

ABSTRACT
PREX2 is a PTEN interacting protein that is significantly mutated in melanoma and pancreatic ductal adenocarcinoma. Recently, we reported the mechanistic basis of melanomagenesis by PREX2 mutations. Truncating PREX2 mutations activate its guanine nucleotide exchange factor activity for its substrate RAC1. This leads to increased PI3K/AKT signaling associated with reduced DNA methylation and increased cell proliferation in NRAS-mutant melanoma. Here, we provide additional data that indicates a reciprocal regulation of PREX2 by PTEN whereby loss of PTEN results in a dramatic increase in expression of PREX2 at the protein level. Pharmacologic studies revealed destabilization of PREX2 by inhibition of PI3K/AKT signaling. Additionally, we provide data to show a selective decrease in a particular histone mark, H4 Lys20 trimethylation, in cells expressing PREX2 E824* truncating mutation globally and at the imprint control region of CDKN1C (also known as p57) and IGF2. The decrease in H4K20 trimethylation coupled with DNA hypomethylation at this particular locus is associated with genomic imprinting and regulation of expression of p57 and IGF2. Taken together, these results demonstrate the complex signaling mechanisms that involve PREX2, PI3K/AKT/PTEN and downstream epigenetic machinery to deregulate expression of key cell cycle regulators.
Keywords: epigenetics, genomic imprinting, mouse models of cancer, PI3K, PREX2, RAC1
Recent next generation DNA sequencing of tumors have provided insight into the complexity of somatic mutations and given us a compendium of novel significantly mutated cancer genes.1 It is expected that functional genomic studies will provide detailed molecular and functional roles for these mutations in tumor development. We identified PREX2 (phosphatidylinositol-3, 4, 5-triphosphate-dependent Rac-exchange factor 2) as being significantly mutated in human melanomas.2 Interestingly, the International Cancer Genomics Consortium (ICGC) recently reported that PREX2 is also significantly mutated in pancreatic ductal adenocarcinoma.3
PREX2 is a guanine nucleotide exchange factor (GEF) for RAC1 and is a known PTEN binding protein.4,5 Mechanistically, PREX2 has been shown to regulate RAC1 mediated cellular invasion in a manner that cross-talks with PTEN signaling and also regulates insulin signaling and glucose homeostasis through the PI3K pathway.6,7 Initial melanoma genome sequencing studies showed various patterns of PREX2 mutations including missense and truncating mutations. Using xenograft models, we were able to show that truncating PREX2 mutations have oncogenic activity.2 However, the exact mechanism behind PREX2 mutations driven melanoma development was unclear.
To study PREX2 mutations in a tissue and time restricted manner, we generated an inducible transgenic mouse model that expresses a truncating PREX2 mutant (TetO-lox-STOP-lox-PREX2E824*) in melanocytes.8 We crossed these transgenic mice with mice that have a melanoma sensitizing background, i.e. lack the tumor suppressor Ink/Arf and inducibly express a constitutively active NRASQ61K to generate a genetically engineered mouse (GEM) model of melanoma. Tumor formation was induced by administration of tamoxifen (which induces cre mediated recombination and removal of the stopper cassette) and doxycycline (which allows expression of PREX2E824% and NRASQ61K transgenes from tet-responsive promoters). Interestingly, we observed increased incidence of melanoma formation in mice harboring the inducible PREX2 transgene. We also generated xenograft tumors by expressing control GFP, PREX2 wild type or various PREX2 truncating mutations in primary immortalized melanocytes. Again, truncating PREX2 mutations induced increased tumor formation. To explore the molecular mechanisms behind the ability of PREX2 mutations to induce increased tumor formation, we performed gene expression profiling of tumors from both xenograft and GEM models. Integrative cross-species analysis revealed regulations of cell cytoskeleton organization, cell cycle and ribosome biogenesis as key biological pathways to be significantly enriched in tumors having PREX2 truncating tumors. The connection of PREX2 to RAC1 can explain the changes in cell cytoskeleton signaling pathway while the known role of PREX2 in PTEN biology is expected to explain the enrichment in ribosome biogenesis. However, it was not clear why cell cycle regulation is perturbed in PREX2 mutant tumors and we investigated this aspect further.
Histologically, we saw that PREX2 mutant tumors are highly proliferative and show increased Ki67 (a commonly used marker of proliferating cells) staining. Further, we observed reduced expression of key negative cell cycle regulators such as CDKN1C (also known as p57) and CDKN1B (also known as p27) and increased IGF2 (insulin like growth factor 2) in PREX2 mutant tumors. So, how are truncating mutations in PREX2 resulting in these biological changes? To answer this question, we next studied the biochemical and signaling consequences of truncating PREX2 mutations. First, we purified recombinant full length PREX2 or an N-terminal truncated PREX2 (M1-R363) from Sf9 cells and performed guanine nucleotide exchange (GEF) activity assays using RAC1 as a substrate. This revealed higher GEF activity of the truncated PREX2 compared to full length PREX2. In support of this data, we also showed that cells having PREX2 truncating mutation (K278*, E824* and Q1430*) have increased GEF activity as demonstrated by increased GTP loaded RAC1.
What is the molecular basis for the increased GEF activity of truncated PREX2? Our results suggest the existence of at least 2 cooperating mechanisms to account for this. First, truncating PREX2 mutations do not bind to PTEN which is known to have inhibitory effect on the GEF activity of PREX2.6 Hence, by avoiding the inhibitory influence of PTEN, PREX2 mutants have an intrinsically higher GEF activity. Second, using structural modeling of PREX2:RAC1 interaction, we demonstrated that the GEF domain of PREX2 behaves similar to the GEF domain of PREX1. It is known that the C-terminus of PREX1 has an auto-inhibitory role on its N-terminal GEF activity.9,10 Our structural model suggests that a similar mechanism exists in PREX2 and PREX2 truncating mutations relieve the auto-inhibition of GEF activity by the C-terminus. Next we asked what the consequences of increased PREX2 GEF activity and RAC1 activation are. Using reverse phase protein array (RPPA) and immunoblotting, we observed increased phosphorylation of AKT at Ser473 and Thr308 in PREX2 mutant tumors. Gain-of-function and loss-of-function experiments revealed that the increased activation of AKT by PREX2 mutations was dependent on activation of RAC1. This is in line with earlier studies that demonstrated RAC1 can directly bind and activate PI3K.11-13 This mechanism adds another layer of complexity in Rac1-PI3K interaction which is known to involve complex feedback loops including actin cytoskeletal and local phosphorylated lipid changes to sustain activation of the pathway.14
Next we wondered whether the PI3K/AKT pathway in turn exerts any regulatory effects on PREX2. To answer this, we first utilized mouse embryonic fibroblasts (MEFs) isolated from Rosa26-CreERT2 and PTENL/L double positive mice. Introduction of 4-hydroxytamoxifen to these cells activated Cre resulting in genomic recombination, depletion of PTEN and activation of the PI3K/AKT signaling pathway. Interestingly, we observed a dramatic increase in PREX2 protein expression upon acute depletion of PTEN in primary MEFs in vitro (Fig. 1A). This increase in PREX2 protein expression was not associated with an increase in mRNA expression, suggesting post-transcriptional mechanisms such as protein stability are responsible for the observation (Fig. 1B). In line with this observation, inhibition of phosphoinositide-3 (PI3) kinase signaling with pan-PI3 kinase inhibitor (NVP-BKM120) or a dual PI3K/mTOR inhibitor (NVP-BEZ235) led to dose dependent reduction in PREX2 expression (Fig. 1C). Importantly, the reduction in PREX2 protein induced by PI3K inhibitor BKM120 was rescued by co-treatment of cells with the proteasome inhibitor MG132 further supporting the notion that the primary regulatory mechanism of PREX2 expression is at the level of protein stability or translation (Fig. 1D). Taken together, these results show the presence of a reciprocal interplay between PREX2 and the PI3K pathway whereby PREX2 truncating mutations activate the PI3K pathway and activation of the PI3K pathway in turn results in stabilization of PREX2. This interplay is expected to amplify signaling once initiated.
Figure 1.
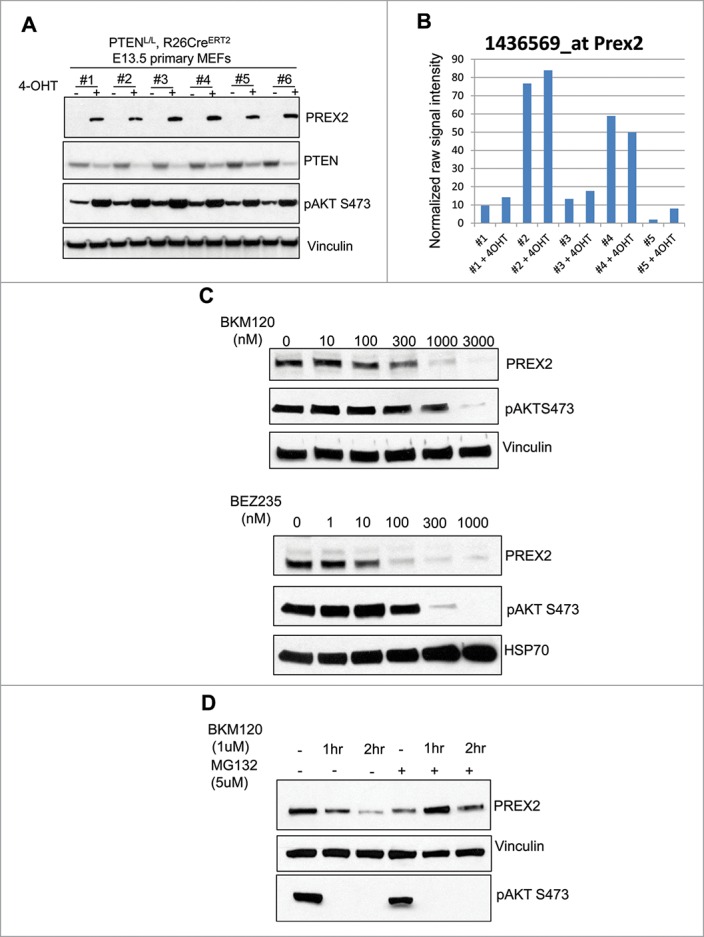
PTEN deletion increases PREX2 expression while PI3K/AKT inhibitors destabilize its expression. (A) Mouse embryonic fibroblasts (MEFs) were isolated from PTENL/L, Rosa26-CreERT2 E14 embryos and treated with 1µM of 4-hydroxytamoxifin (4-OHT) to induce Cre-mediated genetic recombination and deletion of PTEN. Cells were lysed and whole cell extracts were then subjected to blotting with respective antibodies. The experiments were performed twice. (B) MEFs were isolated from PTENL/L, Rosa26-CreERT2 E14 embryos and treated with 1µM of 4-hydroxytamoxifin (4-OHT) 3 times to induce Cre-mediated genetic recombination and deletion of PTEN. RNA was isolated and microarray was done. Plot represents normalized signal intensity of a probe that corresponds to PREX2 across multiple independently generated MEF lines with our without 4-OHT induced PTEN depletion. (C-D) 4-hydroxytamoxifen (4-OHT) treated (hence PTEN deleted) E14 PTENL/L, Rosa26-CreERT2 MEFs were treated with the indicated concentrations of NVP-BKM120 or NVP-BEZ235 for 24 hours. Cells were lysed and immunoblotted as shown (left panels). (D) Alternatively, similar MEFs were left untreated or were pretreated with MG132 (5µM) for 30 minutes prior to addition of NVP-BKM120 (1µM) for the indicated time points. Whole cell lysates were subjected to immunoblotting with the respective antibodies. Experiments were performed twice.
Finally, we investigated how changes in upstream signaling events are related to changes in gene expression regulation. As we have observed strong downregulation of p57 and upregulation of IGF2, we used the gene expression regulation of these genes as read outs to investigate the molecular mechanisms of their perturbation in PREX2 mutant tumors. In our published report, we showed that PREX2 mutation or PTEN deletion induces DNA hypomethylation at the critical imprint control region of p57 and IGF2 which has the well-known effect of downregulating p57 and upregulating IGF2.15-17
Here, we provide further information that support these observations. First, we performed global chromatin immunoprecipitation and sequencing (ChIP-Seq) analysis of an additional epigenetic regulatory mark which is implicated in regulation of genomic imprinting, i.e Histone H4 lysine 20 methylation.16,18,19 We investigated all 3 possible methylation marks, (H4K20) mono-(Me1), di-(Me2) and tri-methylation (Me3), in primary immortalized melanocytes. We observed significant global reduction in H4K20Me3, but not in H4K20Me1 and Me2, in cells expressing PREX2 mutants as compared to GFP control and wildtype PREX2 (Fig. 2A–C). Analysis of H4K20Me3 ChIP-seq profile using a peak-calling algorithm, Scripture,20 showed that H4K20 marks were indeed enriched in the imprint control region as described before,21 and that H4K20Me3 mark was reduced at the imprint control region in cells expressing mutant PREX2 compared to wildtype controls (Fig. 3A). To further quantify these results, we performed qPCR analysis of the H4K20Me3 levels in the ICR locus which showed consistent reduction of this methylation mark in PREX2 mutant samples (Fig. 3B).
Figure 2.
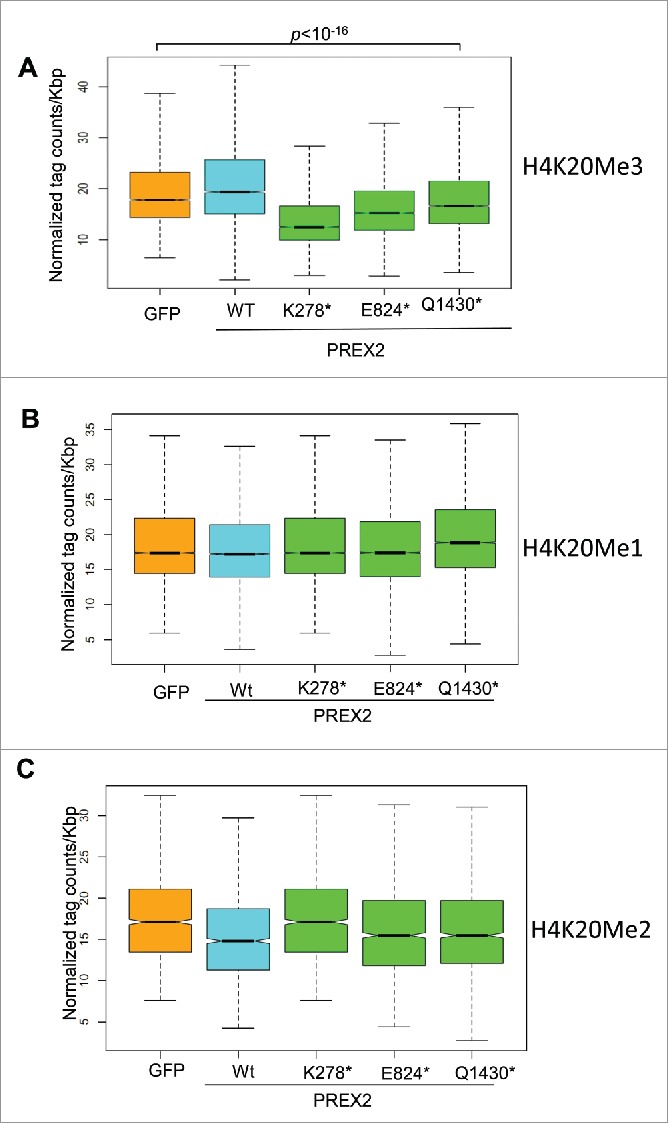
H4K20Me3, but not H4K20ME1 and H4K20Me2, is decreased in PREX2 mutants. (A-C) Quantification of ChIP-Seq data of Histone H4 Lysine 20 (H4K20) tri-methylation (H4K20Me3), H4K20 mono-methylation (H4K20Me1) and H4K20 di-methylation (H4K20ME2). Total read numbers were normalized to 10 million reads for each sample and these values used for the following analyses. Aggregate plots were generated using the NGSplot toolbox around GFP-Me3 binding sites. Box plots were generated calculating RPKM values around GFP-Me3/2/1 enriched regions for each sample. Wilcoxon test was applied to compare conditions. Boxplots depict normalized H4K20Me3 read numbers per kilobase pair (Kbp) for GFP control, WT or respective PREX2 mutants over genome-wide peak regions of H4K20ME3 identified in GFP control melanocytes. ChIP-seq was done once.
Figure 3.
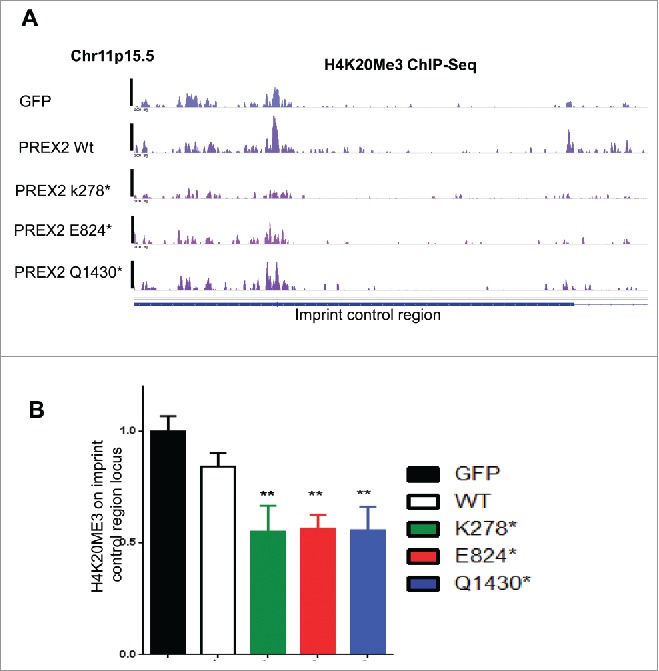
H4K20Me3 is decreased at the imprint control region of CDKN1C in PREX2 mutants. H4K20Me3 comparison in melanocytes expressing control GFP, wild type or various PREX2 mutants. (A) shows H4K20Me3 ChIP-Seq tracks in melanocytes around the imprint control region (ICR) of CDKN1C (at Chr11p.15.5) on chromosome 11 and (B) Quantitative PCR comparing H4K20Me3 levels at the ICR locus. Primers amplify one of the major H4K20ME3 peaks as seen in the ChIP-Seq tracks from (A). ** denotes p <0.05 after comparing to GFP controls by Student's t-test and error bars show standard deviation.
In summary, our study demonstrated the oncogenic capacity of truncating PREX2 mutations in vivo. We identified a direct link between an established oncogenic signaling pathway i.e the PI3K/AKT pathway and provided insights into the downstream regulation of cell growth regulators p57 and IGF2, in melanoma pathogenesis (Fig. 4). However, a number of important outstanding questions remain. Atomic level X-ray structures of full length and truncated PREX2 protein in complex with its substrate will be of great help in elucidating the molecular basis of PREX2 GEF activation. Our understanding of the role of RAC1 in activation of the PI3K/AKT pathway is still rudimentary and need to be explored in greater depth using biochemical, genetic and pharmacologic tools. A further interesting research question is how PREX2 protein stability is affected by the PI3K signaling. What is the ubiquitin ligase that targets PREX2 for degradation? Is it regulated by PI3K/AKT mediated phosphorylation? Finally, even though we have shown 2 major epigenetic marks to be altered in PREX2 mutant cells, we still do not know the direct molecular link between PREX2 and these changes. Are any of the DNA or histone methylatransferases or histone demethylases phosphorylated by PI3K/AKT? Does PI3K, AKT, PTEN or PREX2 interact with H4K20 methyltransferases such as Suv420h1/2? Research to identify the molecular link between changes in the RAC1-PI3K/AKT pathway and downstream epigenetic and gene expression changes is an exciting future endeavor which could have potential translational benefits.
Figure 4.
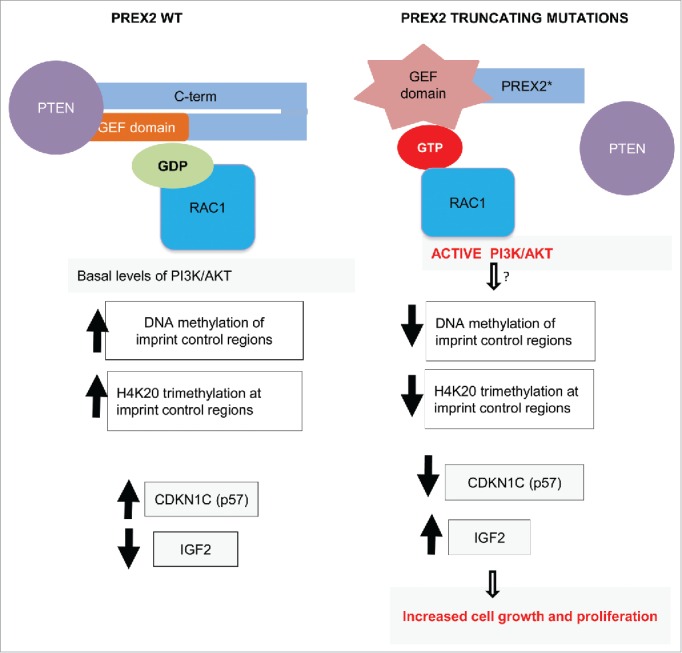
Working model of consequences of PREX2 truncating mutations. PREX2 is bound to PTEN and its guanine nucleotide exchange factor (GEF) domain is kept in inactive state associated with its C-terminus (C-term) in wild type cells. This results in basal levels of downstream signaling events. In tumors with truncating PREX2 mutations, mutant PREX2 is unable to interact with PTEN. Furthermore, its intramolecular inhibitory confirmation is lost resulting in activation of its GEF activity. This results in GTP loading of RAC1 that in turn leads to direct activation of the PI3K/AKT pathway. These alterations (via still unknown links) perturb DNA methylation and H4K20 trimethylation at critical genomic imprint control region leading to downregulation of a key cell cycle regulator CDKN1C (also known as p57) and upregulation of IGF2 which result in increased cell growth and proliferation.
Materials and methods
Cell culture and generation of mouse embryonic fibroblasts
The generation of human primary melanocytes (PMEL/hTERT/CDK4(R24C)/P53DD with either NRASG12D or BRAFV600E have been previously described.22 These cells were grown in Ham's F10 medium (Invitrogen), containing 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. 293FT cells were grown in DMEM (Invitrogen) containing 10% FBS. We generated MEFs from embryonic day 13.5 (E13.5) embryos using standard methods following PTENL/L, Rosa26-CreERT2 mouse intercrosses. MEFs were grown in DMEM (Invitrogen) containing 10% FBS and 1% penicillin/streptomycin.
Deletion of PTEN by 4-hydroxytamoxifen treatment; NVS-BKM120 and NVS-BEZ235 treatment
Freshly isolated PTENL/L, Rosa26-CreERT2 MEFs were treated once with 1µM 4-hydroxytamoxifen. Whole cell extracts and RNA were isolated from these early passage MEFs (passage 3). This results in partial PTEN depletion. MEFs were treated with BKM120, BEZ235 or MG132 at various concentrations in DMSO for indicated times for time course experiments.
Western blotting and antibodies
Cells were lysed in lysis buffer (20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 2 mM EDTA, 1% NP40) containing protease and phosphatase inhibitor cocktail tablets (Roche) on ice. Lysates were subjected to denaturation by boiling in SDS-loading buffer (BioRad). Lysates were then loaded on SDS-PAGE gels (BioRad) and transferred using BioRad Turbo semidry transfer system to nitrocellulose membranes which were probed with respective antibodies: PREX2 antibodies (1:1000, Sigma-Aldrich or monoclonal antibody from Abcam), PTEN (1:1000), phospho-Akt S475 (1:1000,Cell Signaling), vinculin (1:5000)
Chromatin immunoprecipitation and sequencing
Cells (5 million per antibody) were crosslinked using 1% paraformaldehyde for 10 mins at 37 degrees. Reaction was quenched by 0.125 M glycine for 5 mins, cells washed with PBS and stored at −80°C. Next day cells were thawed on ice and lysed with RIPA buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA pH 8.0, 140 mM NaCl, 1% Triton x-100, 0.1%SDS, 0.1% DOC) for 10 min on ice. Sonication was performed using Branson Sonifier 250 to achieve shear length of 300–600 bp. Extracts were then incubated overnight with respective antibody-dynabead mixture that were incubated separately for 1 hr at 4°C earlier [Rabbit IgG, H4K20me1, H4K20me2, H4K20me3 (ALL Abcam)]. Immunecomplexes were then washed 5 times with RIPA buffer, once with RIPA-500 (RIPA with 500 mM NaCl) and once with LiCl wash buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA pH 8.0, 250 mM LiCl ,0.5% NP-40 ,0.5% DOC). Elution and decrosslinking was performed in direct elution buffer (10 mM Tris-Cl pH 8.0, 5 mM EDTA, 300 mM NaCl, 0.5% SDS) by incubating immunecomplxes at 65°C for 4–16 hrs. Proteinase K (20 mg/ml) and RNaseA treatment was performed and DNA cleaned up using SPRI beads (Beckman-Coulter). Library preparation was done using NEBNext ChIP-Seq library preparation kit per manufacturer's instructions. Illumina paired end adapters were used. Sequencing was performed in Hiseq2000 (Illumina).
H4K20Me3 ChIP and qPCR
ChIP was done as described above. Next quantitative PCR for 2 regions (R1 and R2; R1 being test region at KCNQ1OT1 locus and R2 being negative region showing no enrichment in ChIP-Seq) was performed on ChIP DNA (H4K20me3) using SYBR Greener reagent and Stratagene Mx3005p instrument. Relative enrichment on R1 normalized to values on R2 are shown.
Primer information is shown below:
R1 – Test region at imprint control locus
840: TTCCTGACCCACTACTCTGT
841: CAGTGAATCATCTTCGGAGTG
R2 - Negative region
808: GTGTACGATTCTTAACCATGGAG
809: CATAAACTGAGTGAGTTACCAG
ChIP-Seq analysis
H4K20ME ChIP-Seq reads were aligned using Bowtie (version 1.0.0)23 to human genome assembly NCBI Build 37 (UCSC hg19) with the following parameters: -n 1 -m 1 –best –strata (uniquely mapped reads with one mismatch were retained).
Enriched regions were detected by Scripture package20 with varying window sizes (750,1500,5000), and peaks were defined whose coverage at 0.05 significance.24 Collapsed repeat regions in the human genome25 and “ultra-high signal suspect regions” identified by ENCODE project26 (https://docs.google.com/spreadsheet/ccc?key=0Am6FxqAtrFDwdE5LYWh2MkVscmtCWEdtNUN2eEVEYmcandhl=en#gid=5) were masked during the peak calling procedure. Called regions were merged with BEDTools.27 Intervals with less than 350 base pair length and enriched regions in whole cell extract samples were excluded from the final list. Sequences were extended to 200 bp and average read counts were calculated in 25 bp bins. Total read numbers were normalized to 10 million reads for each sample and these values used for the following analyses.
Aggregate plots were generated using the NGSplot toolbox (https://code.google.com/p/ngsplot/) around GFP-Me3 binding sites. Box plots were generated calculating RPKM values around GFP-Me3/2/1 enriched regions for each sample. Wilcoxon test was applied to compare conditions.
Abbreviations
- PI3K
phosphatidylinositol-4,5-bisphosphate 3-kinase
- PREX2
phosphatidylinositol-3, 4, 5-triphosphate-dependent Rac exchange factor 2
- PTEN
phosphatase and tensin homolog
- RAC1
ras-related C3 botulinum toxin substrate 1
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Garraway LA, Lander ES. Lessons from the cancer genome. Cell 2013; 153:17-37; PMID:23540688; http://dx.doi.org/ 10.1016/j.cell.2013.03.002 [DOI] [PubMed] [Google Scholar]
- [2].Berger MF, Hodis E, Heffernan TP, Deribe YL, Lawrence MS, Protopopov A, Ivanova E, Watson IR, Nickerson E, Ghosh P, et al.. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature 2012; 485:502-6; PMID:22622578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, Johns AL, Miller D, Nones K, Quek K, et al.. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015; 518:495-501; PMID:25719666; http://dx.doi.org/ 10.1038/nature14169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Fine B, Hodakoski C, Koujak S, Su T, Saal LH, Maurer M, Hopkins B, Keniry M, Sulis ML, Mense S, et al.. Activation of the PI3K pathway in cancer through inhibition of PTEN by exchange factor P-REX2a. Science 2009; 325:1261-5; PMID:19729658; http://dx.doi.org/ 10.1126/science.1173569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Donald S, Hill K, Lecureuil C, Barnouin R, Krugmann S, John Coadwell W, Andrews SR, Walker SA, Hawkins PT, Stephens LR, et al.. P-Rex2, a new guanine-nucleotide exchange factor for Rac. FEBS Lett 2004; 572:172-6; PMID:15304343; http://dx.doi.org/ 10.1016/j.febslet.2004.06.096 [DOI] [PubMed] [Google Scholar]
- [6].Mense SM, Barrows D, Hodakoski C, Steinbach N, Schoenfeld D, Su W, Hopkins BD, Su T, Fine B, Hibshoosh H, et al.. PTEN inhibits PREX2-catalyzed activation of RAC1 to restrain tumor cell invasion. Sci Signal 2015; 8:ra32; PMID:25829446; http://dx.doi.org/ 10.1126/scisignal.2005840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hodakoski C, Hopkins BD, Barrows D, Mense SM, Keniry M, Anderson KE, Kern PA, Hawkins PT, Stephens LR, Parsons R. Regulation of PTEN inhibition by the pleckstrin homology domain of P-REX2 during insulin signaling and glucose homeostasis. Proc Natl Acad Sci U S A 2014; 111:155-60; PMID:24367090; http://dx.doi.org/ 10.1073/pnas.1213773111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lissanu Deribe Y, Shi Y, Rai K, Nezi L, Amin SB, Wu CC, Akdemir KC, Mahdavi M, Peng Q, Chang QE, et al.. Truncating PREX2 mutations activate its GEF activity and alter gene expression regulation in NRAS-mutant melanoma. Proc Natl Acad Sci U S A 2016; 113(9):E1296–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lucato CM, Halls ML, Ooms LM, Liu HJ, Mitchell CA, Whisstock JC, Ellisdon AM. The Phosphatidylinositol (3,4,5)-Trisphosphate-dependent Rac Exchanger 1.Ras-related C3 Botulinum Toxin Substrate 1 (P-Rex1.Rac1) Complex Reveals the Basis of Rac1 Activation in Breast Cancer Cells. J Biol Chem 2015; 290:20827-40; PMID:26112412; http://dx.doi.org/ 10.1074/jbc.M115.660456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Urano D, Nakata A, Mizuno N, Tago K, Itoh H. Domain-domain interaction of P-Rex1 is essential for the activation and inhibition by G protein betagamma subunits and PKA. Cell Signal 2008; 20:1545-54; PMID:18514484; http://dx.doi.org/ 10.1016/j.cellsig.2008.04.009 [DOI] [PubMed] [Google Scholar]
- [11].Dillon LM, Bean JR, Yang W, Shee K, Symonds LK, Balko JM, McDonald WH, Liu S, Gonzalez-Angulo AM, Mills GB, et al.. P-REX1 creates a positive feedback loop to activate growth factor receptor, PI3K/AKT and MEK/ERK signaling in breast cancer. Oncogene 2015; 34:3968-76; PMID:25284585; http://dx.doi.org/ 10.1038/onc.2014.328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Fritsch R, de Krijger I, Fritsch K, George R, Reason B, Kumar MS, Diefenbacher M, Stamp G, Downward J. RAS and RHO families of GTPases directly regulate distinct phosphoinositide 3-kinase isoforms. Cell 2013; 153:1050-63; PMID:23706742; http://dx.doi.org/ 10.1016/j.cell.2013.04.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Yang HW, Shin MG, Lee S, Kim JR, Park WS, Cho KH, Meyer T, Heo WD. Cooperative activation of PI3K by Ras and Rho family small GTPases. Mol Cell 2012; 47:281-90; PMID:22683270; http://dx.doi.org/ 10.1016/j.molcel.2012.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Campa CC, Ciraolo E, Ghigo A, Germena G, Hirsch E. Crossroads of PI3K and Rac pathways. Small GTPases 2015; 6:71-80; PMID:25942647; http://dx.doi.org/ 10.4161/21541248.2014.989789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Choufani S, Shuman C, Weksberg R. Beckwith-Wiedemann syndrome. Am J Med Genet C Semin Med Genet 2010; 154C:343-54; PMID:20803657; http://dx.doi.org/ 10.1002/ajmg.c.30267 [DOI] [PubMed] [Google Scholar]
- [16].Shin JY, Fitzpatrick GV, Higgins MJ. Two distinct mechanisms of silencing by the KvDMR1 imprinting control region. EMBO J 2008; 27:168-78; PMID:18079696; http://dx.doi.org/ 10.1038/sj.emboj.7601960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhang P, Liegeois NJ, Wong C, Finegold M, Hou H, Thompson JC, Silverman A, Harper JW, DePinho RA, Elledge SJ. Altered cell differentiation and proliferation in mice lacking p57KIP2 indicates a role in Beckwith-Wiedemann syndrome. Nature 1997; 387:151-8; PMID:9144284; http://dx.doi.org/ 10.1038/387151a0 [DOI] [PubMed] [Google Scholar]
- [18].McEwen KR, Ferguson-Smith AC. Distinguishing epigenetic marks of developmental and imprinting regulation. Epigenetics Chromatin 2010; 3:2; PMID:20180964; http://dx.doi.org/ 10.1186/1756-8935-3-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Macdonald WA. Epigenetic mechanisms of genomic imprinting: common themes in the regulation of imprinted regions in mammals, plants, and insects. Genet Res Int 2012; 2012:585024; PMID:22567394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Guttman M, Garber M, Levin JZ, Donaghey J, Robinson J, Adiconis X, Fan L, Koziol MJ, Gnirke A, Nusbaum C, et al.. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat Biotechnol 2010; 28:503-10; PMID:20436462; http://dx.doi.org/ 10.1038/nbt.1633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Pannetier M, Julien E, Schotta G, Tardat M, Sardet C, Jenuwein T, Feil R. PR-SET7 and SUV4-20H regulate H4 lysine-20 methylation at imprinting control regions in the mouse. EMBO Rep 2008; 9:998-1005; PMID:18724273; http://dx.doi.org/ 10.1038/embor.2008.147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Garraway LA, Widlund HR, Rubin MA, Getz G, Berger AJ, Ramaswamy S, Beroukhim R, Milner DA, Granter SR, Du J, et al.. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 2005; 436:117-22; PMID:16001072; http://dx.doi.org/ 10.1038/nature03664 [DOI] [PubMed] [Google Scholar]
- [23].Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 2009; 10:R25; PMID:19261174; http://dx.doi.org/ 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Garber M, Yosef N, Goren A, Raychowdhury R, Thielke A, Guttman M, Robinson J, Minie B, Chevrier N, Itzhaki Z, et al.. A high-throughput chromatin immunoprecipitation approach reveals principles of dynamic gene regulation in mammals. Molecular cell 2012; 47:810-22; PMID:22940246; http://dx.doi.org/ 10.1016/j.molcel.2012.07.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Pickrell JK, Gaffney DJ, Gilad Y, Pritchard JK. False positive peaks in ChIP-seq and other sequencing-based functional assays caused by unannotated high copy number regions. Bioinformatics 2011; 27:2144-6; PMID:21690102; http://dx.doi.org/ 10.1093/bioinformatics/btr354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Consortium EP, Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, Snyder M. An integrated encyclopedia of DNA elements in the human genome. Nature 2012; 489:57-74; PMID:22955616; http://dx.doi.org/ 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010; 26:841-2; PMID:20110278; http://dx.doi.org/ 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]