hexanes; beige powder (54% yield); mp 219 °C. 1H NMR (DMSO-d6) δ 2.92 (t, J = 7.1 Hz, 2H), 3.90 (t, J = 7.2 Hz, 2H), 7.14 (m, 1H), 7.26–7.33 (m, 3H), 11.69 (s, 1H), 12.04 (s, 1H). LCMS: retention time 2.91 min, m/z 268 [M + H]+.
6-Bromo-2-(4-chlorophenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (13l)
Compound 13l was prepared as described for the preparation of 13a, except 1-chloro-4-(2-iodoethyl)benzene (1.6 equiv) was used in place of methyl iodide. The crude product was purified by trituration with cold diethyl ether; yellow solid (42% yield). 1H NMR (DMSO-d6) δ 2.94 (t, J = 7.3 Hz, 2H), 4.03 (t, J = 7.3 Hz, 2H), 7.25 (d, J = 8.3 Hz, 2H), 7.34 (d, J = 8.3 Hz, 2H), 12.52 (s, 1H).
6-(Benzyloxy)-2-(4-chlorophenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (14l)
Compound 14l was prepared from 13l as described for the preparation of 14a, except the reaction was heated over 3 days; white solid (72% yield). 1H NMR (DMSO-d6) δ 2.90 (t, J = 6.7 Hz, 2H), 3.96 (t, J = 6.9 Hz, 2H), 5.05 (s, 2H), 7.15 (d, J = 8.3 Hz, 2H), 7.31 (d, J = 8.3 Hz, 2H), 7.36–7.43 (m, 5H), 12.12 (s, 1H).
2-(4-Chlorophenethyl)-6-hydroxy-1,2,4-triazine-3,5(2H,4H)-dione (11l)
Compound 11l was prepared from 14l as described for the preparation of 11g, with the exception that only 2 equiv of BBr3 were used and that the compound was purified by trituration with EtOAc/hexanes;vwhite solid (52% yield); mp 225 °C. 1H NMR (DMSO-d6) δ 2.90 (t, J = 7.2 Hz, 2H), 3.87 (t, J = 7.3 Hz, 2H), 7.20 (d, J = 8.3 Hz, 2H), 7.33 (d, J = 8.3 Hz, 2H), 11.69 (s, 1H), 12.03 (s, 1H). LCMS (20% acetonitrile/80% water for 0.25 min followed by an increase to 85% acetonitrile/15% water over 1.5 min and continuation of 85% acetonitrile/15% water for 2.25 min; flow rate 1.25 mL/min): retention time 1.39 min, m/z 268 [M + H]+.
6-Bromo-2-(2-fluorophenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (13m)
Compound 13m was prepared as described for the preparation of 13a, except 2-fluoro-4-(2-iodoethyl)benzene (2.0 equiv) was used in place of methyl iodide. The crude product was purified by trituration with cold diethyl ether; yellow solid (34% yield). 1H NMR (DMSO-d6) δ 2.98 (t, J = 7.2 Hz, 2H), 4.05 (t, J = 7.2 Hz, 2H), 7.15 (m, 2H), 7.29 (m, 2H), 12.55 (s, 1H).
6-(Benzyloxy)-2-(2-fluorophenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (14m)
Compound 14m was prepared from 13m as described for the preparation of 14a, with the exception that the reaction was heated for 3 days and that the product was purified by trituration with EtOAc/hexanes; white powder (65% yield). 1H NMR (DMSO-d6): δ 2.96 (t, J = 6.8 Hz, 2H), 3.97 (t, J = 6.6 Hz, 2H), 4.95 (s, 2H), 7.11 (m, 2H), 7.17 (m, 1H), 7.24 (m, 1H), 7.36–7.41 (m, 5H), 12.17 (s, 1H).
2-(2-Fluorophenethyl)-6-hydroxy-1,2,4-triazine-3,5(2H,4H)-dione (11m)
Compound 11m was prepared from 14m as described for the preparation of 11a, with the exception that a mixture of methanol and ethyl acetate (1:1) was used as the solvent and that the hydrogenation was performed at 30 psi for 1.5 h; white solid (83% yield); mp 194 °C. 1H NMR (DMSO-d6) δ 2.95 (t, J = 7.2 Hz, 2H), 3.88 (t, J = 7.2 Hz, 2H), 7.12 (m, 2H), 7.26 (m, 2H), 11.68 (bs, 1H), 11.99 (bs, 1H). LCMS: retention time 1.30 min, m/z 252 [M + H]+.
6-Bromo-2-(3-fluorophenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (13n)
Compound 13n was prepared as described for the preparation of 13a, except 3-fluoro-4-(2-iodoethyl)benzene (2.0 equiv) was used in place of methyl iodide. The crude product was purified by trituration with cold diethyl ether; yellow solid (66% yield). 1H NMR (DMSO-d6) δ 2.96 (t, J = 7.3 Hz, 2H), 4.06 (t, J = 7.3 Hz, 2H), 7.05 (m, 2H), 7.22 (m, 1H), 7.33 (m, 1H), 12.53 (s, 1H).
6-(Benzyloxy)-2-(3-fluorophenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (14n)
Compound 14n was prepared from 13n as described for the preparation of 14a, except the product was purified by trituration with EtOAc/hexanes; tan solid (78% yield). 1H NMR (DMSO-d6) δ 2.94 (t, J = 6.8 Hz, 2H), 3.98 (t, J = 6.9 Hz, 2H), 5.06 (s, 2H), 6.96–7.05 (m, 3H), 7.29 (m, 1H), 7.36–7.43 (m, 5H), 12.13 (s, 1H).
2-(3-Fluorophenethyl)-6-hydroxy-1,2,4-triazine-3,5(2H,4H)-dione (11n)
Compound 11n was prepared from 14n as described for the preparation of 11a, with the exception that a mixture of methanol and ethyl acetate (1:1) was used as a solvent and that the hydrogenation was performed overnight at 30 psi; white solid (79% yield); mp 197 °C. 1H NMR (DMSO-d6) δ 2.93 (t, J = 7.3 Hz, 2H), 3.89 (t, J = 7.2 Hz, 2H), 7.03 (m, 3H), 7.31 (m, 1H), 11.89 (bs, 2H). LCMS: retention time 2.10 min, m/z 252 [M + H]+.
6-Bromo-2-(4-fluorophenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (13o)
Compound 13o was prepared as described for the preparation of 13a, except 1-fluoro-4-(2-iodoethyl)benzene (2.0 equiv) was used in place of methyl iodide. The crude product was purified by trituration with cold diethyl ether; yellow solid (42% yield). 1H NMR (DMSO-d6) δ 2.93 (t, J = 7.1 Hz, 2H), 4.03 (t, J = 7.3 Hz, 2H), 7.11 (m, 2H), 7.26 (m, 2H), 12.52 (s, 1H).
6-(Benzyloxy)-2-(4-fluorophenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (14o)
Compound 14o was prepared from 13o as described for the preparation of 14a; white solid (81% yield). 1H NMR (DMSO-d6) δ 2.90 (t, J = 6.8 Hz, 2H), 3.95 (t, J = 6.9 Hz, 2H), 5.07 (s, 2H), 7.09 (m, 2H), 7.17 (m, 2H), 7.36–7.43 (m, 5H), 12.12 (s, 1H).
2-(4-Fluorophenethyl)-6-hydroxy-1,2,4-triazine-3,5(2H,4H)-dione (11o)
Compound 11o was prepared from 14o as described for the preparation of 11a, except the hydrogenation was performed under an atmospheric pressure of hydrogen for 3 h. The product was purified by trituration with EtOAc/hexanes; light-pink powder (73% yield); mp 193 °C. 1H NMR (DMSO-d6) δ 2.89 (t, J = 7.3 Hz, 2H), 3.87 (t, J = 7.3 Hz, 2H), 7.10 (m, 2H), 7.22 (m, 2H), 11.72 (bs, 1H), 11.97 (bs, 1H). LCMS: retention time 1.95 min, m/z 252 [M + H]+.
6-Bromo-2-(2-methylphenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (13p)
Compound 13p was prepared as described for the preparation of 13a, except 3-methyl-4-(2-iodoethyl)benzene (2.0 equiv) was used in place of methyl iodide. The crude product was purified by trituration with cold diethyl ether; tan solid (52% yield). 1H NMR (DMSO-d6) δ 2.31 (s, 3H), 2.94 (m, 2H), 3.98 (m, 2H), 7.12 (m, 3H), 7.15 (m, 1H), 12.54 (s, 1H).
6-(Benzyloxy)-2-(2-methylphenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (14p)
Compound 14p was prepared from 13p as described for the preparation of 14a, except the product was purified by trituration with EtOAc/hexanes;: beige powder (66% yield). 1H NMR (DMSO-d6) δ 2.30 (s, 3H), 2.90 (t, J = 7.5 Hz, 2H), 3.90 (t, J = 7.8 Hz, 2H), 5.07 (s, 2H), 7.03 (m, 1H), 7.09–7.12 (m, 2H), 7.14 (m, 1H), 7.36–7.45 (m, 5H), 12.16 (s, 1H).
6-Hydroxy-2-(2-methylphenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (11p)
Compound 11p was prepared from 14p as described for the preparation of 11a, with the exception that a mixture of methanol and ethyl acetate (1:1) was used as a solvent and the hydrogenation was performed at 30 psi for 2 h; gray solid (71% yield); mp 207 °C. 1H NMR (DMSO-d6) δ 2.31 (s, 3H), 2.89 (t, J = 7.7 Hz, 2H), 3.81 (t, J = 7.7 Hz, 2H), 7.10 (m, 3H), 7.14 (m, 1H), 11.93 (bs, 2H). LCMS: retention time 2.42 min, m/z 248 [M + H]+.
6-Bromo-2-(3-methylphenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (13q)
Compound 13q was prepared as described for the preparation of 13a, except 3-methyl-4-(2-iodoethyl)benzene (2.0 equiv) was used in place of methyl iodide. The crude product was purified by trituration with cold diethyl ether; tan solid (66% yield). 1H NMR (DMSO-d6) δ 2.27 (s, 3H), 2.89 (t, J = 7.6 Hz, 2H), 4.01 (t, J = 7.6 Hz, 2H), 7.02 (m, 3H), 7.18 (t, J = 7.3 Hz, 1H), 12.50 (s, 1H).
6-(Benzyloxy)-2-(3-methylphenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (14q)
Compound 14q was prepared from 13q as described for the preparation of 14a, except the product was purified by trituration with EtOAc/hexanes; yellow powder (62% yield). 1H NMR (DMSO-d6) δ 2.25 (s, 3H), 2.87 (t, J = 6.8 Hz, 2H), 3.94 (t, J = 7.0 Hz, 2H), 5.07 (s, 2H), 6.98 (m, 3H), 7.16 (t, J = 7.6 Hz, 1H), 7.37–7.45 (m, 5H), 12.14 (s, 1H).
6-Hydroxy-2-(3-methylphenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (11q)
Compound 11q was prepared from 14q as described for the preparation of 11a, with the exception that a mixture of methanol and ethyl acetate (1:1) was used as a solvent and that the hydrogenation was performed overnight at 30 psi; white powder (82% yield); mp 210 °C. 1H NMR (DMSO-d6) δ 2.27 (s, 3H), 2.86 (t, J = 7.3 Hz, 2H), 3.85 (t, J = 7.2 Hz, 2H), 6.99 (m, 3H), 7.18 (t, J = 7.8 Hz, 2H), 11.71 (s, 1H), 12.01 (s, 1H). LCMS: retention time 2.53 min, m/z 248 [M + H]+.
6-Bromo-2-(4-methylphenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (13r)
Compound 13r was prepared as described for the preparation of 13a, except 4-methyl-4-(2-iodoethyl)benzene (2.0 equiv) was used in place of methyl iodide. The crude product was purified by trituration with cold diethyl ether; tan solid (63% yield). 1H NMR (DMSO-d6) δ 2.26 (s, 3H), 2.89 (t, J = 7.5 Hz, 2H), 4.00 (m, 2H), 7.10 (s, 4H), 12.52 (s, 1H).
6-(Benzyloxy)-2-(4-methylphenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (14r)
Compound 14r was prepared from 13r as described for the preparation of 14a; white foam (72% yield). 1H NMR (DMSO-d6) δ 2.24 (s, 3H), 2.86 (t, J = 7.2 Hz, 2H), 3.93 (t, J = 7.1 Hz, 2H), 5.06 (s, 2H), 7.03 (m, 2H), 7.06 (m, 2H), 7.36–7.43 (m, 5H), 12.12 (s, 1H).
6-Hydroxy-2-(4-methylphenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (11r)
Compound 11r was prepared from 14r as described for the preparation of 11a, except the hydrogenation was performed at 30 psi for 2 h; white powder (54% yield); mp 219 °C. 1H NMR (DMSO-d6) δ 2.25 (s, 3H), 2.85 (t, J = 7.5 Hz, 2H), 3.84 (t, J = 7.6 Hz, 2H), 7.08 (q, J = 8.1, 11.4 Hz, 4H), 11.68 (s, 1H), 12.02 (s, 1H). LCMS: retention time 2.49 min, m/z 248 [M + H]+.
6-Bromo-2-(3-(trifluoromethyl)phenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (13s)
Compound 13s was prepared as described for the preparation of 13a, except 1-(2-iodoethyl)-3-(trifluoromethyl)benzene (2.0 equiv) was used in place of methyl iodide. The crude product was purified by trituration with cold diethyl ether; yellow solid (48%). 1H NMR (DMSO-d6) δ 3.06 (t, J = 7.2 Hz, 2H), 4.10 (t, J = 7.1 Hz, 2H), 7.55 (m, 4H), 12.53 (s, 1H).
6-(Benzyloxy)-2-(3-(trifluoromethyl)phenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (14s)
Compound 14s was prepared from 13s as described for the preparation of 14a; white solid (85%). 1H NMR (DMSO-d6) δ 3.02 (t, J = 6.7 Hz, 2H), 4.01 (t, J = 6.8 Hz, 2H), 5.00 (s, 2H), 7.36 (m, 1H), 7.40 (m, 4H), 7.50 (m, 4H), 12.13 (s, 1H).
6-Hydroxy-2-(3-(trifluoromethyl)phenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (11s)
Compound 11s was prepared from 14s as described for the preparation of 11a; white powder (85% yield); mp 201 °C. 1H NMR (DMSO-d6) δ 3.02 (t, J = 7.1 Hz, 2H), 3.93 (t, J = 7.2 Hz, 2H), 7.51 (m, 3H), 7.574 (m, 1H), 11.70 (s, 1H), 12.03 (s, 1H). LCMS: retention time 3.23 min, m/z 302 [M + H]+.
6-Bromo-2-(4-(trifluoromethyl)phenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (13t)
Compound 13t was prepared as described for the preparation of 13a, except 1-(2-iodoethyl)-4-(trifluoromethyl)benzene (2.0 equiv) was used in place of methyl iodide. The crude product was purified using a Biotage Isolera One flash purification system with a silica gel flash cartridge (EtOAc/hexanes); yellow solid (69% yield). 1H NMR (DMSO-d6) δ 3.04 (t, J = 7.2 Hz, 2H), 4.08 (t, J = 7.1 Hz, 2H), 7.46 (d, J = 7.1 Hz, 2H), 7.64 (d, J = 8.1 Hz, 2H), 12.53 (s, 1H).
6-(Benzyloxy)-2-(4-(trifluoromethyl)phenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (14t)
Compound 14t was prepared from 13t as described for the preparation of 14a, except the product was purified by trituration with EtOAc/hexanes; white solid (71% yield). 1H NMR (DMSO-d6) δ 3.01 (t, J = 6.7 Hz, 2H), 4.01 (t, J = 6.8 Hz, 2H), 5.02 (s, 2H), 7.38–7.42 (m, 7H), 7.62 (d, J = 8.1 Hz, 2H), 12.13 (s, 1H).
6-Hydroxy-2-(4-(trifluoromethyl)phenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (11t)
Compound 11t was prepared from 14t as described for the preparation of 11a, with the exception that a mixture of methanol and EtOAc (1:1) was used as a solvent and that the hydrogenation was performed overnight at 30 psi; gray solid (83% yield); mp 222 °C. 1H NMR (DMSO-d6) δ 3.01 (t, J = 7.1 Hz, 2H), 3.92 (t, J = 7.1 Hz, 2H), 7.42 (d, J = 7.8 Hz, 2H), 7.64 (d, J = 8.1 Hz, 2H), 11.71 (s, 1H), 12.02 (s, 1H). LCMS: retention time 2.99 min, m/z 302 [M + H]+.
6-Bromo-2-(2-methoxyphenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (13u)
Compound 13u was prepared as described for the preparation of 13a, with the exception that 1-(2-iodoethyl)-2-methoxybenzene (2.0 equiv) was used in place of methyl iodide. The crude product was purified by trituration with cold diethyl ether; tan solid (57% yield). 1H NMR (DMSO-d6) δ 2.91 (t, J = 6.9 Hz, 2H), 3.73 (s, 3H), 4.01 (t, J = 7.0 Hz, 2H), 6.87 (dt, J = 1.0, 7.6 Hz, 1H), 6.93 (d, J = 7.6 Hz, 1H), 7.11 (m, 1H), 7.21 (m, 1H), 12.52 (s, 1H).
6-(Benzyloxy)-2-(2-methoxyphenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (14u)
Compound 14u was prepared from 13u as described for the preparation of 14a, except the product was purified by trituration with EtOAc/hexanes; white solid (63% yield). 1H NMR (DMSO-d6) δ 2.89 (t, J = 6.7 Hz, 2H), 3.74 (s, 3H), 3.94 (t, J = 6.7 Hz, 2H), 4.89 (s, 2H), 6.84 (t, J = 7.3 Hz, 1H), 6.93 (d, J = 8.1 Hz, 1H), 7.02 (dd, J = 1.8, 7.3 Hz, 1H), 7.20 (m, 1H), 7.35–7.41 (m, 5H), 12.12 (s, 1H).
6-Hydroxy-2-(2-methoxyphenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (11u)
Compound 11u was prepared from 14u as described for the preparation of 11a, with the exception that a mixture of methanol and EtOAc (1:1) was used as a solvent and that the hydrogenation was performed overnight under atmospheric pressure of hydrogen; yellow powder (83% yield); mp 166 °C. 1H NMR (DMSO-d6) δ 2.88 (t, J = 7.2 Hz, 2H), 3.75 (s, 3H), 3.84 (t, J = 7.2 Hz, 2H), 6.85 (dt, J = 1.0, 7.3 Hz, 1H), 6.93 (d, J = 7.8 Hz, 1H), 7.07 (dt, J = 1.5, 7.3 Hz, 1H), 7.20 (dt, J = 1.8, 8.1 Hz, 1H), 11.63 (s, 1H), 12.0 (s, 1H). LCMS: retention time 2.03 min, m/z 264 [M + H]+.
6-Hydroxy-2-(2-hydroxyphenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (15u)
Compound 15u was prepared from 11u as described for the preparation of 11g, with the exception that 3 equiv of BBr3 were used and that the reaction was stirred for 1.5 h at rt; off-white solid (68% yield); mp 205 °C. 1H NMR (DMSO-d6) δ 2.83 (t, J = 7.3 Hz, 2H), 3.83 (t, J = 7.3 Hz, 2H), 6.68 (dt, J = 1.3, 7.6 Hz, 1H), 6.75 (d, J = 7.8 Hz, 1H), 6.97–7.03 (m, 2H), 9.38 (s, 1H), 11.63 (bs, 1H), 11.97 (bs, 1H). LCMS: retention time 0.70 min, m/z 250 [M + H]+.
6-Bromo-2-(3-methoxyphenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (13v)
Compound 13v was prepared as described for the preparation of 13a, except 1-(2-iodoethyl)-3-methoxybenzene (2.0 equiv) was used in place of methyl iodide. The crude product was purified by trituration with cold diethyl ether; tan solid (36% yield). 1H NMR (DMSO-d6) δ 2.91 (t, J = 7.5 Hz, 2H), 3.73 (s, 3H), 4.04 (m, 2H), 6.77 (m, 1H), 6.79 (m, 2H), 7.21 (t, J = 7.7 Hz, 1H), 12.53 (s, 1H).
6-(Benzyloxy)-2-(3-methoxyphenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (14v)
Compound 14v was prepared from 13v as described for the preparation of 14a, except the product was purified by trituration with EtOAc/hexanes; white solid (64% yield). 1H NMR (DMSO-d6) δ 2.89 (t, J = 7.1 Hz, 2H), 3.70 (s, 3H), 3.96 (t, J = 7.2 Hz, 2H), 5.06 (s, 2H), 6.72–6.78 (m, 3H), 7.19 (t, J = 7.8 Hz, 1H), 7.36–7.44 (m, 5H), 12.14 (s, 1H).
6-Hydroxy-2-(3-methoxyphenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (11v)
Compound 11v was prepared from 14v as described for the preparation of 11a, with the exception that a mixture of methanol and EtOAc (1:1) was used as a solvent and that the hydrogenation was performed overnight at 30 psi; off-white solid (63% yield); mp 224 °C. 1H NMR (DMSO-d6) δ 2.88 (t, J = 7.6 Hz, 2H), 3.72 (s, 3H), 3.87 (t, J = 7.6 Hz, 2H), 6.75–6.78 (m, 3H), 7.20 (t, J = 8.0 Hz, 1H), 11.76 (bs, 1H), 11.99 (bs, 1H), 11.97 (bs, 1H). LCMS: retention time 1.80 min, m/z 264 [M + H]+.
6-Hydroxy-2-(3-hydroxyphenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (15v)
Compound 15v was prepared from 11v as described for the preparation of 11g; white solid (53% yield); mp 232 °C. 1H NMR (DMSO-d6) δ 2.80 (t, J = 7.2 Hz, 2H), 3.83 (t, J = 7.2 Hz, 2H), 6.58 (m, 3H), 7.07 (t, J = 7.7 Hz, 1H), 9.32 (s, 1H), 11.69 (s, 1H), 12.04 (s, 1H). LCMS: retention time 0.37 min, m/z 250 [M + H]+.
6-Bromo-2-(4-methoxyphenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (13w)
Compound 13w was prepared as described for the preparation of 13a, except 1-(2-iodoethyl)-4-methoxybenzene (2.0 equiv) was used in place of methyl iodide. The crude product was purified by trituration with cold diethyl ether; tan solid (58% yield). 1H NMR (DMSO-d6) δ 2.87 (t, J = 7.5 Hz, 2H), 3.71 (s, 3H), 3.99 (m, 2H), 6.84 (d, J = 8.6 Hz, 2H), 7.12 (m, 2H), 12.52 (s, 1H).
6-(Benzyloxy)-2-(4-methoxyphenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (14w)
Compound 14w was prepared from 13w as described for the preparation of 14a, except the product was purified by trituration with EtOAc/hexanes; white solid (70% yield). 1H NMR (DMSO-d6) δ 2.84 (t, J = 7.1 Hz, 2H), 3.69 (s, 3H), 3.91 (t, J = 7.1 Hz, 2H), 5.06 (s, 2H), 6.82 (t, J = 8.8 Hz, 2H), 7.05 (t, J = 8.6 Hz, 2H), 7.17 (m, 2H), 7.36–7.44 (m, 5H), 12.12 (s, 1H).
6-Hydroxy-2-(4-methoxyphenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (11w)
Compound 11w was prepared from 14w as described for the preparation of 11a, with the exception that a mixture of methanol and EtOAc (1:1) was used as a solvent and that the hydrogenation was performed at 50 psi for 3 h; off-white solid (62% yield); mp 244 °C. 1H NMR (DMSO-d6) δ 2.83 (t, J = 7.5 Hz, 2H), 3.71 (s, 3H), 3.82 (t, J = 7.5 Hz, 2H), 6.83 (d, J = 8.6 Hz, 2H), 7.09 (d, J = 8.6 Hz, 2H), 11.74 (bs, 1H), 11.98 (bs, 1H). LCMS: retention time 1.75 min, m/z 264 [M + H]+.
6-Hydroxy-2-(4-hydroxyphenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (15w)
Compound 15w was prepared from 11w as described for the preparation of 11g: light-pink solid (54% yield); mp >280 °C (decomp). 1H NMR (DMSO-d6) δ 2.78 (t, J = 7.6 Hz, 2H), 3.80 (t, J = 7.6 Hz, 2H), 6.65 (d, J = 8.3 Hz, 2H), 6.96 (d, J = 8.3 Hz, 2H), 9.22 (s, 1H), 11.66 (s, 1H), 12.01 (s, 1H). LCMS (5% acetonitrile/95% water for 0.5 min followed by an increase to 40% acetonitrile/60% water over 4 min and continuation of 40% acetonitrile/60% water for 3.5 min): retention time 3.20 min, m/z 250 [M + H]+.
6-Bromo-2-(3-phenoxyphenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (13x)
Compound 13x was prepared as described for the preparation of 13a, except 1-(2-iodoethyl)-3-phenoxybenzene (2.0 equiv) was used in place of methyl iodide; yield 21% (white solid). 1H NMR (DMSO-d6): δ 2.93 t, J = 7.1 Hz, 2H), 4.05 (t, J = 7.1 Hz, 2H), 6.85–6.67 (m, 2H), 6.95–7.01 (m, 3H), 7.10–7.15 (m, 1H), 7.29–7.39 (m, 3H), 12.53 (s, 1H).
6-(Benzyloxy)-2-(3-phenoxyphenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (14x)
Compound 14x was prepared from 13x as described for the preparation of 14a; white solid (52% yield). 1H NMR (DMSO-d6): δ 2.88 (t, J = 6.5 Hz, 2H), 3.96 (t, J = 6.5 Hz, 2H), 5.03 (s, 2H), 6.75 (m, 1H), 6.84–6.86 (m, 1H), 6.91–6.96 (m, 3H), 7.09–7.13 (m, 1H), 7.28–7.39 (m, 8H), 12.14, (bs, 1H).
6-Hydroxy-2-(3-phenoxyphenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (11x)
Compound 11x was prepared from 14x as described for the preparation of 11a; tan solid (78% yield); mp 150–153 °C. 1H NMR (DMSO-d6): δ 2.89 (t, J = 7.2 Hz, 2H), 3.86 (t, J = 7.2 Hz, 2H), 6.79 (m, 1H), 6.83–6.86 (m, 1H), 6.95–6.98 (m, 3H), 7.10–7.14 (m, 1H), 7.28–7.40 (m, 3H), 11.86 (bs, 1H). LCMS (20% acetonitrile/80% water for 0.25 min followed by an increase to 85% acetonitrile/15% water over 1.5 min and continuation of 85% acetonitrile/15% water for 2.25 min; flow rate 1.25 mL/min): retention time 1.78 min, m/z 326 [M + H]+.
6-Bromo-2-(4-phenoxyphenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (13y)
Compound 13y was prepared as described for the preparation of 13a, except 1-(2-iodoethyl)-4-phenoxybenzene (2.0 equiv) was used in place of methyl iodide; tan solid (24% yield). 1H NMR (DMSO-d6): δ 2.93 (m, 2H), 4.04 (m, 2H), 6.93–6.98 (m, 4H), 7.10–7.14 (m, 1H), 7.21–7.25 (m, 2H), 7.36–7.40 (m, 2H), 12.54 (s, 1H).
6-(Benzyloxy)-2-(4-phenoxyphenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (14y)
Compound 14y was prepared from 13y as described for the preparation of 14a; white solid (41% yield). 1H NMR (DMSO-d6): δ 2.90 (t, J = 7.0 Hz, 2H), 3.96 (t, J = 7.0 Hz, 2H), 5.08 (s, 2H), 6.91–6.96 (m, 4H), 7.09–7.12 (m, 1H), 7.16–7.18 (m, 2H), 7.33–7.45 (m, 7H), 12.14 (s, 1H).
6-Hydroxy-2-(4-phenoxyphenethyl)-1,2,4-triazine-3,5(2H,4H)-dione (11y)
Compound 11y was prepared from 14y as described for the preparation of 11g; white solid (69% yield); mp 207–209 °C. 1H NMR (DMSO-d6): δ 2.90 (t, J = 7.2 Hz, 2H), 3.88 (t, J = 7.3 Hz, 2H), 6.92–6.98 (m, 4H), 7.10–7.14 (m, 1H), 7.21 (m, 2H), 7.35–7.40 (m, 2H), 11.69 (bs, 1H), 12.04 (bs, 1H). LCMS (20% acetonitrile/80% water for 0.25 min followed by an increase to 85% acetonitrile/15% water over 1.5 min and continuation of 85% acetonitrile/15% water for 2.25 min; flow rate 1.25 mL/min): retention time 1.80 min, m/z 326 [M + H]+.
2-(2-(Biphenyl-4-yl)ethyl)-6-bromo-1,2,4-triazine-3,5(2H,4H)-dione (13z)
Compound 13z was prepared as described for the preparation of 13a, except 4-(2-iodoethyl)biphenyl (2.0 equiv) was used in place of methyl iodide. The crude product was purified by trituration with cold diethyl ether; tan solid (59% yield). 1H NMR (DMSO-d6) δ 2.99 (t, J = 7.5 Hz, 2H), 4.08 (t, J = 7.6 Hz, 2H), 7.31–7.37 (m, 3H), 7.45 (t, J = 7.6 Hz, 2H), 7.59–7.65 (m, 4H), 12.55 (s, 1H).
6-(Benzyloxy)-2-(2-(biphenyl-4-yl)ethyl)-1,2,4-triazine-3,5(2H,4H)-dione (14z)
Compound 14z was prepared from 13z as described for the preparation of 14a; white solid (53% yield). 1H NMR (DMSO-d6) δ 2.96 (t, J = 7.2 Hz, 2H), 4.00 (t, J = 7.1 Hz, 2H), 5.05 (s, 2H),7.25 (d, J = 8.1 Hz, 2H), 7.34–7.46 (m, 8H), 7.58–7.64 (m, 4H), 12.16 (s, 1H).
2-(2-(Biphenyl-4-yl)ethyl)-6-hydroxy-1,2,4-triazine-3,5(2H,4H)-dione (11z)
Compound 11z was prepared from 14z as described for the preparation of 11a, with the exception that a mixture of methanol and EtOAc (2:1) was used as a solvent and that the hydrogenation was performed overnight at 30 psi; white powder (89% yield); mp 254 °C. 1H NMR (DMSO-d6) δ 2.95 (t, J = 7.6 Hz, 2H), 3.92 (t, J = 7.5 Hz, 2H), 7.28 (d, J = 8.1 Hz, 2H), 7.35 (t, J = 6.8 Hz, 1H), 7.45 (t, J = 7.6 Hz, 2H), 7.59 (d, J = 8.3 Hz, 2H), 7.63 (d, J = 7.1 Hz, 2H), 11.71 (s, 1H), 12.06 (s, 1H). LCMS: retention time 3.37 min, m/z 310 [M + H]+.
4-(Benzyloxymethyl)-6-bromo-2-(2-(pyridin-2-yl)ethyl)-1,2,4-triazine-3,5(2H,4H)-dione (17a)
To a solution of 16 (0.30 g, 0.96 mmol), 2-(pyridin-2-yl)ethanol (0.12 g, 1.0 mmol), and triphenylphosphine (0.30 g, 1.1 mmol) in THF (5 mL) was added dropwise diisopropyl azodicarboxylate (0.23 mL, 1.2 mmol) at 0 °C. The mixture was stirred at 0 °C for 10 min, then heated at 66 °C for 6.5 h. The reaction was concentrated and the residual oil was purified using a Biotage Isolera One flash purification system with a silica gel flash cartridge (EtOAc/hexanes) to give 0.34 g of 17a as a clear oil (85% yield). 1H NMR (CDCl3) δ 3.22 (t, J = 7.1 Hz, 2H), 4.37 (t, J = 7.6 Hz, 2H), 4.68 (s, 2H), 5.49 (s, 2H), 7.12–7.19 (m, 2H), 7.29–7.34 (mt, 5H), 7.61 (dt, J = 2.0, 7.8 Hz, 1H), 8.51 (d, J = 4.8 Hz, 1H).
6-(Benzyloxy)-4-(benzyloxymethyl)-2-(2-(pyridin-2-yl)ethyl)-1,2,4-triazine-3,5(2H,4H)-dione (18a)
To a solution of benzyl alcohol (0.13 mL, 1.2 mmol) in DMF (3 mL) was added 60% w/w sodium hydride (60% dispersion in mineral oil, 0.049 g, 1.2 mmol) at 0 °C. The mixture was stirred for 10 min, and a solution of 17a (0.34 g, 0.81 mmol) in DMF (5 mL) was added via syringe. The reaction was stirred at 0 °C for 30 min and quenched by the addition of water and EtOAc. The organic layer was dried over Na2SO4 and concentrated. The residual oil was purified using a Biotage Isolera One flash purification system with a silica gel flash cartridge (EtOAc/hexanes) to give 0.18 g of 18a as an oil (50% yield). 1H NMR (CDCl3) δ 3.17 (t, J = 7.1 Hz, 2H) 4.27 (t, J = 5.6 Hz, 2H), 4.67 (s, 2H), 5.07 (s, 2H), 5.47 (s, 2H), 7.12 (m, 2H), 7.29–7.42 (m, 10H), 7.59 (dt, J = 2.0, 7.8 Hz, 1H), 8.54 (d, J = 5.1 Hz, 1H).
6-Hydroxy-2-(2-(pyridin-2-yl)ethyl)-1,2,4-triazine-3,5(2H,4H)-dione (19a)
To a solution of 18a (0.18 g, 0.40 mmol) in dichloromethane (8 mL) was added a 1.0 M solution of BBr3 in dichloromethane (0.80 mL, 0.80 mmol) at rt. The reaction was stirred for 45 min, concentrated, and purified by preparative HPLC (0% acetonitrile/100% water for 5 min followed by an increase to 20% acetonitrile/80% water over 35 min and an increase to 40% acetonitrile/60% water for 10 min; flow rate 10 mL/min) to give 0.017 g of 19a as a white solid (18% yield); mp 225 °C. 1H NMR (DMSO-d6) δ 3.17 (t, J = 5.8 Hz, 2H), 4.04 (t, J = 6.8 Hz, 2H), 7.59 (m, 2H), 8.07 (m, 1H), 8.63 (m, 1H), 11.64 (bs, 1H), 12.04 (s, 1H). LCMS (5% acetonitrile/95% water for 0.5 min followed by an increase to 40% acetonitrile/60% water over 4 min and continuation of 40% acetonitrile/60% water for 3.5 min; flow rate 0.25 mL/min; column Luna Plus C18, 2.1 mm × 100 mm, 1.8 μm): retention time 1.06 min, m/z 235 [M + H]+.
2-(2-(1H-Pyrazol-1-yl)ethyl)-4-(benzyloxymethyl)-6-bromo-1,2,4-triazine-3,5(2H,4H)-dione (17b)
Compound 17b was prepared as described for the preparation of 17a, except 2-(1H-pyrazol-1-yl)ethanol was used in place of 2-(pyridin-2-yl)ethanol; clear oil (86% yield). 1H NMR (CDCl3) δ 4.36 (t, J = 5.9 Hz, 2H), 4.50 (t, J = 5.9 Hz, 2H), 4.67 (s, 2H), 5.47 (s, 2H), 6.24 (t, J = 2.1 Hz, 1H), 7.29–7.34 (m, 5H), 7.36 (m, 1H), 7.46 (m, 1H).
2-(2-(1H-Pyrazol-1-yl)ethyl)-6-(benzyloxy)-4-(benzyloxymethyl)-1,2,4-triazine-3,5(2H,4H)-dione (18b)
Compound 18b was prepared from 17b as described for the preparation of 18a, except the reaction was stirred at 0 °C and warmed up to rt over a period of 4 h; clear oil (47% yield). 1H NMR (CDCl3): δ 4.27 (t, J = 5.9 Hz, 2H), 4.43 (t, J = 5.9 Hz, 2H), 4.67 (s, 2H), 5.05 (s, 2H), 5.46 (s, 2H), 6.21 (t, J = 2.1 Hz, 1H), 7.20 (m, 1H), 7.28–7.41 (m, 10H), 7.51 (m, 1H).
2-(2-(1H-Pyrazol-1-yl)ethyl)-6-hydroxy-1,2,4-triazine-3,5(2H,4H)-dione (19b)
To a solution of 19b (0.14 g, 0.32 mmol) in methanol (5 mL) was added one spatula tip of 10% Pd/C. The mixture was hydrogenated overnight under an atmospheric pressure of hydrogen and filtered through a pad of Celite. The filtrate was concentrated, and the residual material was triturated with EtOAc/MeOH to give 0.060 g of compound 19b as a white solid (83% yield); mp 275 °C (dec). 1H NMR (DMSO-d6) δ 4.01 (t, J = 6.2 Hz, 2H), 4.36 (t, J = 6.2 Hz, 2H), 6.20 (t, J = 2.1 Hz, 1H), 7.40 (d, J = 1.5 Hz, 1H), 7.69 (d, J = 2.0 Hz, 1H), 11.69 (s, 1H), 12.04 (s, 1H). LCMS (5% acetonitrile/95% water for 0.5 min followed by an increase to 40% acetonitrile/60% water over 4 min and continuation of 40% acetonitrile/60% water for 3.5 min; flow rate 0.25 mL/min; column Luna Plus C18, 2.1 mm × 100 mm, 1.8 μm): retention time 1.32 min, m/z 224 [M + H]+.
2-(2-(1H-Benzo[d]imidazol-1-yl)ethyl)-4-(benzyloxymethyl)-6-bromo-1,2,4-triazine-3,5(2H,4H)-dione (17c)
Compound 17c was prepared as described for the preparation of 17a, except 2-(1H-benzo[d]imidazol-1-yl)ethanol was used in place of 2-(pyridin-2-yl)ethanol and that the compound was purified by trituration with EtOAc/hexanes; beige solid (72% yield). 1H NMR (CDCl3) δ 4.38 (t, J = 6.6 Hz, 2H), 4.56 (t, J = 6.4 Hz, 2H), 4.61 (s, 2H), 5.42 (s, 2H), 7.28–7.34 (m, 7H), 7.39 (m, 1H), 7.80 (m, 1H), 7.91 (s, 1H).
2-(2-(1H-Benzo[d]imidazol-1-yl)ethyl)-6-(benzyloxy)-4-(benzyloxymethyl)-1,2,4-triazine-3,5(2H,4H)-dione (18c)
Compound 18c was prepared from 17c as described for the preparation of 18a, with the exception that the reaction was stirred overnight at rt and that the compound was purified by a silica gel flash chromatography (1% NH4OH/EtOAc,); white solid (34% yield). 1H NMR (CDCl3) δ 4.25 (t, J = 5.9 Hz, 2H), 4.47 (t, J = 5.9 Hz, 2H), 4.60 (s, 2H), 4.75 (s, 2H), 5.40 (s, 2H), 7.27–7.36 (m, 13H), 7.71 (s, 1H), 7.78 (m, 1H).
2-(2-(1H-Benzo[d]imidazol-1-yl)ethyl)-6-hydroxy-1,2,4-triazine-3,5(2H,4H)-dione (19c)
Compound 19c was prepared from 18c as described for the preparation of 19b; beige solid (18% yield); mp 260 °C (dec). 1H NMR (DMSO-d6) δ 3.99 (t, J = 5.8 Hz, 2H), 4.50 (t, J = 5.8 Hz, 2H), 7.18 (m, 1H), 7.23 (m, 1H), 7.51 (d, J = 7.6 Hz, 1H), 7.61 (d, J = 7.3 Hz, 1H), 8.14 (s, 1H). LCMS (5% acetonitrile/95% water for 0.5 min followed by an increase to 40% acetonitrile/60% water over 4 min and continuation of 40% acetonitrile/60% water for 3.5 min; flow rate 0.25 mL/min; column Luna Plus C18, 2.1 mm × 100 mm, 1.8 μm): retention time 3.26 min, m/z 274 [M + H]+.
2-(2-(1H-Pyrrolo[2,3-b]pyridin-1-yl)ethyl)-4-(benzyloxymethyl)-6-bromo-1,2,4-triazine-3,5(2H,4H)-dione (17d)
Compound 17d was prepared as described for the preparation of 17a, with the exception that 2-(1H-pyrrolo[2,3-b]pyridin-1-yl)ethanol was used in place of 2-(pyridin-2-yl)ethanol and that the reaction was stirred overnight: white solid (quantitative yield). 1H NMR (CDCl3) δ 1.27 (d, J = 6.3 Hz, 2H), 4.36 (t, J = 5.3 Hz, 2H), 4.67 (t, J = 5.6 Hz, 2H), 5.44 (s, 2H), 6.49 (d, J = 3.5 Hz, 1H), 7.00 (dd, J = 4.8, 7.8 Hz, 1H), 7.17 ((d, J = 3.5 Hz, 2H), 7.31 (m, 1H), 7.34–7.36 (m, 4H), 7.86 (dd, J = 1.5, 7.8 Hz, 1H), 8.11 (dd, J = 1.5, 4.8 Hz, 1H).
2-(2-(1H-Pyrrolo[2,3-b]pyridin-1-yl)ethyl)-6-(benzyloxy)-4-(benzyloxymethyl)-1,2,4-triazine-3,5(2H,4H)-dione (18d)
Compound 18d was prepared from 17d as described for the preparation of 18a, with the exception that the reaction was stirred for 4 h at rt and that the compound was precipitated from EtOAc; white solid (51% yield). 1H NMR (CDCl3) δ 4.28 (t, J = 5.3 Hz, 2H), 4.52 (s, 2H), 4.63 (t, J = 5.8 Hz, 2H), 4.66 (s, 2H), 5.43 (s, 2H), 6.42 (d, J = 3.5 Hz, 1H),), 7.02 (m, 2H), 7.22 (dd, J = 1.8, 7.1 Hz, 2H), 7.29–7.36 (m, 8H), 7.84 (dd, J = 1.5, 7.8 Hz, 1H), 8.22 (dd, J = 1.5, 4.8 Hz, 1H).
2-(2-(1H-Pyrrolo[2,3-b]pyridin-1-yl)ethyl)-6-hydroxy-1,2,4-triazine-3,5(2H,4H)-dione (19d)
Compound 19d was prepared from 18d as described for the preparation of 19a, with the exception that 4.0 equiv of BBr3 were used and that the reaction was stirred for 2 h at rt. The crude material was purified by preparative HPLC (5% acetonitrile/95% water for 5 min followed by an increase to 35% acetonitrile/65% water over 35 min and an increase to 50% acetonitrile/50% water for 10 min; flow rate 15 mL/min); beige powder (79% yield); mp 259 °C. 1H NMR (DMSO-d6) δ 4.03 (t, J = 4.8 Hz, 2H), 4.51 (t, J = 5.1 Hz, 2H), 6.43 (d, J = 3.3 Hz, 1H), 7.05 (dd, J = 2.8, 7.6 Hz, 1H), 7.46 (d, J = 3.3 Hz, 1H), 7.92 (d, J = 7.8 Hz, 1H), 8.16 (d, J = 4.3 Hz, 1H), 11.89 (bs, 2H). LCMS (5% acetonitrile/95% water for 0.5 min followed by an increase to 40% acetonitrile/60% water over 4 min and continuation of 40% acetonitrile/60% water for 3.5 min): retention time 0.45 min, m/z 274 [M + H]+.
4-(Benzyloxymethyl)-6-bromo-2-(2-phenylpropyl)-1,2,4-triazine-3,5(2H,4H)-dione (17e)
Compound 17e was prepared as described for the preparation of 17a, except 2-phenylpropan-1-ol was used in place of 2-(pyridin-2-yl)ethanol; colorless oil (87% yield). 1H NMR (CDCl3) δ 1.31 (d, J = 6.8 Hz, 3H), 3.34 (m, 1H), 4.03 (dd, J = 7.8, 13.4 Hz, 1H), 4.13 (dd, J = 8.3, 13.4 Hz, 1H), 4.58 (s, 2H), 5.44 (s, 2H), 7.21 (m, 3H), 7.28–7.33 (m, 7H).
6-(Benzyloxy)-4-(benzyloxymethyl)-2-(2-phenylpropyl)-1,2,4-triazine-3,5(2H,4H)-dione (18e)
Compound 18e was prepared from 17e as described for the preparation of 18a; colorless oil (52% yield). 1H NMR (CDCl3) δ 1.27 (d, J = 7.1 Hz, 3H); 3.24 (m, 1H), 3.99 (d, J = 7.1 Hz, 2H), 4.59 (s, 2H), 5.04 (qt, J = 12.4, 23.8 Hz, 2H), 5.43 (s, 2H), 7.14–7.20 (m, 3H), 7.25 (m, 2H), 7.29–7.33 (m, 5H), 7.38–7.41 (m, 5H).
6-Hydroxy-2-(2-phenylpropyl)-1,2,4-triazine-3,5(2H,4H)-dione (19e)
Compound 19e was prepared from 18e as described for the preparation of 19b, with the exception that the product was purified by trituration with EtOAc/hexanes; beige solid (12% yield); mp 217 °C. 1H NMR (DMSO-d6) δ 1.19 (d, J = 7.1 Hz, 3H), 3.22 (m, 1H), 3.78 (d, J = 6.8 Hz, 2H), 7.22 (m, 3H), 7.28 (m, 2H), 11.65 (s, 1H), 11.99 (s, 1H). LCMS (20% acetonitrile/80% water for 0.25 min followed by an increase to 85% acetonitrile/15% water over 1.5 min and continuation of 85% acetonitrile/15% water for 2.25 min; flow rate 1.25 mL/min): retention time 0.93 min, m/z 248 [M + H]+.
4-(Benzyloxymethyl)-6-bromo-2-(2,2-difluoro-2-phenylethyl)-1,2,4-triazine-3,5(2H,4H)-dione (17f)
Compound 17f was prepared as described for the preparation of 17a, except 2,2-difluoro-2-phenylethanol was used in place of 2-(pyridin-2-yl)ethanol: colorless oil (95% yield). 1H NMR (DMSO-d6) δ 4.53 (s, 2H), 4.66 (t, J = 13.9 Hz, 2H), 5.30 (s, 2H), 7.27–7.36 (m, 5H), 7.50 (m, 3H), 7.56 (m, 2H).
6-(Benzyloxy)-4-(benzyloxymethyl)-2-(2,2-difluoro-2-phenylethyl)-1,2,4-triazine-3,5(2H,4H)-dione (18f)
Compound 18f was prepared from 17f as described for the preparation of 18a, with the exception that 1.2 equiv of benzyl alcohol and 1.2 equiv of sodium hydride were used and that the reaction was stirred at rt for 2 h; white solid (59% yield). 1H NMR (DMSO-d6) δ 4.49 (s, 2H), 4.56 (t, J = 12.8 Hz, 2H), 4.83 (s, 2H), 5.27 (s, 2H), 7.26–7.33 (m, 5H), 7.40 (m, 5H), 7.50–7.58 (m, 5H).
2-(2,2-Difluoro-2-phenylethyl)-6-hydroxy-1,2,4-triazine-3,5(2H,4H)-dione (19f)
Compound 19f was prepared from 18f as described for the preparation of 19b, with the exception that a mixture of methanol, EtOAc, and acetic acid (1:1:0.1) was used as solvent. A mixture of compound 19f and its N-hydroxylmethyl derivative was obtained as a beige solid. The residue was redissolved in methanol and treated with a catalytic amount of sodium carbonate and stirred over 2 days. The reaction mixture was acidified with a 10% KHSO4 solution to pH 4, and the precipitate was filtered and then washed thoroughly with water and with 10% EtOAc/hexanes to give compound 19g as a white powder (36% yield); mp 245 °C. 1H NMR (DMSO-d6) δ 4.38 (t, J = 14.0 Hz, 2H), 7.50 (m, 5H), 11.88 (m, 1H), 12.17 (m, 1H). LCMS: retention time 2.25 min, m/z 270 [M + H]+.
4-(Benzyloxymethyl)-6-bromo-2-(2-oxo-2-phenylethyl)-1,2,4-triazine-3,5(2H,4H)-dione (20)
To a suspension of NaH (0.14 g, 3.5 mmol) in DMF (2.5 mL) at rt was slowly added a solution of 16 (1.0 g, 3.2 mmol) in DMF (7 mL) via syringe. The mixture was stirred at rt for 1 h, after which bromoacetophenone (0.70 g, 3.5 mmol) was added in one portion. The reaction was stirred for 3.5 h. The reaction mixture was partitioned between EtOAc and water. The organic layer was dried over Na2SO4 and concentrated. The residual material was purified by a Biotage Isolera One flash purification system with a silica gel flash cartridge (EtOAc/hexanes) to give 1.27 g of 20 as a white solid (92% yield). 1H NMR (CDCl3) δ 4.72 (s, 2H), 5.40 (s, 2H), 5.54 (s, 2H), 7.31–7.39 (m, 5H), 7.54 (t, J = 7.6 Hz, 2H), 7.67 (m, 1H), 7.97 (m, 2H).
6-(Benzyloxy)-4-(benzyloxymethyl)-2-(2-oxo-2-phenylethyl)-1,2,4-triazine-3,5(2H,4H)-dione (21)
Compound 21 was prepared from 20 as described for the preparation of 18a, with the exception that 2.2 equiv of benzyl alcohol and 2.2 equiv of sodium hydride were used; yellow oil (45% yield). 1H NMR (CDCl3) δ 4.72 (s, 2H), 5.16 (s, 2H), 5.29 (s, 2H), 5.53 (s, 2H), 7.29–7.41 (m, 10H), 7.54 (t, J = 7.3 Hz, 2H), 7.67 (m, 1H), 7.99 (m, 2H).
6-Hydroxy-2-(2-oxo-2-phenylethyl)-1,2,4-triazine-3,5(2H,4H)-dione (22)
Compound 22 was prepared from 21 as described for the preparation of 19a, with the exception that 4.0 equiv of BBr3 were used and that the reaction was stirred for 10 min. The crude material was purified by trituration with EtOAc/hexanes; white solid (63% yield); mp 220 °C. 1H NMR (DMSO-d6) δ 5.27 (s, 2H), 7.58 (t, J = 7.8 Hz, 2H), 7.71 (t, J = 7.6 Hz, 1H), 8.02 (d, J = 7.3 Hz, 2H), 11.78 (bs, 1H), 12.30 (s, 1H). LCMS (20% acetonitrile/80% water for 0.25 min followed by an increase to 85% acetonitrile/15% water over 1.5 min and continuation of 85% acetonitrile/15% water for 2.25 min; flow rate 1.25 mL/min): retention time 0.26 min, m/z 248 [M + H]+.
6-Hydroxy-2-(2-hydroxy-2-phenylethyl)-1,2,4-triazine-3,5(2H,4H)-dione (23)
To a solution of 21 (0.17 g, 0.37 mmol) in a 2:1 mixture of methanol and EtOAc (15 mL) were added a small spatula tip of chloro(1,5-cyclooctadiene)rhodium(I) dimer and one drop of triethylamine. The mixture was stirred for 36 h under hydrogen (300 psi) using a mechanical stirrer. The reaction was filtered and concentrated to give a brown oil, which was subsequently dissolved in dichloromethane (5 mL) and treated with a 1.0 M solution of BBr3 in dichloromethane (1.5 mL, 1.5 mmol) for 1.5 h. The reaction was quenched by the addition of water. The mixture was concentrated and purified by preparative HPLC (10% acetonitrile/90% water for 5 min, followed by an increase to 50% acetonitrile/50% water over 35 min and then an increase to 70% acetonitrile/30% water over 10 min; flow rate 15 mL/min) to give 0.024 g of 23 as a beige solid (26% yield); mp 202 °C. 1H NMR (DMSO-d6) δ 3.66 (dd, J = 4.0, 13.1 Hz, 1H), 3.85 (dd, J = 9.1, 13.4 Hz, 1H), 4.91 (dd, J = 4.3, 9.4 Hz, 1H), 5.52 (bs, 1H), 7.27–7.34 (m, 5H), 11.66 (s, 1H), 12.03 (s, 1H). LCMS (5% acetonitrile/95% water for 0.25 min followed by an increase to 40% acetonitrile/60% water over 1.5 min and continuation of 40% acetonitrile/60% water for 2.25 min; flow rate 1.25 mL/min): retention time 1.61 min, m/z 250 [M + H]+.
4-(Benzyloxymethyl)-6-bromo-2-phenyl-1,2,4-triazine-3,5(2H,4H)-dione (24)
To a solution of 16 (0.50 g, 1.6 mmol) in dichloromethane (25 mL) were added pyridine (0.26 mL, 3.20 mmol), phenylboronic acid (0.39 g, 3.20 mmol), and copper(II) acetate (0.44 g, 2.4 mmol). The mixture was stirred at rt overnight and filtered through a pad of Celite. The filtrate was concentrated, and the resulting residue was partitioned between EtOAc and water. The organic layer was washed with a 10% KHSO4 solution, dried over Na2SO4, and concentrated. The residue was purified by flash column chromatography using EtOAc/hexanes as eluent to give 0.62 g of 24 as a white solid (quantitative yield). 1H NMR (CDCl3) δ 4.77 (s, 2H), 5.61 (s, 2H), 7.30–7.37 (m, 5H), 7.42 (m, 1H), 7.46–7.49 (m, 4H).
6-(Benzyloxy)-4-(benzyloxymethyl)-2-phenyl-1,2,4-triazine-3,5(2H,4H)-dione (25)
Compound 25 was prepared from 24 as described for the preparation of 18a; yellow oil (44% yield). 1H NMR (CDCl3) δ 4.76 (s, 2H), 5.25 (s, 2H), 5.58 (s, 2H), 7.29–7.33 (m, 5H), 7.38–7.40 (m, 5H, 7.44–7.47 (m, 5H).
6-Hydroxy-2-phenyl-1,2,4-triazine-3,5(2H,4H)-dione (26)
Compound 26 was prepared from 25 as described for the preparation of 19a, with the exception that 4.0 equiv of BBr3 were used. After completion of the reaction, the reaction mixture was concentrated and the resulting residue was partitioned between EtOAc and water. The organic layer was washed with brine, dried over Na2SO4, and concentrated. The crude product was purified by trituration with EtOAc/hexanes; white powder (33% yield); mp 239 °C. 1H NMR (DMSO-d6) δ 7.32 (t, J = 7.3 Hz, 1H), 7.44 (t, J = 7.8 Hz, 1H), 7.49 (m, 2H), 11.85 (bs, 1H), 12.23 (s, 1H). LCMS (20% acetonitrile/80% water for 0.25 min followed by an increase to 85% acetonitrile/15% water over 1.5 min and continuation of 85% acetonitrile/15% water for 2.25 min; flow rate 1.25 mL/min): retention time 1.08 min, m/z 206 [M + H]+.
1-Phenethylhydrazinecarbothioamide (28)
To a suspension of 27 (5.00 g, 21.3 mmol) in ethanol (50 mL) was added ammonium thiocyanate (1.60 g, 21.0 mmol) at rt. The white suspension was heated at 78 °C for 50 h. The reaction was cooled to rt, and the resultant solid was removed by filtration. The filtrate was concentrated by approximately one-half, and the resultant solid was removed by filtration. This was repeated four times. The remaining filtrate was partitioned between EtOAc (50 mL) and satd NaHCO3 (20 mL). The organic layer was dried over MgSO4 and concentrated. The crude material was purified by silica gel column chromatography (80% EtOAc/hexanes w/0.1% NH4OH) to give 0.66 g of 28 as a white solid (16% yield). 1H NMR (DMSO-d6) δ 2.94 (m, 2H), 4.09 (m, 2H), 4.88 (s, 2H), 7.18–7.33 (m, 5H), 7.44 (br s, 2H).
Methyl 2-(2-Carbamothioyl-2-phenethylhydrazinyl)-2-oxoacetate (29)
To a solution of 28 (0.66 g, 3.4 mmol) in THF (15 mL) was added a solution of methyl chlorooxoacetate (0.41 g, 3.4 mmol) in THF (5 mL) over 2 min at rt. The mixture was stirred for 14 h and concentrated to dryness. The residue was dissolved in EtOAc (50 mL) and washed with satd NaHCO3 (25 mL). The organic layer was dried over MgSO4 and concentrated. The crude material was purified by silica gel column chromatography (5% MeOH/CH2Cl2) to give 0.30 g of 29 as a white solid (31% yield). 1H NMR (CDCl3) δ 3.09 (t, J = 7.03 Hz, 2H), 3.88 (s, 3H), 4.32 (m, 2H), 6.03 (s, 2H), 7.25–7.37 (m, 5H), 8.22 (brs, 1H).
6-Hydroxy-2-phenethyl-3-thioxo-3,4-dihydro-1,2,4-triazin-5(2H)-one (30)
To a solution of 29 (0.30 g, 1.1 mmol) in THF (5 mL) was added dropwise a solution of DBU (0.32 g, 2.1 mmol) in THF (3 mL) at rt. The mixture was stirred at rt for 14 h and then heated to 50 °C for 30 min. The reaction was concentrated to dryness. The residue was partitioned between EtOAc and 5% KHSO4. The organic layer was separated, dried over MgSO4, and concentrated. The crude material was purified by silica gel chromatography (1% AcOH/EtOAc) to give 0.031 g of 30 as a yellow solid (11% yield); mp 190–230 °C. 1H NMR (CD3OD) δ 3.11 (m, 2H), 4.41 (m, 2H), 7.19–7.31 (m, 5H). LCMS: retention time 2.90 min, m/z 250 [M + H]+.
5-Hydroxy-1-phenethylpyrimidine-2,4(1H,3H)-dione (32)
To a solution of 5-hydroxypyrimidine-2,4(1H,3H)-dione 31 (0.30 g, 2.3 mmol) in acetonitrile (15 mL) was added N,O-bis(trimethylsilyl)acetamide (2.0 mL, 8.2 mmol). The mixture was heated at 82 °C for 3 h, after which phenethyl iodide (0.54 g, 2.3 mmol) was added via syringe. The reaction was heated for 48 h at the same temperature, and the reaction mixture was concentrated in vacuo. The resulting residue was dissolved in dichloromethane, and the organic solution was washed twice with water, dried over Na2SO4, and concentrated. The crude material was triturated with cold diethyl ether and filtered to give 0.080 g of 32 as a tan solid (15% yield). 1H NMR (DMSO-d6): δ 2.86 (t, J = 8.0 Hz, 2H), 3.79 (t, J = 8.0 Hz, 2H), 7.10 (s, 1H), 7.22–7.31 (m, 5H), 8.53 (s, 1H), 11.36 (s, 1H). LCMS (20% acetonitrile/80% water for 0.25 min followed by an increase to 85% acetonitrile/15% water over 1.5 min and continuation of 85% acetonitrile/15% water for 2.25 min; flow rate 1.25 mL/min): retention time 0.45 min, m/z 233 [M + H]+.
Human DAAO (hDAAO) Purification
HEK cells expressing hDAAO were grown to confluence, harvested, washed, and resuspended in ice-cold assay buffer (Tris buffer, 50 mM, pH 8.5) containing flavin adenine dinucleotide (FAD, 100 μM) prior to sonication. The resulting cell lysate was centrifuged at 16000g for 10 min at 4 °C, and the hDAAO was purified using an UNO Q6 ion-exchange column pre-equilibrated in diluted assay buffer (10 mM, pH 8.5) on a FPLC system (Bio-Rad; Hercules, CA). The hDAAO was eluted by a linear gradient of 0–0.5 M NaCl, and the fractions with the highest DAAO activity were combined and stored at −80 °C in 20% glycerol until usage. The specific activity with respect to d-serine was determined to be 35.0 ± 0.2 μmol/min/mg, comparable to that of purified protein previously reported.31 The final concentration of hDAAO in the assay described below was adjusted in such a manner that a rate of 2.0 mOD/min can be achieved for the oxidation of d-serine (5 mM) in the absence of a DAAO inhibitor.
In Vitro DAAO Asay
d-Serine was purchased from Bachem Biosciences Inc. (King of Prussia, PA), horse radish peroxidase from Worthington Biochemical Corporation (Freehold, NJ), and o-phenylenediamine from Pierce Biotechnology, Inc. (Rockford, IL). All other chemicals were obtained from Sigma-Aldrich (St. Louis, MO). A reliable 96-well plate d-amino acid oxidase (DAAO) assay was developed based on previously published methods.32 Briefly, d-serine (5 mM) was oxidatively deaminated by hDAAO in the presence of molecular oxygen and flavin adenosine dinucleotide (FAD; 10 μM) to yield the corresponding α-keto acid, ammonia, and hydrogen peroxide. The resulting hydrogen peroxide was quantified using horseradish peroxidase (0.01 mg/mL) and o-phenylenediamine (180 μg/mL), which turns yellowish-brown upon oxidation. DAAO activity was correlated to the rate formation of the colored product, i.e., rate of change of absorbance at 411 nm. All reactions were carried out for 20 min at room temperature in a 100 μL volume in Tris buffer (50 mM, pH 8.5). Additionally, stock solutions and serial dilutions of potential DAAO inhibitors were made in 20:80 DMSO:buffer with a final assay DMSO concentration of 2%.
In Vitro Metabolic Stability Studies
The metabolic stability was evaluated using mouse liver microsomes. For the cytochrome P450 (CYP)-mediated metabolism, the reaction was carried out with 100 mM potassium phosphate buffer, pH 7.4, in the presence of NADPH regenerating system (1.3 mM NADPH, 3.3 mM glucose 6-phosphate, 3.3 mM MgCl2, 0.4 U/mL glucose-6-phosphate dehydrogenase, 50 μM sodium citrate). Reactions, in duplicate, were initiated by addition of the liver microsomes to the incubation mixture (compound final concentration was 5 μM; 0.5 mg/mL microsomes). For the glucuronidation reaction, a test compound were added to TRIS-HCl buffer (50 mM, pH 7.5) with microsomes (0.5 mg/mL), along with MgCl2 (8 mM) and alamethicin (25 μg/mL), and preincubated at 37 °C. The reaction was initiated (in duplicate) with UDPGA (2 mM; final concentration). Controls in the absence cofactors were carried out to determine the specific cofactor-free degradation. After 60 min of incubation, aliquots of the mixture were removed and the reaction quenched by addition of two times the volume of ice-cold acetonitrile spiked with the internal standard. Compound disappearance was monitored over time using a liquid chromatography and tandem mass spectrometry (LC/MS/MS) method.
In Vivo Pharmacokinetics
All procedures involving mice were approved by and conformed to the guidelines of the Institutional Animal Care Committee of the Johns Hopkins University. In vivo pharmacokinetics studies were performed using male CD1 mice (n = 3 for each time point for each group). Approximately 1 mL of whole blood was collected from each animal by cardiac puncture into heparinized microcentrifuge tubes, capped, gently inverted a few times, and stored on wet ice until centrifugation (10 min at 800g, 4 °C). Thereafter, the top layer of each tube (∼400 μL plasma) was aspirated via transfer pipet, dispensed into a clean nonheparinized microcentrifuge tube, and stored at −80 °C until subsequent analyses. Plasma levels of 8b and 11h were analyzed using an Agilent 1290 HPLC system coupled to an Agilent 6520 QTOF-MS system. Plasma d-serine level analyses were carried out using the previously reported method involving derivatization with Marfey’s reagent,33 followed by the quantification of the derivatized d-serine using an Agilent 1290 HPLC system coupled to an Agilent 6520 QTOF-MS system.
Acknowledgments
This work was in part supported by National Institutes of Health (R01MH091387 to T.T.) and the Johns Hopkins Brain Science Institute NeuroTranslational Drug Discovery program. We thank Reiji Tsukamoto for his technical assistance.
Glossary
Abbreviations Used
- DAAO
d-amino acid oxidase
- AUC
area under the curve
- BOM
benzyloxymethyl
- tPSA
topological polar surface area
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.5b00482.
Molecular formula strings (CSV)
The authors declare no competing financial interest.
Supplementary Material
References
- Nunes E. A.; MacKenzie E. M.; Rossolatos D.; Perez-Parada J.; Baker G. B.; Dursun S. M. D-serine and schizophrenia: an update. Expert Rev. Neurother. 2012, 12, 801–812. 10.1586/ern.12.65. [DOI] [PubMed] [Google Scholar]
- Kantrowitz J. T.; Malhotra A. K.; Cornblatt B.; Silipo G.; Balla A.; Suckow R. F.; D'Souza C.; Saksa J.; Woods S. W.; Javitt D. C. High dose D-serine in the treatment of schizophrenia. Schizophrenia Res. 2010, 121, 125–130. 10.1016/j.schres.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganote C. E.; Peterson D. R.; Carone F. A. The nature of D-serine--induced nephrotoxicity. Am. J. Pathol. 1974, 77, 269–282. [PMC free article] [PubMed] [Google Scholar]
- Curti B.; Ronchi S.; Simonetta P. M., D- and L-Amino Acid Oxidases. In Chemistry and Biochemistry of Flavoenzyme; Muller F., Ed. CRC Press: Boca Raton, FL, 1992; Vol. 3, pp 69–94. [Google Scholar]
- Rais R.; Thomas A. G.; Wozniak K.; Wu Y.; Jaaro-Peled H.; Sawa A.; Strick C. A.; Engle S. J.; Brandon N. J.; Rojas C.; Slusher B. S.; Tsukamoto T. Pharmacokinetics of oral D-serine in D-amino acid oxidase knockout mice. Drug Metab. Dispos. 2012, 40, 2067–2073. 10.1124/dmd.112.046482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa M.; Okamura T.; Kasai N.; Hori Y.; Summer K. H.; Konno R. D-amino-acid oxidase is involved in D-serine-induced nephrotoxicity. Chem. Res. Toxicol. 2005, 18, 1678–1682. 10.1021/tx0500326. [DOI] [PubMed] [Google Scholar]
- Sacchi S.; Caldinelli L.; Cappelletti P.; Pollegioni L.; Molla G. Structure-function relationships in human D-amino acid oxidase. Amino Acids 2012, 43, 1833–1850. 10.1007/s00726-012-1345-4. [DOI] [PubMed] [Google Scholar]
- Ferraris D. V.; Tsukamoto T. Recent advances in the discovery of D-amino acid oxidase inhibitors and their therapeutic utility in schizophrenia. Curr. Pharm. Des. 2011, 17, 103–111. 10.2174/138161211795049633. [DOI] [PubMed] [Google Scholar]
- Sacchi S.; Rosini E.; Pollegioni L.; Molla G. D-amino acid oxidase inhibitors as a novel class of drugs for schizophrenia therapy. Curr. Pharm. Des. 2013, 19, 2499–2511. 10.2174/1381612811319140002. [DOI] [PubMed] [Google Scholar]
- Adage T.; Trillat A. C.; Quattropani A.; Perrin D.; Cavarec L.; Shaw J.; Guerassimenko O.; Giachetti C.; Greco B.; Chumakov I.; Halazy S.; Roach A.; Zaratin P. In vitro and in vivo pharmacological profile of AS057278, a selective D-amino acid oxidase inhibitor with potential anti-psychotic properties. Eur. Neuropsychopharmacol. 2008, 18, 200–214. 10.1016/j.euroneuro.2007.06.006. [DOI] [PubMed] [Google Scholar]
- Sparey T.; Abeywickrema P.; Almond S.; Brandon N.; Byrne N.; Campbell A.; Hutson P. H.; Jacobson M.; Jones B.; Munshi S.; Pascarella D.; Pike A.; Prasad G. S.; Sachs N.; Sakatis M.; Sardana V.; Venkatraman S.; Young M. B. The discovery of fused pyrrole carboxylic acids as novel, potent D-amino acid oxidase (DAO) inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 3386–3391. 10.1016/j.bmcl.2008.04.020. [DOI] [PubMed] [Google Scholar]
- Ferraris D.; Duvall B.; Ko Y. S.; Thomas A. G.; Rojas C.; Majer P.; Hashimoto K.; Tsukamoto T. Synthesis and biological evaluation of D-amino acid oxidase inhibitors. J. Med. Chem. 2008, 51, 3357–3359. 10.1021/jm800200u. [DOI] [PubMed] [Google Scholar]
- Duplantier A. J.; Becker S. L.; Bohanon M. J.; Borzilleri K. A.; Chrunyk B. A.; Downs J. T.; Hu L. Y.; El-Kattan A.; James L. C.; Liu S.; Lu J.; Maklad N.; Mansour M. N.; Mente S.; Piotrowski M. A.; Sakya S. M.; Sheehan S.; Steyn S. J.; Strick C. A.; Williams V. A.; Zhang L. Discovery, SAR, and pharmacokinetics of a novel 3-hydroxyquinolin-2(1H)-one series of potent D-amino acid oxidase (DAAO) inhibitors. J. Med. Chem. 2009, 52, 3576–3585. 10.1021/jm900128w. [DOI] [PubMed] [Google Scholar]
- Berry J. F.; Ferraris D. V.; Duvall B.; Hin N.; Rais R.; Alt J.; Thomas A. G.; Rojas C.; Hashimoto K.; Slusher B. S.; Tsukamoto T. Synthesis and SAR of 1-hydroxy-1H-benzo[d]imidazol-2(3H)-ones as inhibitors of D-amino acid oxidase. ACS Med. Chem. Lett. 2012, 3, 839–843. 10.1021/ml300212a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katane M.; Osaka N.; Matsuda S.; Maeda K.; Kawata T.; Saitoh Y.; Sekine M.; Furuchi T.; Doi I.; Hirono S.; Homma H. Identification of novel D-amino acid oxidase inhibitors by in silico screening and their functional characterization in vitro. J. Med. Chem. 2013, 56, 1894–1190. 10.1021/jm3017865. [DOI] [PubMed] [Google Scholar]
- Kawazoe T.; Tsuge H.; Imagawa T.; Aki K.; Kuramitsu S.; Fukui K. Structural basis of D-DOPA oxidation by D-amino acid oxidase: alternative pathway for dopamine biosynthesis. Biochem. Biophys. Res. Commun. 2007, 355, 385–391. 10.1016/j.bbrc.2007.01.181. [DOI] [PubMed] [Google Scholar]
- Raje M.; Hin N.; Duvall B.; Ferraris D. V.; Berry J. F.; Thomas A. G.; Alt J.; Rojas C.; Slusher B. S.; Tsukamoto T. Synthesis of kojic acid derivatives as secondary binding site probes of d-amino acid oxidase. Bioorg. Med. Chem. Lett. 2013, 23, 3910–3913. 10.1016/j.bmcl.2013.04.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hondo T.; Warizaya M.; Niimi T.; Namatame I.; Yamaguchi T.; Nakanishi K.; Hamajima T.; Harada K.; Sakashita H.; Matsumoto Y.; Orita M.; Takeuchi M. 4-Hydroxypyridazin-3(2H)-one derivatives as novel D-amino acid oxidase inhibitors. J. Med. Chem. 2013, 56, 3582–3592. 10.1021/jm400095b. [DOI] [PubMed] [Google Scholar]
- Hopkins S. C.; Heffernan M. L.; Saraswat L. D.; Bowen C. A.; Melnick L.; Hardy L. W.; Orsini M. A.; Allen M. S.; Koch P.; Spear K. L.; Foglesong R. J.; Soukri M.; Chytil M.; Fang Q. K.; Jones S. W.; Varney M. A.; Panatier A.; Oliet S. H.; Pollegioni L.; Piubelli L.; Molla G.; Nardini M.; Large T. H. Structural, kinetic, and pharmacodynamic mechanisms of D-amino acid oxidase inhibition by small molecules. J. Med. Chem. 2013, 56, 3710–3724. 10.1021/jm4002583. [DOI] [PubMed] [Google Scholar]
- Terry-Lorenzo R. T.; Chun L. E.; Brown S. P.; Heffernan M. L.; Fang Q. K.; Orsini M. A.; Pollegioni L.; Hardy L. W.; Spear K. L.; Large T. H. Novel human D-amino acid oxidase inhibitors stabilize an active site lid open conformation. Biosci. Rep. 2014, 34, 487–499. 10.1042/BSR20140071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange J. H.; Venhorst J.; van Dongen M. J.; Frankena J.; Bassissi F.; de Bruin N. M.; Besten C.; de Beer S. B.; Oostenbrink C.; Markova N.; Kruse C. G. Biophysical and physicochemical methods differentiate highly ligand-efficient human D-amino acid oxidase inhibitors. Eur. J. Med. Chem. 2011, 46, 4808–4819. 10.1016/j.ejmech.2011.04.023. [DOI] [PubMed] [Google Scholar]
- Zimmermann S. C.; Rais R.; Alt J.; Burzynski C.; Slusher B. S.; Tsukamoto T. Structure-metabolism relationships in the glucuronidation of D-amino acid oxidase inhibitors. ACS Med. Chem. Lett. 2014, 5, 1251–1253. 10.1021/ml500335z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zajac M.; Muszalska I. Mechanism of ceftriaxone degradation in aqueous solution. Acta Polym. Pharm. 1998, 55, 35–39. [PubMed] [Google Scholar]
- Dudfield P. J.; Le V.-D.; Lindell S. D.; Rees C. W. Synthesis of C-ribosyl imidazo[2,1-f ][1,2,4]triazines as inhibitors of adenosine and AMP deaminases. J. Chem. Soc., Perkin Trans. 1 1999, 2929–2936. 10.1039/a904065j. [DOI] [Google Scholar]
- Pontillo J.; Guo Z.; Wu D.; Struthers R. S.; Chen C. Synthesis of aryl-1,2,4-triazine-3,5-diones as antagonists of the gonadotropin-releasing hormone receptor. Bioorg. Med. Chem. Lett. 2005, 15, 4363–4366. 10.1016/j.bmcl.2005.06.057. [DOI] [PubMed] [Google Scholar]
- Leroy I.; Dupont-Passelaigue E.; Valeille K.; Rival Y.; Junquero D.. Derivatives of triazines and uracils, their preparation and their application in human therapeutics. U.S. Patent 8,618,287, Dec31, 2013.
- Jensen K. A.; Anthoni U.; Kägi B.; Larsen C.; Pedersen C. T.; Rosen U. Studies of thioacids and their derivatives. IX. Thiosemicarbazides. Acta Chem. Scand. 1968, 22, 1–50. 10.3891/acta.chem.scand.22-0001. [DOI] [Google Scholar]
- Branch C. L.; Eggleston D. S.; Haltiwanger R. C.; Kaura A. C.; Tyler J. W. Synthesis of 6-Hydroxy-2-methyl-3-thioxo-2H-1,2,4-triazin-5-one. Synth. Commun. 1996, 26, 2075–2084. [Google Scholar]
- Nagata Y.; Shimojo T.; Akino T. D-amino acid oxidase in mouse liver—II. Comp. Biochem. Physiol. B 1988, 91, 503–504. 10.1016/0305-0491(88)90012-0. [DOI] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawazoe T.; Tsuge H.; Pilone M. S.; Fukui K. Crystal structure of human D-amino acid oxidase: context-dependent variability of the backbone conformation of the VAAGL hydrophobic stretch located at the si-face of the flavin ring. Protein Sci. 2006, 15, 2708–2717. 10.1110/ps.062421606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook S. P.; Galve-Roperh I.; Martinez del Pozo A.; Rodriguez-Crespo I. Direct calcium binding results in activation of brain serine racemase. J. Biol. Chem. 2002, 277, 27782–27792. 10.1074/jbc.M111814200. [DOI] [PubMed] [Google Scholar]
- Berna M. J.; Ackermann B. L. Quantification of serine enantiomers in rat brain microdialysate using Marfey’s reagent and LC/MS/MS. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2007, 846, 359–363. 10.1016/j.jchromb.2006.08.029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.