

ABSTRACT
The mechanical properties of the ECM strongly influence the behavior of all cell types within a given tissue. Increased matrix tension promotes epithelial cell proliferation by engaging mitogenic mechanotransduction signaling including the Salvador/Warts/Hippo, PI 3-kinase, Rho, Wnt and MAP kinase pathways. The Rho signaling pathways in particular are capable of increasing intra-cellular tension by elevating the production and contractility of the actomyosin cytoskeleton, which counteracts tension changes within the matrix in a process termed mechano-reciprocity. We have discovered that Rho-ROCK signaling increases the production of ECM through paracrine signaling between the epithelium and fibroblasts and also the remodeling of the ECM by regulating focal adhesion dynamics in fibroblasts. These two phenomena together cause increased ECM tension. Enhanced mechano-reciprocity results in ever-increasing intra- and extra-cellular tension in a vicious cycle that promotes cell proliferation and tumor progression. These insights reveal that inhibiting mechano-reciprocity, reducing ECM tension and targeting cancer-associated fibroblasts in a coordinated fashion has potential as cancer therapy.
KEYWORDS: 14-3-3ζ, cancer, extra-cellular matrix, fibroblasts, mechanoreciprocity, mechanotransduction, MYPT, Rho, ROCK, wound healing
Introduction
Mechanical forces in biology
The capacity to exert mechanical force upon an object is a fundamental requirement for physical interaction with the environment. Flowing from this is the need to detect and measure external forces exerted upon the body. Everyday activities like sitting, standing and handling objects require not only the exertion of mechanical force, but the ability to detect mechanical signals and coordinate a timely response. This is facilitated by the sense of proprioception, which is mediated by mechanoreceptors, nerve endings specialized for the detection of forces. However, it has been clear for some time that mechanical force can be detected not only by specialized nerve receptors, but also by all other cell types. Indeed, the generation and detection of mechanical force is a key aspect of cell and developmental biology. Cells interact with their environment by exerting and sensing mechanical forces between themselves and the extra-cellular matrix (ECM). These mechanical forces greatly influence cell behavior, by guiding decisions about cell migration, growth, division and differentiation.
Mechanical signaling and cancer
Whereas the biochemical aspects of unrestrained cell growth and proliferation in cancer have been extensively studied over many decades, an understanding of the biophysical mechanisms underlying how force and mechanical stress influence tumor development is only just emerging. However, as early as 1972, reports appeared that mechanical force has the capacity to influence tumor growth and metastasis.1 Throughout the late 70s and early 80s, reports emerged that the application of mechanical stress upon cells is capable of eliciting distinct phenotypic responses.2-4 The first clues into how mechanical force may be converted to a biochemical signal for propagation within the cell were provided by Carvalho et al.5 who demonstrated that integrin expression and subcellular distribution is markedly altered upon the application of external force on cells. As integrins were well known to link cells to the extra-cellular matrix6 and were dysregulated in cancers, they then became the prime candidates for the mechanosensing (Fig. 1) receptors transducing mechanical signals to the intra-cellular biochemical machinery. This pioneering work caused a flurry of papers providing evidence for a link between extra-cellular mechanical stresses and changes in actin cytoskeletal structure, adhesion complex composition and number, cell migration and spreading, and proliferation via integrin ligation (reviewed in ref. 7), thereby linking extra-cellular biophysical signals to intra-cellular biochemical changes. This process became known as mechanotransduction (Fig. 1) and several signaling pathways including those mediated by Salvador/Warts/Hippo,8 PI 3-kinase,9 Rho small GTPases,10 Wnts11,12 and MAP kinases10 have been shown to be activated downstream of changes in the mechanical properties of the ECM.
Figure 1.

Mechanosensing through engagement of integrins with the ECM causes mechanotransduction, by activation of Talin and autophosphorylation of FAK, which initiates several intra-cellular signal transduction pathways, including PI 3-kinase/Akt and ROCK signaling. Mechanotransduction gives rise to increased actin polymerization and actomyosin contractility, establishing mechano-reciprocity, which in turn leads to paracrine signaling between the parenchyma and the stroma, increasing ECM production and remodeling.
It therefore follows that changes in the mechanical properties of the ECM have implications for tumor progression, as mitogenic pathways are directly linked to changes in ECM stiffness via mechanotransduction. However, the observation that ECM was increased in many different cancers and directly influenced tumor growth and spread13,14 suggested that tumors themselves were capable of strongly promoting changes in the ECM that facilitated their growth and spread. Several growth factors of tumor origin, including TGFβ, CTGF, IL6 and LIF are known to directly act upon tumor-associated fibroblasts and other stromal cells to increase the production and remodeling of ECM molecules to increase ECM stiffness.15-18 While some of the mechanisms underlying mechano-sensation, mechanotransduction and conversely the direct involvement of tumors in regulating ECM stiffness are beginning to be uncovered, a precise understanding of how these processes are integrated and coordinated still remains elusive.
The Rho family of small GTPases and the regulation of intracellular tension
Rho (Ras homology) GTPases are monomeric GTP-binding proteins and are a subset of the Ras superfamily, comprising 22 members of which the best characterized are Rho, Rac and Cdc42. Rho family members act as molecular regulators of signal transduction by switching between GDP-bound inactive and GTP-bound active states, and have numerous molecular targets including kinases, transcription factors and scaffold proteins that mediate diverse cellular processes such as migration, proliferation, adhesion and apoptosis in cell type and temporal context-dependent ways (reviewed in ref. 19). The regulation of actomyosin cytoskeletal dynamics by these proteins is the best studied aspect of their biology.
Dynamic remodeling of the actin cytoskeleton to generate intra-cellular force is key to cell migration. The extension of actin filament networks by actin polymerization generates intra-cellular forces, as filaments in the leading edge are compressed between transient associations with the cell membrane and the bulk of the actin cytoskeletal network behind them. As protrusions grow and retract, individual actin filaments undergo tension from transient bonds with the membrane, and are bent or compressed depending on their orientation.20 Myosin-mediated contraction of the actin cytoskeleton results in a reduction in plasma membrane surface area and causes clustering of cell surface integrins, which are responsible for attachment to the ECM. Integrin-mediated ECM adhesion activates guanine nucleotide exchange factors (GEFs), which enhance RhoA activity.21 During ameboid cell movement, Rac interacts with lamellipodin at the leading edge to extend actin filaments and generate lamellipodia, sheet-like cell protrusions supported by short actin filaments in a branched network, which adhere to substrates and determine the direction of cell movement.22 Cdc42 controls the formation of filopodia, thin protrusions from the cell membrane that contain parallel actin bundles for mechanosensing the environment in order to determine cell polarity—another mechanism by which the direction of movement is determined.23 In order to direct migration, actin polymerization must be restricted to a portion of the plasma membrane. Cdc42 directs this polarity, by acting through the Par polarity complex, localizing Rac activity and stabilizing microtubules within the cytoskeleton. At the trailing edge of cells exhibiting mesenchymal motility, and to a lesser extent those exhibiting ameboid motility, RhoA controls retraction through activation of effector kinases such as Rho-associated kinase (ROCK).24,25 Furthermore, Rho-mediated activation of ROCK is required for the formation of membrane ruffles and lamellae and in particular membrane blebs. Membrane blebs are a feature of ameboid motility (also termed blebbing motility), a mode of cell migration that is independent of lamellipodia and frequently observed in cancer cells.26 Rho signaling is also responsible for the generation of intra-cellular tension, which is required for the morphological changes associated with other cellular processes such as apoptosis27,28 and cytokinesis.29
The effector proteins of Rho signaling, Rho-associated kinases 1 and 2 (ROCK1 and ROCK2), are serine-threonine kinases containing a Rho-binding domain and are activated upon interaction with Rho-GTP.30 Activation of ROCK by Rho induces stress fiber assembly in a number of ways. ROCK directly phosphorylates and activates the regulatory myosin light chain (MLC),31 it indirectly increases MLC phosphorylation by phosphorylating and inhibiting the myosin targeting subunit of MLC phosphatase (MYPT)32 and directly phosphorylates and activates the LIM kinases (LIMK1 and LIMK2), which subsequently phosphorylate and inhibit cofilin thereby stabilizing actin structures.33 In cultured cells, parallel bundles of actin filaments form stress fibers that link sites of focal adhesions, permitting tension to be transmitted between focal adhesions and to the ECM. As well as being the product of intra-cellular tension, stress fibers themselves are able to exert force upon focal adhesions, permitting forces exerted by the external environment to be counteracted.21
Rho-ROCK signaling and the regulation of extra-cellular tension
Not surprisingly, cells are not merely passive reactors to changes in the mechanical properties of the ECM, but are active players in the remodeling of their environments. Protein components of the ECM can be degraded by a variety of proteinases such as the families of matrix metalloproteinases (MMPs)34 and a disintegrin and metalloproteinases (ADAMs)35 that are produced by cancer cells to remodel the ECM. While most proteinases acting on the ECM are secreted, a subset of MMPs are membrane tethered, permitting cells to exert a greater degree of spatial control over their deployment.36 The ECM may also be remodeled by changes to its composition and level of cross-linking. Lysyl oxidases and lysyl hydroxylases are produced by cells to crosslink collagen chains, thereby increasing ECM stiffness.37 The mechanical properties of the ECM are also significantly altered in diseased states, such as chronic wounds, cancer and fibrosis, by changes to ECM composition.38,39
Recently, we demonstrated a mechanism by which epithelial cells and tumor cells of epithelial origin are capable of increasing the mechanical stiffness of the ECM by elevating the production of ECM components.14,39,40 Activation of ROCK in the context of tumor cells or the hyper-proliferation of epidermal cells in the context of wound healing results in paracrine signals arising from the proliferating epithelia that act on stromal fibroblasts, increasing their production of collagen, fibronectin, periostin and tenascin C, components of the ECM, and thereby increasing matrix stiffness.14,39 While the nature of the specific paracrine signals caused by activation of ROCK remains to be uncovered, cytokines and other secreted molecules of tumor origin have been previously implicated in the recruitment and activation of cancer-associated fibroblasts, the key regulators of ECM composition and stiffness in the tumor context.15-17 Nevertheless, ROCK activation in tumor cells results in increased ECM stiffness of the magnitude frequently observed in epithelial cancers including cutaneous squamous cell carcinoma,14 breast cancer13 and pancreatic cancer41 and promotes tumor progression. We therefore propose that ROCK integrates inputs from growth factor and mechanotransduction signaling to produce the appropriate cellular response, be it migration, proliferation, ECM production or remodeling.
Biochemical changes arising from increased extra-cellular tension
External forces drive clustering of integrin molecules at the cell membrane, stabilizing focal adhesions and linking the intra-cellular actin cytoskeleton to the ECM. The stiffer the ECM or substrate to which a cell is attached, the greater the size and strength of those focal adhesions, and the greater the cellular response, as intra-cellular forces are generated to balance the extra-cellular forces. Upon integrin activation, talin and vinculin are recruited to the complex, and focal adhesion kinase (FAK) is auto-phosphorylated at Y397 and/or Y925 and activated. FAK phosphorylation results in the recruitment of Src-homology-2 (SH2) domain-containing proteins Src and Shc that link FAK to the Ras pathway and Rho-ROCK pathway and, via PI 3-kinase and Akt activation, to the stabilization of β-catenin and the transcriptional activity of TCF/LEF transcription factors. Extra-cellular signal-regulated kinase (ERK) is also regulated by the FAK-Src complex, and is responsible for mechanical signal transduction from the microenvironment, to regulate intra-cellular processes.10,14,42
Responses to cell-matrix interactions are governed by a complex interplay between signaling pathways involving FAK, ERK, β-catenin, Rho and others, and cells are therefore able to respond to extra-cellular forces in myriad ways. For example, in the mammary gland, where development and homeostasis occur within the context of a pliable ECM, Rho activity in fibroblasts was increased upon heightened matrix rigidity. This led to an increase in focal adhesion assembly and growth factor-dependent ERK activation, suggesting that a mechano-regulatory “circuit” amalgamates stimulatory extra-cellular tension with focal adhesion formation through ERK and Rho-dependent cytoskeletal changes, to maintain homeostasis.13
Mechano-reciprocity
Mechano-reciprocity (Fig. 1) is the ability of cells to enhance or moderate intra-cellular tension in order to adapt to increased or reduced extra-cellular stiffness respectively43 (termed outside-in signaling), but conversely may also refer to their ability to remodel the extra-cellular matrix and modify its mechanical properties in order to offset changes in intra-cellular tension (inside-out signaling). In a situation where sustained growth factor stimulation or oncogenic mutation causes increased mechanotransduction signaling such as via the Rho, YAP, β-catenin and/or MAP kinase pathways, the combination of inside-out and outside-in signaling has the potential to establish a vicious cycle by which ever-escalating ECM stiffness causes uncontrolled cell proliferation. Indeed, this is observed in mechano-responsive cancers such as those of the skin, breast and intestine14,44 and is characterized by persistent activation of the ROCK protein. We have shown that enhanced mechano-reciprocity of this kind promotes tumor progression.14 Targeting runaway mechano-reciprocity is therefore a novel way in which diseases of cellular homeostasis characterized by increased ECM stiffness such as cancer or fibrosis may be treated.
Mechano-reciprocity in diseases of cellular homeostasis
It is becoming increasingly evident that persistent mechanical signaling can advance cancer progression. Overexpression of Rho GTPases has been associated with progression of disease in a number of malignancies, and Rho, Rac, Cdc42 and ROCK45 have all been reported to be mutated in various cancers to confer tumor promoting functions and inhibit tumor suppressive functions (reviewed in ref. 46).
The role of mechanical signaling in driving tumorigenesis has been well characterized in the mammary gland. A known mechano-responsive tissue,44 the mammary epithelium is subjected to a multitude of external forces throughout development and cycling, such as ductal morphogenesis, lactation, and involution to remodel the gland and degrade the ECM.47 A pliable ECM is best suited to these morphological changes. Compression analysis of normal and tumor tissue of murine mammary origin demonstrated that although normal mammary tissue was soft, tumor and peri-tumoral tissue was significantly stiffer.10 Matrix stiffness is promoted by a greater amount of collagen cross-linking.48 This ECM stiffening is sensed by cellular integrins, which in turn activate RhoA signaling, resulting in generation of focal adhesions and influencing tumor cell invasion, cell proliferation, and changes in cytoskeletal organization. Thus, a persistent and high level of mechano-reciprocity is potentially able to create a feed-forward mechanism, promoting further cancer development. This phenotype is often seen in patients with a high mammographic density and it is becoming clearer that mammographic density in the normal breast is an important prognostic marker for breast cancer, with a higher density indicating a 4-to-6 fold higher likelihood of developing breast cancer.49
We have found that skin in which ROCK had been hyper-activated exhibited increased MLC2 phosphorylation and elevated ECM production resulting in greater tissue stiffness. Increased tissue stiffness gave rise to mechano-reciprocal activation of the PtdIns 3-kinase signaling pathway which led to the stabilization of β-catenin and epidermal hyper-proliferation. Interestingly, in the context of the multi-step chemical carcinogenesis model, mechano-reciprocity cooperated with oncogenic transformation to promote tumor progression in a manner that was dependent on ROCK-induced tissue stiffness. Conversely, inhibition of ROCK signaling by treatment with the inhibitor Y-27632 lowered ECM stiffness and impeded tumor formation and growth.14,50 These observations clearly show that tumor cells have a key role in establishing a stiff ECM that promotes their own proliferation and growth.
A number of parallels exist between normal and malignant tissue development and wound healing. Rapid cell proliferation and mechanisms mediating cytoskeletal changes during cell migration are similar in acute wound healing to those involved in tumor progression. At all stages of wound healing, including hemostasis, inflammation, proliferation and remodeling, examples of mechano-reciprocity exist between the ECM and a number of cell types including platelets, immune cells, fibroblasts, endothelial cells and keratinocytes.51 We have recently reported that enhanced signaling through ROCK, occurring during either cutaneous squamous cell carcinoma progression or acute skin wound healing and which causes increased ECM production in both contexts, is negatively regulated by the phospho-serine binding molecular adaptor protein 14-3-3ζ.39 14-3-3ζ bound to the ROCK antagonist MYPT1, promoting its MLC phosphatase function and antagonizing ROCK-mediated phosphorylation of MLC. Mice lacking 14-3-3ζ exhibited increased signal flux through ROCK leading to enhanced mechano-reciprocity, causing rapid wound healing. This observation was corroborated using a novel pharmacological inhibitor of 14-3-3ζ, which accelerated acute wound healing in wild-type mice.39
Interestingly, this negative regulatory mechanism that moderated signaling through ROCK and thereby limited mechano-reciprocity, was lost in the majority of patient samples of cutaneous squamous cell carcinoma, which exhibited little or no 14-3-3ζ compared to normal skin. Accordingly, 14-3-3ζ-deficient mice on the multi-step chemical carcinogenesis protocol formed larger tumors than wild-type mice of the same strain background. Taken together, these observations lead us to the conclusion that 14-3-3ζ-mediated promotion of MLC dephosphorylation is a mechanism by which mechano-reciprocity is maintained within manageable limits such that normal wound healing is facilitated while tumor formation is prevented (Fig. 2).
Figure 2.
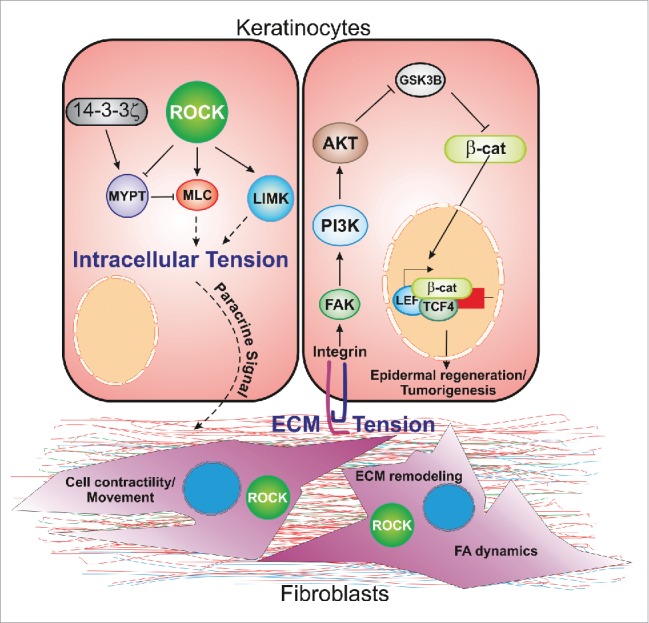
ROCK activation enhances intracellular tension in epidermal cells by activating MLC and LIM kinases. It also increases extracellular tension by elevating ECM production by dermal fibroblasts. ROCK activation in dermal fibroblasts promotes ECM remodeling by regulating focal adhesion dynamics and fibroblast migration. In both contexts, 14-3-3ζ limits signal flux through ROCK, thereby maintaining mechano-reciprocity between normal physiological boundaries, permitting normal wound healing and protecting against tumor formation. This figure has been adapted from ref. 50.
We do not believe that 14-3-3ζ is the only negative regulator of mechano-reciprocity. Under steady-state conditions, 14-3-3ζ-deficient mice exhibit thinner skin than wild-type mice,39 suggesting that compensatory mechanisms as yet undiscovered and not involving the seven other 14-3-3 family members are capable of maintaining normal cellular homeostasis at near-physiological levels.
Targeting mechano-reciprocity and the extra-cellular matrix as therapy
Given the highly tumor-promoting environment caused by increased mechano-reciprocity, targeting the players that facilitate it (ROCK signaling, fibroblasts and their activation) and enhancing the activities of its negative regulators (14-3-3ζ) are potentially novel approaches to cancer therapy. However, an important consideration is the effect that inhibition of pleiotropic proteins like ROCK or 14-3-3ζ may have on cellular homeostasis and indeed normal development. For instance, tumor capillary endothelial cells subjected to stress exhibited cytoskeletal rearrangements, and exerted stronger Rho-ROCK-mediated traction compared to non-cancer cells. Pre-treatment with the ROCK inhibitor Y-27632 before the application of stress restored normal actin behavior in tumor cells, but caused significant cytoskeletal disruption in normal cells.52 This indicates that normal and tumor cells differ in sensitivity to external mechanical force as governed by ROCK signaling and illustrates the challenges accompanying attempts to target tumors via the mechanotransduction machinery.
The most common current therapy regimes involve targeting tumor cells directly via inhibition of cell-intrinsic functions such as their proliferative capacity or their ability to evade apoptosis, whereas the therapies that offer the most promise, such as immunotherapy or hormone therapy, target non-intrinsic functions that require cell to cell communication. Their need for cell to cell communication could be viewed as a vulnerability of cancers. The cell to ECM communication mechanisms that enhance mechano-reciprocity are mediated by communication between cancer cells and normal (fibroblast) cells lacking oncogenic mutations, a particularly serious vulnerability. Normalizing the ECM by normalizing tumor-associated fibroblasts therefore holds out the tantalizing possibility of therapies that halt or even reverse tumor progression. This approach may also provide new opportunities for combination therapies where the tumor and its ECM are both targeted concurrently.
Concluding remarks
In conclusion, analyzing Rho-ROCK pathway activation in the in vivo context has revealed its function in augmenting mechano-reciprocity via enhanced ECM stiffness. These observations have also highlighted the importance of mechano-reciprocity in normal tissue homeostasis and demonstrated that negative regulators of this process have significant therapeutic utility.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was funded by the National Health and Medical Research Council, the Cancer Council South Australia's Beat Cancer Project and the Royal Adelaide Hospital Research Fund. MSS was supported by a Future Fellowship from the Australian Research Council.
References
- [1].Cohen HJ, Laszlo J. Influence of trauma on the unusual distribution of metastases from carcinoma of the larynx. Cancer 1972; 29:466-71 [DOI] [PubMed] [Google Scholar]
- [2].De Witt MT, Handley CJ, Oakes BW, Lowther DA. In vitro response of chondrocytes to mechanical loading. The effect of short term mechanical tension. Connect Tissue Res 1984; 12:97-109; PMID:6327186; http://dx.doi.org/ 10.3109/03008208408992775 [DOI] [PubMed] [Google Scholar]
- [3].Franke RP, Grafe M, Schnittler H, Seiffge D, Mittermayer C, Drenckhahn D. Induction of human vascular endothelial stress fibres by fluid shear stress. Nature 1984; 307:648-9; PMID:6537993; http://dx.doi.org/ 10.1038/307648a0 [DOI] [PubMed] [Google Scholar]
- [4].Takeuchi S. Wound healing in the cornea of the chick embryo. IV. Promotion of the migratory activity of isolated corneal epithelium in culture by the application of tension. Dev Biol 1979; 70:232-40; PMID:456741; http://dx.doi.org/ 10.1016/0012-1606(79)90019-8 [DOI] [PubMed] [Google Scholar]
- [5].Carvalho RS, Scott JE, Yen EH. The effects of mechanical stimulation on the distribution of β 1 integrin and expression of β 1-integrin mRNA in TE-85 human osteosarcoma cells. Arch Oral Biol 1995; 40:257-64; PMID:7541624; http://dx.doi.org/ 10.1016/0003-9969(95)98814-F [DOI] [PubMed] [Google Scholar]
- [6].Schwartz MA, Ingber DE. Integrating with integrins. Mol Biol Cell 1994; 5:389-93; PMID:8054683; http://dx.doi.org/ 10.1091/mbc.5.4.389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Banes AJ, Tsuzaki M, Yamamoto J, Fischer T, Brigman B, Brown T, Miller L. Mechanoreception at the cellular level: the detection, interpretation, and diversity of responses to mechanical signals. Biochem Cell Biol 1995; 73:349-65; PMID:8703408; http://dx.doi.org/ 10.1139/o95-043 [DOI] [PubMed] [Google Scholar]
- [8].Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, et al.. Role of YAP/TAZ in mechanotransduction. Nature 2011; 474:179-83; PMID:21654799; http://dx.doi.org/ 10.1038/nature10137 [DOI] [PubMed] [Google Scholar]
- [9].Chaudhuri O, Koshy ST, Branco da Cunha C, Shin JW, Verbeke CS, Allison KH, Mooney DJ. Extracellular matrix stiffness and composition jointly regulate the induction of malignant phenotypes in mammary epithelium. Nat Mater 2014; 13:970-8; PMID:24930031; http://dx.doi.org/ 10.1038/nmat4009 [DOI] [PubMed] [Google Scholar]
- [10].Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, et al.. Tensional homeostasis and the malignant phenotype. Cancer Cell 2005; 8:241-54; PMID:16169468; http://dx.doi.org/ 10.1016/j.ccr.2005.08.010 [DOI] [PubMed] [Google Scholar]
- [11].Burkhalter RJ, Symowicz J, Hudson LG, Gottardi CJ, Stack MS. Integrin regulation of β-catenin signaling in ovarian carcinoma. J Biol Chem 2011; 286:23467-75; PMID:21518759; http://dx.doi.org/ 10.1074/jbc.M110.199539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Case N, Ma M, Sen B, Xie Z, Gross TS, Rubin J. Beta-catenin levels influence rapid mechanical responses in osteoblasts. J Biol Chem 2008; 283:29196-205; PMID:18723514; http://dx.doi.org/ 10.1074/jbc.M801907200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Provenzano PP, Inman DR, Eliceiri KW, Knittel JG, Yan L, Rueden CT, White JG, Keely PJ. Collagen density promotes mammary tumor initiation and progression. BMC Med 2008; 6:11; PMID:18442412; http://dx.doi.org/ 10.1186/1741-7015-6-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Samuel MS, Lopez JI, McGhee EJ, Croft DR, Strachan D, Timpson P, Munro J, Schröder E, Zhou J, Brunton VG, et al.. Actomyosin-mediated cellular tension drives increased tissue stiffness and β-catenin activation to induce epidermal hyperplasia and tumor growth. Cancer Cell 2011; 19:776-91; PMID:21665151; http://dx.doi.org/ 10.1016/j.ccr.2011.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Albrengues J, Bourget I, Pons C, Butet V, Hofman P, Tartare-Deckert S, Feral CC, Meneguzzi G, Gaggioli C. LIF mediates proinvasive activation of stromal fibroblasts in cancer. Cell reports 2014; 7:1664-78; PMID:24857661; http://dx.doi.org/ 10.1016/j.celrep.2014.04.036 [DOI] [PubMed] [Google Scholar]
- [16].Depner S, Lederle W, Gutschalk C, Linde N, Zajonz A, Mueller MM. Cell type specific interleukin-6 induced responses in tumor keratinocytes and stromal fibroblasts are essential for invasive growth. Int J Cancer 2014; 135:551-62; PMID:23165423; http://dx.doi.org/ 10.1002/ijc.27951 [DOI] [PubMed] [Google Scholar]
- [17].Quante M, Tu SP, Tomita H, Gonda T, Wang SS, Takashi S, Baik GH, Shibata W, Diprete B, Betz KS, et al.. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell 2011; 19:257-72; PMID:21316604; http://dx.doi.org/ 10.1016/j.ccr.2011.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Okada H, Kikuta T, Kobayashi T, Inoue T, Kanno Y, Takigawa M, Sugaya T, Kopp JB, Suzuki H. Connective tissue growth factor expressed in tubular epithelium plays a pivotal role in renal fibrogenesis. J Am Soc Nephrol 2005; 16:133-43; PMID:15574513; http://dx.doi.org/ 10.1681/ASN.2004040339 [DOI] [PubMed] [Google Scholar]
- [19].Ridley AJ. Historical overview of Rho GTPases. Methods Mol Biol 2012; 827:3-12; PMID:22144264; http://dx.doi.org/ 10.1007/978-1-61779-442-1_1 [DOI] [PubMed] [Google Scholar]
- [20].Romet-Lemonne G, Jegou A. Mechanotransduction down to individual actin filaments. Eur J Cell Biol 2013; 92:333-8; PMID:24252518; http://dx.doi.org/ 10.1016/j.ejcb.2013.10.011 [DOI] [PubMed] [Google Scholar]
- [21].Burridge K, Wittchen ES. The tension mounts: stress fibers as force-generating mechanotransducers. J Cell Biol 2013; 200:9-19; PMID:23295347; http://dx.doi.org/ 10.1083/jcb.201210090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Krause M, Gautreau A. Steering cell migration: lamellipodium dynamics and the regulation of directional persistence. Nat Rev Mol Cell Biol 2014; 15:577-90; PMID:25145849; http://dx.doi.org/ 10.1038/nrm3861 [DOI] [PubMed] [Google Scholar]
- [23].Farhan H, Hsu VW. Cdc42 and Cellular Polarity: Emerging Roles at the Golgi. Trends Cell Biol 2015; 26(4):241-8; PMID:26704441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ridley AJ. Rho GTPase signalling in cell migration. Curr Opin Cell Biol 2015; 36:103-12; PMID:26363959; http://dx.doi.org/ 10.1016/j.ceb.2015.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zegers MM, Friedl P. Rho GTPases in collective cell migration. Small GTPases 2014; 5:e28997; PMID:25054920; http://dx.doi.org/ 10.4161/sgtp.28997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].O'Connor K, Chen M. Dynamic functions of RhoA in tumor cell migration and invasion. Small GTPases 2013; 4:141-7; PMID:24025634; http://dx.doi.org/ 10.4161/sgtp.25131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Coleman ML, Sahai EA, Yeo M, Bosch M, Dewar A, Olson MF. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat Cell Biol 2001; 3:339-45; PMID:11283606; http://dx.doi.org/ 10.1038/35070009 [DOI] [PubMed] [Google Scholar]
- [28].Croft DR, Coleman ML, Li S, Robertson D, Sullivan T, Stewart CL, Olson MF. Actin-myosin-based contraction is responsible for apoptotic nuclear disintegration. J Cell Biol 2005; 168:245-55; PMID:15657395; http://dx.doi.org/ 10.1083/jcb.200409049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kosako H, Goto H, Yanagida M, Matsuzawa K, Fujita M, Tomono Y, Okigaki T, Odai H, Kaibuchi K, Inagaki M. Specific accumulation of Rho-associated kinase at the cleavage furrow during cytokinesis: cleavage furrow-specific phosphorylation of intermediate filaments. Oncogene 1999; 18:2783-8; PMID:10348354; http://dx.doi.org/ 10.1038/sj.onc.1202633 [DOI] [PubMed] [Google Scholar]
- [30].Samuel MS, Olson MF. ROCK Kinases (Rho-associated coiled-coil containing protein kinase) In: Choi S, ed. Encyclopedia of Signaling Molecules. New York: Springer; 2012:1686-90. [Google Scholar]
- [31].Amano M, Ito M, Kimura K, Fukata Y, Chihara K, Nakano T, Matsuura Y, Kaibuchi K. Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). J Biol Chem 1996; 271:20246-9; PMID:8702756; http://dx.doi.org/ 10.1074/jbc.271.34.20246 [DOI] [PubMed] [Google Scholar]
- [32].Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, Yamamori B, Feng J, Nakano T, Okawa K, et al.. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science 1996; 273:245-8; PMID:8662509; http://dx.doi.org/ 10.1126/science.273.5272.245 [DOI] [PubMed] [Google Scholar]
- [33].Maekawa M, Ishizaki T, Boku S, Watanabe N, Fujita A, Iwamatsu A, Obinata T, Ohashi K, Mizuno K, Narumiya S. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science 1999; 285:895-8; PMID:10436159; http://dx.doi.org/ 10.1126/science.285.5429.895 [DOI] [PubMed] [Google Scholar]
- [34].Verma RP, Hansch C. Matrix metalloproteinases (MMPs): chemical-biological functions and (Q)SARs. Bioorg Med Chem 2007; 15:2223-68; PMID:17275314; http://dx.doi.org/ 10.1016/j.bmc.2007.01.011 [DOI] [PubMed] [Google Scholar]
- [35].Edwards DR, Handsley MM, Pennington CJ. The ADAM metalloproteinases. Mol Aspects Med 2008; 29:258-89; PMID:18762209; http://dx.doi.org/ 10.1016/j.mam.2008.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Page-McCaw A, Ewald AJ, Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat Rev Mol Cell Biol 2007; 8:221-33; PMID:17318226; http://dx.doi.org/ 10.1038/nrm2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Cox TR, Gartland A, Erler JT. Lysyl Oxidase, a Targetable Secreted Molecule Involved in Cancer Metastasis. Cancer Res 2016; 76:188-92; PMID:26732355; http://dx.doi.org/ 10.1158/0008-5472.CAN-15-2306 [DOI] [PubMed] [Google Scholar]
- [38].Oskarsson T. Extracellular matrix components in breast cancer progression and metastasis. Breast 2013; 22 Suppl 2:S66-72; PMID:24074795; http://dx.doi.org/ 10.1016/j.breast.2013.07.012 [DOI] [PubMed] [Google Scholar]
- [39].Kular J, Scheer KG, Pyne NT, Allam AH, Pollard AN, Magenau A, Wright RL, Kolesnikoff N, Moretti PA, Wullkopf L, et al.. A Negative Regulatory Mechanism Involving 14-3-3zeta Limits Signaling Downstream of ROCK to Regulate Tissue Stiffness in Epidermal Homeostasis. Dev Cell 2015; 35:759-74; PMID:26702834; http://dx.doi.org/ 10.1016/j.devcel.2015.11.026 [DOI] [PubMed] [Google Scholar]
- [40].Ibbetson SJ, Pyne NT, Pollard AN, Olson MF, Samuel MS. Mechanotransduction pathways promoting tumor progression are activated in invasive human squamous cell carcinoma. Am J Pathol 2013; 183:930-7; PMID:23830873; http://dx.doi.org/ 10.1016/j.ajpath.2013.05.014 [DOI] [PubMed] [Google Scholar]
- [41].Shields MA, Dangi-Garimella S, Redig AJ, Munshi HG. Biochemical role of the collagen-rich tumour microenvironment in pancreatic cancer progression. Biochem J 2012; 441:541-52; PMID:22187935; http://dx.doi.org/ 10.1042/BJ20111240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Provenzano PP, Inman DR, Eliceiri KW, Keely PJ. Matrix density-induced mechanoregulation of breast cell phenotype, signaling and gene expression through a FAK-ERK linkage. Oncogene 2009; 28:4326-43; PMID:19826415; http://dx.doi.org/ 10.1038/onc.2009.299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lopez JI, Mouw JK, Weaver VM. Biomechanical regulation of cell orientation and fate. Oncogene 2008; 27:6981-93; PMID:19029939; http://dx.doi.org/ 10.1038/onc.2008.348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Schedin P, Keely PJ. Mammary gland ECM remodeling, stiffness, and mechanosignaling in normal development and tumor progression. Cold Spring Harb Perspect Biol 2011; 3:a003228; PMID:20980442; http://dx.doi.org/ 10.1101/cshperspect.a003228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lochhead PA, Wickman G, Mezna M, Olson MF. Activating ROCK1 somatic mutations in human cancer. Oncogene 2010; 29:2591-8; PMID:20140017; http://dx.doi.org/ 10.1038/onc.2010.3 [DOI] [PubMed] [Google Scholar]
- [46].Orgaz JL, Herraiz C, Sanz-Moreno V. Rho GTPases modulate malignant transformation of tumor cells. Small GTPases 2014; 5:e29019; PMID:25036871; http://dx.doi.org/ 10.4161/sgtp.29019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Paszek MJ, Weaver VM. The tension mounts: mechanics meets morphogenesis and malignancy. J Mammary Gland Biol Neoplasia 2004; 9:325-42; PMID:15838603; http://dx.doi.org/ 10.1007/s10911-004-1404-x [DOI] [PubMed] [Google Scholar]
- [48].Keely PJ. Mechanisms by which the extracellular matrix and integrin signaling act to regulate the switch between tumor suppression and tumor promotion. J Mammary Gland Biol Neoplasia 2011; 16:205-19; PMID:21822945; http://dx.doi.org/ 10.1007/s10911-011-9226-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].McCormack VA, dos Santos Silva I. Breast density and parenchymal patterns as markers of breast cancer risk: a meta-analysis. Cancer Epidemiol Biomarkers Prev 2006; 15:1159-69; PMID:16775176; http://dx.doi.org/ 10.1158/1055-9965.EPI-06-0034 [DOI] [PubMed] [Google Scholar]
- [50].Samuel MS, Olson MF. Actomyosin contractililty: force power drives tumor growth. Cell Cycle 2011; 10:3409-10; PMID:22067650; http://dx.doi.org/ 10.4161/cc.10.20.17722 [DOI] [PubMed] [Google Scholar]
- [51].Schultz GS, Davidson JM, Kirsner RS, Bornstein P, Herman IM. Dynamic reciprocity in the wound microenvironment. Wound Repair Regen 2011; 19:134-48; PMID:21362080; http://dx.doi.org/ 10.1111/j.1524-475X.2011.00673.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Ghosh K, Thodeti CK, Dudley AC, Mammoto A, Klagsbrun M, Ingber DE. Tumor-derived endothelial cells exhibit aberrant Rho-mediated mechanosensing and abnormal angiogenesis in vitro. Proc Natl Acad Sci U S A 2008; 105:11305-10; PMID:18685096; http://dx.doi.org/ 10.1073/pnas.0800835105 [DOI] [PMC free article] [PubMed] [Google Scholar]