

Abstract
EZH2 or EZH1 (enhancer of zeste homologue 2 or 1) is the catalytic subunit of polycomb repressive complex 2 (PRC2) that catalyzes methylation of histone H3 lysine 27 (H3K27). PRC2 hyperactivity and/or hypertrimethylation of H3K27 are associated with numerous human cancers, therefore inhibition of PRC2 complex has emerged as a promising therapeutic approach. Recent studies have shown that EZH2 and EZH1 are not functionally redundant and inhibition of both EZH2 and EZH1 is necessary to block the progression of certain cancers such as mixed-lineage leukemia (MLL)-rearranged leukemias. Despite the significant advances in discovery of EZH2 inhibitors, there has not been a systematic structure-activity relationship (SAR) study to investigate the selectivity between EZH2 and EZH1 inhibition. Here, we report our SAR studies that focus on modifications to various regions of the EZH2/1 inhibitor UNC1999 (5) to investigate the impact of the structural changes on EZH2 and EZH1 inhibition and selectivity.
Graphical abstract
INTRODUCTION
EZH2 (enhancer of zeste homologue 2, also known as KMT6 (lysine methyltransferase 6) or KMT6A) or EZH1 (enhancer of zeste homologue 1, also known as KMT6B) is the catalytic subunit of polycomb repressive complex 2 (PRC2) that catalyzes methylation of histone H3 lysine 27 (H3K27).1,2 PRC2 is a crucial, highly conserved chromatin-modifying complex that contains three core subunits: (1) EZH1 or EZH2, (2) SUZ12 (suppressor of zeste 12), (3) EED (embryonic ectoderm development), and additional subunits such as histone and DNA interacting proteins RbAp46/48 and AEBP2.3–5 While EZH2 or EZH1 is the catalytic subunit, their methyltransferase activity can only be activated as part of PRC2 complex via interactions with EED and the VEFS domain of SUZ12 (SUZ12–VEFS).6–10 The importance of these interactions is supported by the fact that the isolated SET domain of EZH2 lacks H3K27 peptide and cofactor SAM (S-adenosyl-L-methionine) recognition and it is inactive.11,12 These interactions have also been clearly demonstrated by the recent crystal structures of the PRC2 complex from a thermophilic fungus, Chaetomium hermophilum (Ct),13 and inhibitor-bound American chameleon (Anolis carolinensis (Ac))/human PRC2 complex14 as well as human PRC2 in complex with the H3K27M mutant peptide.15
The trimethylation of H3K27 (H3K27me3) catalyzed by PRC2 is a transcriptionally repressive epigenetic mark that regulates gene expression, differentiation, and development.3 PRC2 hyperactivity and/or hypertrimethylation of H3K27 have been associated with a number of human cancers16,17 such as breast,18,19 prostate,20 lymphoma,21,22 myeloma,23 and leukemia.24 Gain-of-function point mutations at tyrosine 641 in the C-terminal SET domain of EZH2 have been identified and are observed in 7% of follicular lymphomas and 22% of germinal center B-cell (GCB) and diffuse large B-cell lymphomas (DLBCLs).22 It is not yet determined whether EZH1 is also overexpressed in B-cell malignancies. It has also been demonstrated that EZH2 and EZH1 compensate for one another, and both of them are critical for sustaining aggressive MLL-rearranged leukemias by enforcing cell proliferation and suppressing cell differentiation programs.25–27
EZH2 and EZH1 are highly homologous with 76% overall sequence identity and 96% sequence identity in their catalytic SET domains.28 However, EZH2 and EZH1 have different expression patterns. While EZH1 is ubiquitously expressed and present in both dividing and differentiated cells, EZH2 is only expressed in actively dividing cells.29–31 EZH1 containing PRC2 complex (PRC2–EZH1) functions directly in chromatin compaction, while EZH2 containing PRC2 complex (PRC2–EZH2) does not.29 Given these differences, it was proposed that PRC2 complexes differ in their differentiation roles.29,32 Therefore, depending upon the cellular context, the loss of either EZH2 or EZH1 catalytic activity or the inactivation of their methyltransferase activity together might be needed to block the proliferation of tumor cells. For example, inhibition of EZH2 alone is sufficient to exert robust antiproliferative activity in DLBCLs.33,34 On the other hand, inhibition of both EZH2 and EZH1 is needed to block the progression of MLL-rearranged leukemias in cell-based and animal models.27
Numerous highly potent and selective small-molecule inhibitors of PRC2 have recently been discovered. Most of these inhibitors contain a common pyridonemethyl-amide core constituting a highly optimized moiety for binding to EZH2 and EZH1 in a cofactor-competitive manner (Figure 1). Some of these small molecules also feature an indazole ring attached to pyridonemethyl-amide core where the others contain an indole or aniline ring. The first two published EZH2 inhibitors are EPZ005687 (1)33 and GSK126 (2),34 which are highly selective for EZH2 over a range of other methyltransferases (50 and 150-fold selective for EZH2 over EZH1, respectively). GSK343 (3) and EI1 (4)35 are also selective toward EZH2 over EZH1 (60- and 140-fold, respectively). UNC1999 (5) was the first reported orally bioavailable inhibitor of EZH2 and EZH1 and is a potent dual inhibitor of both enzymes with around 5-fold selectivity for EZH2 over EZH1.36 EPZ-6438 (6),37 an EZH2 inhibitor (35-fold selective over EZH1), as well as compound 2 are being evaluated in the clinic for the treatment of advanced solid tumors or B-cell lymphomas.38–41 A new chemotype represented by inhibitor 7, which contains a 4-amino-2,2′,6,6′-tetramethylpiperidine core, was discovered.42 However, the inhibitors featuring this new core have not demonstrated good pharmacokinetic (PK) properties and any in vivo activity.43 Replacing the core with dimethylpyridone and subsequent SAR studies resulted in the discovery of CPI-360 (8), which was 100-fold selective for EZH2 over EZH1.43,44 Recently, CPI-1205, an analogue of 8 has entered phase 1 clinical trials for evaluation in patients with B-cell lymphomas.45 EPZ011989 (9), a potent and orally bioavailable EZH2 inhibitor, was obtained by replacing one of the phenyl rings with acetylene and modifying the pyran substituent of compound 6.46 Again, on the basis of 6, ZLD1039 (10), which is 12-fold selective for EZH2 over EZH1, has been discovered.47,48 Most recently, compound 11, a potent EZH2 inhibitor, was cocrystallized with the active Ac/human PRC2 complex.14
Figure 1.

Reported small-molecule inhibitors of EZH2 and EZH1.
Although there have been very significant advances in discovery of EZH2 inhibitors as we discussed above, a systematic structure–activity relationship (SAR) study to investigate EZH2 and EZH1 selectivity has not been reported. Herein, we report our SAR studies that focus on modifications to several regions of our inhibitor 5 and investigate the impact of structural changes on EZH2 and EZH1 potency and selectivity. We describe the design, synthesis, and biochemical evaluation of novel compounds against both PRC2–EZH2 and PRC2–EZH1 and discuss structural features that lead to selectivity between EZH2 and EZH1.
RESULTS AND DISCUSSION
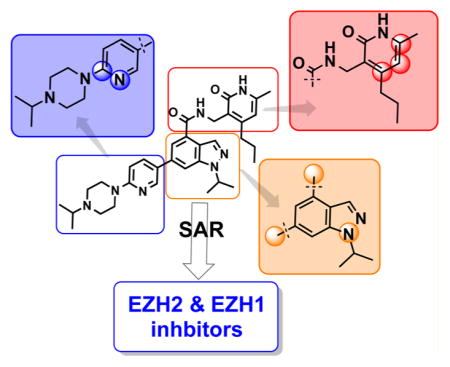
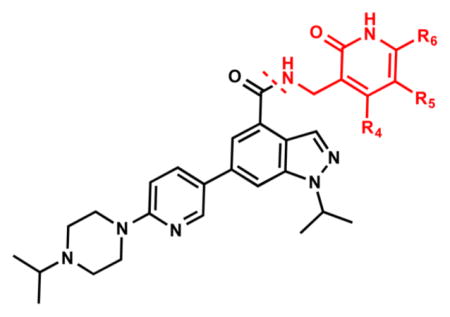
We investigated the following three regions of the chemical series represented by compound 5: (1) the crucial pyridonemethyl-amide core (Figure 2, red box), mainly focusing on the substitutions at positions 4, 5, and 6 of the pyridone ring (2-pyridone numbering); (2) the indazole moiety (Figure 2, orange box), including changes in connectivity of this ring to the pyridonemethyl-amide core and 3-pyridine moiety, as well as the replacement ring systems and various alkyl substituents on the indazole N-1 (indazole numbering); and (3) the pyridine-2-yl-piperazine substituent at the 6-position of the indazole ring (Figure 2, blue box), including different ring systems and substitutions at this position.
Figure 2.
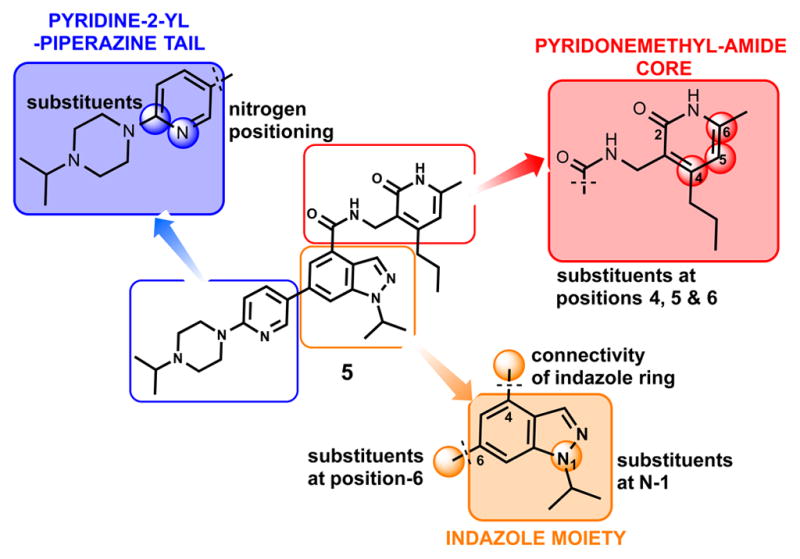
Regions of compound 5 investigated for EZH2 and EZH1 selectivity.
We previously reported 5 (Figure 1) as a very potent and selective pyridone containing dual inhibitor of EZH2 and EZH1 (IC50 < 10 nM and 69 ± 14 nM, respectively).36 The recently published cocrystal structure of inhibitor 11 in complex with the Ac/human PRC2 (PDB: 5IJ7) has clearly demonstrated the significance of the pyridonemethyl-amide core in binding to EZH2.14 We docked inhibitor 5 into the human PRC2 crystal structure (PDB: 5HYN)15 based on the binding mode of inhibitor 11 in complex with the Ac/human PRC2.14 The docked structure of compound 5 (Figure 3A,B) displays that the pyridonemethyl-amide core is anchored via hydrogen-bond interactions with backbone oxygen and nitrogen atoms of W624 and makes van der Waals contacts with N688, F665, and R685. In addition, the carbonyl oxygen of the amide makes a hydrogen-bond with the backbone NH of Y111 of the activation loop. The indazole moiety interacts with the I-SET helix of EZH2 and I109, M110 with van der Waals contacts. The pyridine moiety interacts with Y111 (activation loop) and Y661 (I-SET of EZH2) via π–π stacking while the piperazinium is involved in an ionic interaction with D237 (Figure 3A,B). W624 is part of the conserved GXG motif of the SET domain and this region is occupied by the homocysteine moiety of the cofactor product SAH (S-adenosyl-L-homocysteine) in the absence of a ligand. The inhibitor and SAH partly share this binding site, resulting in cofactor-competitive inhibition (Figure 3C). In addition, Y111 from the activation loop and Y661 from the I-SET region are in contact with each other and create an unanticipated groove where the activation loop forms the lid of this pocket.14 The pyridine2-yl-piperazine tail of compound 5 interacts with this closed pocket to create further contacts with the complex (Figure 3D).
Figure 3.

Docked pose of compound 5 in the human PRC2 structure. (A) Docked pose of compound 5 (pink). (B) A 2D representation of molecular interactions between compound 5 and the residues of the binding site. (C) Superimposition of compound 5 and SAH (aligned from PDB entry 5HYN). The pyridone moiety of compound 5 mimics the homocysteine moiety of SAH (green). (D) Binding site of PRC2 complex where compound 5 binds through a groove formed by the SET domain and the activation loop of EZH2 (aqua) and the EED complex (gray).
As described above, we focused on three regions of inhibitor 5 for SAR investigation. The general syntheses of key intermediates and derivatives of 5 are shown in Scheme 1. The pyridone-methylamine derivatives (15) were prepared by reacting corresponding α,β-unsaturated ketones (12) with the cyanoacetamide 13 in the presence of a base followed by air oxidation to give corresponding nitriles (14), which were then reduced to obtain the desired amines (15) (Scheme 1A). The commercially available indazole 16 reacted with alkyl bromides to give a mixture of N-1 and N-2 substituted indazoles that were separated via column chromatography (Scheme 1B). The desired alkylated indazoles (17) were then hydrolyzed to yield 6-bromo-indazole-4-carboxylates (18). Suzuki coupling of 17 with the boronic acid 20, which was prepared from the bromide 19 (Scheme 1C), and subsequent hydrolysis resulted in carboxylic acid intermediates 21, which were further converted to the desired derivatives (23) by amide coupling reactions with pyridone-methylamine derivatives (15) (Scheme 1D). Alternatively, amide coupling between 15 and 18 gave intermediates 22, which were subjected to Suzuki coupling with the boronic acid 20 to yield the desired derivatives (23) (Scheme 1E).
Scheme 1.

General Syntheses of Key Intermediates and Derivatives of 5 for SAR Studies
Because the pyridone moiety forms critical interactions with the PRC2 complex, we first explored the substitutions at the 4, 5, and 6 positions (2-pyridone numbering) of the pyridone ring and synthesized the new derivatives outlined in Table 1. Because most of the known inhibitors in Figure 1 possess 4,6-alkyl substitution, we started with 4,6-dimethyl pyridone and we kept the 6-methyl substituent constant, as we altered the substituent at the 4-position (compounds 24–29, Table 1). 4,6-Dimethyl (24) and 4-ethyl-6-methyl (25) substituted compounds inhibited EZH2 with an IC50 of 12 ± 2 nM. While 24 did not display appreciable inhibitory activity against EZH1 (IC50 > 2500 nM), 25 inhibited it with an IC50 of 810 ± 70 nM with more than 3-fold increase in potency from 24. Our inhibitor 5, which contains 4-propyl-6-methyl substitution, inhibited EZH2 with an IC50 of less than 10 nM, it was also potent against EZH1 (IC50 = 69 ± 14 nM) with 5–8-fold selectivity favoring EZH2. As the hydrophobic interactions increased with the longer chain alkyl group at 4-position, the potency for EZH2 remained high around 10 nM while potency for EZH1 increased around 50-fold from methyl to propyl group. Further elongation of the carbon chain (n-butyl, 26) or branched alkyl groups such as iso-propyl (27), cyclopentane (28), and iso-butyl (29) groups at this position were also tolerated: EZH1 potency varied between 86 to 320 nM while EZH2 potency remained below 10 nM. These results have indicated that the 4-position of the pyridone ring can accommodate a longer or larger hydrophobic group, which is most likely involved with van der Waals contacts with R685, F686, and F665 (Figure 4A). Interestingly, changes at this position had more pronounced effects on EZH1 potency. It should be noted that most of these compounds have IC50 values below the measurable limit of our assay conditions for EZH2. With these results in hand, we then kept the 4-methyl substitution constant and modified the substituent at 6-position (compounds 30–33). While 6-ethyl (30) and 6-sec-butyl (31) substituents were tolerated by EZH2 albeit with reduced potency (IC50 of 73 ± 15 and 32 ± 9 nM, respectively), these two compounds did not exhibit appreciable inhibitory activity against EZH1 (Table 1). On the other hand, 6-tert-butyl (32) and 6-phenyl (33) substituents resulted in a complete loss of potency for both enzymes. Our docking model (Figure 4) suggests that there is enough space for a primary or secondary alkyl group, but a bulkier and less flexible substituent (e.g., t-Bu or Ph) creates steric clash with the protein and abolishes the EZH2 inhibitory activity (Figure 4B). Overall, modifications at this position decreased EZH1 inhibitory activity. Removal of both substitutions (34, Table 1) from the pyridone ring resulted in >500-fold loss of potency. Comparison of the potency of compounds 24 and 34 clearly shows the significance of these pyridone substituents on inhibitory activity. A previous study on the effects of methyl groups in ligand–protein binding suggested that a methyl group positioned in a hydrophobic environment instead of a hydrogen may result in a 10-fold increase in potency.49 However, to observe an effect beyond that, the methyl group should also prompt a more favorable conformational change.49 A possible structural explanation for the pronounced effects of an alkyl substituent at the 4-and 6-positons of the pyridone ring on EZH2 inhibition is that the addition of more van der Waals contacts leads to positioning the pyridonemethyl-amide core in an optimal geometry for the hydrogen bond interactions with W624 and Y111 (Figure 3A), which in turn lowers enthalpic contribution to the free energy of binding. These observations are also in line with the literature precedent, showing the effects of 4, 6-methyl groups for EZH2 inhibition.50 Adding a substituent at the 5-position of the pyridone ring largely diminishes the inhibitor activity of the compounds for both EZH2 and EZH1 (compounds 35–37, Table 1). As it can be seen from our docking model (Figure 4B), substitution at the 5-position will clash with R685 backbone and F665. The side chain of F665 interacts with the pyridone moiety through hydrophobic contacts and altering the cofactor binding site such as to obstruct the methionine moiety of the cofactor to be anchored at that location (see Supporting Information, Figure S1). The sterically less demanding and relatively more strained cyclopentyl ring (38), however, retains some potency toward EZH2 but not EZH1 with IC50 of 32 ± 7 and >2500 nM, respectively. We have also investigated other modifications to the pyridone ring (Table 2). The 4-hydroxyl group (39) replacing the 4-propyl group (5) resulted in a total loss of potency for both enzymes, most likely because the favorable hydrophobic interactions are abolished. The 2-methyl-pyrimidine-4-one (40), pyrimidine-4-one (41), and 6-methyl-pyrimidine-2,4-dione (42) groups completely abrogated inhibitory activity against EZH2 and EZH1. Similarly, replacing the pyridone ring with the 4-amino-2,2′,6,6′-tetramethylpiperidine group (43, Table 2) led to a complete loss of potency for EZH2 and EZH1.
Table 1.
In Vitro Potencies of Newly Synthesized Pyridone Derivatives for EZH2 and EZH1
| |||
|---|---|---|---|
| Cmpd | Structure | IC50(nM) | |
| EZH2 | EZH1 | ||
| 24 |

|
12 ± 2 | >2500 |
| 25 |
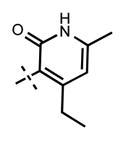
|
12 ± 2 | 810 ± 71 |
| 5 |
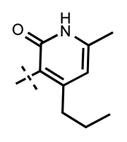
|
<10 | 69 ± 14 |
| 26 |
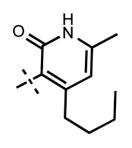
|
<10 | 160 ± 14 |
| 27 |
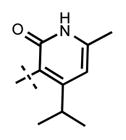
|
<10 | 320 ± 58 |
| 28 |
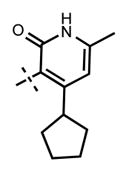
|
<10 | 86 ± 12 |
| 29 |
|
<10 | 211 ± 46 |
| 30 |
|
73 ± 15 | >5000 |
| 31 |
|
32 ± 9 | >1250 |
| 32 |

|
>10000 | >10000 |
| 33 |
|
>10000 | >10000 |
| 34 |

|
>5000 | >5000 |
| 35 |
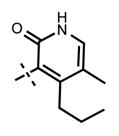
|
1016 ± 68 | >5000 |
| 36 |

|
328 ± 74 | >5000 |
| 37 |

|
>2500 | >2500 |
| 38 |
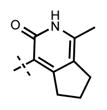
|
32 ± 7 | >2500 |
Figure 4.

Proposed binding site in the SET domain of EZH2. (A) Close view on the pyridone moiety of compound 5 from its docked pose in human PRC2 complex. Substitution at 4-position of the pyridone ring allows the compound to make more van der Waals contacts with F665, R685 and F686. (B) Close view on the pyridone moiety of compound 31 from its docked pose in human PRC2 complex. White surface, EZH2 binding pocket; purple, van der Waals surface of the ligand.
Table 2.
In Vitro Potencies of Newly Synthesized Inhibitors with Modified Pyridone Core
| |||
|---|---|---|---|
| Cmpd | Structure | IC50(nM) | |
| EZH2 | EZH1 | ||
| 5 |
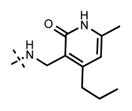
|
<10 | 69 ± 14 |
| 39 |
|
>10000 | >10000 |
| 40 |

|
>2300 | >10000 |
| 41 |
|
>10000 | >10000 |
| 42 |
|
>10000 | >10000 |
| 43 |
|
>10000 | >10000 |
Next, we turned our attention to the indazole ring of 5 and investigated various bicycles and connectivities to the pyridonemethyl-amide moiety and the pyridine-piperazine tail as well as various substituents at the N-1 (indazole numbering) of the indazole ring (Table 3). Moving the i-Pr group from N-1 to N-2 of the indazole (44) decreased the potency of the inhibitor more than 30-fold for both enzymes. Varying the connectivity of the indazole ring to the amide group from the 4-position to the 3-position (45) obliterated the inhibitory activity. In addition, simultaneously moving the pyridine-2-yl-piperazine tail connection from the 6- to the 5-position and the amide group from the 4-position to the 3-position (46) led to a complete loss of potency. Switching the amide and pyridine-tail connection (47) reduced the potency for EZH2 (IC50 = 540 ± 91 nM) and abolished all EZH1 inhibition. Imidazo[1,2-a]pyridine compounds (48 and 49) did not display any inhibitory activity for EZH2 or EZH1, and pyrazolo[1,5-a]pyridine derivative (50) was inactive as well. With these results in hand, we chose to use the indazole ring system with the 4-amide and 6-pyridine connection for the rest of our SAR studies.
Table 3.
In Vitro Potencies of Newly Synthesized Inhibitors with Modified Indazole Motif
| |||
|---|---|---|---|
| Cmpd | Structure | IC50(nM) | |
| EZH2 | EZH1 | ||
| 5 |
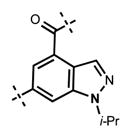
|
<10 | 69 ± 14 |
| 44 |
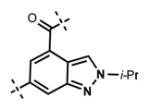
|
384 ± 70 | >2500 |
| 45 |
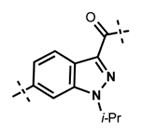
|
>5000 | >5000 |
| 46 |
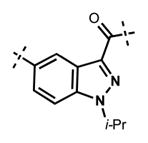
|
>5000 | >5000 |
| 47 |
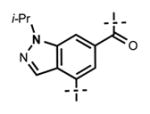
|
540 ± 91 | >5000 |
| 48 |
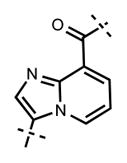
|
>5000 | >5000 |
| 49 |
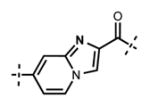
|
>5000 | >5000 |
| 50 |
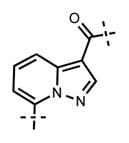
|
>10000 | >10000 |
Next, we probed the role of the substituent at the indazole N-1 (Table 4). While isopropyl (5), t-butyl (51), 3-pentyl (52), cyclopentyl (53), and cyclohexyl (54) substituted inhibitors were very potent (IC50 < 10 nM) for EZH2, they also had consistent potency for EZH1 (between 69 and 127 nM). The smaller methyl (55) and larger cycloheptyl (56) derivatives were 10–20-fold less potent for EZH2 and did not have appreciable inhibitory activity against EZH1. Compounds 57–61 displayed decreased potency with increasing size of the substituent (from cyclobutyl to cyclohexyl (57–59) and to phenyl (60) and a branched large alkyl (61)) for EZH2, and they were inactive against EZH1. Changing the cyclohexyl group (54) to the cyclohexylmethyl group (59) and further to the cyclohexylethyl group (62) led to a significant drop in potency. Similarly, replacing the cyclopentyl group (53) to the cyclopentylmethyl group (58) resulted in a > 15-fold potency loss. Overall, α-branched groups at this position displayed the best potency, and structural modifications at this position affected EZH1 inhibition to a greater extent than that of EZH2. Interestingly, the hexanol group (63) exhibited high potency for both EZH2 and EZH1 (IC50 = 25 ± 5 nM and IC50 = 394 ± 55 nM, respectively). Compound 63 might adopt two different conformations to interact with the PRC2 complex according to our docking poses. In the first pose, the indazole moiety rotates 180° as compared to compound 5 (Figure 5A) and the hexanol group interacts through hydrogen bonds with Y658 and R679. The second pose suggests that the hexanol group is outside the catalytic binding site and interacts with K211 of EED (Figure 5B). In both poses, the hexanol tail is implicated in additional hydrogen bonds that enhance potency for EZH2.
Table 4.
In Vitro Potencies of Newly Synthesized Inhibitors with Indazole N-1 Modifications
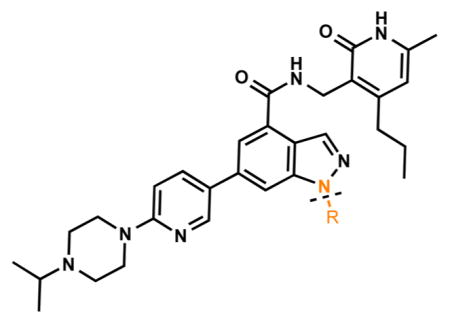
| |||
|---|---|---|---|
| Cmpd | Structure | IC50(nM) | |
| EZH2 | EZH1 | ||
| 5 |
|
<10 | 69 ± 14 |
| 51 |
|
<10 | 127 ± 29 |
| 52 |
|
<10 | 126 ± 22 |
| 53 |
|
<10 | 108 ±23 |
| 54 |

|
<10 | 315 ± 19 |
| 55 |
|
190 ± 33 | >2500 |
| 56 |
|
117 ± 17 | >2500 |
| 57 |
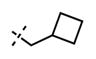
|
57 ±3 | >2500 |
| 58 |
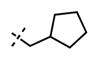
|
149 ± 34 | >2500 |
| 59 |
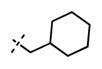
|
697 ± 42 | >2500 |
| 60 |
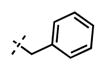
|
280 ± 64 | >5000 |
| 61 |
|
1111 ± 35 | >5000 |
| 62 |
|
>1250 | >2500 |
| 63 |
|
25 ± 5 | 394 ± 55 |
Figure 5.

Two possible docking poses for compound 63. (A) The indazole moiety rotates 180° in relation to compound 5, and the hexanol interacts through hydrogen bonds with Y658 and R679. (B) The hexanol group is outside the catalytic binding site and interacts with K211 of EED.
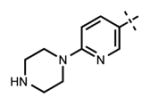
We have also investigated the pyridine-2-yl-piperizine tail of compound 5 that is substituted at 6-position of the indazole ring (Table 5). We believe that this part of compound 5 interacts with the closed pocket formed by the activation loop of EZH2 (Figure 3D). We made modifications including the replacement of the pyridine ring as well as truncation of this tail. Compounds 64 and 65 that replace the isopropyl group on the piperazine outer nitrogen with an ethyl group and hydrogen did not affect the potency for either of the proteins. We then shortened the tail by removing the piperazine ring all together and replaced the pyridine group with the phenyl group (66, Table 5). Compounds 67–68 contain different phenyl substitutions (para-fluoro (67) and para-trifluoromethyl (68)). While these compounds maintained relatively good potency for EZH2, they did not display appreciable inhibitory activity against EZH1 (IC50 > 1250 nM). 2-Pyridine (69) and para-substituted pyridine derivatives (para-fluoro (70) and para-trifluoromethyl (71)) displayed a similar trend as the larger substituents led to the loss of EZH1 potency while maintaining the relatively good potency for EZH2. We also explored various nitrogen containing rings such as piperidine (72), piperazine (73), and homopiperazine (74), which are directly attached to the 6-position of the indazole ring. Again, the EZH2 potencies of the piperidine and piperazine analogues were high (IC50 = 11 ± 1 and 17 ± 3 nM, respectively). On the other hand, their EZH1 potencies dropped more than 10-fold (IC50 = 613 ± 29 and 880 ± 173 nM, respectively). The homopiperazine (74) and piperidine-4-amino (75) derivatives exhibited similar behavior. Interestingly, the 6-bromo analogue (76), which is the intermediate used for the synthesis of aforementioned compounds, showed good potency with only 3–4-fold drop in potency (IC50 = 35 ± 8 nM) for EZH2. However, this compound did not display appreciable inhibitory activity against EZH1 (IC50 > 5000 nM). Our in silico studies suggest that compound 76 can adopt two different bound conformations in the PRC2-EZH2 complex. In the first conformation (Figure 6A), the bromo group is an acceptor of hydrogen bond from C66351,52 and the benzene ring of the indazole stacks with Y111 of the activation loop. In the second conformation (Figure 6B), a 180° rotation around the benzamide bond places both indazole rings to stack with Y111. Overall, these results emphasized that modifications to this tail are particularly important to EZH1 inhibition. The truncation of the tail does not significantly affect EZH2 potency but all truncated compounds suffered a drastic reduction in EZH1 potency.
Table 5.
In Vitro Potencies of Newly Synthesized Inhibitors with Modifications to the Pyridine-2-yl-piperizine Tail Region

| |||
|---|---|---|---|
| Cmpd | Structure | IC50 (nM) | |
| EZH2 | EZH1 | ||
| 5 |
|
<10 | 69 ± 14 |
| 64 |
|
<10 | 88 ± 22 |
| 65 |
|
<10 | 60 ± 9 |
| 66 |
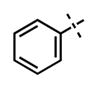
|
41 ± 10 | >1250 |
| 67 |
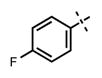
|
48 ± 17 | >2500 |
| 68 |
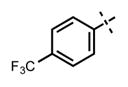
|
66 ± 4 | >1250 |
| 69 |
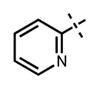
|
24 ± 4 | 681 ± 90 |
| 70 |
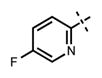
|
22 ± 5 | 773 ± 152 |
| 71 |

|
64 ± 20 | >5000 |
| 72 |
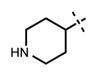
|
11 ± 1 | 613 ± 29 |
| 73 |
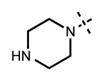
|
17 ± 3 | 880 ± 173 |
| 74 |
|
42 ± 7 | >2500 |
| 75 |
|
26 ± 4 | >1250 |
| 76 |
|
35 ± 8 | >5000 |
Figure 6.

Two possible docking poses for compound 76. (A) The bromo group is an acceptor of hydrogen bond from C663 and the benzene ring of the indazole stacks with Y111 of the activation loop. (B) A 180° rotation around the benzamide bond leads to both indazole rings stacking with Y111.
CONCLUSION
In this study, we explored three main regions of our EZH2/1 inhibitor 5 in a systematic manner to identify moieties that affect EZH2 and EZH1 selectivity. We first investigated the core pyridone ring, namely the substitutions at positions 4, 5, and 6 of this ring. The potency difference between compounds 24 and 34, caused by the removal of the two methyl groups at the 4 and 6 positions, clearly showed the importance of the methyl groups for inhibiting these two enzymes, especially EZH2. Interestingly, further changes from the methyl substitution (24) to the propyl substitution (5) at the 4-position of the pyridone ring affected the EZH1 potency to a greater extent than EZH2. These results underscore the importance of the subtle structural modifications in ligand structure. While the 6-methyl group is essential for potency, further modifications at this position did not improve the potency for either enzyme. In addition, structural alterations at the 5-position were not promising for improving the inhibitory activity as shown by our experimental results and supported by our docking studies. We next focused on the indazole ring after establishing that this ring was the best among the heterocyclic rings we explored and examined the effects of various substituents at the N-1 position of this ring system. We found that structural modifications at this position also had stronger effects on EZH1 potency than EZH2 potency. In general, these derivatives displayed high potency for EZH2. Furthermore, we probed the pyridinyl-piperazine tail of the inhibitor scaffold. This pendant tail group occupies the ligand binding site partially formed by the activation loop and is probably the most diverged moiety among the reported pyridonemethyl-amide based inhibitors. Therefore, on the basis of our results, one may speculate that this region can perhaps be exploited further to achieve better EZH1 potency and possibly selectivity.
The cocrystal structure of PRC2 in complex with inhibitor 11 has established that the pyridone core mimics the amino acid moiety of the cofactor SAM rather than the adenine moiety and the inhibitor binding site overlaps only partially with the cofactor binding site.14 Thus, earlier EZH2 homology models predicting that the inhibitor binding site completely overlaps with the cofactor binding site were incorrect. Because the binding pocket of EZH2 is nearly identical to that of EZH1, in the absence of structural data for EZH1, it is very difficult to predict the structural basis for the observed SAR differences for these two catalytic subunits. Any structural information on the PRC2-EZH1 complex would be extremely useful for developing EZH1 selective chemical tools.
In summary, our systematic SAR studies identified multiple structural features that affect EZH2 and EZH1 potency and selectivity. Taking advantage of the structural insight revealed by the recently published cocrystal structure of PRC2-EZH2 in complex with a pyridone-based inhibitor, we have conducted in silico analyses of our EZH2 SAR results and provided structural explanation for key SAR findings. We anticipate that this work will facilitate further development of EZH2 and/or EZH1 selective inhibitors and chemical tools.
EXPERIMENTAL SECTION
Chemistry: General Procedures
HPLC spectra for all compounds were acquired using an Agilent 1200 series system with DAD detector. Chromatography was performed on a 2.1 mm × 150 mm Zorbax 300SB-C18 5 μm column with water containing 0.1% formic acid as solvent A and acetonitrile containing 0.1% formic acid as solvent B at a flow rate of 0.4 mL/min. The gradient program was as follows: 1% B (0–1 min), 1–99% B (1–4 min), and 99% B (4–8 min). High-resolution mass spectra (HRMS) data were acquired in positive ion mode using an Agilent G1969A API-TOF with an electrospray ionization (ESI) source. Nuclear magnetic resonance (NMR) spectra were acquired on a Bruker DRX-600 spectrometer with 600 MHz for proton (1H NMR) and 150 MHz for carbon (13C NMR); chemical shifts are reported in (δ). Data are reported as follows: chemical shifts (δ), multiplicity (br = broad, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet); coupling constant(s) (J) in Hz; integration. Unless otherwise noted, NMR data were collected at 25 °C. Flash column chromatography was performed using a TeledyneISCO Rf+ system. Preparative HPLC was performed on Agilent Prep 1200 series with UV detector set to 254 nm. Samples were injected onto a Phenomenex Luna 75 mm × 30 mm, 5 mm, C18 column at room temperature. The flow rate was 40 mL/min. A linear gradient was used with 10% (or 50%) of MeOH (A) in H2O (with 0.1% TFA) (B) to 100% of MeOH (A). All final compounds had >95% purity by either UV absorbance at 254 nm during tandem liquid chromatography/mass spectrometry (LCMS) or by the HPLC methods described above.
General Procedure A: Synthesis of Compounds 24–38 (Table 1) and 39–43 (Table 2)
The crude 3-(aminomethyl)-pyridin-2(1H)-one analogues (15) (0.1 mmol), 1-isopropyl-6-(6-(4-isopropylpiperazin-1-yl)pyridin-3-yl)-1H-indazole-4-carboxylic acid (21) (0.05–0.08 mmol), NMM (0.15 mmol), HOAt (0.1 mmol), and EDCI (0.1 mmol) were dissolved in DMSO (2 mL). The contents were stirred at room temperature overnight, monitored by LC/MS, and the crude product was then purified by HPLC to yield the desired compound (24, 26, 32, 33, 34, 39, 40, 41, 42, 43) as solid, or crude 3-(aminomethyl)pyridin-2(1H)-one analogues (15) (0.055 mmol), TBTU (0.075 mmol), and 1-isopropyl-6-(6-(4-isopropylpiperazin-1-yl)pyridin-3-yl)-1H-indazole-4-carboxylic acid (21) (0.05 mmol) were dissolved in DMF (2 mL) prior to the addition of DIPEA (20 μL). The contents were stirred at room temperature overnight, monitored by LC/MS, and the crude product was purified by HPLC to yield the desired compound (25, 27, 28, 29, 30, 31, 35, 36, 37, 38) as solid.
N-((4,6-Dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-1-iso-propyl-6-(6-(4-isopropylpiperazin-1-yl)pyridin-3-yl)-1H-indazole-4-carboxamide (24)
Yield 4.6 mg (16%). 1H NMR (600 MHz, methanol-d4) δ 8.53 (s, 1H), 8.38 (s, 1H), 8.25 (d, 1H, J = 9.1 Hz), 7.99 (s, 1H), 7.78 (s, 1H), 7.21 (d, 1H, J = 9.1 Hz), 6.24 (s, 1H), 5.09 (td, 1H, J = 6.6 Hz, J = 13.2 Hz), 4.58 (s, 2H), 4.56 (br s, 4H), 3.63 (td, 1H, J = 6.6 Hz, J = 13.2 Hz), 3.47 (br s, 4H), 2.45 (s, 3H), 2.28 (s, 3H), 1.57 (d, 6H, J = 6.6 Hz), 1.43 (d, 6H, J = 6.6 Hz). HRMS (m/z) for C31H40N7O2+ [M + H]+: calculated 542.3238, found 542.3247.
N-((4-Ethyl-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-1-isopropyl-6-(6-(4-isopropylpiperazin-1-yl)pyridin-3-yl)-1H-indazole-4-carboxamide (25)
Yield 9.4 mg (22%). 1H NMR (600 MHz, methanol-d4) δ 8.59 (d, 1H, J = 2.4 Hz), 8.39 (d, 1H, J = 6.2 Hz), 8.14 (dd, 1H, J = 2.5 Hz, J = 8.9 Hz), 7.98 (s, 1H), 7.79 (dd, 1H, J = 1.1 Hz, J = 3.8 Hz), 7.11 (d, 1H, J = 8.9 Hz), 6.22 (s, 1H), 5.13–5.09 (m, 1H), 4.62 (s, 2H), 4.61 (br s, 4H), 3.66–3.61 (m, 5H), 2.82 (q, 2H, J = 7.6 Hz), 2.30 (s, 3H), 1.59 (d, 6H, J = 6.6 Hz), 1.45 (d, 6H, J = 6.7 Hz), 1.26 (t, 3H, J = 7.7 Hz). HRMS (m/z) for C32H42N7O2+ [M + H]+: calculated 556.3395, found 556.3394.
N-((4-Butyl-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-1-isopropyl-6-(6-(4-isopropylpiperazin-1-yl)pyridin-3-yl)-1H-indazole-4-carboxamide (26)
Yield 52 mg (81%). 1H NMR (600 MHz, methanol-d4) δ 8.57 (d, 1H, J = 2.4 Hz), 8.36 (s, 1H), 8.03 (dd, 1H, J = 2.6 Hz, J = 8.8 Hz), 7.93 (s, 1H), 7.77 (d, 1H, J = 1.1 Hz), 6.99 (d, 1H, J = 8.9 Hz), 6.15 (s, 1H), 5.12–5.07 (m, 1H), 4.60 (s, 2H), 3.77 (br s, 4H), 3.21–3.17 (m, 1H), 3.08 (br s, 4H), 2.75 (t, 2H, J = 7.8 Hz), 2.26 (s, 3H), 1.59–1.55 (m, 2H), 1.57 (d, 6H, J = 6.6 Hz), 1.46–1.40 (m, 2H), 1.28 (d, 6H, J = 6.6 Hz), 0.92 (t, 3H, J = 7.4 Hz). HRMS (m/z) for C34H46N7O2+ [M + H]+: calculated 584.3708, found 584.3710.
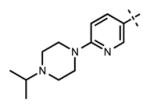
1-Isopropyl-N-((4-isopropyl-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-6-(6-(4-isopropylpiperazin-1-yl)pyridin-3-yl)-1H-inda-zole-4-carboxamide (27)
Yield 28.5 mg (55%). 1H NMR (400 MHz, chloroform-d) δ 12.60 (s, 1H), 8.48 (d, 1H, J = 2.3 Hz), 8.38 (s, 1H), 7.89 (t, 1H, J = 5.7 Hz), 7.76 (dd, 1H, J = 2.5 Hz, J = 8.8 Hz), 7.70 (d, 1H, J = 0.9 Hz), 7.57 (s, 1H), 6.69 (d, 1H, J = 8.9 Hz), 6.04 (s, 1H), 4.90–4.83 (m, 1H), 4.71 (d, 2H, J = 5.8 Hz), 3.59 (t, 4H, J = 5.0 Hz), 3.57–3.50 (m, 1H), 2.75–2.69 (m, 1H), 2.63 (t, 4H, J = 5.0 Hz), 2.19 (s, 3H), 1.58 (d, 6H, J = 6.6 Hz), 1.21 (d, 6H, J = 6.8 Hz), 1.08 (d, 6H, J = 6.6 Hz). HRMS (m/z) for C33H44N7O2+ [M + H]+: calculated 570.3551, found 570.3557.
N-((4-Cyclopentyl-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)-methyl)-1-isopropyl-6-(6-(4-isopropylpiperazin-1-yl)pyridin-3-yl)-1H-indazole-4-carboxamide (28)
Yield 26.0 mg (56%). 1H NMR (600 MHz, chloroform-d) δ 12.37 (s, 1H), 8.44 (d, 1H, J = 1.3 Hz), 8.41 (s, 1H), 8.02 (s, 1H), 7.75 (d, 1H, J = 7.2 Hz), 7.66 (s, 1H), 7.57 (s, 1H), 6.59 (d, 1H, J = 8.7 Hz), 6.06 (s, 1H), 4.91–4.86 (m, 1H), 4.72 (d, 2H, J = 4.8 Hz), 3.87 (br s, 4H), 3.59–3.50 (m, 1H), 3.45–3.44 (m, 1H), 3.09 (br s, 4H), 2.23 (s, 3H), 2.08–2.06 (m, 2H), 1.83–1.76 (m, 4H), 1.59 (d, 6H, J = 6.6 Hz), 1.56–1.53 (m, 2H), 1.34 (d, 6H, J = 6.4 Hz). HRMS (m/z) for C35H46N7O2+ [M + H]+: calculated 596.3708, found 596.3713.
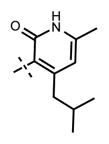
N-((4-Isobutyl-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-1-isopropyl-6-(6-(4-isopropylpiperazin-1-yl)pyridin-3-yl)-1H-indazole-4-carboxamide (29)
Yield 30.4 mg (62%). 1H NMR (600 MHz, chloroform-d) δ 12.97 (s, 1H), 8.50 (d, 1H, J = 1.5 Hz), 8.40 (s, 1H), 8.06 (t, 1H, J = 5.0 Hz), 7.78 (dd, 1H, J = 2.0 Hz, J = 8.7 Hz), 7.74 (s, 1H), 7.60 (s, 1H), 6.69 (d, 1H, J = 8.8 Hz), 5.92 (s, 1H), 4.91–4.86 (m, 1H), 4.68 (d, 2H, J = 5.4 Hz), 3.61 (br s, 4H), 2.77–2.74 (m, 1H), 2.66 (br s, 4H), 2.63 (d, 2H, J = 7.2 Hz), 2.17 (s, 3H), 1.93–1.88 (m, 1H), 1.60 (d, 6H, J = 6.5 Hz), 1.11 (d, 6H, J = 6.4 Hz), 0.99 (d, 6H, J = 6.4 Hz). HRMS (m/z) for C34H46N7O2+ [M + H]+: calculated 584.3708, found 584.3713.
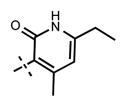
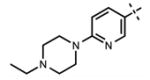
N-((6-Ethyl-4-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-1-isopropyl-6-(6-(4-isopropylpiperazin-1-yl)pyridin-3-yl)-1H-indazole-4-carboxamide (30)
Yield 20.3 mg (61%). 1H NMR (600 MHz, methanol-d4) δ 8.55 (d, 1H, J = 2.4 Hz), 8.38 (s, 1H), 8.00 (dd, 1H, J = 2.5 Hz, J = 8.8 Hz), 7.92 (s, 1H), 7.78 (d, 1H, J = 0.8 Hz), 6.97 (d, 1H, J = 8.9 Hz), 6.18 (s, 1H), 5.10–5.06 (m, 1H), 4.60 (s, 2H), 3.80 (br s, 4H), 3.16 (br s, 4H), 2.55 (q, 2H, J = 7.6 Hz), 2.45 (s, 3H), 1.57 (d, 6H, J = 6.6 Hz), 1.33 (d, 6H, J = 6.5 Hz), 1.24 (t, 3H, J = 7.6 Hz). HRMS (m/z) for C32H42N7O2+ [M + H]+: calculated 556.3395, found 556.3408.
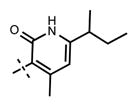
N-((6-(sec-Butyl)-4-methyl-2-oxo-1,2-dihydropyridin-3-yl)-methyl)-1-isopropyl-6-(6-(4-isopropylpiperazin-1-yl)pyridin-3-yl)-1H-indazole-4-carboxamide (31)
Yield 24.5 mg (63%). 1H NMR (600 MHz, chloroform-d) δ 11.99 (s, 1H), 8.49 (s, 1H), 8.41 (s, 1H), 7.83 (s, 1H), 7.78 (dd, 1H, J = 1.7 Hz, J = 8.6 Hz), 7.71 (s, 1H), 7.60 (s, 1H), 6.69 (d, 1H, J = 8.7 Hz), 5.93 (s, 1H), 4.91–4.87 (m, 1H), 4.67 (d, 2H, J = 5.6 Hz), 3.66 (br s, 4H), 2.82 (br s, 1H), 2.71 (br s, 4H), 2.49–2.45 (m, 4H), 1.65–1.60 (m, 1H), 1.60 (d, 6H, J = 6.6 Hz), 1.52–1.45 (m, 1H), 1.17–1.14 (m, 9H), 0.73 (t, 3H, J = 7.3 Hz). HRMS (m/z) for C34H46N7O2+ [M + H]+: calculated 584.3708, found 584.3713.
N-((6-(tert-Butyl)-4-methyl-2-oxo-1,2-dihydropyridin-3-yl)-methyl)-1-isopropyl-6-(6-(4-isopropylpiperazin-1-yl)pyridin-3-yl)-1H-indazole-4-carboxamide (32)
Yield 14 mg (81%). 1H NMR (600 MHz, methanol-d4) δ 8.56 (s, 1H), 8.39 (s, 1H), 8.24 (d, 1H, J = 8.9 Hz), 7.99 (s, 1H), 7.79 (s, 1H), 7.21 (d, 1H, J = 8.9 Hz), 6.29 (s, 1H), 5.12–5.06 (m, 1H), 4.59 (s, 2H), 3.56 (br s, 4H), 3.66–3.60 (m, 5H), 2.48 (s, 3H), 1.57 (d, 6H, J = 6.5 Hz), 1.43 (d, 6H, J = 6.5 Hz), 1.32 (s, 9H). HRMS (m/z) for C34H46N7O2+ [M + H]+: calculated 584.3708, found 584.3710.
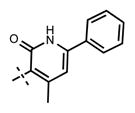
1-Isopropyl-6-(6-(4-isopropylpiperazin-1-yl)pyridin-3-yl)-N-((4-methyl-2-oxo-6-phenyl-1,2-dihydropyridin-3-yl)methyl)-1H-inda-zole-4-carboxamide (33)
Yield 15 mg (71%). 1H NMR (600 MHz, methanol-d4) δ 8.57 (d, 1H, J = 2.4 Hz), 8.40 (s, 1H), 8.21 (dd, 1H, J = 2.4 Hz, J = 9.0 Hz), 7.98 (s, 1H), 7.81 (d, 1H, J = 1.0 Hz), 7.67–7.66 (m, 2H), 7.51–7.48 (m, 3H), 7.17 (d, 1H, J = 9.0 Hz), 6.61 (s, 1H), 5.12–5.07 (m, 1H), 4.65 (s, 2H), 4.56 (br s, 4H), 3.64–3.59 (m, 5H), 2.55 (s, 3H), 1.57 (d, 6H, J = 6.6 Hz), 1.42 (d, 6H, J = 6.7 Hz). HRMS (m/z) for C36H42N7O2+ [M + H]+: calculated 604.3395, found 604.3399.
1-Isopropyl-6-(6-(4-isopropylpiperazin-1-yl)pyridin-3-yl)-N-((2-oxo-1,2-dihydropyridin-3-yl)methyl)-1H-indazole-4-carboxamide (34)
Yield: 16 mg (65%). 1H NMR (600 MHz, methanol-d4) δ 8.56 (d, 1H, J = 2.3 Hz), 8.39 (s, 1H), 8.23 (dd, 1H, J = 2.4 Hz, J = 9.0 Hz), 8.00 (s, 1H), 7.84 (d, 1H, J = 1.1 Hz), 7.64 (d, 1H, J = 5.8 Hz), 7.40 (dd, 1H, J = 1.8 Hz, J = 6.5 Hz), 7.20 (d, 1H, J = 9.1 Hz), 6.41 (t, 1H, J = 6.7 Hz), 5.10 (td, 1H, J = 6.6 Hz, J = 13.3 Hz), 4.60 (br s, 4H), 4.51 (s, 2H), 3.42 (br s, 4H), 3.63 (td, 1H, J = 6.6 Hz, J = 13.3 Hz), 1.57 (d, 6H, J = 6.6 Hz), 1.43 (d, 6H, J = 6.7 Hz). HRMS (m/z) for C29H36N7O2+ [M + H]+: calculated 514.2925, found 514.2927.
N-((4-Ethyl-5-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-1-isopropyl-6-(6-(4-isopropylpiperazin-1-yl)pyridin-3-yl)-1H-indazole-4-carboxamide (35)
Yield: 32.9 mg (72%). 1H NMR (600 MHz, methanol-d4) δ 8.61 (d, 1H, J = 2.4 Hz), 8.40 (s, 1H), 8.19 (dd, 1H, J = 2.4 Hz, J = 9.2 Hz), 8.00 (s, 1H), 7.81 (s, 1H), 7.27 (s, 1H), 7.17 (d, 1H, J = 8.8 Hz), 5.14–5.09 (m, 1H), 4.69 (s, 2H), 4.61 (br s, 4H), 3.66–3.61 (m, 5H), 2.93 (q, 2H, J = 7.7 Hz), 2.18 (s, 3H), 1.60 (d, 6H, J = 6.4 Hz), 1.45 (d, 6H, J = 6.5 Hz), 1.24 (t, 3H, J = 7.6 Hz). HRMS (m/z) for C32H42N7O2+ [M + H]+: calculated 556.3395, found 556.3403.
1-Isopropyl-6-(6-(4-isopropylpiperazin-1-yl)pyridin-3-yl)-N-((4,5,6-trimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-1H-inda-zole-4-carboxamide (36)
Yield: 18.7 mg (56%). 1H NMR (600 MHz, methanol-d4) δ 8.57 (d, 1H, J = 2.4 Hz), 8.38 (s, 1H), 8.02 (dd, 1H, J = 2.5 Hz, J = 8.8 Hz), 7.93 (s, 1H), 7.78 (d, 1H, J = 1.0 Hz), 6.98 (d, 1H, J = 8.8 Hz), 5.11–5.07 (m, 1H), 4.65 (s, 2H), 3.66–3.56 (m, 5H), 3.23 (br s, 4H), 2.39 (s, 3H), 2.28 (s, 3H), 2.05 (s, 3H), 1.58 (d, 6H, J = 6.6 Hz), 1.43 (d, 6H, J = 6.7 Hz). HRMS (m/z) for C32H42N7O2+ [M + H]+ calculated 556.3395, found 556.3390.
1-Isopropyl-6-(6-(4-isopropylpiperazin-1-yl)pyridin-3-yl)-N-((1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-1H-in-dazole-4-carboxamide (37)
Yield: 12.8 mg (42%). 1H NMR (600 MHz, methanol-d4) δ 8.57 (d, 1H, J = 2.4 Hz), 8.38 (s, 1H), 8.03 (dd, 1H, J = 2.5 Hz, J = 8.8 Hz), 7.93 (s, 1H), 7.78 (s, 1H), 7.00 (d, 1H, J = 8.8 Hz), 5.11–5.06 (m, 1H), 4.61 (s, 2H), 3.88 (br s, 4H), 3.53–3.44 (m, 1H), 3.32 (br s, 4H), 2.96 (t, 2H, J = 5.9 Hz), 2.50 (t, 2H, J = 5.6 Hz), 2.22 (s, 3H), 1.80–1.74 (m, 4H), 1.57 (d, 6H, J = 6.6 Hz), 1.39 (d, 6H, J = 6.6 Hz). HRMS (m/z) for C34H44N7O2+ [M + H]+ calculated 582.3551, found 582.3555.
1-Isopropyl-6-(6-(4-isopropylpiperazin-1-yl)pyridin-3-yl)-N-((1-methyl-3-oxo-3,5,6,7-tetrahydro-2H-cyclopenta[c]pyridin-4-yl)-methyl)-1H-indazole-4-carboxamide (38)
Yield: 11.8 mg (42%). 1H NMR (400 MHz, methanol-d4) δ 8.57 (s, 1H), 8.39 (s, 1H), 8.17 (dd, 1H, J = 2.5 Hz, J = 9.0 Hz), 7.99 (s, 1H), 7.81 (d, 1H, J = 1.3 Hz), 7.15 (d, 1H, J = 8.0 Hz), 5.12–5.05 (m, 1H), 4.54 (s, 2H), 3.65–3.57 (m, 1H), 3.48 (br s, 8H), 3.13 (t, 2H, J = 7.6 Hz), 2.77 (t, 2H, J = 7.3 Hz), 2.28 (s, 3H), 2.16–2.08 (m, 2H), 1.58 (d, 6H, J = 6.6 Hz), 1.43 (d, 6H, J = 6.7 Hz). HRMS (m/z) for C33H42N7O2+ [M + H]+ calculated 568.3395, found 568.3397.
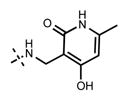
N-((4-Hydroxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-1-isopropyl-6-(6-(4-isopropylpiperazin-1-yl)pyridin-3-yl)-1H-inda-zole-4-carboxamide (39)
Yield: 7.8 mg (39%). 1H NMR (600 MHz, methanol-d4) δ 8.59 (d, 1H, J = 2.1 Hz), 8.40 (s, 1H), 8.15 (dd, 1H, J = 2.3 Hz, J = 9.0 Hz), 8.00 (s, 1H), 7.85 (s, 1H), 7.13 (d, 1H, J = 8.7 Hz), 6.13 (s, 1H), 5.12–5.07 (m, 1H), 4.65 (br s, 4H), 4.54 (s, 2H), 3.65–3.58 (m, 5H), 2.29 (s, 3H), 1.58 (d, 6H, J = 6.8 Hz), 1.43 (d, 6H, J = 6.7 Hz). HRMS (m/z) for C30H38N7O3+ [M + H]+: calculated 544.3031, found 544.3030.
1-Isopropyl-6-(6-(4-isopropylpiperazin-1-yl)pyridin-3-yl)-N-((2-methyl-6-oxo-1,6-dihydropyrimidin-5-yl)methyl)-1H-indazole-4-carboxamide (40)
Yield: 12.0 mg (63%). 1H NMR (600 MHz, methanol-d4) δ 8.61 (s, 1H), 8.39 (s, 1H), 8.20 (d, 1H, J = 10.2 Hz), 8.02 (s, 1H), 7.99 (s, 1H), 7.86 (s, 1H), 7.18 (d, 1H, J = 9.0 Hz), 5.13–5.08 (m, 1H), 4.59 (br s, 4H), 4.45 (s, 2H), 3.66–3.58 (m, 5H), 2.55 (s, 3H), 1.58 (d, 6H, J = 6.6 Hz), 1.43 (d, 6H, J = 6.6 Hz). HRMS (m/z) for C29H37N8O2 + [M + H]+ calculated 529.3034, found 529.3033.
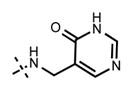
1-Isopropyl-6-(6-(4-isopropylpiperazin-1-yl)pyridin-3-yl)-N-((6-oxo-1,6-dihydropyrimidin-5-yl)methyl)-1H-indazole-4-carboxamide (41)
Yield: 11.2 mg (60%). 1H NMR (600 MHz, methanol-d4) δ 8.59 (d, 1H, J = 2.3 Hz), 8.54 (br s, 1H), 8.39 (s, 1H), 8.23 (dd, 1H, J = 2.4 Hz, J = 9.0 Hz), 8.12 (br s, 1H), 8.02 (s, 1H), 7.85 (d, 1H, J = 0.9 Hz), 7.21 (d, 1H, J = 9.0 Hz), 5.13–5.08 (m, 1H), 4.50 (br s, 4H), 4.48 (s, 2H) 3.65–3.60 (m, 1H), 3.50 (br s, 4H), 2.66 (s, 3H), 1.58 (d, 6H, J = 6.6 Hz), 1.43 (d, 6H, J = 6.7 Hz). HRMS (m/z) for C28H35N8O2+ [M + H]+: calculated 515.2877, found 515.2878.
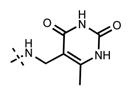
1-Isopropyl-6-(6-(4-isopropylpiperazin-1-yl)pyridin-3-yl)-N-((6-methyl-2,4-dioxo-1,2,3,4-tetrahydropyrimidin-5-yl)methyl)-1H-in-dazole-4-carboxamide (42)
Yield: 5.9 mg (30%). 1H NMR (600 MHz, methanol-d4) δ 8.60 (s, 1H), 8.35 (s, 1H), 8.11 (dd, 1H, J = 1.3 Hz, J = 8.9 Hz), 7.95 (s, 1H), 7.76 (s, 1H), 7.09 (d, 1H, J = 8.8 Hz), 5.11–5.06 (m, 1H), 4.60 (br s, 4H), 4.38 (s, 2H), 3.63–3.57 (m, 5H), 2.39 (s, 3H), 1.57 (d, 6H, J = 6.5 Hz), 1.43 (d, 6H, J = 6.6 Hz). HRMS (m/z) for C29H37N8O3+ [M + H]+: calculated 545.2983, found 545.2980.
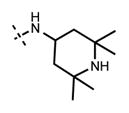
1-Isopropyl-6-(6-(4-isopropylpiperazin-1-yl)pyridin-3-yl)-N-(2,2,6,6-tetramethylpiperidin-4-yl)-1H-indazole-4-carboxamide (43)
Yield: 33.0 mg (78%). 1H NMR (600 MHz, methanol-d4) δ 8.61 (d, 1H, J = 2.3 Hz), 8.38 (s, 1H), 8.32 (dd, 1H, J = 2.4 Hz, J = 9.1 Hz), 8.05 (s, 1H), 7.84 (d, 1H, J = 1.0 Hz), 7.30 (d, 1H, J = 9.1 Hz), 5.15–5.08 (m, 1H), 4.68–4.58 (m, 5H), 3.67–3.56 (m, 5H), 2.20 (dd, 2H, J = 3.6 Hz, J = 13.8 Hz), 1.76 (t, 2H, J = 13.0 Hz), 1.61 (s, 6H), 1.58 (d, 6H, J = 6.6 Hz), 1.49 (s, 6H), 1.42 (d, 6H, J = 6.7 Hz). HRMS (m/z) for C32H48N7O+ [M + H]+: calculated 546.3915, found 546.3922.
General Procedure B: Synthesis of compounds 44–50 (Table 3) and 51–63 (Table 4), and 66, 68, 69, and 71–76 (Table 5)
The crude 3-(aminomethyl)pyridin-2(1H)-one analogues (15) (0.1 mmol), 6-bromo-1-alkyl-1H-indazole-4-carboxylic acid (18) (0.05–0.08 mmol), NMM (0.15 mmol), HOAt (11 mg, 0.10 mmol), and EDCI (0.10 mmol) were dissolved in DMSO (2 mL). The contents were stirred at room temperature overnight, monitored by LC/MS, quenched with water, and the desired product (22) was separated out and filtered to get the compound (22) as solid. Then compound (22) (0.10 mmol, 1.0 equiv), (6-(4-isopropylpiperazin-1-yl)pyridin-3-yl)boronic acid (20) (0.12 mmol, 1.2 equiv), and potassium acetate (0.30 mmol, 3.0 equiv) were mixed with 1,4-dioxane (50 mL) and water (10 mL) in a flask. To this mixture, 1,1′-bis(diphenylphosphino)ferrocene-palladium(II)dichloride dichloro-methane complex (10–20 wt %) was added under argon atmosphere at room temperature. The mixture was heated to 80 °C for overnight and cooled to room temperature. The crude product was then purified by flash column chromatography (gradient from 100% hexane to 100% ethyl acetate, then from 100% ethyl acetate to 10% methanol) to yield the desired compound (44–63, 66, 68, 69, 71, 72). For the synthesis of compounds 73–75, intermediate 22 (Scheme 1) (0.10 mmol, 1.0 equiv), corresponding amines (0.15 mmol, 1.5 equiv), t-BuONa (0.15 mmol, 1.5 equiv), and BINAP (0.02 mmol, 0.2 equiv) were mixed with 1,4-dioxane (50 mL) in a flask. To this mixture, Pd2(dba)3 (0.01 mmol, 0.1 equiv) was added under argon atmosphere at room temperature. The mixture was heated to 80 °C for overnight and cooled to room temperature. The crude product was then purified by flash column chromatography (gradient from 100% hexane to 100% ethyl acetate, then from 100% ethyl acetate to 10% methanol) or purified by HPLC to yield the desired product (73, 74, and 75). Synthesis and full characterization of compounds 5, 64, 65, 67, and 70 have been previously published.36
2-Isopropyl-6-(6-(4-isopropylpiperazin-1-yl)pyridin-3-yl)-N-((6-methyl-2-oxo-4-propyl-1,2-dihydropyridin-3-yl)methyl)-2H-inda-zole-4-carboxamide (44)
Yield: 4.6 mg (7%). 1H NMR (600 MHz, methanol-d4) δ 8.60 (s, 1H), 8.56 (s, 1H), 8.09 (dd, 1H, J = 1.5 Hz, J = 8.9 Hz), 7.93 (s, 1H), 7.79 (s, 1H), 7.10 (d, 1H, J = 9.0 Hz), 6.17 (s, 1H), 4.93–4.89 (m, 1H), 4.60 (br s, 4H), 4.59 (s, 2H), 3.64–3.57 (m, 5H), 2.75 (t, 2H, J = 7.8 Hz), 2.26 (s, 3H), 1.67 (d, 6H, J = 6.7 Hz), 1.66–1.61 (m, 2H), 1.42 (d, 6H, J = 6.6 Hz), 1.02 (t, 3H, J = 7.3 Hz). HRMS (m/z) for C33H44N7O2+ [M + H]+: calculated 570.3551, found 570.3542.
1-Isopropyl-6-(6-(4-isopropylpiperazin-1-yl)pyridin-3-yl)-N-((6-methyl-2-oxo-4-propyl-1,2-dihydropyridin-3-yl)methyl)-1H-inda-zole-3-carboxamide (45)
Yield: 50 mg (86%). 1H NMR (600 MHz, methanol-d4) δ 8.45 (d, 1H, J = 2.1 Hz), 7.92 (dd, 1H, J = 2.2 Hz, J = 8.7 Hz), 7.83 (d, 1H, J = 8.7 Hz), 7.80 (s, 1H), 7.42 (d, 1H, J = 8.2 Hz), 6.96 (d, 1H, J = 8.8 Hz), 6.14 (s, 1H), 5.59–5.53 (m, 1H), 4.59 (s, 2H), 3.72 (br s, 4H), 3.11–3.06 (m, 1H), 2.98 (br s, 4H), 2.74 (t, 2H, J = 7.8 Hz), 2.26 (s, 3H), 1.68–1.60 (m, 2H), 1.61 (d, 6H, J = 6.6 Hz), 1.24 (d, 6H, J = 6.5 Hz), 1.03 (t, 3H, J = 7.3 Hz). HRMS (m/z) for C33H44N7O2+ [M + H]+: calculated 570.3551, found 570.3553.
1-Isopropyl-5-(6-(4-isopropylpiperazin-1-yl)pyridin-3-yl)-N-((6-methyl-2-oxo-4-propyl-1,2-dihydropyridin-3-yl)methyl)-1H-inda-zole-3-carboxamide (46)
Yield: 58 mg (64%). 1H NMR (600 MHz, methanol-d4) δ 8.55 (d, 1H, J = 2.4 Hz), 8.00–7.98 (m, 2H), 7.74 (d, 1H, J = 9.0 Hz), 7.60 (dd, 1H, J = 1.5 Hz, J = 9.0 Hz), 6.98 (d, 1H, J = 8.8 Hz), 6.13 (s, 1H), 5.62–5.56 (m, 1H), 4.59 (s, 2H), 3.75 (br s, 4H), 3.22–3.18 (m, 1H), 3.09 (t, 4H, J = 4.8 Hz), 2.74 (t, 2H, J = 7.8 Hz), 2.26 (s, 3H), 1.68–1.61 (m, 2H), 1.61 (d, 6H, J = 6.6 Hz), 1.29 (d, 6H, J = 6.7 Hz), 1.02 (t, 3H, J = 7.3 Hz). HRMS (m/z) for C33H44N7O2+ [M + H]+: calculated 570.3551, found 570.3554.
1-Isopropyl-4-(6-(4-isopropylpiperazin-1-yl)pyridin-3-yl)-N-((6-methyl-2-oxo-4-propyl-1,2-dihydropyridin-3-yl)methyl)-1H-inda-zole-6-carboxamide (47)
Yield: 71 mg (79%). 1H NMR (600 MHz, methanol-d4) δ 8.50 (d, 1H, J = 2.3 Hz), 8.16 (s, 1H), 8.04 (s, 1H), 7.97 (dd, 1H, J = 2.5 Hz, J = 8.8 Hz), 7.61 (d, 1H, J = 1.1 Hz), 6.99 (d, 1H, J = 8.9 Hz), 6.14 (s, 1H), 5.07–5.03 (m, 1H), 4.57 (s, 2H), 3.67 (br s, 4H), 2.86–2.81 (m, 1H), 2.78 (t, 4H, J = 4.5 Hz), 2.72 (t, 2H, J = 8.1 Hz), 2.26 (s, 3H), 1.66–1.60 (m, 2H), 1.58 (d, 6H, J = 6.7 Hz), 1.17 (d, 6H, J = 6.5 Hz), 1.00 (t, 3H, J = 7.3 Hz). HRMS (m/z) for C33H44N7O2+ [M + H]+: calculated 570.3551, found 570.3555.
3-(6-(4-Isopropylpiperazin-1-yl)pyridin-3-yl)-N-((6-methyl-2-oxo-4-propyl-1,2-dihydropyridin-3-yl)methyl)imidazo[1,2-a]pyridine-8-carboxamide (48)
Yield: 42 mg (40%). 1H NMR (600 MHz, methanol-d4) δ 8.72 (d, 1H, J = 6.8 Hz), 8.47 (s, 1H), 8.37 (d, 1H, J = 7.3 Hz), 8.08 (s, 1H), 7.94 (d, 1H, J = 9.8 Hz), 7.49 (t, 1H, J = 7.0 Hz), 7.16 (d, 1H, J = 8.9 Hz), 6.17 (s, 1H), 4.74–4.72 (d, 2H, J = 12.0 Hz), 4.61 (s, 2H), 3.65–3.59 (m, 2H), 3.31–3.20 (m, 4H), 2.72 (t, 2H, J = 7.8 Hz), 2.27 (s, 3H), 1.67–1.61 (m, 2H), 1.43 (d, 6H, J = 6.6 Hz), 1.01 (t, 3H, J = 7.3 Hz). HRMS (m/z) for C30H38N7O2+ [M + H]+: calculated 528.3081, found 528.3079.
7-(6-(4-Isopropylpiperazin-1-yl)pyridin-3-yl)-N-((6-methyl-2-oxo-4-propyl-1,2-dihydropyridin-3-yl)methyl)pyrazolo[1,5-a]pyridine-3-carboxamide (49)
Yield: 50 mg (48%). 1H NMR (600 MHz, methanol-d4) δ 8.63 (s, 1H), 8.40 (s, 1H), 8.22 (d, 1H, J = 8.8 Hz), 8.14 (d, 1H, J = 8.9 Hz), 7.50 (t, 1H, J = 7.8 Hz), 7.09 (d, 1H, J = 6.7 Hz), 6.94 (d, 1H, J = 9.0 Hz), 6.14 (s, 1H), 4.53 (s, 2H), 3.69 (br s, 4H), 2.80–2.66 (m, 7H), 2.26 (s, 3H), 1.65–1.58 (m, 2H), 1.14 (d, 6H, J = 6.3 Hz), 0.99 (t, 3H, J = 7.3 Hz). HRMS (m/z) for C30H38N7O2+ [M + H]+: calculated 528.3081, found 528.3082.
7-(6-(4-Isopropylpiperazin-1-yl)pyridin-3-yl)-N-((6-methyl-2-oxo-4-propyl-1,2-dihydropyridin-3-yl)methyl)imidazo[1,2-a]pyridine-2-carboxamide (50)
Yield: 53 mg (50%). 1H NMR (600 MHz, methanol-d4) δ 8.78 (d, 1H, J = 7.0 Hz), 8.69 (d, 1H, J = 1.8 Hz), 8.56 (s, 1H), 8.15 (dd, 1H, J = 2.1 Hz, J = 8.9 Hz), 7.98 (s, 1H), 7.79 (d, 1H, J = 7.0 Hz), 7.11 (d, 1H, J = 9.0 Hz), 6.19 (s, 1H), 4.70 (br s, 4H), 4.56 (s, 2H), 3.64–3.58 (m, 5H), 2.67 (m, 2H), 2.28 (s, 3H), 1.63 (m, 2H), 1.42 (d, 6H, J = 6.6 Hz), 1.00 (t, 3H, J = 7.3 Hz). HRMS (m/z) for C30H38N7O2+ [M + H]+: calculated 528.3081, found 528.3088.
1-(tert-Butyl)-6-(6-(4-isopropylpiperazin-1-yl)pyridin-3-yl)-N-((6-methyl-2-oxo-4-propyl-1,2-dihydropyridin-3-yl)methyl)-1H-inda-zole-4-carboxamide (51)
Yield: 25 mg (83%). 1H NMR (600 MHz, methanol-d4) δ 8.50 (d, 1H, J = 2.5 Hz), 8.28 (s, 1H), 8.03 (s, 1H), 7.98 (dd, 1H, J = 2.6 Hz, J = 8.8 Hz), 7.72 (d, 1H, J = 1.1 Hz), 6.98 (d, 1H, J = 8.9 Hz), 6.15 (s, 1H), 4.59 (s, 2H), 3.71 (br s, 4H), 3.02–2.98 (m, 1H), 2.92 (br s, 4H), 2.75–2.73 (m, 2H), 2.26 (s, 3H), 1.81 (s, 9H), 1.68–1.61 (m, 2H), 1.22 (d, 6H, J = 6.5 Hz), 1.02 (t, 3H, J = 7.4 Hz). HRMS (m/z) for C34H46N7O2+ [M + H]+: calculated 584.3708, found 584.3709.
6-(6-(4-Isopropylpiperazin-1-yl)pyridin-3-yl)-N-((6-methyl-2-oxo-4-propyl-1,2-dihydropyridin-3-yl)methyl)-1-(pentan-3-yl)-1H-inda-zole-4-carboxamide (52)
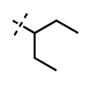
Yield: 40 mg (61%). 1H NMR (600 MHz, methanol-d4) δ 8.56 (d, 1H, J = 1.8 Hz), 8.41 (s, 1H), 8.03 (dd, 1H, J = 2.3 Hz, J = 8.8 Hz), 7.94 (s, 1H), 7.76 (s, 1H), 7.00 (d, 1H, J = 8.8 Hz), 6.15 (s, 1H), 4.60 (s, 2H), 4.59–4.55 (m, 1H), 3.81 (br s, 4H), 3.17 (br s, 4H), 2.74 (t, 2H, J = 7.8 Hz), 2.26 (s, 3H), 2.11–2.03 (m, 2H), 1.98–1.91 (m, 3H), 1.67–1.61 (m, 2H), 1.32 (d, 6H, J = 6.6 Hz), 1.02 (t, 3H, J = 7.3 Hz), 0.70 (t, 6H, J = 7.3 Hz). HRMS (m/z) for C35H48N7O2+ [M + H]+: calculated 598.3864, found 598.3868.
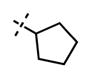
1-Cyclopentyl-6-(6-(4-isopropylpiperazin-1-yl)pyridin-3-yl)-N-((6-methyl-2-oxo-4-propyl-1,2-dihydropyridin-3-yl)methyl)-1H-inda-zole-4-carboxamide (53)
Yield: 78 mg (62%). 1H NMR (600 MHz, methanol-d4) δ 8.49 (s, 1H), 8.33 (s, 1H), 7.94 (d, 1H, J = 8.7 Hz), 7.89 (s, 1H), 7.75 (s, 1H), 6.87 (d, 1H, J = 8.7 Hz), 6.12 (s, 1H), 5.24–5.20 (m, 1H), 4.58 (s, 2H), 3.59 (br s, 4H), 2.76–2.70 (m, 7H), 2.24 (s, 3H), 2.21–2.18 (m, 2H), 2.12–2.08 (m, 2H), 1.98–1.94 (m, 2H), 1.79–1.75 (m, 2H), 1.65–1.61 (m, 2H), 1.13 (d, 6H, J = 6.1 Hz), 1.00 (t, 3H, J = 7.1 Hz). HRMS (m/z) for C35H46N7O2+ [M + H]+: calculated 596.3708, found 596.3711.
1-Cyclohexyl-6-(6-(4-isopropylpiperazin-1-yl)pyridin-3-yl)-N-((6-methyl-2-oxo-4-propyl-1,2-dihydropyridin-3-yl)methyl)-1H-inda-zole-4-carboxamide (54)
Yield: 10 mg (16%). 1H NMR (600 MHz, methanol-d4) δ 8.52 (d, 1H, J = 2.4 Hz), 8.34 (s, 1H), 7.99 (dd, 1H, J = 2.6 Hz, J = 8.9 Hz), 7.91 (s, 1H), 7.75 (d, 1H, J = 1.2 Hz), 6.92 (d, 1H, J = 8.9 Hz), 6.14 (s, 1H), 4.70–4.64 (m, 1H), 4.59 (s, 2H), 3.61 (t, 4H, J = 4.5 Hz), 2.78–2.70 (m, 7H), 2.25 (s, 3H), 2.03–1.92 (m, 6H), 1.80–1.77 (m, 1H), 1.67–1.56 (m, 4H), 1.40–1.32 (m, 1H), 1.14 (d, 6H, J = 6.5 Hz), 1.01 (t, 3H, J = 7.4 Hz). HRMS (m/z) for C36H48N7O2+ [M + H]+: calculated 610.3864, found 610.3865.
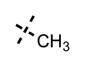
6-(6-(4-Isopropylpiperazin-1-yl)pyridin-3-yl)-1-methyl-N-((6-methyl-2-oxo-4-propyl-1,2-dihydropyridin-3-yl)methyl)-1H-inda-zole-4-carboxamide (55)
Yield: 50 mg (77%). 1H NMR (600 MHz, methanol-d4) δ 8.55 (d, 1H, J = 2.1 Hz), 8.31 (s, 1H), 7.99 (dd, 1H, J = 2.3 Hz, J = 8.8 Hz), 7.86 (s, 1H), 7.76 (s, 1H), 6.95 (d, 1H, J = 8.8 Hz), 6.14 (s, 1H), 4.58 (s, 2H), 4.11 (s, 3H), 3.81 (br s, 4H), 3.36–3.34 (m, 1H), 3.20 (br s, 4H), 2.73 (t, 2H, J = 7.8 Hz), 2.25 (s, 3H), 1.66–1.60 (m, 2H), 1.34 (d, 6H, J = 6.4 Hz), 1.01 (t, 3H, J = 7.3 Hz). HRMS (m/z) for C31H40N7O2+ [M + H]+: calculated 542.3238, found 542.3246.
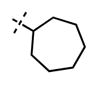
1-Cycloheptyl-6-(6-(4-isopropylpiperazin-1-yl)pyridin-3-yl)-N-((6-methyl-2-oxo-4-propyl-1,2-dihydropyridin-3-yl)methyl)-1H-inda-zole-4-carboxamide (56)
Yield: 53 mg (84%). 1H NMR (600 MHz, methanol-d4) δ 8.56 (s, 1H), 8.34 (s, 1H), 8.02 (dd, 1H, J = 2.3 Hz, J = 8.8 Hz), 7.90 (s, 1H), 7.75 (s, 1H), 6.97 (d, 1H, J = 8.8 Hz), 6.14 (s, 1H), 4.91–4.89 (m, 1H), 4.59 (s, 2H), 3.79 (br s, 4H), 3.28–3.24 (m, 1H), 3.13 (br s, 4H), 2.73 (t, 2H, J = 7.8 Hz), 2.25 (s, 3H), 2.20–2.15 (m, 2H), 2.08–2.06 (m, 2H), 1.89–1.87 (m, 2H), 1.77–1.61 (m, 8H), 1.30 (d, 6H, J = 6.6 Hz), 1.01 (t, 3H, J = 7.3 Hz). HRMS (m/z) for C37H50N7O2+ [M + H]+: calculated 624.4021, found 624.4027.
1-(Cyclobutylmethyl)-6-(6-(4-isopropylpiperazin-1-yl)pyridin-3-yl)-N-((6-methyl-2-oxo-4-propyl-1,2-dihydropyridin-3-yl)methyl)-1H-indazole-4-carboxamide (57)
Yield: 60 mg (86%). 1H NMR (600 MHz, methanol-d4) δ 8.52 (s, 1H), 8.32 (s, 1H), 7.99 (d, 1H, J = 8.2 Hz), 7.91 (s, 1H), 7.76 (s, 1H), 6.94 (d, 1H, J = 8.7 Hz), 6.14 (s, 1H), 4.59 (s, 2H), 4.50 (d, 2H, J = 3.3 Hz), 3.64 (br s, 4H), 2.96–2.88 (m, 1H), 2.83–2.80 (m, 1H), 2.76–2.72 (m, 6H), 2.25 (s, 3H), 2.00–1.88 (m, 6H), 1.66–1.62 (m, 2H), 1.16 (d, 6H, J = 6.1 Hz), 1.01 (t, 3H, J = 7.1 Hz). HRMS (m/z) for C35H46N7O2+ [M + H]+: calculated 596.3708, found 596.3716.
1-(Cyclopentylmethyl)-6-(6-(4-isopropylpiperazin-1-yl)pyridin-3-yl)-N-((6-methyl-2-oxo-4-propyl-1,2-dihydropyridin-3-yl)methyl)-1H-indazole-4-carboxamide (58)
Yield: 54 mg (85%). 1H NMR (600 MHz, methanol-d4) δ 8.54 (d, 1H, J = 2.2 Hz), 8.34 (s, 1H), 8.00 (dd, 1H, J = 2.4 Hz, J = 8.8 Hz), 7.89 (s, 1H), 7.76 (s, 1H), 6.98 (d, 1H, J = 8.8 Hz), 6.13 (s, 1H), 4.59 (s, 2H), 4.40 (d, 2H, J = 7.4 Hz), 3.83 (br s, 4H), 3.40–3.36 (m, 1H), 3.22 (t, 4H, J = 4.5 Hz), 2.72 (t, 2H, J = 7.8 Hz), 2.56–2.51 (m, 1H), 2.25 (s, 3H), 1.70–1.50 (m, 8H), 1.34 (d, 8H, J = 6.6 Hz), 1.00 (t, 3H, J = 7.3 Hz). HRMS (m/z) for C36H48N7O2+ [M + H]+: calculated 610.3864, found 610.3870.
1-(Cyclohexylmethyl)-6-(6-(4-isopropylpiperazin-1-yl)pyridin-3-yl)-N-((6-methyl-2-oxo-4-propyl-1,2-dihydropyridin-3-yl)methyl)-1H-indazole-4-carboxamide (59)
Yield: 97 mg (76%). 1H NMR (600 MHz, methanol-d4) δ 8.48 (d, 1H, J = 1.3 Hz), 8.34 (s, 1H), 7.92 (d, 1H, J = 6.9 Hz), 7.83 (s, 1H), 7.75 (s, 1H), 6.85 (d, 1H, J = 8.8 Hz), 6.10 (s, 1H), 4.58 (s, 2H), 4.28 (d, 2H, J = 7.0 Hz), 3.59 (br s, 4H), 2.81–2.72 (m, 1H), 2.72–2.68 (m, 6H), 2.23 (s, 3H), 1.99–1.95 (m, 1H), 1.68–1.51 (m, 8H) 1.19–1.15 (m, 2H), 1.13 (d, 6H, J = 6.4 Hz), 1.08–1.03 (m, 2H), 1.00 (t, 3H, J = 7.3 Hz). HRMS (m/z) for C37H50N7O2+ [M + H]+: calculated 624.4021, found 624.4028.
1-Benzyl-6-(6-(4-isopropylpiperazin-1-yl)pyridin-3-yl)-N-((6-methyl-2-oxo-4-propyl-1,2-dihydropyridin-3-yl)methyl)-1H-inda-zole-4-carboxamide (60)
Yield: 68 mg (77%). 1H NMR (600 MHz, methanol-d4) δ 8.42 (s, 1H), 8.39 (s, 1H), 7.88 (d, 1H, J = 7.2 Hz), 7.81 (s, 1H), 7.76 (s, 1H), 7.29–7.20 (m, 5H), 6.86 (d, 1H, J = 8.8 Hz), 6.13 (s, 1H), 5.70 (s, 2H), 4.58 (s, 2H), 3.58 (br s, 4H), 2.75–2.66 (m, 7H), 2.24 (s, 3H), 1.66–1.60 (m, 2H), 1.13 (d, 6H, J = 6.4 Hz), 1.01 (t, 3H, J = 7.3 Hz). HRMS (m/z) for C37H44N7O2+ [M + H]+: calculated 618.3551, found 618.3550.
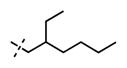
1-(2-Ethylhexyl)-6-(6-(4-isopropylpiperazin-1-yl)pyridin-3-yl)-N-((6-methyl-2-oxo-4-propyl-1,2-dihydropyridin-3-yl)methyl)-1H-in-dazole-4-carboxamide (61)
Yield: 53 mg (85%). 1H NMR (600 MHz, methanol-d4) δ 8.55 (d, 1H, J = 1.9 Hz), 8.35 (s, 1H), 8.01 (dd, 1H, J = 2.2 Hz, J = 8.8 Hz), 7.86 (s, 1H), 7.77 (s, 1H), 6.99 (d, 1H, J = 8.8 Hz), 6.14 (s, 1H), 4.59 (s, 2H), 4.38 (d, 2H, J = 6.9 Hz), 3.81 (br s, 4H), 3.30–3.28 (m, 1H), 3.16 (br s, 4H), 2.73 (t, 2H, J = 7.8 Hz), 2.26 (s, 3H), 2.06–2.03 (m, 1H), 1.70–1.61 (m, 2H), 1.32–1.20 (m, 14H), 1.01 (t, 3H, J = 7.3 Hz), 0.91 (t, 3H, J = 7.4 Hz), 0.83 (t, 3H, J = 6.9 Hz). HRMS (m/z) for C38H54N7O2+ [M + H]+: calculated 640.4334, found 640.4336.
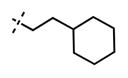
1-(2-Cyclohexylethyl)-6-(6-(4-isopropylpiperazin-1-yl)pyridin-3-yl)-N-((6-methyl-2-oxo-4-propyl-1,2-dihydropyridin-3-yl)methyl)-1H-indazole-4-carboxamide (62)
Yield: 53 mg (85%). 1H NMR (600 MHz, methanol-d4) δ 8.53 (d, 1H, J = 2.3 Hz), 8.34 (s, 1H), 7.98 (dd, 1H, J = 2.4 Hz, J = 8.8 Hz), 7.85 (s, 1H), 7.76 (s, 1H), 6.95 (d, 1H, J = 8.8 Hz), 6.13 (s, 1H), 4.58 (s, 2H), 4.49 (t, 2H, J = 7.1 Hz), 3.79 (s, 4H), 3.30–3.26 (m, 1H), 3.14 (t, 4H, J = 4.7 Hz), 2.72 (t, 2H, J = 7.8 Hz), 2.25 (s, 3H), 1.79–1.74 (m, 4H), 1.68–1.60 (m, 5H), 1.30 (d, 6H, J = 6.6 Hz), 1.20–1.12 (m, 4H), 1.00 (t, 3H, J = 7.3 Hz), 1.00–0.93 (m, 2H). HRMS (m/z) for C38H52N7O2+ [M + H]+: calculated 638.4177, found 638.4177.
1-(6-Hydroxyhexyl)-6-(6-(4-isopropylpiperazin-1-yl)pyridin-3-yl)-N-((6-methyl-2-oxo-4-propyl-1,2-dihydropyridin-3-yl)methyl)-1H-in-dazole-4-carboxamide (63)
1H NMR (400 MHz, methanol-d4) δ 8.48 (d, 1H, J = 2.0 Hz), 8.32 (s, 1H), 7.94 (dd, 1H, J = 2.3 Hz, J = 8.9 Hz), 7.84 (s, 1H), 7.74 (s, 1H), 6.88 (d, 1H, J = 8.9 Hz), 6.10 (s, 1H), 4.57 (s, 2H), 4.46 (t, 2H, J = 6.8 Hz), 3.59 (t, 4H, J = 4.8 Hz), 3.46 (t, 2H, J = 6.4 Hz), 2.80–2.71 (m, 7H), 2.23 (s, 3H), 1.94–1.87 (m, 2H), 1.66–1.51 (m, 2H), 1.49–1.42 (m, 2H), 1.39–1.25 (m, 4H), 1.12 (d, 6H, J = 6.5 Hz), 0.99 (t, 3H, J = 7.3 Hz). MS (m/z) [M + H]+: 628.2.
1-Isopropyl-N-((6-methyl-2-oxo-4-propyl-1,2-dihydropyridin-3-yl)methyl)-6-phenyl-1H-indazole-4-carboxamide (66)
Yield: 37 mg (75%). 1H NMR (400 MHz, chloroform-d) δ 8.42 (s, 1H), 7.83 (s, 1H), 7.75 (s, 1H), 7.68 (s, 1H), 7.64 (d, 2H, J = 8.0 Hz), 7.43 (t, 2H, J = 6.0 Hz), 7.36 (t, 1H, J = 6.0 Hz), 6.07 (s, 1H), 4.94–4.87 (m, 1H), 4.68 (d, 2H, J = 4.0 Hz), 2.78 (t, 2H, J = 8.0 Hz), 2.20 (s, 3H), 1.68–1.61 (m, 2H), 1.60 (d, 6H, J = 6.0 Hz), 1.03 (t, 3H, J = 6.0 Hz). MS (m/z) [M + H]+: 443.2.
1-Isopropyl-N-((6-methyl-2-oxo-4-propyl-1,2-dihydropyridin-3-yl)methyl)-6-(4-(trifluoromethyl)phenyl)-1H-indazole-4-carboxa-mide (68)
Yield: 10 mg (18%). 1H NMR (400 MHz, chloroform-d) δ 8.42 (s, 1H), 7.74 (m, 7H), 6.14 (s, 1H), 4.94–4.90 (m, 1H), 4.69 (d, 2H, J = 4.0 Hz), 2.81 (t, 2H, J = 8.0 Hz), 2.26 (s, 3H), 1.69–1.62 (m, 2H), 1.61 (d, 6H, J = 4.0 Hz), 1.03 (t, 3H, J = 6.0 Hz). MS (m/z) [M + H]+: 511.2.
1-Isopropyl-N-((6-methyl-2-oxo-4-propyl-1,2-dihydropyridin-3-yl)methyl)-6-(pyridin-2-yl)-1H-indazole-4-carboxamide (69)
Yield: 20 mg (63%). 1H NMR (400 MHz, chloroform-d) δ 8.94 (d, 1H, J = 4.9 Hz), 8.46 (s, 1H), 8.31 (s, 1H), 8.12 (t, 1H, J = 7.3 Hz), 8.02 (d, 1H, J = 8.0 Hz), 7.97 (s, 1H), 7.59–7.56 (m, 2H), 6.48 (s, 1H), 5.04–4.97 (m, 1H), 4.70 (d, 2H, J = 5.5 Hz), 2.92 (t, 2H, J = 8.0 Hz), 2.44 (s, 3H), 1.73–1.64 (m, 2H), 1.62 (d, 6H, J = 6.7 Hz), 1.05 (t, 3H, J = 7.3 Hz). MS (m/z) [M + H]+: 444.3.
1-Isopropyl-N-((6-methyl-2-oxo-4-propyl-1,2-dihydropyridin-3-yl)methyl)-6-(5-(trifluoromethyl)pyridin-2-yl)-1H-indazole-4-carbox-amide (71)
Yield: 38 mg (67%). 1H NMR (400 MHz, chloroform-d) δ 13.02 (s, 1H), 8.92 (s, 1H), 8.46 (s, 1H), 8.36 (s, 1H), 8.17 (s, 1H), 7.99 (m, 3H), 6.03 (s, 1H), 5.03–4.96 (m, 1H), 4.69 (d, 2H, J = 4.0 Hz), 2.75 (t, 2H, J = 8.0 Hz), 2.22 (s, 3H), 1.64–1.60 (m, 2H), 1.61 (d, 6H, J = 8.0 Hz), 1.02 (t, 3H, J = 6.0 Hz). MS (m/z) [M + H]+: 512.3.
1-Isopropyl-N-((6-methyl-2-oxo-4-propyl-1,2-dihydropyridin-3-yl)methyl)-6-(piperidin-4-yl)-1H-indazole-4-carboxamide (72)
To a solution of 1-isopropyl-N-((6-methyl-2-oxo-4-propyl-1,2-dihydropyr-idin-3-yl)methyl)-6-(1,2,3,6-tetrahydropyridin-4-yl)-1H-indazole-4-carboxamide in methanol (30 mL) was added Raney Ni (20 mg) under nitrogen atmosphere. The contents were purged and kept under H2 overnight. The reaction was monitored by LC/MS. After completion, the mixture was concentrated in vacuo and then purifed by HPLC to yield the title compound (3 mg, 12%) as solid. 1H NMR (600 MHz, methanol-d4) δ 8.29 (s, 1H), 7.63 (s, 1H), 7.46 (s, 1H), 6.16 (s, 1H), 5.02–4.97 (m, 1H), 4.58 (s, 2H), 3.54 (d, 2H, J = 12.7 Hz), 3.18 (dt, 2H, J = 2.3 Hz, J = 12.8 Hz), 3.14–3.09 (m, 1H), 2.75 (t, 2H, J = 7.8 Hz), 2.26 (s, 3H), 2.17 (d, 2H, J = 14.2 Hz), 2.04–1.96 (m, 2H), 1.67–1.61 (m, 2H), 1.55 (d, 6H, J = 6.6 Hz), 1.02 (t, 3H, J = 7.3 Hz). HRMS (m/z) for C26H36N5O2+ [M + H]+: calculated 450.2864, found 450.2868.
1-Isopropyl-N-((6-methyl-2-oxo-4-propyl-1,2-dihydropyridin-3-yl)methyl)-6-(piperazin-1-yl)-1H-indazole-4-carboxamide (73)
Yield: 12.8 mg (15%). 1H NMR (600 MHz, methanol-d4) δ 8.21 (s, 1H), 7.36 (d, 1H, J = 1.6 Hz), 7.17 (s, 1H), 6.18 (s, 1H), 4.97–4.90 (m, 1H), 4.56 (s, 2H), 3.53 (t, 4H, J = 5.1 Hz), 3.41 (t, 4H, J = 4.8 Hz), 2.73 (t, 2H, J = 6.0 Hz), 2.27 (s, 3H), 1.66–1.60 (m, 2H), 1.52 (d, 6H, J = 6.6 Hz), 1.01 (t, 3H, J = 7.3 Hz). HRMS (m/z) for C25H35N6O2+ [M + H]+: calculated 451.2816, found 451.2826.
6-(1,4-Diazepan-1-yl)-1-isopropyl-N-((6-methyl-2-oxo-4-propyl-1,2-dihydropyridin-3-yl)methyl)-1H-indazole-4-carboxamide (74)
Yield: 17.7 mg (20%). 1H NMR (600 MHz, methanol-d4) δ 8.14 (s, 1H), 7.19 (d, 1H, J = 1.7 Hz), 6.90 (s, 1H), 6.22 (s, 1H), 4.95–4.89 (m, 1H), 4.57 (s, 2H), 3.91 (t, 2H, J = 5.4 Hz), 3.71 (t, 2H, J = 6.1 Hz), 3.46 (t, 2H, J = 5.1 Hz), 3.31–3.29 (m, 2H), 2.75 (t, 2H, J = 7.8 Hz), 2.28 (s, 3H), 2.27–2.23 (m, 2H), 1.67–1.60 (m, 2H), 1.50 (d, 6H, J = 6.6 Hz), 1.02 (t, 3H, J = 7.3 Hz). HRMS (m/z) for C26H37N6O2+ [M + H]+: calculated 465.2973, found 465.2974.
1-Isopropyl-N-((6-methyl-2-oxo-4-propyl-1,2-dihydropyridin-3-yl)methyl)-6-(piperidin-4-ylamino)-1H-indazole-4-carboxamide (75)
Yield: 3.7 mg (4.5%). 1H NMR (600 MHz, methanol-d4) δ 8.07 (s, 1H), 6.98 (s, 1H), 6.75 (s, 1H), 6.20 (s, 1H), 4.92–4.84 (m, 1H), 4.56 (s, 2H), 3.78–3.74 (m, 1H), 3.48–3.44 (m, 2H), 3.23–3.17 (m, 2H), 2.74 (t, 2H, J = 7.8 Hz), 2.30–2.25 (m, 5H), 1.76–1.55 (m, 4H), 1.50 (d, 6H, J = 6.6 Hz), 1.02 (t, 3H, J = 7.3 Hz). HRMS (m/z) for C26H37N6O2+ [M + H]+: calculated 465.2973, found 465.2978.
6-Bromo-1-isopropyl-N-((6-methyl-2-oxo-4-propyl-1,2-dihydro-pyridin-3-yl)methyl)-1H-indazole-4-carboxamide (76)
Yield: 380 mg (96%). 1H NMR (600 MHz, methanol-d4) δ 8.34 (s, 1H), 8.03 (s, 1H), 7.63 (d, 1H, J = 1.4 Hz), 6.14 (s, 1H), 4.98–4.94 (m, 1H), 4.55 (s, 2H), 2.72 (t, 2H, J = 7.4 Hz), 2.26 (s, 3H), 1.66–1.60 (m, 2H), 1.53 (d, 6H, J = 6.6 Hz), 1.01 (t, 3H, J = 7.4 Hz). HRMS (m/z) for C21H26BrN4O2+ [M + H]+: calculated 445.1234 and 447.1213 found 445.1237 and 447.1219.
Biochemical Assays
Methyltransferase activity assays for EZH2 trimeric complex (EZH2:EED:SUZ12) and EZH1 pentameric complex (EZH1:EED:SUZ12:RBBP4:AEBP2) were performed by monitoring the incorporation of tritium-labeled methyl group to lysine 27 of H3 (21–44) peptide using the Scintillation Proximity Assay (SPA). The enzymatic reactions were performed at 23 °C with 1 h incubation of 10 μL of reaction mixture in 20 mM Tris-HCl, pH 8, 5 mM DTT, and 0.01% Triton X-100 containing 2 and 4 μM of 3H-SAM (catalogue no. NET155 V250UC; PerkinElmer; www.perkinelmer.com) for reactions including EZH2 and EZH1 complexes, respectively, and 1 μM of biotinylated H3 (21–44). In all assays, 20 nM EZH2 complex or 10 nM EZH1complex was used. Compounds were titrated from 0.3 nM to 5 μM. To stop the reactions, 10 μL of 7.5 M guanidine hydrochloride was added, followed by 50 μL of buffer (20 mM Tris, pH 8.0), mixed, and transferred to a 384-well streptavidin coated flash plate (catalogue no. SMP410A001PK, PerkinElmer, http://www.perkinelmer.ca). After mixing, the mixtures in flash-plate were incubated for 2 h and the CPM counts were measured using Topcount plate reader (PerkinElmer, www.perkinelmer.com). The CPM counts in the absence of compound for each data set were defined as 100% activity. In the absence of the enzyme, the CPM counts in each data set were defined as background (0%). All enzymatic reactions were performed in triplicate and IC50 values were determined by fitting the data to the Four Parameter Logistic equation using GraphPad Prism 7 software.
Structure of PRC2-EZH2 Complex
The complex structure of the human PRC2-EZH2 (PDB: 5HYN) was used for molecular docking. The coordinates of the I-SET helix (I646 to N668) of this structure were modified according to the I-SET helix of the complex PRC2-AcEZH2 (PDB: 5IJ7, cocrystallized with a pyridone inhibitor) in order to take into account the induced fit effect by the inhibitor on the positioning of this helix. Both crystal structures were prepared using the structure preparation tool available in Schrödinger 2015–3 (Schrödinger Release 2015–3: Maestro, version 10.3, Schrödinger, LLC, New York, NY, 201.) with hydrogen optimization and default settings following their download from the PDB.
Molecular Docking
Prior to docking, all compounds were ionized using the fixpka program of OpenEye QUACPAC 1.5.0 (OpenEye Scientific Software, Santa Fe, NM, http://www.eyesopen.com.). Their three-dimensional structures were generated using LigPrep (Schrödinger Release 2015–3: LigPrep, version 3.5, Schrödinger, LLC, New York, NY, 201.) with OPLS_2005 force field. The softwares PLANTS53 and Glide54 were used to dock the inhibitors to the modified structure of 5HYN (modified I-SET helix). The PDB structure 5IJ7 was superimposed with the modified 5HYN structure, and the centroid of “Inhibitor 1” (5IJ7) was defined as a center of the binding site for PLANTS docking. The binding site radius was set to 12 Å, and hydrogen bond constraints were set on backbone oxygen and nitrogen atoms of W624 and the nitrogen atom of backbone Y111 in order to place the pyridonemethyl-amide in the proper region. Similarly, the centroid of “Inhibitor 1” was defined as a center of the Glide receptor grid. The pyridonemethyl-amide scaffold was constrained with a tolerance of 0.6 Å, and the Glide standard precision (SP) mode with its default parameters was set. A maximum number of 10 different poses per docking software was used for each ligand. A Tanimoto coefficient was applied to compute the similarity of the interaction fingerprints55 generated for each pose with the reference “Inhibitor 1”. For each ligand, we kept the pose that exhibits the most similar interactions (highest Tanimoto coefficient) as “Inhibitor 1”. Each kept complex was refined using Prime by selecting OPLS_3 as force field, VSGB as solvation mode, and atoms within 5 Å of any heavy ligand atoms were optimized using the local optimization protocol.54
Supplementary Material
Acknowledgments
The research described here was supported by the grant R01GM103893 (to J.J.) from the U.S. National Institutes of Health. The Structural Genomics Consortium (SGC) is a registered charity (no. 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Innovative Medicines Initiative (EU/EFPIA) [ULTRA-DD grant no. 115766], Janssen, Merck & Co., Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, Sao Paulo Research Foundation-FAPESP, Takeda, and the Wellcome Trust. X.Y. and A.M. were supported by a postdoctoral fellowship from the SGC. K.D.K. was supported by a predoctoral fellowship from the Medicinal Chemistry Division of the American Chemical Society. This work was supported in part through the computational resources provided by Scientific Computing at the Icahn School of Medicine at Mount Sinai. We thank OpenEye Scientific Software for providing us with a free academic license.
ABBREVIATIONS USED
- EZH2
enhancer of zeste 2 polycomb repressive complex 2 subunit
- KMT6
lysine methyltransferase 6
- EZH1
enhancer of zeste homologue 1
- PRC2
polycomb repressive complex 2
- EED
embryonic ectoderm development
- SUZ12
suppressor of zeste 12
- RbAp46
retinoblastoma binding protein 7
- RbAp48
retinoblastoma binding protein 4
- AEBP2
adipocyte enhancer-binding protein 2
- H3K27
histone 3, lysine 27
- PRC2-EZH1
EZH1 containing PRC2 complex
- PRC2-EZH2
EZH2 containing PRC2 complex
- SAM
S-5′-adenosyl-L-methionine
- H3K27me3
trimethylation of H3K27
- SET
Su(var.)3–9 (the suppressor of position-effect variegation 3–9), en(zeste) (an enhancer of the eye color mutant zeste), and trithorax (the homeotic gene regulator)
- GCB
germinal center B-cell
- DLBCLs
diffuse large B-cell lymphomas
- MLL
mixed lineage leukemia
- SAR
structure-activity relationship
- PK
pharmaco-kinetic
- PDB
Protein Data Bank
- SAH
S-5′-adenosyl-L-homocysteine
- IC50
half-minimal (50%) inhibitory concentration
- nM
nanomolar
- Ph
phenyl
- t-Bu
tertiary butyl
- HPLC
high-performance liquid chromatography
- NMM
N-methylmorpholine
- HOAt
1-hydroxy-7-azabenzotriazole
- EDCI
1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide hydrochloride
- DMSO
dimethyl sulfoxide
- DMF
N,N-dimethylformamide
- DIPEA
N,N-diisopropylethylamine
- BINAP
2,2′-bis-(diphenylphosphino)-1,1′-binaphthyl
- Pd2(dba)3
tris-(dibenzylideneacetone)dipalladium(0)
Footnotes
Author Contributions
All authors have given approval to the final version of the manuscript.
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmed-chem.6b00855.
Synthetic methods and conditions for the synthesis of intermediates and spectral data for these intermediates (PDF)
References
- 1.Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–1043. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- 2.Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002;16:2893–2905. doi: 10.1101/gad.1035902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–349. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Di Croce L, Helin K. Transcriptional regulation by polycomb group proteins. Nat Struct Mol Biol. 2013;20:1147–1155. doi: 10.1038/nsmb.2669. [DOI] [PubMed] [Google Scholar]
- 5.Vizan P, Beringer M, Ballare C, Di Croce L. Role of PRC2-associated factors in stem cells and disease. FEBS J. 2015;282:1723–1735. doi: 10.1111/febs.13083. [DOI] [PubMed] [Google Scholar]
- 6.Cao R, Zhang Y. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol Cell. 2004;15:57–67. doi: 10.1016/j.molcel.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 7.Montgomery ND, Yee D, Chen A, Kalantry S, Chamberlain SJ, Otte AP, Magnuson T. The murine polycomb group protein Eed is required for global histone H3 lysine-27 methylation. Curr Biol. 2005;15:942–947. doi: 10.1016/j.cub.2005.04.051. [DOI] [PubMed] [Google Scholar]
- 8.Margueron R, Justin N, Ohno K, Sharpe ML, Son J, Drury WJ, III, Voigt P, Martin SR, Taylor WR, De Marco V, Pirrotta V, Reinberg D, Gamblin SJ. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature. 2009;461:762–767. doi: 10.1038/nature08398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pasini D, Bracken AP, Hansen JB, Capillo M, Helin K. The polycomb group protein Suz12 is required for embryonic stem cell differentiation. Mol Cell Biol. 2007;27:3769–3779. doi: 10.1128/MCB.01432-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pasini D, Bracken AP, Jensen MR, Lazzerini Denchi E, Helin K. Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J. 2004;23:4061–4071. doi: 10.1038/sj.emboj.7600402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu H, Zeng H, Dong A, Li F, He H, Senisterra G, Seitova A, Duan S, Brown PJ, Vedadi M, Arrowsmith CH, Schapira M. Structure of the catalytic domain of EZH2 reveals conformational plasticity in cofactor and substrate binding sites and explains oncogenic mutations. PLoS One. 2013;8:e83737. doi: 10.1371/journal.pone.0083737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Antonysamy S, Condon B, Druzina Z, Bonanno JB, Gheyi T, Zhang F, MacEwan I, Zhang A, Ashok S, Rodgers L, Russell M, Gately Luz J. Structural context of disease-associated mutations and putative mechanism of autoinhibition revealed by X-ray crystallographic analysis of the EZH2-SET domain. PLoS One. 2013;8:e84147. doi: 10.1371/journal.pone.0084147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiao L, Liu X. Structural basis of histone H3K27 trimethylation by an active polycomb repressive complex 2. Science. 2015;350:aac4383. doi: 10.1126/science.aac4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brooun A, Gajiwala KS, Deng YL, Liu W, Bolanos B, Bingham P, He YA, Diehl W, Grable N, Kung PP, Sutton S, Maegley KA, Yu X, Stewart AE. Polycomb repressive complex 2 structure with inhibitor reveals a mechanism of activation and drug resistance. Nat Commun. 2016;7:11384. doi: 10.1038/ncomms11384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Justin N, Zhang Y, Tarricone C, Martin SR, Chen S, Underwood E, De Marco V, Haire LF, Walker PA, Reinberg D, Wilson JR, Gamblin SJ. Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat Commun. 2016;7:11316. doi: 10.1038/ncomms11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003;22:5323–5335. doi: 10.1093/emboj/cdg542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simon JA, Lange CA. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat Res, Fundam Mol Mech Mutagen. 2008;647:21–29. doi: 10.1016/j.mrfmmm.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 18.Ding L, Erdmann C, Chinnaiyan AM, Merajver SD, Kleer CG. Identification of EZH2 as a molecular marker for a precancerous state in morphologically normal breast tissues. Cancer Res. 2006;66:4095–4099. doi: 10.1158/0008-5472.CAN-05-4300. [DOI] [PubMed] [Google Scholar]
- 19.Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, Ghosh D, Sewalt RG, Otte AP, Hayes DF, Sabel MS, Livant D, Weiss SJ, Rubin MA, Chinnaiyan AM. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci U S A. 2003;100:11606–11611. doi: 10.1073/pnas.1933744100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, Ghosh D, Pienta KJ, Sewalt RG, Otte AP, Rubin MA, Chinnaiyan AM. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624–629. doi: 10.1038/nature01075. [DOI] [PubMed] [Google Scholar]
- 21.Velichutina I, Shaknovich R, Geng H, Johnson NA, Gascoyne RD, Melnick AM, Elemento O. EZH2-mediated epigenetic silencing in germinal center B cells contributes to proliferation and lymphomagenesis. Blood. 2010;116:5247–5255. doi: 10.1182/blood-2010-04-280149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, Paul JE, Boyle M, Woolcock BW, Kuchenbauer F, Yap D, Humphries RK, Griffith OL, Shah S, Zhu H, Kimbara M, Shashkin P, Charlot JF, Tcherpakov M, Corbett R, Tam A, Varhol R, Smailus D, Moksa M, Zhao Y, Delaney A, Qian H, Birol I, Schein J, Moore R, Holt R, Horsman DE, Connors JM, Jones S, Aparicio S, Hirst M, Gascoyne RD, Marra MA. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42:181–185. doi: 10.1038/ng.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hernando H, Gelato KA, Lesche R, Beckmann G, Koehr S, Otto S, Steigemann P, Stresemann C. EZH2 Inhibition blocks multiple myeloma mell mrowth through upregulation of epithelial tumor suppressor genes. Mol Cancer Ther. 2016;15:287–298. doi: 10.1158/1535-7163.MCT-15-0486. [DOI] [PubMed] [Google Scholar]
- 24.Shih AH, Abdel-Wahab O, Patel JP, Levine RL. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer. 2012;12:599–612. doi: 10.1038/nrc3343. [DOI] [PubMed] [Google Scholar]
- 25.Neff T, Sinha AU, Kluk MJ, Zhu N, Khattab MH, Stein L, Xie H, Orkin SH, Armstrong SA. Polycomb repressive complex 2 is required for MLL-AF9 leukemia. Proc Natl Acad Sci U S A. 2012;109:5028–5033. doi: 10.1073/pnas.1202258109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shi J, Wang E, Zuber J, Rappaport A, Taylor M, Johns C, Lowe SW, Vakoc CR. The Polycomb complex PRC2 supports aberrant self-renewal in a mouse model of MLL-AF9; Nras(G12D) acute myeloid leukemia. Oncogene. 2013;32:930–938. doi: 10.1038/onc.2012.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu B, On DM, Ma A, Parton T, Konze KD, Pattenden SG, Allison DF, Cai L, Rockowitz S, Liu S, Liu Y, Li F, Vedadi M, Frye SV, Garcia BA, Zheng D, Jin J, Wang GG. Selective inhibition of EZH2 and EZH1 enzymatic activity by a small molecule suppresses MLL-rearranged leukemia. Blood. 2015;125:346–357. doi: 10.1182/blood-2014-06-581082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Verma SK, Tian X, LaFrance LV, Duquenne C, Suarez DP, Newlander KA, Romeril SP, Burgess JL, Grant SW, Brackley JA, Graves AP, Scherzer DA, Shu A, Thompson C, Ott HM, Aller GSV, Machutta CA, Diaz E, Jiang Y, Johnson NW, Knight SD, Kruger RG, McCabe MT, Dhanak D, Tummino PJ, Creasy CL, Miller WH. Identification of potent, selective, cell-active inhibitors of the histone lysine methyltransferase EZH2. ACS Med Chem Lett. 2012;3:1091–1096. doi: 10.1021/ml3003346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Margueron R, Li G, Sarma K, Blais A, Zavadil J, Woodcock CL, Dynlacht BD, Reinberg D. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol Cell. 2008;32:503–518. doi: 10.1016/j.molcel.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laible G, Wolf A, Dorn R, Reuter G, Nislow C, Lebersorger A, Popkin D, Pillus L, Jenuwein T. Mammalian homologues of the Polycomb-group gene Enhancer of zeste mediate gene silencing in Drosophila heterochromatin and at S. cerevisiae telomeres. EMBO J. 1997;16:3219–3232. doi: 10.1093/emboj/16.11.3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Visser HP, Gunster MJ, Kluin-Nelemans HC, Manders EM, Raaphorst FM, Meijer CJ, Willemze R, Otte AP. The Polycomb group protein EZH2 is upregulated in proliferating, cultured human mantle cell lymphoma. Br J Haematol. 2001;112:950–958. doi: 10.1046/j.1365-2141.2001.02641.x. [DOI] [PubMed] [Google Scholar]
- 32.Son J, Shen SS, Margueron R, Reinberg D. Nucleosome-binding activities within JARID2 and EZH1 regulate the function of PRC2 on chromatin. Genes Dev. 2013;27:2663–2677. doi: 10.1101/gad.225888.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, Sacks JD, Raimondi A, Majer CR, Song J, Scott MP, Jin L, Smith JJ, Olhava EJ, Chesworth R, Moyer MP, Richon VM, Copeland RA, Keilhack H, Pollock RM, Kuntz KW. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol. 2012;8:890–896. doi: 10.1038/nchembio.1084. [DOI] [PubMed] [Google Scholar]
- 34.McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, AD, Diaz E, Lafrance LV, Mellinger M, Duquenne C, Tian X, Kruger RG, McHugh CF, Brandt M, Miller WH, Dhanak D, Verma SK, Tummino PJ, Creasy CL. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492:108–112. doi: 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- 35.Qi W, Chan H, Teng L, Li L, Chuai S, Zhang R, Zeng J, Li M, Fan H, Lin Y, Gu J, Ardayfio O, Zhang JH, Yan X, Fang J, Mi Y, Zhang M, Zhou T, Feng G, Chen Z, Li G, Yang T, Zhao K, Liu X, Yu Z, Lu CX, Atadja P, Li E. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc Natl Acad Sci U S A. 2012;109:21360–21365. doi: 10.1073/pnas.1210371110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Konze KD, Ma A, Li F, Barsyte-Lovejoy D, Parton T, MacNevin CJ, Liu F, Gao C, Huang XP, Kuznetsova E, Rougie M, Jiang A, Pattenden SG, Norris JL, James LI, Roth BL, Brown PJ, Frye SV, Arrowsmith CH, Hahn KM, Wang GG, Vedadi M, Jin J. An orally bioavailable chemical probe of the lysine methyltransferases EZH2 and EZH1. ACS Chem Biol. 2013;8:1324–1334. doi: 10.1021/cb400133j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, Porter Scott M, Chesworth R, Moyer MP, Copeland RA, Richon VM, Pollock RM, Kuntz KW, Keilhack H. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A. 2013;110:7922–7927. doi: 10.1073/pnas.1303800110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.ClinicalTrials.gov Identifier: NCT02082977. A study to investigate the safety, pharmacokinetics, pharmacodynamics and clinical activity of GSK2816126 in subjects with relapsed/refractory diffuse large B cell and transformed follicular lymphoma. [accessed July 8, 2016]. [Google Scholar]
- 39.ClinicalTrials.gov Identifier: NCT01897571. An open-label, multicenter, phase 1/2 study of E7438 (EZH2 histone methyl transferase [HMT] inhibitor) as a single agent in subjects with advanced solid tumors or with B-cell lymphomas. [accessed July 8, 2016]. [Google Scholar]
- 40.Kaniskan HÜ, Konze KD, Jin J. Selective inhibitors of protein methyltransferases. J Med Chem. 2015;58:1596–1629. doi: 10.1021/jm501234a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaniskan HÜ, Jin J. Chemical probes of histone lysine methyltransferases. ACS Chem Biol. 2015;10:40–50. doi: 10.1021/cb500785t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garapaty-Rao S, Nasveschuk C, Gagnon A, Chan EY, Sandy P, Busby J, Balasubramanian S, Campbell R, Zhao F, Bergeron L, Audia JE, Albrecht BK, Harmange JC, Cummings R, Trojer P. Identification of EZH2 and EZH1 small molecule inhibitors with selective impact on diffuse large B cell lymphoma cell growth. Chem Biol. 2013;20:1329–1339. doi: 10.1016/j.chembiol.2013.09.013. [DOI] [PubMed] [Google Scholar]
- 43.Gehling VS, Vaswani RG, Nasveschuk CG, Duplessis M, Iyer P, Balasubramanian S, Zhao F, Good AC, Campbell R, Lee C, Dakin LA, Cook AS, Gagnon A, Harmange JC, Audia JE, Cummings RT, Normant E, Trojer P, Albrecht BK. Discovery, design, and synthesis of indole-based EZH2 inhibitors. Bioorg Med Chem Lett. 2015;25:3644–3649. doi: 10.1016/j.bmcl.2015.06.056. [DOI] [PubMed] [Google Scholar]
- 44.Bradley WD, Arora S, Busby J, Balasubramanian S, Gehling VS, Nasveschuk CG, Vaswani RG, Yuan CC, Hatton C, Zhao F, Williamson KE, Iyer P, Mendez J, Campbell R, Cantone N, Garapaty-Rao S, Audia JE, Cook AS, Dakin LA, Albrecht BK, Harmange JC, Daniels DL, Cummings RT, Bryant BM, Normant E, Trojer P. EZH2 inhibitor efficacy in non-Hodgkin’s lymphoma does not require suppression of H3K27 monomethylation. Chem Biol. 2014;21:1463–1475. doi: 10.1016/j.chembiol.2014.09.017. [DOI] [PubMed] [Google Scholar]
- 45.ClinicalTrials.gov Identifier: NCT02395601. A study evaluating CPI-1205 in patients with B-cell lymphomas. [accessed July 8, 2016]. [Google Scholar]
- 46.Campbell JE, Kuntz KW, Knutson SK, Warholic NM, Keilhack H, Wigle TJ, Raimondi A, Klaus CR, Rioux N, Yokoi A, Kawano S, Minoshima Y, Choi HW, Porter Scott M, Waters NJ, Smith JJ, Chesworth R, Moyer MP, Copeland RA. EPZ011989; A potent, orally-available EZH2 inhibitor with robust in vivo activity. ACS Med Chem Lett. 2015;6:491–495. doi: 10.1021/acsmedchemlett.5b00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Song X, Gao T, Wang N, Feng Q, You X, Ye T, Lei Q, Zhu Y, Xiong M, Xia Y, Yang F, Shi Y, Wei Y, Zhang L, Yu L. Selective inhibition of EZH2 by ZLD1039 blocks H3K27methylation and leads to potent anti-tumor activity in breast cancer. Sci Rep. 2016;6:20864. doi: 10.1038/srep20864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gao TT, Zhang LD, Zhu YX, Song XJ, Feng Q, Lei Q, Shi SX, Deng HX, Xiong MH, You XY, Zuo WQ, Liu L, Peng CT, Wang NY, Ye TH, Xia Y, Yu LT. ZLD1122; a novel EZH2 and EZH1 small molecular inhibitor, blocks H3K27 methylation and diffuse large B cell lymphoma cell growth. RSC Adv. 2016;6:28512–28521. [Google Scholar]
- 49.Leung CS, Leung SS, Tirado-Rives J, Jorgensen WL. Methyl effects on protein-ligand binding. J Med Chem. 2012;55:4489–4500. doi: 10.1021/jm3003697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kuntz KW, Campbell JE, Keilhack H, Pollock RM, Knutson SK, Porter-Scott M, Richon VM, Sneeringer CJ, Wigle TJ, Allain CJ, Majer CR, Moyer MP, Copeland RA, Chesworth R. The importance of being me: magic methyls, methyltransferase inhibitors, and the siscovery of Tazemetostat. J Med Chem. 2016;59:1556–1564. doi: 10.1021/acs.jmedchem.5b01501. [DOI] [PubMed] [Google Scholar]
- 51.Wilcken R, Zimmermann MO, Lange A, Joerger AC, Boeckler FM. Principles and applications of halogen bonding in medicinal chemistry and chemical biology. J Med Chem. 2013;56:1363–1388. doi: 10.1021/jm3012068. [DOI] [PubMed] [Google Scholar]
- 52.Scholfield MR, Zanden CM, Carter M, Ho PS. Halogen bonding (X-bonding): a biological perspective. Protein Sci. 2013;22:139–152. doi: 10.1002/pro.2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Korb O, Stutzle T, Exner TE. PLANTS: Application of ant colony optimization to structure-based drug design. Lect Notes Comput Sci. 2006;4150:247–258. [Google Scholar]
- 54.Small-Molecule Drug Discovery Suite 2015–4: Glide, version 6.9. Schrödinger, LLC; New York: 2015. [Google Scholar]
- 55.Marcou G, Rognan D. Optimizing fragment and scaffold docking by use of molecular interaction fingerprints. J Chem Inf Model. 2007;47:195–207. doi: 10.1021/ci600342e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.