

Abstract
Phenotypic cell-based screening is a powerful approach to small-molecule discovery, but a major challenge of this strategy lies in determining the intracellular target and mechanism of action (MoA) for validated hits. Here, we show that the small-molecule BRD0476, a novel suppressor of pancreatic β-cell apoptosis, inhibits interferon-gamma (IFN-γ)-induced Janus kinase 2 (JAK2) and signal transducer and activation of transcription 1 (STAT1) signaling to promote β-cell survival. However, unlike common JAK-STAT pathway inhibitors, BRD0476 inhibits JAK-STAT signaling without suppressing the kinase activity of any JAK. Rather, we identified the deubiquitinase ubiquitin-specific peptidase 9X (USP9X) as an intracellular target, using a quantitative proteomic analysis in rat β cells. RNAi-mediated and CRISPR/Cas9 knockdown mimicked the effects of BRD0476, and reverse chemical genetics using a known inhibitor of USP9X blocked JAK-STAT signaling without suppressing JAK activity. Site-directed mutagenesis of a putative ubiquitination site on JAK2 mitigated BRD0476 activity, suggesting a competition between phosphorylation and ubiquitination to explain small-molecule MoA. These results demonstrate that phenotypic screening, followed by comprehensive MoA efforts, can provide novel mechanistic insights into ostensibly well-understood cell signaling pathways. Furthermore, these results uncover USP9X as a potential target for regulating JAK2 activity in cellular inflammation.
Graphical abstract
INTRODUCTION
Janus kinases (JAKs) are an important class of tyrosine kinases involved in STAT signaling,1–3 which is dysregulated in many inflammatory diseases (e.g., type 1 diabetes and rheumatoid arthritis) and cancers (e.g., various myeloproliferative disorders and acute lymphoblastic leukemia).4,5 Enormous efforts have been made over the past decade to target dysregulated JAK-STAT signaling pharmacologically, but selective JAK inhibitors remain difficult to develop. Current clinically used JAK inhibitors have numerous side effects and are not always efficacious.6–8 Further, many signaling activities of JAK involve scaffolding and protein–protein interactions, which may not be targeted by kinase inhibition. These observations suggest that novel kinase-independent mechanisms to inhibit JAK-STAT signaling may provide an attractive alternative to classical kinase inhibition.
Phenotypic cell-based screening is a powerful approach to uncover novel mechanisms to modulate dysregulated signaling.9 We previously used this approach to identify BRD0476,10,11 a compound derived from diversity-oriented synthesis (DOS).12–15 Here, we describe a systematic approach to mechanism-of-action (MoA) studies and show that BRD0476 inhibits interferon-gamma (IFN-γ)-induced JAK2 and signal transducer and activator of transcription 1 (STAT1) signaling to promote β-cell survival in an in vitro model of type 1 diabetes, consistent with STAT1−/− mice being protected from autoimmune diabetes.16,17 Unlike clinically used JAK-STAT pathway inhibitors, BRD0476 does not have kinase inhibitory activity. In parallel, quantitative proteomics experiments18 in rat β cells revealed the deubiquitinase ubiquitin-specific peptidase 9X (USP9X) as an intracellular target. Our results suggest that a competition between phosphorylation and ubiquitination on JAK2 explains the ability of BRD0476 to protect β cells from death. These results demonstrate that comprehensive MoA efforts can provide novel mechanistic insights into ostensibly well-understood cell signaling pathways.
RESULTS AND DISCUSSION
BRD0476 Protects Human β Cells from Cytokine-Induced Apoptosis
We developed BRD0476 (NIH probe ML-187; Figure 1a) based on a stereoselective compound identified in a phenotypic screen for suppressors of inflammatory cytokine-induced β-cell apoptosis.11,19 BRD0476 had no effect on cell death induced by endoplasmic reticulum stress, high glucose levels, or the fatty acid palmitate in the rat INS-1E insulinoma cell line20 (Figure S1), suggesting that its activity is specific to the cytokine cocktail (IL-1β, IFN-γ, TNF-α) used to induce apoptosis. We then treated dissociated primary human islet cells21 from multiple donors to determine the human physiological relevance of these results. After a six day treatment with the cytokine cocktail, caspase-3 activity was elevated approximately 2-fold, while addition of BRD0476 reduced this activity in a dose-dependent manner (Figure 1b, c, Figure S2). Glucose-stimulated insulin secretion was also impaired by treatment with the cytokine cocktail; similarly, co-incubation with BRD0476 restored insulin secretion in these cells (Figure 1d, e, Figure S2). Thus, we concluded that BRD0476 inhibits cytokine-induced apoptosis and preserves β-cell function in human islets.
Figure 1.
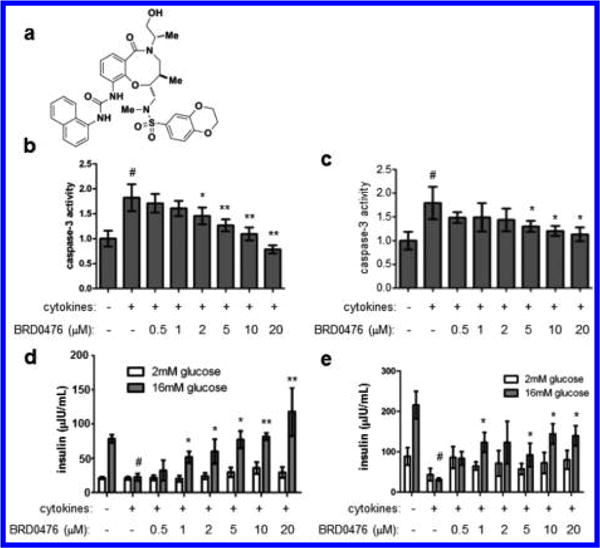
BRD0476 suppresses inflammatory cytokine-induced human β-cell apoptosis. (a) Chemical structure of BRD0476. (b, c) Caspase-3 activity in dissociated human islets from two independent donor preparations, treated for 6 days with cytokines (a combination of IL-1β, IFN-γ, and TNF-α) and the indicated concentration of BRD0476. (d, e) Glucose-stimulated insulin secretion in dissociated human islets from two independent donor preparations, treated for 6 days with cytokines and the indicated concentration of BRD0476. Data represent the mean ± standard deviation from four individual islet donors; donor information is available in Figure S2. # p < 0.001 compared to no treatment, * p < 0.001, ** p < 0.0001 compared to cytokine treatment, Student’s t test.
BRD0476 Inhibits JAK-STAT Signaling
Because these effects were identified using a phenotypic cell-based assay, we took a comprehensive approach to determine the MoA22 of BRD0476. We first performed gene-expression profiling on INS-1E cells treated with the cytokine cocktail for 6 h in the absence or presence of 10 μM BRD0476. The compound on its own had very little effect on gene expression, with only ~10 genes up- or downregulated >2-fold (Figure S3). Gene-set enrichment analysis23 revealed that the genes most affected by the addition of BRD0476, in the presence of cytokines, were involved in IFN-γ-induced JAK-STAT signaling (Figure 2a,b), with coordinate downregulation upon treatment with BRD0476. We used quantitative PCR to confirm that the effects of BRD0476 primarily converged on genes involved in signaling induced by IFN-γ (Figure 2c), including Stat1, Irf1, and Cxcl9. Further, the top leading-edge genes, resident in 25–50% of significantly enriched gene sets, were IFN-γ-regulated genes, such as Stat1, Cxcl10, Ccl5, and Ifit3 (Figure 2d). Consistent with gene-expression effects, BRD0476 directly inhibited IFN-γ signaling, as measured by STAT1 reporter gene activity (Figure 2e). STAT1 transcriptional activity is regulated by phosphorylation by members of the JAK family.24 Cytokine treatment rapidly increased STAT1 phosphorylation at Tyr701 in INS-1E cells, and total STAT1 protein levels also increased over 24 h treatment (Figure 2f).25 Simultaneous treatment with BRD0476 nearly completely abolished STAT1 phosphorylation, in as little as 1 h (Figure 2f). We also observed a partial decrease in total STAT1 protein, further suggesting that STAT1-dependent transcription was halted. Accordingly, translocation of STAT1 to the nucleus induced by cytokines was inhibited by BRD0476 (Figure S4).26 However, BRD0476 had no suppressive effect on IL-6-induced STAT3 phosphorylation in HepG2 cells (Figure S5), suggesting a specificity for STAT1 signaling. In light of the central role of STAT1 in autoimmune diabetes,17,25 these results indicate that inhibition of IFN-γ-induced STAT1 signaling by BRD0476 is sufficient to suppress β-cell apoptosis induced by inflammatory cytokines.
Figure 2.
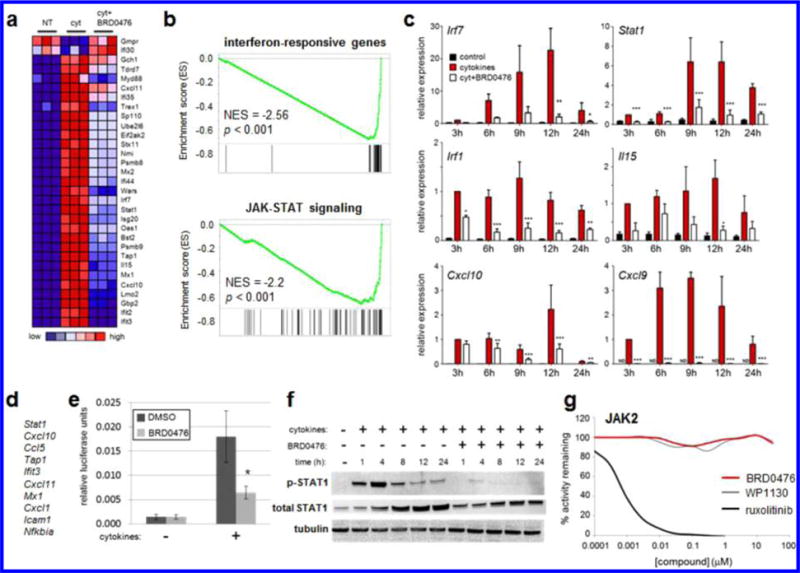
BRD0476 inhibits JAK-STAT signaling without inhibiting the kinase activity of JAK. (a) Heat map representing individual members of the top gene set, as measured by gene-set enrichment analysis, modulated by cytokines and BRD0476 after 6 h treatment in INS-1E cells. (b) Enrichment plots and scores for gene sets representing IFN-γ-responsive genes and the JAK-STAT signaling pathway. (c) Quantitative PCR of IFN-γ-responsive genes after treatment with cytokines and 10 μM BRD0476 for the indicated times in INS-1E cells. * p < 0.05, ** p < 0.01, *** p < 0.001 versus treatment, ANOVA with Tukey corrected t test. (d) Genes present in the leading edge of >25% enriched gene sets. (e) Gamma-activated sequence-driven luciferase activity in INS-1E cells treated for 18 h with cytokines and 10 μM BRD0476. * p < 0.0001 compared to cytokine treatment with DMSO, Student’s t test. (f) Phosphorylation of STAT1 and total STAT1 protein, as measured by Western blot, in INS-1E treated for the indicated times with cytokines and 10 μM BRD0476. Tubulin was included as a loading control. (g) Biochemical kinase activity of JAK2 in the presence of the indicated concentrations of BRD0476, WP1130, or ruxolitinib.
BRD0476 Is Not a Kinase Inhibitor
One of the simpler explanations for these results is that BRD0476 inhibits the kinase activity of JAK. To investigate this possibility, we profiled a panel of 96 human kinases for inhibition by 10 μM BRD0476. Even at this high concentration, no kinases were inhibited by more than 40% (Figure S6). In particular, BRD0476 had no effect on JAK1, JAK2, or JAK3 activities (Figures 2g and S7). These results suggest that BRD0476 inhibits JAK2 and STAT1 phosphorylation in a kinase-independent manner.
BRD0476 Target Identification Using SILAC
Next, we employed a quantitative proteomic strategy to uncover the direct cellular targets of BRD0476 and to complement our gene-expression analysis. To that end, we synthesized an analog of BRD0476 containing a PEG-amine linker at the alcohol side chain (Figure S8) and confirmed that this modification preserved suppressive activity against β-cell apoptosis (Figure S9). We then performed stable isotope labeling of amino acids in cell culture (SILAC) followed by mass spectrometry18 to identify enriched putative binding partners from INS-1E cells, comparing immobilized compound in the absence or presence of excess soluble competitor. This experiment resulted in 16 candidate protein binders of BRD0476 (Figure S10), including the deubiquitinase ubiquitin-specific peptidase 9, X-linked isoform (USP9X) (Figure 3a).
Figure 3.
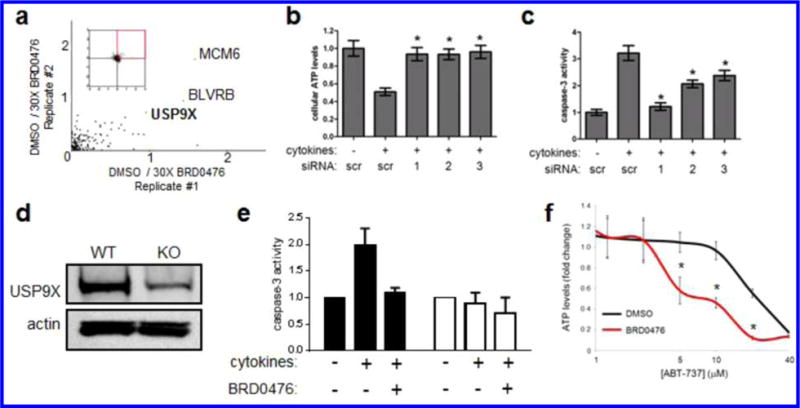
BRD0476 interacts with USP9X in both β cells and cancer cells. (a) Log2 ratios of changes of protein abundance for cells incubated with immobilized compound in the absence or presence of soluble competitor. Proteins in INS-1E cells were metabolically labeled with light and heavy amino acids lysine and arginine using SILAC methodology. Cell lysates were incubated either with BRD0476-loaded beads and 30× soluble BRD0476 or BRD0476-beads alone. Each dot represents a distinct protein. (b) Cellular ATP levels in cytokine-treated INS-1E cells following knock-down with three individual siRNA constructs for Usp9x. (c) Caspase-3 activity in cytokine-treated INS-1E cells following knock-down with three individual siRNA constructs for Usp9x. (d) Immunoblot for USP9X and actin for INS-1E wild-type and CRISPR/Cas9-knockout clone. (e) Caspase-3 activity in INS-1E wild type (black bars) and CRISPR/Cas9-knockout clone (white bars) treated for 1 day with cytokines and BRD0476. (f) Cellular ATP levels in DLD-1 colon carcinoma cells treated for 48 h with the indicated concentration of the BCL2 inhibitor ABT-737 and 10 μM BRD0476. * p < 0.0001 compared to DMSO treatment, Student’s t test.
Disruption of Usp9x by siRNA and CRISPR/Cas9 System Mimics the Protective Effect of BRD0476
In order to confirm a direct involvement of USP9X in BRD0476 activity, we used siRNA and the CRISPR/Cas9 system to disrupt the Usp9x gene in INS-1E cells. siRNA knockdown of USP9X nearly completely suppressed apoptosis induced by cytokine treatment (Figure 3b,c). Further, through the nonhomologous end-joining DNA repair mechanism, treatment of cells with our CRISPR/Cas9 construct resulted in the insertion of a single thymidine residue at position 4740 of the gene, which introduced a stop codon at the beginning of the catalytic domain. At the protein level, expression of USP9X was strongly reduced (63 ± 6%) compared to the control (Figure 3d). The strong reduction of USP9X levels was mirrored by inhibition of cytokine-induced apoptosis, as measured by caspase-3 activity (Figure 3e), mimicking the effect of BRD0476 treatment. Knockdown of other SILAC candidates did not rescue β cells from the cytokine-induced apoptosis (Figure S11).
BRD0476 Is a Selective but Moderate Inhibitor of USP9X Activity
Biochemical testing of the purified catalytic domain of human USP9X revealed that BRD0476 had no direct effect on deubiquitinase activity, but the compound did show some activity (~50% inhibition) against purified full-length FLAG-tagged protein (Figure S12). To determine BRD0476 specificity, we performed deubiquitinase profiling and found no inhibition of 11 other enzymes in this superfamily (Figure S13). These results suggest that BRD0476 is relatively selective for USP9X and may interact with USP9X at an allosteric site distant from the catalytic domain.
To further confirm interaction between BRD0476 and USP9X, we used differential scanning fluorimetry.27,28 We observed a trend toward a decrease in USP9X melting temperature compared to vehicle control (Figure S14). WP1130, an inhibitor of multiple deubiquitinases, including USP9X,29 also decreased the melting temperature of binding to USP9X (Figure S14), suggesting that these compounds may decrease the stability of USP9X. The relatively low signal observed for both compounds may be due to the large size of USP9X (~290 kDa) relative to these compounds and the difficulty in detecting large shifts resulting from binding. Remarkably, treatment of USP9X with a combination of both compounds resulted in a larger shift in melting temperature than either alone (Figure S14), suggesting that BRD0476 and WP1130 bind to different portions of the protein.
BRD0476 Shows Efficacy in a Known Cancer-Cell Drug Resistance Model
To show further evidence of USP9X engagement by BRD0476, we used a cancer model where USP9X has been reported to have a role in cancer-cell drug resistance. USP9X-overexpressing tumors are relatively resistant to BCL2 inhibitors, such as navitoclax and ABT-737, due to deubiquitination and stabilization of the antiapoptotic protein MCL1.30 We reasoned that if BRD0476 inhibits human USP9X, it should also increase cellular sensitivity to BCL2 inhibition, similar to that seen with genetic knock-down30 and treatment with WP1130.31 To test this hypothesis, we treated the DLD-1 colon cancer cell line with a combination of BRD0476 and ABT-737 and found that BRD0476 enhanced the toxicity of ABT-737 in a dose-dependent manner (Figure 3f). However, treatment with BRD0476 alone had no effect on these cells (Figure S15). These results are consistent with the role of BRD0476 as a modulator of USP9X.
BRD0476 Activity Suggests Involvement of USP9X in JAK-STAT Signaling
To establish a link between JAK-STAT signaling and USP9X activity, we synthesized a biotinylated version of BRD0476 (Figure S8) for cellular pull-down experiments. As expected, USP9X was isolated from INS-1E cells; further, JAK2 was recovered from the protein complex (Figure 4a), indicating that both proteins reside in a complex targeted by BRD0476. Indeed, JAK2 and USP9X do interact, as measured by co-immunoprecipitation (Figure 4b). Next, we adopted a reverse chemical-genetic strategy by comparing the activity of BRD0476 to the known USP9X inhibitor, WP1130. Like BRD0476, WP1130 decreased cytokine-induced phosphorylation of STAT1 in INS-1E cells after 30 min treatment (Figure 4c). Importantly, neither BRD6283, an inactive analog of BRD0476,26 nor VM001, an inactive analog of WP1130,32 had any effect on STAT1 phosphorylation (Figure 4c). WP1130 induced significant cellular toxicity at the same concentrations at which it inhibited STAT1 phosphorylation (data not shown), probably due to targeting other deubiquitinases, precluding us from determining its suppressive effects on β-cell apoptosis.
Figure 4.
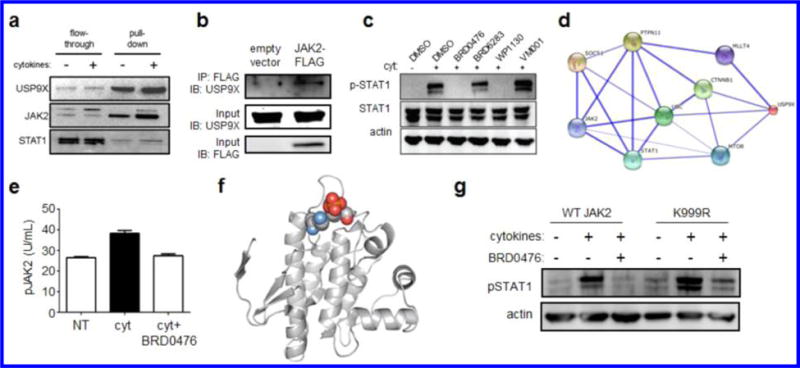
USP9X interacts with JAK2 to promote cytokine signaling, which is inhibited in a kinase-independent manner by BRD0476. (a) Affinity pull-down of USP9X and associated proteins by biotinylated BRD0476. (b) Co-immunoprecipitation of USP9X and JAK2 in INS-1E cells transfected with empty vector or JAK2-FLAG. Immunoblots of input material for total USP9X and FLAG are included. (c) Phosphorylation of STAT1 and total STAT1 protein in INS-1E cells treated 30 min with cytokines and the indicated compound. Actin was included as a loading control. (d) Network graph indicating proteins connecting JAK2 to USP9X. Weight of edges is correlated with confidence level. (e) Phosphorylation of JAK2 in INS-1E cells treated with the cytokine cocktail and 10 μM BRD0476. Data represent the mean ± standard deviation. * p < 0.0001 versus control treatment, # p < 0.001 versus cytokine treatment, Student’s t test. (f) Structural analysis of JAK2 (pdb: 4GL9),41 with the proximity of Lys999 (blue) and Tyr1007 (red) indicated. (g) Phosphorylation of STAT1 in INS-1E cells transfected with wild-type JAK2 or K999R, followed by 30 min treatment with the cytokine cocktail and 10 μM BRD0476.
BRD0476 Affects JAK2 Phosphorylation but Not Its Degradation
An examination of the protein interaction network surrounding human USP9X revealed that both STAT1 and JAK2 are connected to USP9X (Figure 4d), providing further evidence that JAK2 is functionally related to USP9X. Consistent with these observations, JAK2 phosphorylation induced by cytokine treatment was decreased by co-treatment with BRD0476 in INS-1E cells (Figure 4e). These effects were independent of any changes in total JAK2 levels and were also observed in the presence of the proteasome inhibitor lactacystin (Figure S16), indicating that protein degradation is not involved in the activity of BRD0476 on JAK-STAT signaling.
BRD0476 Induces Competition between Phosphorylation at Tyr1007 and Ubiquitination at Lys999 on JAK2
Despite the involvement of the deubiquitinase USP9X, the overall ubiquitination status of JAK2 did not change upon treatment with cytokines or BRD0476 (Figure S17). We reasoned that a monoubiquitination event might regulate JAK2 activity, thus escaping detection by either Western blot or mass spectrometry. Therefore, we evaluated the predicted ubiquitination sites on JAK2 (Figure S18)33,34 and found that the close proximity of Lys999 to Tyr1007, phosphorylation of which is required for activity (Figure 4f), provides a potential site of competition between phosphorylation and ubiquitination for JAK2 signaling competence. Ubiquitination of JAK2 has been shown to regulate its signaling activity.35 Transient transfection of INS-1E cells with JAK2-K999R resulted in sustained STAT1 phosphorylation, even in the presence of BRD0476 (Figure 4g). Some decrease in STAT1 phosphorylation was still observed with BRD0476, presumably due to the limited transfection efficiency of INS-1E cells. Mutation of Lys850, another predicted site of ubiquitination, had no effect on BRD0476 activity (Figure S19). These results suggest that the inability of this JAK2 lysine to be ubiquitinated enables persistent JAK-STAT signaling in response to IFN-γ stimulation.
CONCLUSIONS
This study provides an example of mechanism-of-action (MoA) determination for a compound identified by phenotypic screening and provides the novel mechanistic insight that extremely rapid inhibition of IFN-γ-induced JAK-STAT signaling may be achieved without kinase inhibition. Furthermore, BRD0476 possesses a pair of highly novel phenotypes: it suppresses cell death in β cells on the one hand and enhances cancer cell death on the other. The effectiveness of JAK-STAT inhibition on β-cell death is consistent with the observation that STAT1−/− mice are protected from autoimmune diabetes.16,17 The metabolic instability of BRD0476 in mouse and human liver microsomes made challenging its testing in animal models. We did, however, confirm that incubation of BRD0476 with cells in culture did not substantially alter the chemical structure (Figure S20), indicating that the compound was not metabolized in the time frame of the experiments presented here. Further medicinal chemistry studies will enable the evaluation of next-generation compounds to fully validate USP9X as a potential therapeutic target for type 1 diabetes and cancer. As a general strategy for inhibiting JAK2-mediated signaling, this approach may be an alternative way to treat inflammatory diseases being explored for FDA-approved kinase inhibitors, such as ruxolitinib and tofacitinib.36–39 This study illustrates the utility of DOS as a tool to uncover compounds with novel biological activity12,40 and demonstrates that a comprehensive approach to MoA studies provides a powerful complement to phenotypic screening.
Supplementary Material
Acknowledgments
These studies were supported by a NIH-NIDDK Type 1 Diabetes Pathfinder Award (DP2-DK083048, to B.K.W.), the NIH Molecular Libraries Program (MLP), the William F. Milton Fund (Harvard University, to A.C.), and Genomics Based Drug Discovery-Target ID Project grant RL1HG004671, which was administratively linked to the NIH grants RL1CA133834, RL1GM084437, and UL1RR024924. A.C. was supported by a career award from the Burroughs-Wellcome Fund and the Society of Fellows, Harvard University. R.G. was supported by NIH-5T34GM008751. S.L.S. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Supporting Information
Experimental details, additional figures, and data. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b04284.
Notes
The authors declare no competing financial interest.
References
- 1.Aaronson DS, Horvath CM. Science. 2002;296:1653–1655. doi: 10.1126/science.1071545. [DOI] [PubMed] [Google Scholar]
- 2.Shuai K, Liu B. Nat Rev Immunol. 2003;3:900–911. doi: 10.1038/nri1226. [DOI] [PubMed] [Google Scholar]
- 3.Stark GR, Darnell JE., Jr Immunity. 2012;36:503–514. doi: 10.1016/j.immuni.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bowman T, Garcia R, Turkson J, Jove R. Oncogene. 2000;19:2474–2488. doi: 10.1038/sj.onc.1203527. [DOI] [PubMed] [Google Scholar]
- 5.O’Shea JJ, Kontzias A, Yamaoka K, Tanaka Y, Laurence A. Ann Rheum Dis. 2013;72(Suppl 2):ii111–115. doi: 10.1136/annrheumdis-2012-202576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pardanani A. Leukemia. 2012;26:1449–1451. doi: 10.1038/leu.2012.21. [DOI] [PubMed] [Google Scholar]
- 7.Schonberg K, Rudolph J, Vonnahme M, Parampalli Yajnanarayana S, Cornez I, Hejazi M, Manser A, Uhrberg M, Verbeek W, Koschmieder S, Brummendorf TH, Brossart P, Heine A, Wolf D. Cancer Res. doi: 10.1158/0008-5472.CAN-14-3198. [Online early access] Published online April 1, 2015. [DOI] [PubMed] [Google Scholar]
- 8.Verstovsek S, Kantarjian H, Mesa RA, Pardanani AD, Cortes-Franco J, Thomas DA, Estrov Z, Fridman JS, Bradley EC, Erickson-Viitanen S, Vaddi K, Levy R, Tefferi A. N Engl J Med. 2010;363:1117–1127. doi: 10.1056/NEJMoa1002028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Swinney DC, Anthony J. Nat Rev Drug Discovery. 2011;10:507–519. doi: 10.1038/nrd3480. [DOI] [PubMed] [Google Scholar]
- 10.Chou DH, Bodycombe NE, Carrinski HA, Lewis TA, Clemons PA, Schreiber SL, Wagner BK. ACS Chem Biol. 2010;5:729–734. doi: 10.1021/cb100129d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Faloon PW, Chou DHC, Forbeck EM, Walpita D, Morgan B, Buhrlage S, Ting A, Perez J, MacPherson LJ, Duvall JR, Dandapani S, Marcaurelle LA, Munoz B, Palmer M, Foley M, Wagner B, Schreiber SL. Probe Reports from the NIH Molecular Libraries Program. National Center for Biotchnology Information; Bethesda, MD: 2010. [PubMed] [Google Scholar]
- 12.Schreiber SL. Science. 2000;287:1964–1969. doi: 10.1126/science.287.5460.1964. [DOI] [PubMed] [Google Scholar]
- 13.Marcaurelle LA, Johannes CW. Prog Drug Res. 2008;66:187–216. doi: 10.1007/978-3-7643-8595-8_3. [DOI] [PubMed] [Google Scholar]
- 14.Nielsen TE, Schreiber SL. Angew Chem, Ent Ed Engl. 2008;47:48–56. doi: 10.1002/anie.200703073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marcaurelle LA, Johannes C, Yohannes D, Tillotson BP, Mann D. Bioorg Med Chem Lett. 2009;19:2500–2503. doi: 10.1016/j.bmcl.2009.03.037. [DOI] [PubMed] [Google Scholar]
- 16.Callewaert HI, Gysemans CA, Ladriere L, D’Hertog W, Hagenbrock J, Overbergh L, Eizirik DL, Mathieu C. Diabetes. 2007;56:2169–2173. doi: 10.2337/db07-0052. [DOI] [PubMed] [Google Scholar]
- 17.Kim S, Kim HS, Chung KW, Oh SH, Yun JW, Im SH, Lee MK, Kim KW, Lee MS. Diabetes. 2007;56:2561–2568. doi: 10.2337/db06-1372. [DOI] [PubMed] [Google Scholar]
- 18.Ong SE, Schenone M, Margolin AA, Li X, Do K, Doud MK, Mani DR, Kuai L, Wang X, Wood JL, Tolliday NJ, Koehler AN, Marcaurelle LA, Golub TR, Gould RJ, Schreiber SL, Carr SA. Proc Natl Acad Sci U S A. 2009;106:4617–4622. doi: 10.1073/pnas.0900191106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chou DH, Duvall JR, Gerard B, Liu H, Pandya BA, Suh BC, Forbeck EM, Faloon P, Wagner BK, Marcaurelle LA. ACS Med Chem Lett. 2011;2:698–702. doi: 10.1021/ml200120m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Merglen A, Theander S, Rubi B, Chaffard G, Wollheim CB, Maechler P. Endocrinology. 2004;145:667–678. doi: 10.1210/en.2003-1099. [DOI] [PubMed] [Google Scholar]
- 21.Walpita D, Hasaka T, Spoonamore J, Vetere A, Takane KK, Fomina-Yadlin D, Fiaschi-Taesch N, Shamji A, Clemons PA, Stewart AF, Schreiber SL, Wagner BK. J Biomol Screen. 2012;17:509–518. doi: 10.1177/1087057111430253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schenone M, Dancik V, Wagner BK, Clemons PA. Nat Chem Biol. 2013;9:232–240. doi: 10.1038/nchembio.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shuai K, Ziemiecki A, Wilks AF, Harpur AG, Sadowski HB, Gilman MZ, Darnell JE. Nature. 1993;366:580–583. doi: 10.1038/366580a0. [DOI] [PubMed] [Google Scholar]
- 25.Moore F, Naamane N, Colli ML, Bouckenooghe T, Ortis F, Gurzov EN, Igoillo-Esteve M, Mathieu C, Bontempi G, Thykjaer T, Orntoft TF, Eizirik DL. J Biol Chem. 2011;286:929–941. doi: 10.1074/jbc.M110.162131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scully SS, Tang AJ, Lundh M, Mosher CM, Perkins KM, Wagner BK. J Med Chem. 2013;56:4125–4129. doi: 10.1021/jm400397x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Niesen FH, Berglund H, Vedadi M. Nat Protoc. 2007;2:2212–2221. doi: 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- 28.Al-Hakim AK, Zagorska A, Chapman L, Deak M, Peggie M, Alessi DR. Biochem J. 2008;411:249–260. doi: 10.1042/BJ20080067. [DOI] [PubMed] [Google Scholar]
- 29.Bartholomeusz GA, Talpaz M, Kapuria V, Kong LY, Wang S, Estrov Z, Priebe W, Wu J, Donato N. J Blood. 2007;109:3470–3478. doi: 10.1182/blood-2006-02-005579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schwickart M, Huang X, Lill JR, Liu J, Ferrando R, French DM, Maecker H, O’Rourke K, Bazan F, Eastham-Anderson J, Yue P, Dornan D, Huang DC, Dixit VM. Nature. 2010;463:103–107. doi: 10.1038/nature08646. [DOI] [PubMed] [Google Scholar]
- 31.Peddaboina C, Jupiter D, Fletcher S, Yap JL, Rai A, Tobin RP, Jiang W, Rascoe P, Rogers MK, Smythe WR, Cao X. BMC Cancer. 2012;12:541. doi: 10.1186/1471-2407-12-541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perry JW, Ahmed M, Chang KO, Donato NJ, Showalter HD, Wobus CE. PLoS Pathog. 2012;8:e1002783. doi: 10.1371/journal.ppat.1002783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen Z, Chen YZ, Wang XF, Wang C, Yan RX, Zhang Z. PLoS One. 2011;6:e22930. doi: 10.1371/journal.pone.0022930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen Z, Zhou Y, Song J, Zhang Z. Biochim Biophys Acta. 2013;1834:1461–1467. doi: 10.1016/j.bbapap.2013.04.006. [DOI] [PubMed] [Google Scholar]
- 35.Kapuria V, Levitzki A, Bornmann WG, Maxwell D, Priebe W, Sorenson RJ, Showalter HD, Talpaz M, Donato N. J Cell Signalling. 2011;23:2076–2085. doi: 10.1016/j.cellsig.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 36.Fridman JS, Scherle PA, Collins R, Burn T, Neilan CL, Hertel D, Contel N, Haley P, Thomas B, Shi J, Collier P, Rodgers JD, Shepard S, Metcalf B, Hollis G, Newton RC, Yeleswaram S, Friedman SM, Vaddi K. J Invest Dermatol. 2011;131:1838–1844. doi: 10.1038/jid.2011.140. [DOI] [PubMed] [Google Scholar]
- 37.Punwani N, Scherle P, Flores R, Shi J, Liang J, Yeleswaram S, Levy R, Williams W, Gottlieb A. J Am Acad Dermatol. 2012;67:658–664. doi: 10.1016/j.jaad.2011.12.018. [DOI] [PubMed] [Google Scholar]
- 38.Sandborn WJ, Ghosh S, Panes J, Vranic I, Su C, Rousell S, Niezychowski W, Study AI. N Engl J Med. 2012;367:616–624. doi: 10.1056/NEJMoa1112168. [DOI] [PubMed] [Google Scholar]
- 39.Yarilina A, Xu K, Chan C, Ivashkiv LB. Arthritis Rheumatol. 2012;64:3856–3866. doi: 10.1002/art.37691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eberhardt L, Kumar K, Waldmann H. Curr Drug Targets. 2011;12:1531–1546. doi: 10.2174/138945011798109482. [DOI] [PubMed] [Google Scholar]
- 41.Pymol. Schrodinger, LLC; Cambridge, MA: 2010. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.