

Abstract
Recent advances in the genetics of amyotrophic lateral sclerosis (ALS) have provided key mechanistic insights to the pathogenesis of this devastating neurodegenerative disease. Among many etiologies for ALS, the identification of mutations and proteinopathies in two RNA binding proteins, TDP-43 (TARDBP or TAR DNA binding protein 43) and its closely related RNA/DNA binding protein FUS (fused in sarcoma), raises the intriguing possibility that perturbations to the RNA homeostasis and metabolism in neurons may contribute to the pathogenesis of these diseases. Although the similarities between TDP-43 and FUS suggest that mutations and proteinopathy involving these two proteins may converge on the same mechanisms leading to neurodegeneration, there is increasing evidence that FUS mutations target distinct mechanisms to cause early disease onset and aggressive progression of disease. This review focuses on the recent advances on the molecular, cellular and genetic approaches to uncover the mechanisms of wild type and mutant FUS proteins during development and in neurodegeneration. These findings provide important insights to understand how FUS mutations may perturb the maintenance of dendrites through fundamental processes in RNA splicing, RNA transport and DNA damage response/repair. These results contribute to the understanding of phenotypic manifestations in neurodegeneration related to FUS mutations, and to identify important directions for future investigations.
Keywords: RNA Binding Protein, Fused in sarcoma (FUS), Prion-like Property, Low Complexity Domain, DNA Damage Repair, RNA Splicing, Amyotrophic Lateral Sclerosis (ALS), Frontotemporal Dementia (FTD)
1. Introduction
1.1. The Expanding Genetic Landscape of Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis (ALS) is an adult-onset neurodegenerative disease that affects upper and lower motor neurons. As initially described by Jean-Martin Charcot more than 140 years ago, the key clinical features in ALS patients include muscle wasting, and progressive loss of spinal motor neurons and upper motor neurons and their axons in the lateral columns of the spinal cord. Recent advances in human genetics have identified many genetic loci that are mutated in patients with famililal ALS (FALS). Among a growing number of genes involved in FALS, mutations in four genes account for the majority of cases. These mutations include missense mutations in superoxide dismutase 1 (SOD1), two genes encoding RNA/DNA binding proteins, TDP-43 (TARDBP or TAR-DNA-binding protein-43) and FUS/TLS (fused in sarcoma/translocation in liposarcoma or FUS), and the GGGGCC hexanucleotide expansions in C9ORF72 gene (Cirulli et al., 2015; Lee et al., 2012; Ling et al., 2013). The discovery of TDP-43 as a major component in the ubiquitin-positive, tau-negative insoluble protein aggregates in neurons and glia represents a major breakthrough in FTD (frontotemporal dementia) and ALS research (Arai et al., 2006; Neumann et al., 2006). Moreover, the impact of this discovery goes beyond the identification of a single disease gene and essentially ushers in a new era of research that focuses on the potential contributions of transcription, RNA splicing and RNA metabolism on neurodegenerative diseases.
TDP-43 is originally identified to bind to the TAR DNA sequence in HIV-1 genome to regulate viral gene expression (Ou et al., 1995). Under physiological conditions, TDP-43 is a ubiquitous nuclear protein, however, in FTD patients, TDP-43 aggregates are present predominantly in neuronal cytoplasm and dystrophic neuronal processes (Arai et al., 2006; Neumann et al., 2006). This distinct feature, defined as TDP-43 proteinopathy, constitutes a major neuropathological diagnosis entity in sporadic ALS (ALS-TDP). Several subsequent studies show that dominant mutations in the TARDBP gene can also be identified in FALS patients (Lattante et al., 2013). The identification of autosomal dominant mutations in the FUS gene in a large kindred of familial ALS (FALS) further expanded the genetic and neuropathological landscape of ALS (Kwiatkowski et al., 2009; Vance et al., 2009). Similar to TDP-43, FUS proteins reside primarily in the neuronal nuclei, but in ALS-FUS patients FUS proteins form large aggregates in the cytoplasm. The morphology of FUS proteinopathy in FALS ranges from diffuse and dense cytoplasmic aggregate present in late onset cases, to basophilic inclusions commonly found in juvenile FALS with FUS-P525L mutation. Finally, in 2011 two groups independently reported the GGGGCC hexanucleotide repeat expansions in the noncoding region of the C9ORF72 gene as causal links to ALS and FTD (DeJesus-Hernandez et al., 2011; Renton et al., 2011). Although TDP-43 proteinopathy can be detected in FTD and ALS patients with C9ORF72 mutations, the neuropathological features in these cases are quite heterogeneous and also include prominent ubiquitin and p62 positive, but TDP-43 negative intracytoplasmic and intranuclear inclusions (Bigio, 2012; Mackenzie et al., 2014).
1.2. Early Disease Onset in FALS Caused by FUS Mutations
It is estimated that mutations in TARDBP and FUS each account for ∼5% of FALS, whereas the GGGGCC expansion mutations in C9ORF72 account for 20-40% of ALS and FTD-ALS cases, depending on the population studied (Cirulli et al., 2015). One important feature noted in a recent study indicates that the age of disease onset for FALS caused by FUS, TARDBP and C9ORF72 mutations differ quite drastically. Mutations in FUS account for ∼35% of FALS in patients younger than 40 years old, whereas mutations in C9ORF72 are much more common in patients older than 50 years of age (Millecamps et al., 2012). Indeed, meta-analyses of 154 ALS cases with FUS mutations (including FALS and SALS with de novo FUS mutations) show an average disease onset of 43.8 ± 17.4 years (Figure 1)(Deng et al., 2014a; Lattante et al., 2013). More than 60% of cases with FUS mutations show disease onset before 45 years of age, with many juvenile ALS cases presenting with disease onset in late teens and early 20's (Figure 1)(Baumer et al., 2010; Huang et al., 2010). These findings are similar to those from another study using smaller sample size, and show that the average disease onset for FUS, SOD1 or TARDBP mutations is 43.6 ± 15.8, 47.7 ± 13.0 and 54.7 ± 15.3, respectively (Yan et al., 2010). Kaplan-Meier survival analysis shows statistically significant differences in the age of onset among these three mutations. This distinctive feature of FUS mutations raises the intriguing hypothesis that mutations in FUS may target divergent mechanisms that perturb the development, maintenance and homeostasis of the nervous system in early postnatal life and in the aging process.
Figure 1. The Age of Disease Onset in FALS Patients with FUS Mutations.

(A) Meta-analyses of 154 FALS patients (either familial ALS inherited mutations or sporadic ALS with de novo mutations) show a predilection early disease onset. Compared to all FUS mutations and the most common mutations that occur in amino acid 521 (FUS-R521C), FUS-P525L mutation tends to occur in late teens and early 20's and represents a much more aggressive form of disease. (B) For sporadic ALS (SALS), about 35.9% and 34.9% of patients show disease onset in the range of 41-55 and 56-65 years old. In contrast, more than 60% of ALS patients with FUS mutations show disease onset before 40 years old. *Statistics for SALS have been adapted from the study by Testa and colleagues (Testa et al., 2004).
This review focuses on the recent progress on the molecular, cellular and genetic approaches to uncover the mechanisms of wild type and mutant FUS proteins. These findings provide important insights to understand how FUS mutations may perturb the fundamental processes in DNA damage response/repair, RNA splicing, and RNA transport, to interpret the phenotypic manifestations in neurodegeneration related to FUS mutations, and to identify important directions for future investigations.
2. Physical Properties of FUS and Their Implications in RNA Metabolism
2.1. RNA Binding Properties of FUS
FUS is identified as an oncogene that undergoes chromosomal translocation in myxoid liposarcoma, in which the N-terminal transcriptional activation domain of FUS is fused to CHOP (CAAT enhancer-binding homologous protein), a growth arrest and DNA-damage inducible member of the C/EBP family of transcription factors (Crozat et al., 1993; Rabbitts et al., 1993). Subsequent studies further reveal that chromosomal translocations involving FUS can be identified in several other human cancers, including acute myeloid leukemia, where the N-terminus of FUS gene is translocated to the ERG gene, a member of the ETS transcription factor family (Ichikawa et al., 1994; Prasad et al., 1994). Structurally, FUS belongs to a family of FET RNA binding proteins, including FUS, Ewing's sarcoma RNA binding protein 1 (EWSR1) and Tata-binding protein-associated factor 2N (TAF-15), that are known to interact with the C-terminal domain of RNA polymerase II (RNAP II) and general transcription factor TFIID (Das et al., 2007; Kwon et al., 2013; Schwartz et al., 2012; Tan and Manley, 2009). Full length human FUS protein contains 526 amino acids that can be divided into the N-terminal “prion-like” or low complexity (LC) Q/G/S/Y domain (amino acids 1-165) and Gly-rich region (amino acids 166-267)(Figure 2). The C-terminal half of FUS protein contains an RNA recognition motif (RRM)(amino acids 285-371), two Arg-Gly-Gly (RGG)-repeat regions (amino acids 371-422 and 453-501), interrupted by a Cys2-Cys2 zinc-finger motif (ZNF)(amino acids 422-453), and a non-conventional nuclear localization signal (NLS)(amino acids 510-526), which interacts with the nuclear transport receptor Transportin 1 (Figure 2)(Dormann et al., 2012; Dormann et al., 2010; Iko et al., 2004). Most of the FALS-associated FUS mutations cluster in the N-terminal LC domain, the second RGG domain and NLS in the C-terminal (Figure 2).
Figure 2. Schematic Diagrams of Genomic Organization of the Human FUS Gene, FUS Mutations Identified in ALS, and Functional Domains in FUS Proteins.

The human FUS gene consists of 15 exons, spanning ∼14.9 Kb, and is located on chromosome 16p11.2. The FUS mRNA transcripts are predicted to contain a 3,433 bp 3′UTR, which has been recently shown to contain 4 disease-related variants. The full length human FUS protein contains 526 amino acids that can be further divided into several functional domains, including the “prion-like” or low complexity (LC) domain that contains the Q/G/S/Y-rich region (amino acids 1-165) and the G-rich region (amino acids 165-267), the Arginine-rich motif (RRM, amino acids 285-371), two Arg-Gly-Gly (RGG)-repeat regions (amino acids 371-422 and 453-501), interrupted by a Cys2-Cys2 zinc-finger motif (ZNF)(amino acids 422-453), and a non-conventional nuclear localization signal (NLS)(amino acids 510-526). The majority of FALS-related mutations are more commonly found in (1) the G-rich region, (2) the 2nd RGG region and (3) the NLS. Additional structural and functional domains in FUS include the prion-like domains and the HDAC1-interacting domains.
Following its discovery as an oncogene involved in chromosomal translocation in malignant tumors, several studies have elucidated the biochemical properties of FUS as an RNA binding protein that regulates splicing. First, by UV cross-linking, it has been shown that FUS can bind to RNA. The binding seems not to depend on the RRM in the C-terminus, but rather on the zinc finger (ZnF) motif (Iko et al., 2004; Zinszner et al., 1997). Second, FUS is an abundant nuclear protein that can form stable complex with many members of the heterogeneous ribonuclear protein (hnRNP) family and can be co-purified from nuclear extracts by single-stranded DNA affinity chromatography (Calvio et al., 1995; Zinszner et al., 1994; Zinszner et al., 1997). One study suggests that the stability of the FUS-hnRNP complex is dependent on the integrity of its constituent RNA (Zinszner et al., 1994). While these results do not prove that FUS can directly interact with RNA, they suggest that at least some component(s) of the FUS-hnRNP complex has RNA-binding activity.
To further determine if FUS directly binds to specific RNA sequence, Lerga and colleagues use an in vitro selection assay and identify a common GGUG motif in RNA oligoribonucleotides that bind to recombinant FUS protein (Lerga et al., 2001). These results are verified using UV cross-linking combined with competition and immunoprecipitation in nuclear extracts. The ability of FUS to directly interact with RNA has been further examined using in FUS antibody immunoprecipitates from mouse and human brain tissues, followed by CLIP (cross-linking immunoprecipitation)-RNA sequencing (CLIP-Seq)(Lagier-Tourenne et al., 2012). This approach shows that both mouse and human FUS proteins bind to RNAs that contain an enriched GUGGU motif, different from the GU-rich binding sequence reported for TDP-43 (Polymenidou et al., 2011; Tollervey et al., 2011). However, several studies using similar CLIP-seq technology do not find similar consensus RNA binding sequences for FUS (Colombrita et al., 2012; Hoell et al., 2011; Ishigaki et al., 2012; Rogelj et al., 2012). Instead, the results from these studies support the idea that FUS binding sites in RNA tend to form stable secondary structures, such as the stem-and-loop structure (Hoell et al., 2011; Ishigaki et al., 2012). Similarly, FUS has also been shown to interact with the short RNA repeats r(UUAGGG) in the G-quadruplex telomeric repeat-containing RNA (TERRA) by forming unique secondary and tertiary structures (Takahama et al., 2013). In a recent study, Wang and colleagues examine the specificity of the putative FUS-binding RNA motifs using electrophoretic mobility shift assays (EMSA), and show that FUS binds to all the repeats with Kd values within a 10-fold range (Wang et al., 2015). Surprisingly, RNAs without any of the reported binding motifs also bind to FUS with similar affinity. Together, these results support that the nucleic acid binding property in FUS can be rather generic or “promiscuous”, and is dictated by the secondary or tertiary structure of RNA.
2.2. Roles of FUS in RNA Splicing
Given the nature of FUS-RNA interactions, what would be the physiological role of FUS in RNA metabolism? Previous CLIP-seq studies show that most RNAs that bind to FUS contain intronic sequences. Perhaps the most unique feature is that in genes with long intron, FUS-RNA binding exhibits a distinct “sawtooth” CLIP pattern (Lagier-Tourenne et al., 2012; Rogelj et al., 2012), with substantially higher FUS cluster density at the beginning of introns and a gradual decrease toward the 3′ sequence. These results suggest that FUS is co-transcriptionally deposited onto the nascent RNA transcripts. In addition, FUS binding has been identified around the alternatively spliced exons and in the promoter antisense strands in several genes implicated in neurodevelopmental and neurodegenerative diseases (Ishigaki et al., 2012), suggesting that FUS may involve in alternative splicing and transcription. The support for FUS in RNA splicing is further underscored by the identification of FUS as an direct interacting partner with splicing factors, SC35 and SRSF10 (serine/arginine-rich splicing factor 10), and as one of the ∼50 non-snRNP proteins in the pre-spliceosome (Behzadnia et al., 2007; Wahl et al., 2009; Yang et al., 1998). While these results provide physical evidence of FUS in RNA splicing, it remains unclear how FUS regulates the recognition of the 5′ splice junction, the formation and stability of Complex A, and the efficiency of splicing. Finally, two studies use epitope-tagged FUS to identify FUS interactome in nuclear extracts from HeLa cells and show that FUS can also interact with SMN and U1 snRNP (Sun et al., 2015; Yamazaki et al., 2012). The results that FUS can interact with SMN is intriguing because SMN is implicated in fatal childhood motor neuron disease spinal muscular atrophy (SMA) and the organization of Gemini of Cajal bodies (Gems). Consistent with these findings, knocking down FUS or expression of FALS-associated FUS mutant proteins severely compromise the formation of Gems in HeLa cells and fibroblasts from FALS patients, respectively. In primary neurons, FALS-associated FUS mutant proteins promote SMN protein aggregation in the cytoplasm and axons of primary neurons (Groen et al., 2013; Yamazaki et al., 2012). In a recent study, Sun and colleagues further show that the RGG domain in FUS and the Tudor domain in SMN are required for direct interaction. Surprisingly, FALS-associated FUS mutations enhance the interaction between FUS and SMN, and thereby affecting the normal functions of SMN by reducing Gems bodies and changing the state steady level of snRNA in transgenic mouse tissues and in fibroblasts from patients expressing mutant FUS proteins (Sun et al., 2015). Global analysis of RNA splicing reveals that mutant FUS-dependent splicing changes mimic partial FUS loss of activity, independent of cytosolic mislocalization. These results provide evidence for both gain and loss of function caused by ALS-linked mutations in FUS and the potential convergence in pathological pathways of ALS and SMA.
2.3. Prion-like Property of FUS and Its Implication in Liquid-to-Solid Phase Transition
The presence of TDP-43 and FUS proteinopathy in ALS raises the intriguing possibility that TDP-43 and FUS may have a high propensity to form protein aggregates. Indeed, using a bioinformatics algorithm designed to identify proteins with “prion-like” or low complexity (LC) domain (Alberti et al., 2009), it has been shown that the N-terminal Q/G/S/Y domain and part of the Gly-rich region in FUS (amino acids 1-239) and the C-terminal Gly-rich region of TDP-43 (amino acids 277-414) respectively rank 15th and 69th among 27,879 proteins in the human proteome for their prion-like property (Cushman et al., 2010; Gitler and Shorter, 2011)(Figure 2). Consistent with this idea, expression of TDP-43 and FUS in the baker's yeast Saccharomyces cerevisiae shows that both proteins are more prone to form protein aggregates that resemble proteinopathy in human diseases and that the protein aggregate formation is dependent on the prion-like domains (Fushimi et al., 2011; Sun et al., 2011). These findings underscore the value of yeasts as a model organism that can be used for genetic screens to identify modifiers that can alleviate FUS proteinopathy. Indeed, such screens reveal many potential candidates that are implicated in RNA metabolic process, ribosome biogenesis, response to cellular stress, etc (Sun et al., 2011). Similar approach has also been exploited as an effective screen to identify the causal links between two other FET family members, EWSR1 and TAF15, to the pathogenesis of ALS (Couthouis et al., 2012; Couthouis et al., 2011). Several other RNA-binding proteins with similar prion-like properties have been implicated in the organization of stress granules and perturbations to this process may also contribute to ALS and other neurodegenerative diseases (Li et al., 2013).
One major advance in understanding the biophysical property of FUS and its role in the formation of RNA granules comes from the observation of a small molecule chemical 5-aryl-isoxazole-3-carboxyamide, which when biotinylated acquires the unique property to aggregate and disaggregate RNA granules in a soluble, cell-free system (Han et al., 2012; Kato et al., 2012). Using biotinylated isoxazole (b-isox), McKnight and colleagues use an elegant and highly efficient hydrogel formation assay and identify that several RNA binding proteins, including FUS, TDP-43, and hnRNPA1, have a high propensity to co-precipitate with b-isox using their LC domain. Many of these proteins have been implicated in the formation of stress granules. Consistent with this idea, mutations that alter the highly conserved [G/S]Y[G/S] motif within the LC domain of FUS completely abolish its ability to form stress granules in cells. The authors use transmission electron microscopy (TEM) and X-ray diffraction analyses to show that FUS form amyloid-like filamentous protein aggregates with prominent filamentous cross-β structure that resembles amyloid proteins. Indeed, both biophysical measurements and ultrastructural analyses show that the fibrillary FUS proteins in the hydrogel resemble FUS aggregates identified in the cytoplasm of spinal motor neurons in a patient with FUS-P525L mutation using immunogold EM (Huang et al., 2010; Kato et al., 2012).
Knowing the unique property of FUS in hydrogel formation, one critical and intuitive question is how FALS-associated mutations in FUS might alter the biophysical properties of FUS and thereby affects the function of FUS in RNA-protein complex formation. To investigate this, Alberti and colleagues show that when cells experience DNA damage or heat stress, FUS rapidly accumulates in distinct compartments in the nucleus and cytoplasm, respectively. They then use an imaging technique, known as “half-bleach” (Brangwynne et al., 2009), to show that FUS redistributes rapidly within stress granules in the cytoplasm and in nuclear FUS assemblies. Their results show that FUS granules undergo frequent fusion, and as soon as they interact, these granules undergo rapid relaxation into a spherical shape (Patel et al., 2015). These results indicate that the FUS-containing compartments, which exist in liquid droplets and hydrogel states, are reversible and extremely dynamic. Interestingly, FUS mutations associated with FALS promote a conversion of FUS-containing liquid droplets to fiber state, which results in impaired protein synthesis in axons and leads to neurotoxicity (Murakami et al., 2015; Patel et al., 2015).
2.4. LC Domain of FUS in High-Order Assembly of Protein-RNA Complex
The ability of FUS to form RNA-protein complexes is further revealed using a 48 nucleotide (nt)-long RNA (prD RNA) from the promoter region of DNMT3b gene (Schwartz et al., 2012; Schwartz et al., 2013). It is important to note that this prD RNA does not contain any of the previously identified FUS binding motifs (Lagier-Tourenne et al., 2012; Lerga et al., 2001), yet exhibit robust binding to recombinant FUS proteins in electrophoretic mobility shift assays (Schwartz et al., 2013). This provides a convenient tool to characterize the essential role of the LC domain in FUS and its mutual interactions with RNA to form high-order protein-RNA complexes. Similar high-order assemblies have been reported using recombinant FUS proteins and synthetic RNA from the intron-exon boundary and 3′UTR of the bdnf gene (Qiu et al., 2014). Many of these bdnf RNA probes do not contain the reported FUS binding motif, again supporting the notion that the RNA structure is perhaps more important for FUS interaction. Interestingly, RNA-FUS assemblies appear to be critical for its interaction with the CTD of RNA polymerase II (Schwartz et al., 2013).
Using the FUS-bdnf RNA interactions, Qiu and colleagues show that mutant FUS-R521C proteins form more stable and higher order protein-RNA assemblies, which are more difficult to dissociate in competition assays (Qiu et al., 2014). Interestingly, in transgenic mice expressing FUS-R521C, the majority of mutant FUS proteins are in the nuclei of spinal motor neurons, suggesting that the presence of high-order mutant FUS-RNA assemblies may interfere with the transcription and/or RNA splicing. Consistent with these results, expression of mutant FUS proteins or siRNA knockdown of FUS in fibroblasts alters the distribution of RNA polymerase II within the nuclei. These results are further confirmed using fibroblasts derived from FALS patients with FUS mutations (Nomura et al., 2014; Schwartz et al., 2014).
3. FUS Mutations and Neurodegeneration in Model Organisms
3.1. Dendrite and Synaptic Defects in Rodent Models of FUS Mutations
To characterize the consequences of expressing mutant FUS proteins in the nervous system, several groups have used a number of transgenic strategies in mice or rats to model disease conditions caused by FUS mutations. Results from these studies show that expressing mutant FUS proteins causes consistent neurodegenerative phenotypes. For instance, both the transgenic mouse and rat models expressing mutant FUS-R521C proteins develop early onset ALS-like symptoms, including hindlimb paralysis, muscle wasting, and reduced innervation at the neuromuscular junction (NMJ)(Huang et al., 2011; Qiu et al., 2014; Sharma et al., 2016). The cardinal phenotypes include age-dependent reductions in dendritic arborization and synaptic density in the spinal motor neurons and cortical neurons in the sensorimotor cortex of the FUS-R521C transgenic mice (Figure 3)(Qiu et al., 2014). Similar dendritic arborization defects have also been reported in neurons in the entorhinal cortex of Camk2a-tTA transgenic rats (Huang et al., 2012), and in the spinal motor neurons and cortical neurons of Cre-inducible transgenic mouse lines that express FUS-R521G in the nervous system (Sephton et al., 2014). The dendritic phenotype caused by FUS-R521C can be recapitulated in cultured cortical neurons, and can be partially rescued by exogenous BDNF (Qiu et al., 2014). In side-by-side comparisons, FUS-R521C and FUS-P525L cause more severe dendritic growth defects compared to wild type FUS. These results support the notion that wild type and mutant FUS affect dendritic growth in gene dosage-dependent manner. In light of these findings, it is interesting to note that transgenic mice expressing higher level of wild type FUS also show early onset motor neuron degeneration in a dosage-dependent manner (Mitchell et al., 2013). Consistent with these results, mutations in the 3′ UTR of the FUS gene have been identified in several FALS patients. These mutations drastically increase the FUS protein expression in the patients' fibroblasts, at levels higher than that in FUS-R521C fibroblasts (Sabatelli et al., 2013), supporting the notion that wild type FUS expressed at exceedingly high levels can be pathogenic.
Figure 3. Dendritic and Synaptic Phenotypes Caused by FUS Mutations.

Neurolucida tracing shows that the dendritic arbors in control motor neurons, highlighted by Golgi staining techniques, had 6 to 8 intersections per radial distance within 100 μm from the cell body, followed by a gradual reduction in the number of dendritic arbors from 100 to 250 μm. Compared to the control, the number of dendritic arbors in the FUS-R521C motor neurons shows no change within the first 50 μm from the cell body, but a significant reduction is noted from 50 to 250 μm, resulting in reduced cumulative dendritic area. To determine if the dendritic phenotype in FUS-R521C spinal motor neurons affected synaptic connectivity, we use ChAT (green) and FUS immunostaining to characterize the density of synapses surrounding motor neurons (Betley et al., 2009). Our results show that FUS proteins are present primarily in neuronal nuclei, but also show extensive colocalization with ChAT+ boutons and synaptophysin-immunoreactive presynaptic terminals. Remarkably, the density of ChAT+ boutons and SIPT showed significant reductions in the anterior horn of FUS-R521C spinal cord. To further characterize the synaptic defects, we use electron microscopy (EM) to ascertain the morphology and density of synapses within 100 μm radius of the cell body of spinal motor neurons, and show that the cell bodies of control motor neurons are surrounded by synaptic terminals arranged as rosette-like structures (Betley et al., 2009). In contrast, the size of post-synaptic density and the number of synapse per unit area are reduced in FUS-R521C motor neurons. Similar dendrite and synaptic defects are also noted in the apical and basal dendrites of the pyramidal neurons in layer IV-V of the sensorimotor cortex (Qiu et al., 2014), and neurons in the entorhinal cortex (Huang et al., 2012).
Unlike the severe loss of spinal motor neurons in the SOD1G93A mice, the neuron loss phenotype in different FUS transgenic models appears to be more modest. At end stage, FUS-R521C transgenic mice and rats, and transgenic mice expressing FUS-R521G or a truncated FUS mutant protein (amino acids 1-359), show greater than 50% preservation of spinal motor neurons (Table 1)(Huang et al., 2011; Qiu et al., 2014; Sephton et al., 2014; Sharma et al., 2016; Shelkovnikova et al., 2013). The majority of mutant FUS-R521C proteins are located within the nuclei of spinal motor neurons in these transgenic animals, with few neurons showing evidence of FUS-R521C protein aggregates in the cytoplasm. The lack of prominent cytoplasmic FUS inclusion in the FUS-R521C transgenic models is quite different from the pathology observed in patients with FALS caused by FUS mutations (Huang et al., 2010; Kwiatkowski et al., 2009; Vance et al., 2009). Another interesting observation is that few neurons in FUS-R521C transgenic rats and wild type FUS transgenic mice show accumulation of ubiquitin-positive inclusions in the cytoplasm. Curiously, however, most of the ubiquitin-positive cytoplasmic inclusions do not contain mutant or wild type FUS proteins (Huang et al., 2011; Mitchell et al., 2013).
Table 1. Summary of FUS loss-of-function and ALS-associated FUS mutations in model organisms.
| Mutation | Species | Targeting methods | Phenotypes | References |
|---|---|---|---|---|
| LOF | Mouse | Knockout |
|
Kuroda et al., 2000 |
| Knockout |
|
Hicks et al., 2000 | ||
| Conditional knockout |
|
Sharma et al., 2016 | ||
| Drosophila | Deletion in caz gene |
|
Wang et al., 2011 | |
| Xenopus | Antisense MO knock-down |
|
Dichmann & Hartland, 2012 | |
| LOF/GOF | Zebrafish | Antisense MO knock-down |
|
Kabashi et al., 2011 |
| Antisense MO |
|
Armstrong & Drapeau, 2013 | ||
| GOF | Zebrafish | mRNA injection WT FUS, FUS-H517Q-R521G, -R495X or -G515X |
|
Bosco et al., 2010 |
| GOF | Rat | Tet-inducible system WT FUS and FUS-R521C |
|
Huang et al., 2011 |
| Camk2a-tTA inducible WT FUS and FUS-R521C |
|
Huang et al., 2012 | ||
| GOF | C. elegans | Pan-neuronal promoter FUS-R514G, -R521G -R522G, R524S, P525L |
|
Murakami et al., 2012 |
| GOF | Mouse | Mouse PrP promoter WT human FUS |
|
Mitchell et al., 2013 |
| GOF | Mouse | Thy1 promoter FUS 1-359 |
|
Shelkovnikova et al., 2013 & 2014 |
| GOF | Mouse | FLAG-FUS-R521C transgenic expression by Syrian hamster PrP promoter |
|
Qiu et al., 2014 |
| GOF | Mouse | FUS-WT or FUS-R521G transgenic expression by ubiquitous Cre promoter |
|
Sephton et al., 2014 |
| GOF | Mouse | FUS-WT, FUS-R521C and FUS-P525L transgenic expression from MAPT locus |
|
Sharma et al., 2016 |
| GOF | Drosophila | Global or neural expression of human FUS |
|
Lanson et al., 2011 Daigle et al., 2013 Shahidullah et al., 2013 |
Abbreviations: LOF: loss of function; GOF: gain of function; MO: morpholino; PrP: prion.
The discrepancy of neuropathology in the rodent models and human patients raise several intriguing questions regarding the cause and significance of FUS-positive cytoplasmic inclusions in FALS. One possible explanation for the lack of FUS+ cytoplasmic inclusions in transgenic rodent models is that the cytoplasmic aggregation of wild type or mutant FUS proteins may be age- and dosage-dependent. Depending on the efficiency of nucleus-to-cytoplasm translocation for wild type and mutant FUS proteins, the early postnatal lethality in most of the transgenic mice or rats may not have given FUS proteins sufficient time to accumulate in the neuronal cytoplasm. Alternatively, it is possible that mouse spinal motor neurons may develop inherent homeostatic mechanisms to maintain the certain level FUS expression (Dini Modigliani et al., 2014). In this regard, only when expressed at exceedingly high level using viral vectors, such as AAV1, will the FUS proteins begin to accumulate in the neuronal cytoplasm (Verbeeck et al., 2012). Regardless of the mechanism, the observations that transgenic mice and rats develop severe neurodegenerative phenotypes even in the absence of prominent FUS proteinopathy in neuronal cytoplasm suggest that increase of wild type FUS proteins or the presence of mutant FUS proteins in nucleus is sufficient to cause disease, most likely through the perturbations of DNA damage repair/response and RNA splicing machinery (Qiu et al., 2014; Wang et al., 2013). Interestingly, FALS-related mutation FUS-R521G exhibits a drastic shift in binding preference from the intronic sequences to sequences in the 3′UTR (Hoell et al., 2011).
While the results from the murine models suggest that mutant FUS proteins may acquire gain-of-function properties to interact with wild type FUS proteins and new RNA targets, we are still at the very early stage of uncovering the mechanism(s) of FUS mutations that contribute to impairments in neuronal survival and defects in dendritic growth and synaptic connectivity. In Sections 4 and 5, we summarize the recent findings on the effects of FUS mutations in DNA damage repair and RNA splicing, which are likely to have synergistic contributions to the dendritic and synaptic defects. These results not only provide important insights on the potential target genes, which might be preferentially affected by the DNA damage repair and RNA splicing defects caused by FUS mutations, they also provide future directions to establish both in vitro and in vivo approaches to characterize how accumulation of mutant FUS proteins in neuronal cytoplasm affects ribosomal functions and RNA transport in dendrites and axons.
3.2. FUS-mediated Neurodegeneration in Other Model Organisms
In addition to the rodent models of FUS mutations, results from other model organisms, including yeasts, Xenopus, and rodents, have revealed a wealth of information regarding the in vivo functions of FUS during organismal development, and how FUS mutations may disrupt these functions and contribute to the neurodegenerative process. There are several additional studies performed in other model organisms, including Drosophila melanogaster, C. elegans and zebrafish (Danio rerio), which provide novel insights on the genetic interactions between FUS and other FALS-related genes, such as TARDBP and SOD1.
Drosophila has a single homolog of FUS, encoded by the cabeza (caz) gene on X chromosome, that shares 53% amino acid identity to its mammalian counterpart (Stolow and Haynes, 1995). The Drosophila Caz protein contains 399 amino acids and is expressed in neurons, glia and muscle cells. Loss-of-function analyses show that only 14% of male caz mutant larvae successfully undergo pupation and eclose to become adults (Wang et al., 2011). The caz mutants that do survive into adulthood exhibit severe locomotion phenotype and a markedly reduced survival in postnatal life. The eclosion phenotype in caz mutants can be rescued by neuron-specific transgenic expression of Caz, wild type human FUS (hFUSWT), hFUSR522H or hFUSP525L at the same expression level, suggesting that hFUSWT, hFUSR522H and hFUSP525L can functionally restore the role of Caz during eclosion. However, neuron-specific transgenic expression of Caz or hFUSWT only partially restores the locomotion and longevity phenotype, whereas neither hFUSR522H nor hFUSP525L is capable of restoring the locomotion or longevity phenotype. Interestingly, Drosophila tbph mutants lacking TDP-43 homolog TBPH also show similar phenotype in eclosion, locomotion and longevity. Whereas expression of TBPH in caz mutants cannot rescue loss of Caz phenotype, overexpression of Caz in tbph mutants restores eclosion, locomotion and longevity. These results support the model that caz and tbph genetically interact in Drosophila to regulate neuronal development and longevity (Wang et al., 2011). Several other studies using Drosophila as a model system also show that expressing mutant human FUS proteins, hFUSR518K, hFUSR521H or hFUSR521C, in the eye, motor neurons or the nervous system leads to eye degeneration, defects in locomotion and increase in mortality (Lanson et al., 2011). Detailed analyses of the locomotion defects indicate that mutant FUS proteins cause synaptic defects before the degeneration of motor neurons. The synaptic defects include disorganization of the presynaptic active zone protein brunchpilot, reduced quantal contents and miniature presynaptic currents, and reduced synaptic currents in the postsynaptic muscle cells (Shahidullah et al., 2013).
The effects of FUS in synaptic functions have also been investigated using the zebrafish larvae as a model system. Antisense morpholino (AMO) knockdown of FUS in zebrafish causes motor behavioral defects reflected as reduced touch-evoked escape response (TEER) and marked reductions in the branching and length of motor axons (Kabashi et al., 2011). Similar to the observations in Drosophila, AMO knockdown of FUS and expressing mutant human FUS proteins in zebrafish also cause defects in synaptic transmission at NMJ by reducing the presynaptic quantal contents (Armstrong and Drapeau, 2013).
4. Mechanism of FUS Mutations in Neurodegeneration: DNA Damage Defects
4.1. DNA Damage & Neurodegeneration
Both prokaryotic and eukaryotic organisms use highly evolutionarily conserved mechanisms to repair DNA damages caused by radiation from the environment or by endogenous sources, such as free radicals produced within the cells. Mutations in DNA repair machinery have been linked to hereditary neurodegenerative diseases (Jackson and Bartek, 2009; McKinnon, 2009; Rass et al., 2007). For instance, ataxia telangiectasia (AT) is an autosomal recessive, early onset neurodegenerative disease caused by mutations in the ATM gene, which encodes a protein kinase that regulates the cellular response to DNA double strand breaks (DSBs). Patients with mutations in the components of the DNA damage sensor complex MRN (MRE11-RAD50-NBS1) also develop severe neurological symptoms, with hypersensitivity to ionizing radiation and genome instability. Another DNA damage repair machinery involves base excision repair (BER), which is the primary mechanism that handles spontaneous DNA damage caused by free radicals and reactive oxygen species (ROS). Patients with mutations in critical components of the BER machinery, including CSA (also known as excision repair cross-complementing rodent repair deficiency, complementation group 6 or ERCC6), CSB (ERCC8), XPD (ERCC2) and XPG (ERCC5), develop Cockyane syndrome, characterized by retinal degeneration, microcephaly, deafness and loss of Purkinje cells in the cerebellum (Cleaver et al., 2009). In addition to the inherited forms of neurodegenerative diseases, DNA damage and genomic instability have also been linked to late-onset neurodegenerative diseases, such as Alzheimer's disease and Parkinson's disease (Anderson et al., 1996; Bender et al., 2006; Lu et al., 2004).
4.2. Roles of FUS in DNA Damage Response and Repair
Several studies have implicated FUS in the DNA damage response and repair machinery during development and in postnatal life. For instance, two groups have independently generated mice lacking FUS (FUS-/- mutants) and show that FUS deficiency consistently leads to a marked increase in DNA damage that affect a wide range of cell types during perinatal development and in postnatal life (Table 1)(Hicks et al., 2000; Kuroda et al., 2000). When maintained in the C57Bl6 background, FUS–/– mice are perinatal lethal and exhibit severe deficiency in B lymphocyte development. In contrast, FUS–/– mice maintained in the mixed 129svev;CD1 genetic background survive into adulthood and show male sterility. Regardless of the genetic background, however, one consistent phenotype in both independent FUS-/- mouse lines are the presence of genomic instability and increased vulnerability to ionizing irradiation. Consequently, FUS–/– mice present with increased numbers of unpaired and mispaired chromosomal axes in pre-meiotic spermatocytes and in B lymphocytes (Hicks et al., 2000; Kuroda et al., 2000).
The robust genomic instability and increased DNA damage phenotype in FUS–/– mice raise the intriguing possibility that FUS might be involved in DNA damage response and repair machinery. Indeed, FUS has been suggested to be involved in the formation of D-loops, an essential step in homologous recombination, and normally presents in chromosome pairing, DNA repair and telomeres (Baechtold et al., 1999; Bertrand et al., 1999). Interestingly, wild type FUS, but not FUS-CHOP fusion protein can be phosphorylated by ATM (ataxia-telangiectasia mutated) in response to DNA double strand breaks (DSBs)(Gardiner et al., 2008). In addition, FUS has been shown to promote homologous DNA pairing, a key step in homologous recombination (HR), whereas the oncogenic fusion protein FUS-CHOP, in which the C-terminal domain of FUS was replaced by the DNA-binding and leucine zipper dimerization domain of CHOP (Crozat et al., 1993), is unable to promote DNA pairing. Since the Gly-rich domain is retained in the FUS-CHOP protein, these data suggest that the N-terminal domain of FUS may be involved to DNA repair through interaction with other proteins in the DNA repair machinery. Consistent with this notion, FUS has been shown to directly interact with CBP/p300, an acetyltransferase, through its N-terminal domain, and leads to the inhibition of CCND1 transcription following DNA damage (Wang et al., 2008), suggesting that FUS may play multiple roles in response to DNA damage. Surprisingly, the ability of FUS to respond to DNA damage depends on the allosteric interaction with single-stranded, low copy number long noncoding RNA transcripts (Wang et al., 2008). Together, these results underscore the unique feature of FUS as RNA and DNA binding protein in regulating the DNA damage response and repair process.
To determine how FUS affects DSB repair, Wang and colleagues use siRNA to knockdown FUS and show that loss of FUS affects homologous recombination and nonhomologous end joint (NHEJ)-mediated DSB repairs in both U2OS cells and primary neurons (Wang et al., 2013). Using γH2AX immunoreactivity as an early marker of DNA damage and a prerequisite marker for DSB repair (Fillingham et al., 2006; Pilch et al., 2003), they further demonstrate that FUS is rapidly recruited to DNA damage sites, which precedes the accumulation of γH2AX. These results, independently confirmed by other groups (Mastrocola et al., 2013; Rulten et al., 2014), suggest that recruitment of FUS to damaged chromatin is required to elicit an effective DDR. The study by Rulten and colleagues further show that FUS recruitment to DNA damage foci is dependent on poly (ADP-ribose) polymerase 1 (PARP1). However, it is unclear if these results indicate the presence of hierarchy of protein complex formation involving PARP1 and FUS in the assembly of DNA response/repair machinery, or the requirement of PARP1 is a cell type-specific phenomenon (Rulten et al., 2014; Wang et al., 2013). Finally, DSB can induce nucleus-to-cytoplasm translocation of FUS and causes phosphorylation of FUS in the C-terminus by DNA-dependent protein kinase (DNA-PK)(Deng et al., 2014b). These studies provide the first molecular clue for the previous observations that FUS–/– mice exhibit enhanced radiation sensitivity, growth retardation, immunodeficiency, and increased genomic instability (Hicks et al., 2000; Kuroda et al., 2000).
4.3. FUS Mutations & HDAC-dependent DNA Damage Response/Repair
Given the critical role of FUS in DNA damage response/repair, it is interesting to note that FALS-associated mutations in FUS do not affect the recruitment of mutant FUS proteins to DNA damage foci. Rather, the mechanism by which FUS regulates DNA damage repair machinery depends on its ability to directly interact with chromatin remodeling factor histone deacetylase 1 (HDAC1), which plays fundamental role in DNA repair and the maintenance of genomic stability (Figure 4A). Deficiency in HDAC1 and the closely related HDAC2 causes severe hypersensitivity to DNA damaging agents and persistent phenotypes related to DNA repair defects, including dysregulation of histone acetylation, abnormalities in heterochromatin formation, and aberrant expansion and recondensing of the chromatin structure in DNA repair process (Dinant et al., 2008; Lukas et al., 2011; Miller et al., 2010). Interestingly, loss of HDAC1 has been reported to sensitize neurons to DNA damage and induce aberrant cell cycle re-entry, while the overexpression of HDAC1 protects neurons from genotoxic agents (Dobbin et al., 2013; Kim et al., 2008). Indeed, both neurons with FUS deficiency and HDAC1–/– neurons exhibit increased DNA damage following etoposide treatment (Dobbin et al., 2013; Wang et al., 2013), supporting the notion that the interaction between FUS and HDAC1 plays an important role in maintaining genome stability and integrity in neurons.
Figure 4. Mechanisms of Wild Type and Mutant FUS in DNA Damage Repair/Response and RNA Splicing.
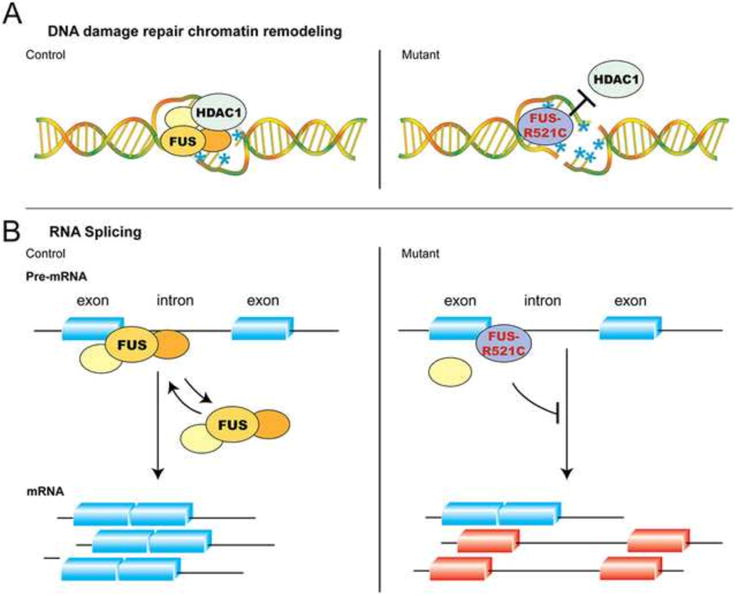
(A) Wild type FUS is rapidly recruited to DNA damage foci caused by double-stranded breaks, where it interacts with chromatin remodeling factor HDAC1. Although FUS-R521C can still be recruited to DNA damage foci, it fails to interact with HDAC1 and PARP1. Due to the defects in DNA repair/response machinery, neurons in FUS-R521C transgenic mice show increased DNA damage (indicated by blue asterisks and the presence of double-stranded breaks). (B) In addition to its role in DNA damage repair, several lines of evidence indicate that FUS can also regulate pre-mRNA splicing. Results from CLIP-RT-PCR and protein-RNA interactions in EMSA assays show that both wild type FUS and FUS-R521C can interact with selective oligoribonucleotides from Bdnf exon-intron boundaries. Whereas the equilibrium of wild type FUS-RNA interactions appears to be more dynamic, FUS-R521C tends to form more stable protein-RNA complexes that are more difficult to dissociate. (Figure adapted from Qiu et al., 2014, with permissions from the Journal of Clinical Investigation.)
The fact that FALS mutations are transmitted in an autosomal dominant manner and FALS mutations do not affect FUS recruitment to DNA damage foci lead to the hypothesis that mutant FUS proteins may have dominant-negative effect that interferes with its interaction with HDAC1 and the subsequent assembly of DNA repair machinery (Figure 4A). Indeed, structure-function analyses show that the Glycine-rich domain (amino acids 156-262) and C-terminal domain (amino acids 450-526) of FUS are required for FUS-HDAC1 interaction (Wang et al., 2013). Remarkably, these two domains in FUS harbor most of the FALS mutations, and FUS mutations in these two domains, including FUS-R244C, FUS-R514S and FUS-R521C, show impaired interaction with HDAC1 and reduced DSB repair efficiency when expressed in cells. Consistent with these results, wild type FUS can be detected in a protein complex with HDAC1 in the control spinal cord tissues. In contrast, protein extracts from FUS-R521C transgenic mice show no detectable complex formation between FUS-R521C and HDAC1 (Qiu et al., 2014). Interestingly, the presence of FUS-R521C almost completely abolishes the protein-protein interaction between wild type FUS and HDAC1 in FUS-R521C transgenic mice. This dominant effect of FUS-R521C is due to the abnormal gain-of-function property of the mutant FUS-R521C protein in forming more stable complex with wild type FUS protein in both HEK293T cells and in FUS-R521C transgenic mice. Consistent with these results, spinal motor neurons and cortical neurons in FUS-R521C transgenic mice and in patients with FUS-R521C or FUS-P525L mutation show a robust increase of γH2AX staining (Qiu et al., 2014; Wang et al., 2013).
The demonstration that DNA damage repair defects contribute to the pathogenesis of neurodegeneration caused by FUS mutations further underscores the critical role of DNA damage repair in neurodegenerative conditions. Indeed, several previous studies have shown that increased levels of 8-hydroxydeoxyguanosine (8-OHdG) residues, a marker of oxidative DNA damage, can be identified in the spinal cord of both sporadic and familial ALS patients (Ferrante et al., 1997). Age-related motor neuron degeneration has been observed in mice lacking the DNA repair protein ERCC1, suggesting that the accumulation of DNA damage contributes to the motor neuron vulnerability (de Waard et al., 2010). To determine whether FUS mutations cause widespread or selective target genes, Qiu and colleagues performed a quantitative PCR-based formamidopyrimidine-DNA glycosylase (FPG), a based excision repair enzyme, assay to identify oxidized purine residues in a highly selected group of neural genes (Qiu et al., 2014). Their results show that signatures of DNA damage can be detected in the 5′ and 3′ UTR of genes that involve in synaptic transmission (NR2A and GluR2) and dendritic growth (Bdnf). One interesting caveat is that the DNA damage in these genes appears to be more prominent in the cortex than in the spinal cord. Hence, future experiments are needed to reveal additional targets at the genome-wide level. While these results suggest that perturbations to multiple signaling pathways may converge on the DNA damage repair defects leading to neurodegeneration, it is important to note that DNA damage due to defects in oxidative stress and nucleotide excision repair is quite different from that caused by double stranded DNA breaks or defects in the ATM pathways. Finally, it is unclear why motor neurons are more susceptible to FUS mutations despite the fact that almost all neurons express mutant FUS proteins and the evidence of DNA damage can also be detected in cortical neurons of FUS-R521C transgenic mice and human disease tissues (Qiu et al., 2014; Wang et al., 2013). Such “selective vulnerability” is a common theme in neurodegenerative diseases. One potential mechanism is that motor neurons may produce excessively higher amount of mitochondrial reactive oxygen species, which may create a vicious cycle that further promotes the accumulation of DNA damage repair defects (Cleaver et al., 2013; Rulten et al., 2014).
5. Mechanism of FUS Mutations in Neurodegeneration: RNA Splicing Defects
5.1. Effects of FUS Mutations in RNA Transcription/Splicing Defects
The causal link between FUS mutations and DNA damage defects provides critical mechanistic insights to neurodegeneration because the process to repair DNA damage is tightly coupled to transcription through regulating the activity of RNA polymerase II and the subsequent RNA processing, including RNA splicing and transport (Cleaver et al., 2009; Kornblihtt et al., 2004; Munoz et al., 2009). Furthermore, a plethora of evidence support that RNA transcription in the eukaryotic cells is a highly dynamic and tightly regulated process that involves multiple intricately connected steps, including splicing of pre-mRNA and transport of mature mRNA to its final destinations (Moore and Proudfoot, 2009; Reed and Hurt, 2002). In the nervous system, these regulatory mechanisms are known to generate a vast diversity of RNA transcripts that control cell fate determination, axon guidance, dendritic growth and synaptic functions (Li et al., 2007; Martin and Ephrussi, 2009). Perturbations to these critical mechanisms have been implicated in neuromuscular diseases, neurodevelopmental disorders and neurodegenerative diseases (Cooper et al., 2009).
Consistent with these findings, two recent studies show that both human patients with FUS-R521C or FUS-P525L mutation, and FUS-R521C transgenic mice exhibited evidence of DNA damage in cortical neurons and spinal motor neurons (Qiu et al., 2014; Wang et al., 2013). These results indicate that the FUS-R521C transgenic mice provide an invaluable system to identify neural genes implicated in DNA damage during neurodegeneration (Graff et al., 2012; Lu et al., 2004). Indeed, a PCR-based screening approach shows that the 5′ non-coding exons of the mouse Bdnf gene, which contain transcriptional start sites and are required splicing of long intronic sequences to generate mature Bdnf mRNA, consistently exhibit evidence of DNA damage. These results lead to the identification of retentions of 5′ splice junctions in the Bdnf mRNA and defects in transporting Bdnf mRNA to distal dendrites. Using electrophoretic mobility shift assays (EMSA), it is further demonstrated that, compared to wild type FUS, mutant FUS-R521C proteins form more stable protein-RNA complex to 5′ splice junction and the 3′UTR sequences of Bdnf pre-mRNA (Qiu et al., 2014)(Figure 4B). These results support the idea that FALS-associated FUS mutation FUS-R521C exhibits aberrant gain-of-function properties, including forming more stable protein-protein interactions with the endogenous wild type FUS and more stable protein-RNA complex, which most likely alter the ability of FUS to recruit DNA damage repair machinery and the equilibrium of the interactions between FUS and RNA in the splicing machinery, respectively (Figure 4)(Qiu et al., 2014; Wang et al., 2013). Similar phenotypes of FUS mutations in DNA damage repair and RNA splicing machinery have been reported in several other studies using biochemical, cell biology and bioinformatics analyses (Hoell et al., 2011; Mastrocola et al., 2013; Zhou et al., 2014).
Given the highly efficient process in RNA splicing, the observations that FUS-R521C can form more stable protein-RNA complex raise the intriguing question as to whether this gain-of-function property may have more widespread intron retention effects on the transcriptomes or only affect a selective subset of target genes. To distinguish these two possibilities, Qiu and colleagues perform genome-wide survey in the transcriptomes of FUS-R521C spinal cord using RNA-seq and show two primary defects involving the transcription and RNA splicing in selective genes that are critical for dendritic growth and synaptic functions (Qiu et al., 2014). For instance, RNA-seq results in the spinal cord of FUS-R521C mutants show perturbations in the expression or splicing of genes involved in the organization of extracellular matrix, including members of the collagen and cadherin gene families that regulate the specificity of axonal projection and target innervation (Robles and Baier, 2012; Sanes and Yamagata, 2009). Interestingly, similar targets have also been identified in the RNA-seq analyses of FUS MO-treated Xenopus morphants (Dichmann and Harland, 2012), suggesting that FUS-R521C phenotype may recapitulate certain transcriptional and RNA splicing defects in FUS loss-of-function mutants. Another intriguing feature of the RNA-seq results in FUS-R521C spinal cord is that many target genes in the extracellular matrix assembly GO categories (GO:0005581, GO:0005201, GO:0005578 and GO:0031012) have also been shown to be transcriptional targets of DNA damage response gene Cockyane syndrome B (CSB) and HDAC1 (Newman et al., 2006), and are frequently misregulated and misspliced in the motor neurons of SALS patients (Rabin et al., 2010). While these results are correlative, they raise the interesting possibility that the recruitment of FUS, HDAC1 and CSB may constitute a critical step in the repair of damaged DNA in FALS caused by FUS mutations and in SALS.
Finally, one remarkable feature in the spinal cord of FUS-R521C transgenic mice is the up-regulation of genes that are functionally related to immune response, complement activation and chemotaxis (Qiu et al., 2014). Consistent with these findings, the FUS-R521C spinal cord show pronounced microgliosis. Since neither wild type FUS or FUS-R521C proteins can be detected in the microglia, these results support the idea that non-cell autonomous mechanisms, triggered by damaged neurons or reactive astroglia, may activate microglia and contribute to the neurodegeneration in ALS. Interestingly, similar non-cell autonomous mechanisms have been reported in the mutant SOD1 models (Boillee et al., 2006a; Boillee et al., 2006b). Alternatively, the defects in DNA damage repair and RNA splicing caused by mutant FUS-R521C may occur in glia cells, which promotes astroglial activation and/or degeneration of oligodendroglia, further contributing to the dendritic loss and synaptic degeneration of spinal motor neurons in FUS-R521C mice.
5.2. Mechanisms of FUS Mutations in RNA Spicing Machinery
Several studies indicate that FUS can physically interact with SMN and U1 snRNP, and that loss of FUS or expressing FALS-associated FUS mutations disrupts the organization of Gemini of Cajal bodies (Gems), where the presence of TDP-43, FUS and another fatal motor neuron disease gene product SMN are required to regulate the assembly of the Gems in several different cell types (Battle et al., 2006; Sun et al., 2015; Yamazaki et al., 2012). Consistent with these findings, FUS-/- hippocampal neurons show a near complete loss of Gems. Interestingly, the integrity of Gems and splicesome is severely perturbed in the spinal motor neurons of patients with sporadic ALS, which most likely is due to the loss of nuclear TDP-43 and prominent up-regulations of U snRNAs and snRNPs (Tsuiji et al., 2013). In addition to its roles in the organization of splicesome, FUS is implicated in the integrity of paraspeckles, which are subnuclear structures that regulate gene expression by nuclear retention of RNA (Bond and Fox, 2009). The core paraspeckle proteins include DBHS (Drosophila melanogaster behavior, human splicing) proteins, PSF/SFPQ, P45NRB/NONO, and PSPC1. In addition, a long noncoding RNA (lncRNA) NEAT1 is also required to maintain the integrity of paraspeckles. In a recent study, FUS and TDP-43 are shown to interact with NEAT1 (Nishimoto et al., 2013), raising the possibility that perturbations to both proteins may disrupt the integrity or maintenance of paraspeckles. Consistent with this idea, spinal motor neurons in transgenic mice expressing truncated FUS mutant protein (amino acids 1-359) show cytoplasmic aggregates of P45NRB/NONO. Although confocal images from these transgenic neurons indicate that P45NRB/NONO proteins and mutant FUS proteins are in close proximity, it is unclear whether the presumed complex between P45NRB/NONO and FUS are disrupted by the presence of mutant FUS proteins (Shelkovnikova et al., 2013). It is also unclear if the number and distribution of paraspeckles are disrupted in the spinal motor neurons of these transgenic mice and in patients with FALS or SALS. Another alternative mechanism for mutant FUS proteins to interfere with the integrity of paraspeckles is by altering the expression of lncRNA NEAT1. Indeed, RNA-seq analyses of spinal cord from FUS-R521C transgenic mice show that the NEAT1 levels are 2-3 folds higher than that in the age-matched controls (Qiu et al., 2014).
6. Future Directions
6.1. Hierarchy of FUS Mutations and Proteinopathy
The discovery of dominant FUS mutations as one of the major causal links for FALS has opened up new windows to understanding the pathogenesis of ALS. Judging from the biophysical properties of FUS, it is tempting to propose that the FALS-associated FUS mutations alter the liquid-solid phase transition in FUS and thereby dominantly interfere with the ability of wild type FUS in DNA damage repair and RNA splicing, leading to both structural and functional defects in dendritic/axonal growth and synaptic transmission. The fundamental effects of mutant FUS proteins can be attributed to its aberrant gain-of-function properties that alter the homeostasis of the interactions of wild type FUS and its the interacting partners, including proteins, pre-mRNAs and lncRNAs, in the DNA damage response/repair and RNA splicing machinery. In many aspects, these gain-of-function properties truly reflect broad and essential roles of wild type FUS in the embryonic and postnatal development, and in the maintenance of the organisms and the nervous system.
Notwithstanding these new insights into FUS mutations, it remains a challenge to determine how these new mechanisms help in understanding the pathogenesis of FALS and SALS. With the availability of transgenic models for FUS and TDP-43, we may begin to determine whether mutations in FUS or TDP-43 target similar or divergent mechanisms that eventually lead to neurodegeneration. This is especially important given that TDP-43 proteinopathy is a major neuropathological feature not only in FALS, but also in SALS. Several directions for future research include (1) whether FUS and TDP-43 proteins use similar or different intracellular trafficking mechanisms for their transport in and out of the neuronal nucleus, (2) how FUS and TDP-43 proteinopathies promote degeneration in spinal motor neurons and other types of neurons, (3) how FUS and TDP-43 mutations might cause non-cell autonomous mechanisms to promote glial pathology, and (4) whether patient-specific induced pluripotent stem cell (iPSC)-derived neurons can provide a new model system more closely related to human disease. Indeed, the combined strengths of using iPSC-derived neurons and model organism Drosophila have led to the identification of novel functions of TDP-43 in mRNA transport in axons (Alami et al., 2014). Future studies using similar approaches can also provide more insights to unravel the mechanisms and their hierarchical interactions in the pathogenesis of human diseases. The iPSC approach will also provide important tools to identify potential therapeutic targets that are specific for different mutations in FUS-mediated FALS.
6.2. The Expanding Repertoire of RNA Machinery in Neurodegeneration
The identification of mutations in FUS, TARDBP and C9ORF72 in ALS and FTD-ALS has expanded the landscape of neurodegenerative diseases caused by defects in RNA metabolism machinery. These studies raise the intriguing questions that FUS, TARDBP and C9ORF72 may be just tips of an iceberg, and that dysregulations in RNA machinery may have broader roles in other neurodegenerative diseases? Indeed, results from several recent studies indicate that the answers to both questions are positive. For instance, mutations in the prion-like domains of hnRNPA2B1 and hnRNAPA1 accelerate filamentous protein aggregate formation, increase the propensity of stress granule formation in neuronal cytoplasm and can contribute to the pathology in patients with multisystem proteinopathy and ALS (Kim et al., 2013; Molliex et al., 2015). Furthermore, chemical mutagenesis screens for recessive mutations (Neuroscience Mutagenesis Facility) have reported two mutations, nmf291 and nmf205, that result in neurological phenotypes. The nmf291 allele is caused by a 5-nucleotide deletion in the U2 snRNA, which leads to profound dysregulation in RNA splicing and an age-dependent, progressive degeneration of the cerebellum (An et al., 2008). In contrast, the nmf205 mutation results in loss of GTPBP2 due to a point mutation in the consensus splice donor site of intron 6 of Gtpbp2, leading to missplicing of Gtpbp2 mRNA and a premature stop codon. Since GTPBP2 is an essential binding partner for the ribosome recycling protein pelota, loss of GTPBP2 results in widespread ribosomal stalling and profound age-dependent neurodegeneration in the cerebellum of nmf205 mutants (Ishimura et al., 2014). Finally, two recent studies show that mutations in the human cleavage and polyadenylation factor subunit 1 (CLP1) gene, which encodes a multifunctional kinase implicated in the maturation of tRNA, mRNA and siRNA, cause severe neurodegeneration in the cerebellum (Karaca et al., 2014; Schaffer et al., 2014). Consistent with these findings, mouse mutants that express kinase-dead mutant CLP1 show neurodegeneration, characterized by a progressive loss of spinal motor neurons, axonal degeneration in the peripheral nerves, and denervation of neuromuscular junctions, leading to impaired motor function, muscle weakness, paralysis and fatal respiratory failure (Hanada et al., 2013). Together, these findings further expand and reinforce the critical role of RNA metabolism in maintaining the normal functions of the nervous system.
Highlights.
RNA/DNA binding protein FUS (fused in sarcoma) regulates RNA homeostasis and DNA damage repair
Prion-like or low complexity domain in FUS regulates liquid to solid phase transition.
FALS-related mutations in FUS cause dendritic and synaptic defects in model organisms
FUS mutations affect RNA splicing and DNA damage response/repair via a dominant negative mechanism.
Acknowledgments
Grant Sponsors: Veterans Administrations BLR&D Merit Review Award, Grant numbers: I01 BX001108-05 and I01 RX002213-01.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alami NH, Smith RB, Carrasco MA, Williams LA, Winborn CS, Han SS, Kiskinis E, Winborn B, Freibaum BD, Kanagaraj A, et al. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron. 2014;81:536–543. doi: 10.1016/j.neuron.2013.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberti S, Halfmann R, King O, Kapila A, Lindquist S. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell. 2009;137:146–158. doi: 10.1016/j.cell.2009.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An JJ, Gharami K, Liao GY, Woo NH, Lau AG, Vanevski F, Torre ER, Jones KR, Feng Y, Lu B, et al. Distinct role of long 3′ UTR BDNF mRNA in spine morphology and synaptic plasticity in hippocampal neurons. Cell. 2008;134:175–187. doi: 10.1016/j.cell.2008.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson AJ, Su JH, Cotman CW. DNA damage and apoptosis in Alzheimer's disease: colocalization with c-Jun immunoreactivity, relationship to brain area, and effect of postmortem delay. J Neurosci. 1996;16:1710–1719. doi: 10.1523/JNEUROSCI.16-05-01710.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–611. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- Armstrong GA, Drapeau P. Loss and gain of FUS function impair neuromuscular synaptic transmission in a genetic model of ALS. Hum Mol Genet. 2013;22:4282–4292. doi: 10.1093/hmg/ddt278. [DOI] [PubMed] [Google Scholar]
- Baechtold H, Kuroda M, Sok J, Ron D, Lopez BS, Akhmedov AT. Human 75-kDa DNA-pairing protein is identical to the pro-oncoprotein TLS/FUS and is able to promote D-loop formation. J Biol Chem. 1999;274:34337–34342. doi: 10.1074/jbc.274.48.34337. [DOI] [PubMed] [Google Scholar]
- Battle DJ, Kasim M, Yong J, Lotti F, Lau CK, Mouaikel J, Zhang Z, Han K, Wan L, Dreyfuss G. The SMN complex: an assembly machine for RNPs. Cold Spring Harb Symp Quant Biol. 2006;71:313–320. doi: 10.1101/sqb.2006.71.001. [DOI] [PubMed] [Google Scholar]
- Baumer D, Hilton D, Paine SM, Turner MR, Lowe J, Talbot K, Ansorge O. Juvenile ALS with basophilic inclusions is a FUS proteinopathy with FUS mutations. Neurology. 2010;75:611–618. doi: 10.1212/WNL.0b013e3181ed9cde. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behzadnia N, Golas MM, Hartmuth K, Sander B, Kastner B, Deckert J, Dube P, Will CL, Urlaub H, Stark H, et al. Composition and three-dimensional EM structure of double affinity-purified, human prespliceosomal A complexes. Embo J. 2007;26:1737–1748. doi: 10.1038/sj.emboj.7601631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet. 2006;38:515–517. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- Bertrand P, Akhmedov AT, Delacote F, Durrbach A, Lopez BS. Human POMp75 is identified as the pro-oncoprotein TLS/FUS: both POMp75 and POMp100 DNA homologous pairing activities are associated to cell proliferation. Oncogene. 1999;18:4515–4521. doi: 10.1038/sj.onc.1203048. [DOI] [PubMed] [Google Scholar]
- Betley JN, Wright CV, Kawaguchi Y, Erdelyi F, Szabo G, Jessell TM, Kaltschmidt JA. Stringent specificity in the construction of a GABAergic presynaptic inhibitory circuit. Cell. 2009;139:161–174. doi: 10.1016/j.cell.2009.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigio EH. Motor neuron disease: the C9orf72 hexanucleotide repeat expansion in FTD and ALS. Nature reviews Neurology. 2012;8:249–250. doi: 10.1038/nrneurol.2012.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boillee S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006a;52:39–59. doi: 10.1016/j.neuron.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006b;312:1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- Bond CS, Fox AH. Paraspeckles: nuclear bodies built on long noncoding RNA. J Cell Biol. 2009;186:637–644. doi: 10.1083/jcb.200906113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brangwynne CP, Eckmann CR, Courson DS, Rybarska A, Hoege C, Gharakhani J, Julicher F, Hyman AA. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science. 2009;324:1729–1732. doi: 10.1126/science.1172046. [DOI] [PubMed] [Google Scholar]
- Calvio C, Neubauer G, Mann M, Lamond AI. Identification of hnRNP P2 as TLS/FUS using electrospray mass spectrometry. Rna. 1995;1:724–733. [PMC free article] [PubMed] [Google Scholar]
- Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, Couthouis J, Lu YF, Wang Q, Krueger BJ, et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science. 2015;347:1436–1441. doi: 10.1126/science.aaa3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleaver JE, Bezrookove V, Revet I, Huang EJ. Conceptual developments in the causes of Cockayne syndrome. Mech Ageing Dev. 2013;134:284–290. doi: 10.1016/j.mad.2013.02.005. [DOI] [PubMed] [Google Scholar]
- Cleaver JE, Lam ET, Revet I. Disorders of nucleotide excision repair: the genetic and molecular basis of heterogeneity. Nat Rev Genet. 2009;10:756–768. doi: 10.1038/nrg2663. [DOI] [PubMed] [Google Scholar]
- Colombrita C, Onesto E, Megiorni F, Pizzuti A, Baralle FE, Buratti E, Silani V, Ratti A. TDP-43 and FUS RNA-binding proteins bind distinct sets of cytoplasmic messenger RNAs and differently regulate their post-transcriptional fate in motoneuron-like cells. J Biol Chem. 2012;287:15635–15647. doi: 10.1074/jbc.M111.333450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper TA, Wan L, Dreyfuss G. RNA and disease. Cell. 2009;136:777–793. doi: 10.1016/j.cell.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couthouis J, Hart MP, Erion R, King OD, Diaz Z, Nakaya T, Ibrahim F, Kim HJ, Mojsilovic-Petrovic J, Panossian S, et al. Evaluating the role of the FUS/TLS-related gene EWSR1 in amyotrophic lateral sclerosis. Hum Mol Genet. 2012;21:2899–2911. doi: 10.1093/hmg/dds116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couthouis J, Hart MP, Shorter J, DeJesus-Hernandez M, Erion R, Oristano R, Liu AX, Ramos D, Jethava N, Hosangadi D, et al. A yeast functional screen predicts new candidate ALS disease genes. Proc Natl Acad Sci U S A. 2011;108:20881–20890. doi: 10.1073/pnas.1109434108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crozat A, Aman P, Mandahl N, Ron D. Fusion of CHOP to a novel RNA-binding protein in human myxoid liposarcoma. Nature. 1993;363:640–644. doi: 10.1038/363640a0. [DOI] [PubMed] [Google Scholar]
- Cushman M, Johnson BS, King OD, Gitler AD, Shorter J. Prion-like disorders: blurring the divide between transmissibility and infectivity. J Cell Sci. 2010;123:1191–1201. doi: 10.1242/jcs.051672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R, Yu J, Zhang Z, Gygi MP, Krainer AR, Gygi SP, Reed R. SR proteins function in coupling RNAP II transcription to pre-mRNA splicing. Mol Cell. 2007;26:867–881. doi: 10.1016/j.molcel.2007.05.036. [DOI] [PubMed] [Google Scholar]
- de Waard MC, van der Pluijm I, Zuiderveen Borgesius N, Comley LH, Haasdijk ED, Rijksen Y, Ridwan Y, Zondag G, Hoeijmakers JH, Elgersma Y, et al. Age-related motor neuron degeneration in DNA repair-deficient Ercc1 mice. Acta Neuropathol. 2010;120:461–475. doi: 10.1007/s00401-010-0715-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H, Gao K, Jankovic J. The role of FUS gene variants in neurodegenerative diseases. Nature reviews Neurology. 2014a;10:337–348. doi: 10.1038/nrneurol.2014.78. [DOI] [PubMed] [Google Scholar]
- Deng Q, Holler CJ, Taylor G, Hudson KF, Watkins W, Gearing M, Ito D, Murray ME, Dickson DW, Seyfried NT, et al. FUS is phosphorylated by DNA-PK and accumulates in the cytoplasm after DNA damage. J Neurosci. 2014b;34:7802–7813. doi: 10.1523/JNEUROSCI.0172-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dichmann DS, Harland RM. fus/TLS orchestrates splicing of developmental regulators during gastrulation. Genes Dev. 2012;26:1351–1363. doi: 10.1101/gad.187278.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinant C, Houtsmuller AB, Vermeulen W. Chromatin structure and DNA damage repair. Epigenetics & chromatin. 2008;1:9. doi: 10.1186/1756-8935-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dini Modigliani S, Morlando M, Errichelli L, Sabatelli M, Bozzoni I. An ALS-associated mutation in the FUS 3′-UTR disrupts a microRNA-FUS regulatory circuitry. Nature communications. 2014;5:4335. doi: 10.1038/ncomms5335. [DOI] [PubMed] [Google Scholar]
- Dobbin MM, Madabhushi R, Pan L, Chen Y, Kim D, Gao J, Ahanonu B, Pao PC, Qiu Y, Zhao Y, et al. SIRT1 collaborates with ATM and HDAC1 to maintain genomic stability in neurons. Nat Neurosci. 2013;16:1008–1015. doi: 10.1038/nn.3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dormann D, Madl T, Valori CF, Bentmann E, Tahirovic S, Abou-Ajram C, Kremmer E, Ansorge O, Mackenzie IR, Neumann M, et al. Arginine methylation next to the PY-NLS modulates Transportin binding and nuclear import of FUS. Embo J. 2012;31:4258–4275. doi: 10.1038/emboj.2012.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, Hruscha A, Than ME, Mackenzie IR, Capell A, Schmid B, et al. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. Embo J. 2010;29:2841–2857. doi: 10.1038/emboj.2010.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante RJ, Browne SE, Shinobu LA, Bowling AC, Baik MJ, MacGarvey U, Kowall NW, Brown RH, Jr, Beal MF. Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J Neurochem. 1997;69:2064–2074. doi: 10.1046/j.1471-4159.1997.69052064.x. [DOI] [PubMed] [Google Scholar]
- Fillingham J, Keogh MC, Krogan NJ. GammaH2AX and its role in DNA double-strand break repair. Biochemistry and cell biology = Biochimie et biologie cellulaire. 2006;84:568–577. doi: 10.1139/o06-072. [DOI] [PubMed] [Google Scholar]
- Fushimi K, Long C, Jayaram N, Chen X, Li L, Wu JY. Expression of human FUS/TLS in yeast leads to protein aggregation and cytotoxicity, recapitulating key features of FUS proteinopathy. Protein & cell. 2011;2:141–149. doi: 10.1007/s13238-011-1014-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner M, Toth R, Vandermoere F, Morrice NA, Rouse J. Identification and characterization of FUS/TLS as a new target of ATM. The Biochemical journal. 2008;415:297–307. doi: 10.1042/BJ20081135. [DOI] [PubMed] [Google Scholar]
- Gitler AD, Shorter J. RNA-binding proteins with prion-like domains in ALS and FTLD-U. Prion. 2011;5:179–187. doi: 10.4161/pri.5.3.17230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff J, Rei D, Guan JS, Wang WY, Seo J, Hennig KM, Nieland TJ, Fass DM, Kao PF, Kahn M, et al. An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature. 2012;483:222–226. doi: 10.1038/nature10849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groen EJ, Fumoto K, Blokhuis AM, Engelen-Lee J, Zhou Y, van den Heuvel DM, Koppers M, van Diggelen F, van Heest J, Demmers JA, et al. ALS-associated mutations in FUS disrupt the axonal distribution and function of SMN. Hum Mol Genet. 2013;22:3690–3704. doi: 10.1093/hmg/ddt222. [DOI] [PubMed] [Google Scholar]
- Han TW, Kato M, Xie S, Wu LC, Mirzaei H, Pei J, Chen M, Xie Y, Allen J, Xiao G, et al. Cell-free formation of RNA granules: bound RNAs identify features and components of cellular assemblies. Cell. 2012;149:768–779. doi: 10.1016/j.cell.2012.04.016. [DOI] [PubMed] [Google Scholar]
- Hanada T, Weitzer S, Mair B, Bernreuther C, Wainger BJ, Ichida J, Hanada R, Orthofer M, Cronin SJ, Komnenovic V, et al. CLP1 links tRNA metabolism to progressive motor-neuron loss. Nature. 2013;495:474–480. doi: 10.1038/nature11923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks GG, Singh N, Nashabi A, Mai S, Bozek G, Klewes L, Arapovic D, White EK, Koury MJ, Oltz EM, et al. Fus deficiency in mice results in defective B-lymphocyte development and activation, high levels of chromosomal instability and perinatal death. Nat Genet. 2000;24:175–179. doi: 10.1038/72842. [DOI] [PubMed] [Google Scholar]
- Hoell JI, Larsson E, Runge S, Nusbaum JD, Duggimpudi S, Farazi TA, Hafner M, Borkhardt A, Sander C, Tuschl T. RNA targets of wild-type and mutant FET family proteins. Nat Struct Mol Biol. 2011;18:1428–1431. doi: 10.1038/nsmb.2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Tong J, Bi F, Wu Q, Huang B, Zhou H, Xia XG. Entorhinal cortical neurons are the primary targets of FUS mislocalization and ubiquitin aggregation in FUS transgenic rats. Hum Mol Genet. 2012;21:4602–4614. doi: 10.1093/hmg/dds299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Zhou H, Tong J, Chen H, Liu YJ, Wang D, Wei X, Xia XG. FUS transgenic rats develop the phenotypes of amyotrophic lateral sclerosis and frontotemporal lobar degeneration. PLoS Genet. 2011;7:e1002011. doi: 10.1371/journal.pgen.1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EJ, Zhang J, Geser F, Trojanowski JQ, Strober JB, Dickson DW, Brown RH, Jr, Shapiro BE, Lomen-Hoerth C. Extensive FUS-immunoreactive pathology in juvenile amyotrophic lateral sclerosis with basophilic inclusions. Brain Pathol. 2010;20:1069–1076. doi: 10.1111/j.1750-3639.2010.00413.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa H, Shimizu K, Hayashi Y, Ohki M. An RNA-binding protein gene, TLS/FUS, is fused to ERG in human myeloid leukemia with t(16;21) chromosomal translocation. Cancer Res. 1994;54:2865–2868. [PubMed] [Google Scholar]
- Iko Y, Kodama TS, Kasai N, Oyama T, Morita EH, Muto T, Okumura M, Fujii R, Takumi T, Tate S, et al. Domain architectures and characterization of an RNA-binding protein, TLS. J Biol Chem. 2004;279:44834–44840. doi: 10.1074/jbc.M408552200. [DOI] [PubMed] [Google Scholar]
- Ishigaki S, Masuda A, Fujioka Y, Iguchi Y, Katsuno M, Shibata A, Urano F, Sobue G, Ohno K. Position-dependent FUS-RNA interactions regulate alternative splicing events and transcriptions. Sci Rep. 2012;2:529. doi: 10.1038/srep00529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimura R, Nagy G, Dotu I, Zhou H, Yang XL, Schimmel P, Senju S, Nishimura Y, Chuang JH, Ackerman SL. RNA function. Ribosome stalling induced by mutation of a CNS-specific tRNA causes neurodegeneration. Science. 2014;345:455–459. doi: 10.1126/science.1249749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashi E, Bercier V, Lissouba A, Liao M, Brustein E, Rouleau GA, Drapeau P. FUS and TARDBP but not SOD1 interact in genetic models of amyotrophic lateral sclerosis. PLoS Genet. 2011;7:e1002214. doi: 10.1371/journal.pgen.1002214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaca E, Weitzer S, Pehlivan D, Shiraishi H, Gogakos T, Hanada T, Jhangiani SN, Wiszniewski W, Withers M, Campbell IM, et al. Human CLP1 mutations alter tRNA biogenesis, affecting both peripheral and central nervous system function. Cell. 2014;157:636–650. doi: 10.1016/j.cell.2014.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Han TW, Xie S, Shi K, Du X, Wu LC, Mirzaei H, Goldsmith EJ, Longgood J, Pei J, et al. Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell. 2012;149:753–767. doi: 10.1016/j.cell.2012.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Frank CL, Dobbin MM, Tsunemoto RK, Tu W, Peng PL, Guan JS, Lee BH, Moy LY, Giusti P, et al. Deregulation of HDAC1 by p25/Cdk5 in neurotoxicity. Neuron. 2008;60:803–817. doi: 10.1016/j.neuron.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B, Li S, Molliex A, et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. 2013;495:467–473. doi: 10.1038/nature11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornblihtt AR, de la Mata M, Fededa JP, Munoz MJ, Nogues G. Multiple links between transcription and splicing. Rna. 2004;10:1489–1498. doi: 10.1261/rna.7100104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda M, Sok J, Webb L, Baechtold H, Urano F, Yin Y, Chung P, de Rooij DG, Akhmedov A, Ashley T, et al. Male sterility and enhanced radiation sensitivity in TLS(-/-) mice. Embo J. 2000;19:453–462. doi: 10.1093/emboj/19.3.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski TJ, Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- Kwon I, Kato M, Xiang S, Wu L, Theodoropoulos P, Mirzaei H, Han T, Xie S, Corden JL, McKnight SL. Phosphorylation-regulated binding of RNA polymerase II to fibrous polymers of low-complexity domains. Cell. 2013;155:1049–1060. doi: 10.1016/j.cell.2013.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Polymenidou M, Hutt KR, Vu AQ, Baughn M, Huelga SC, Clutario KM, Ling SC, Liang TY, Mazur C, et al. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat Neurosci. 2012;15:1488–1497. doi: 10.1038/nn.3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanson NA, Jr, Maltare A, King H, Smith R, Kim JH, Taylor JP, Lloyd TE, Pandey UB. A Drosophila model of FUS-related neurodegeneration reveals genetic interaction between FUS and TDP-43. Hum Mol Genet. 2011;20:2510–2523. doi: 10.1093/hmg/ddr150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattante S, Rouleau GA, Kabashi E. TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: summary and update. Hum Mutat. 2013;34:812–826. doi: 10.1002/humu.22319. [DOI] [PubMed] [Google Scholar]
- Lee EB, Lee VM, Trojanowski JQ. Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat Rev Neurosci. 2012;13:38–50. doi: 10.1038/nrn3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerga A, Hallier M, Delva L, Orvain C, Gallais I, Marie J, Moreau-Gachelin F. Identification of an RNA binding specificity for the potential splicing factor TLS. J Biol Chem. 2001;276:6807–6816. doi: 10.1074/jbc.M008304200. [DOI] [PubMed] [Google Scholar]
- Li Q, Lee JA, Black DL. Neuronal regulation of alternative pre-mRNA splicing. Nat Rev Neurosci. 2007;8:819–831. doi: 10.1038/nrn2237. [DOI] [PubMed] [Google Scholar]
- Li YR, King OD, Shorter J, Gitler AD. Stress granules as crucibles of ALS pathogenesis. J Cell Biol. 2013;201:361–372. doi: 10.1083/jcb.201302044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79:416–438. doi: 10.1016/j.neuron.2013.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, Yankner BA. Gene regulation and DNA damage in the ageing human brain. Nature. 2004;429:883–891. doi: 10.1038/nature02661. [DOI] [PubMed] [Google Scholar]
- Lukas J, Lukas C, Bartek J. More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nature cell biology. 2011;13:1161–1169. doi: 10.1038/ncb2344. [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, Frick P, Neumann M. The neuropathology associated with repeat expansions in the C9ORF72 gene. Acta Neuropathol. 2014;127:347–357. doi: 10.1007/s00401-013-1232-4. [DOI] [PubMed] [Google Scholar]
- Martin KC, Ephrussi A. mRNA localization: gene expression in the spatial dimension. Cell. 2009;136:719–730. doi: 10.1016/j.cell.2009.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastrocola AS, Kim SH, Trinh AT, Rodenkirch LA, Tibbetts RS. The RNA-binding protein fused in sarcoma (FUS) functions downstream of poly(ADP-ribose) polymerase (PARP) in response to DNA damage. J Biol Chem. 2013;288:24731–24741. doi: 10.1074/jbc.M113.497974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinnon PJ. DNA repair deficiency and neurological disease. Nat Rev Neurosci. 2009;10:100–112. doi: 10.1038/nrn2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millecamps S, Boillee S, Le Ber I, Seilhean D, Teyssou E, Giraudeau M, Moigneu C, Vandenberghe N, Danel-Brunaud V, Corcia P, et al. Phenotype difference between ALS patients with expanded repeats in C9ORF72 and patients with mutations in other ALS-related genes. J Med Genet. 2012;49:258–263. doi: 10.1136/jmedgenet-2011-100699. [DOI] [PubMed] [Google Scholar]
- Miller KM, Tjeertes JV, Coates J, Legube G, Polo SE, Britton S, Jackson SP. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat Struct Mol Biol. 2010;17:1144–1151. doi: 10.1038/nsmb.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell JC, McGoldrick P, Vance C, Hortobagyi T, Sreedharan J, Rogelj B, Tudor EL, Smith BN, Klasen C, Miller CC, et al. Overexpression of human wild-type FUS causes progressive motor neuron degeneration in an age- and dose-dependent fashion. Acta neuropathologica. 2013;125:273–288. doi: 10.1007/s00401-012-1043-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ, Mittag T, Taylor JP. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell. 2015;163:123–133. doi: 10.1016/j.cell.2015.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MJ, Proudfoot NJ. Pre-mRNA processing reaches back to transcription and ahead to translation. Cell. 2009;136:688–700. doi: 10.1016/j.cell.2009.02.001. [DOI] [PubMed] [Google Scholar]
- Munoz MJ, Perez Santangelo MS, Paronetto MP, de la Mata M, Pelisch F, Boireau S, Glover-Cutter K, Ben-Dov C, Blaustein M, Lozano JJ, et al. DNA damage regulates alternative splicing through inhibition of RNA polymerase II elongation. Cell. 2009;137:708–720. doi: 10.1016/j.cell.2009.03.010. [DOI] [PubMed] [Google Scholar]
- Murakami T, Qamar S, Lin JQ, Schierle GS, Rees E, Miyashita A, Costa AR, Dodd RB, Chan FT, Michel CH, et al. ALS/FTD Mutation-Induced Phase Transition of FUS Liquid Droplets and Reversible Hydrogels into Irreversible Hydrogels Impairs RNP Granule Function. Neuron. 2015;88:678–690. doi: 10.1016/j.neuron.2015.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Newman JC, Bailey AD, Weiner AM. Cockayne syndrome group B protein (CSB) plays a general role in chromatin maintenance and remodeling. Proc Natl Acad Sci U S A. 2006;103:9613–9618. doi: 10.1073/pnas.0510909103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimoto Y, Nakagawa S, Hirose T, Okano HJ, Takao M, Shibata S, Suyama S, Kuwako K, Imai T, Murayama S, et al. The long non-coding RNA nuclear-enriched abundant transcript 1_2 induces paraspeckle formation in the motor neuron during the early phase of amyotrophic lateral sclerosis. Molecular brain. 2013;6:31. doi: 10.1186/1756-6606-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura T, Watanabe S, Kaneko K, Yamanaka K, Nukina N, Furukawa Y. Intranuclear aggregation of mutant FUS/TLS as a molecular pathomechanism of amyotrophic lateral sclerosis. J Biol Chem. 2014;289:1192–1202. doi: 10.1074/jbc.M113.516492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou SH, Wu F, Harrich D, Garcia-Martinez LF, Gaynor RB. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. Journal of virology. 1995;69:3584–3596. doi: 10.1128/jvi.69.6.3584-3596.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, Stoynov S, Mahamid J, Saha S, Franzmann TM, et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell. 2015;162:1066–1077. doi: 10.1016/j.cell.2015.07.047. [DOI] [PubMed] [Google Scholar]
- Pilch DR, Sedelnikova OA, Redon C, Celeste A, Nussenzweig A, Bonner WM. Characteristics of gamma-H2AX foci at DNA double-strand breaks sites. Biochemistry and cell biology = Biochimie et biologie cellulaire. 2003;81:123–129. doi: 10.1139/o03-042. [DOI] [PubMed] [Google Scholar]
- Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, Ling SC, Sun E, Wancewicz E, Mazur C, et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci. 2011;14:459–468. doi: 10.1038/nn.2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad DD, Ouchida M, Lee L, Rao VN, Reddy ES. TLS/FUS fusion domain of TLS/FUS-erg chimeric protein resulting from the t(16;21) chromosomal translocation in human myeloid leukemia functions as a transcriptional activation domain. Oncogene. 1994;9:3717–3729. [PubMed] [Google Scholar]
- Qiu H, Lee S, Shang Y, Wang WY, Au KF, Kamiya S, Barmada S, Finkbeiner S, Lui H, Carlton CE, et al. ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J Clin Invest. 2014;124 doi: 10.1172/JCI72723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabbitts TH, Forster A, Larson R, Nathan P. Fusion of the dominant negative transcription regulator CHOP with a novel gene FUS by translocation t(12;16) in malignant liposarcoma. Nat Genet. 1993;4:175–180. doi: 10.1038/ng0693-175. [DOI] [PubMed] [Google Scholar]
- Rabin SJ, Kim JM, Baughn M, Libby RT, Kim YJ, Fan Y, La Spada A, Stone B, Ravits J. Sporadic ALS has compartment-specific aberrant exon splicing and altered cell-matrix adhesion biology. Hum Mol Genet. 2010;19:313–328. doi: 10.1093/hmg/ddp498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rass U, Ahel I, West SC. Defective DNA repair and neurodegenerative disease. Cell. 2007;130:991–1004. doi: 10.1016/j.cell.2007.08.043. [DOI] [PubMed] [Google Scholar]
- Reed R, Hurt E. A conserved mRNA export machinery coupled to pre-mRNA splicing. Cell. 2002;108:523–531. doi: 10.1016/s0092-8674(02)00627-x. [DOI] [PubMed] [Google Scholar]
- Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robles E, Baier H. Assembly of synaptic laminae by axon guidance molecules. Curr Opin Neurobiol. 2012 doi: 10.1016/j.conb.2012.04.014. [DOI] [PubMed] [Google Scholar]
- Rogelj B, Easton LE, Bogu GK, Stanton LW, Rot G, Curk T, Zupan B, Sugimoto Y, Modic M, Haberman N, et al. Widespread binding of FUS along nascent RNA regulates alternative splicing in the brain. Sci Rep. 2012;2:603. doi: 10.1038/srep00603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rulten SL, Rotheray A, Green RL, Grundy GJ, Moore DA, Gomez-Herreros F, Hafezparast M, Caldecott KW. PARP-1 dependent recruitment of the amyotrophic lateral sclerosis-associated protein FUS/TLS to sites of oxidative DNA damage. Nucleic Acids Res. 2014;42:307–314. doi: 10.1093/nar/gkt835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatelli M, Moncada A, Conte A, Lattante S, Marangi G, Luigetti M, Lucchini M, Mirabella M, Romano A, Del Grande A, et al. Mutations in the 3′ untranslated region of FUS causing FUS overexpression are associated with amyotrophic lateral sclerosis. Hum Mol Genet. 2013;22:4748–4755. doi: 10.1093/hmg/ddt328. [DOI] [PubMed] [Google Scholar]
- Sanes JR, Yamagata M. Many paths to synaptic specificity. Annu Rev Cell Dev Biol. 2009;25:161–195. doi: 10.1146/annurev.cellbio.24.110707.175402. [DOI] [PubMed] [Google Scholar]
- Schaffer AE, Eggens VR, Caglayan AO, Reuter MS, Scott E, Coufal NG, Silhavy JL, Xue Y, Kayserili H, Yasuno K, et al. CLP1 founder mutation links tRNA splicing and maturation to cerebellar development and neurodegeneration. Cell. 2014;157:651–663. doi: 10.1016/j.cell.2014.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz JC, Ebmeier CC, Podell ER, Heimiller J, Taatjes DJ, Cech TR. FUS binds the CTD of RNA polymerase II and regulates its phosphorylation at Ser2. Genes Dev. 2012;26:2690–2695. doi: 10.1101/gad.204602.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz JC, Podell ER, Han SS, Berry JD, Eggan KC, Cech TR. FUS is sequestered in nuclear aggregates in ALS patient fibroblasts. Mol Biol Cell. 2014;25:2571–2578. doi: 10.1091/mbc.E14-05-1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz JC, Wang X, Podell ER, Cech TR. RNA seeds higher-order assembly of FUS protein. Cell reports. 2013;5:918–925. doi: 10.1016/j.celrep.2013.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sephton CF, Tang AA, Kulkarni A, West J, Brooks M, Stubblefield JJ, Liu Y, Zhang MQ, Green CB, Huber KM, et al. Activity-dependent FUS dysregulation disrupts synaptic homeostasis. Proc Natl Acad Sci U S A. 2014;111:E4769–4778. doi: 10.1073/pnas.1406162111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahidullah M, Le Marchand SJ, Fei H, Zhang J, Pandey UB, Dalva MB, Pasinelli P, Levitan IB. Defects in synapse structure and function precede motor neuron degeneration in Drosophila models of FUS-related ALS. J Neurosci. 2013;33:19590–19598. doi: 10.1523/JNEUROSCI.3396-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Lyashchenko AK, Lu L, Nasrabady SE, Elmaleh M, Mendelsohn M, Nemes A, Tapia JC, Mentis GZ, Shneider NA. ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nature communications. 2016;7:10465. doi: 10.1038/ncomms10465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelkovnikova TA, Peters OM, Deykin AV, Connor-Robson N, Robinson H, Ustyugov AA, Bachurin SO, Ermolkevich TG, Goldman IL, Sadchikova ER, et al. Fused in sarcoma (FUS) protein lacking nuclear localization signal (NLS) and major RNA binding motifs triggers proteinopathy and severe motor phenotype in transgenic mice. J Biol Chem. 2013;288:25266–25274. doi: 10.1074/jbc.M113.492017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolow DT, Haynes SR. Cabeza, a Drosophila gene encoding a novel RNA binding protein, shares homology with EWS and TLS, two genes involved in human sarcoma formation. Nucleic Acids Res. 1995;23:835–843. doi: 10.1093/nar/23.5.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Ling SC, Qiu J, Albuquerque CP, Zhou Y, Tokunaga S, Li H, Qiu H, Bui A, Yeo GW, et al. ALS-causative mutations in FUS/TLS confer gain and loss of function by altered association with SMN and U1-snRNP. Nature communications. 2015;6:6171. doi: 10.1038/ncomms7171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Diaz Z, Fang X, Hart MP, Chesi A, Shorter J, Gitler AD. Molecular determinants and genetic modifiers of aggregation and toxicity for the ALS disease protein FUS/TLS. PLoS Biol. 2011;9:e1000614. doi: 10.1371/journal.pbio.1000614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahama K, Takada A, Tada S, Shimizu M, Sayama K, Kurokawa R, Oyoshi T. Regulation of telomere length by G-quadruplex telomere DNA- and TERRA-binding protein TLS/FUS. Chem Biol. 2013;20:341–350. doi: 10.1016/j.chembiol.2013.02.013. [DOI] [PubMed] [Google Scholar]
- Tan AY, Manley JL. The TET family of proteins: functions and roles in disease. Journal of molecular cell biology. 2009;1:82–92. doi: 10.1093/jmcb/mjp025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testa D, Lovati R, Ferrarini M, Salmoiraghi F, Filippini G. Survival of 793 patients with amyotrophic lateral sclerosis diagnosed over a 28-year period. Amyotrophic lateral sclerosis and other motor neuron disorders : official publication of the World Federation of Neurology, Research Group on Motor Neuron Diseases. 2004;5:208–212. [PubMed] [Google Scholar]
- Tollervey JR, Curk T, Rogelj B, Briese M, Cereda M, Kayikci M, Konig J, Hortobagyi T, Nishimura AL, Zupunski V, et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci. 2011;14:452–458. doi: 10.1038/nn.2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuiji H, Iguchi Y, Furuya A, Kataoka A, Hatsuta H, Atsuta N, Tanaka F, Hashizume Y, Akatsu H, Murayama S, et al. Spliceosome integrity is defective in the motor neuron diseases ALS and SMA. EMBO molecular medicine. 2013;5:221–234. doi: 10.1002/emmm.201202303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbeeck C, Deng Q, Dejesus-Hernandez M, Taylor G, Ceballos-Diaz C, Kocerha J, Golde T, Das P, Rademakers R, Dickson DW, et al. Expression of Fused in sarcoma mutations in mice recapitulates the neuropathology of FUS proteinopathies and provides insight into disease pathogenesis. Mol Neurodegener. 2012;7:53. doi: 10.1186/1750-1326-7-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl MC, Will CL, Luhrmann R. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009;136:701–718. doi: 10.1016/j.cell.2009.02.009. [DOI] [PubMed] [Google Scholar]
- Wang JW, Brent JR, Tomlinson A, Shneider NA, McCabe BD. The ALS-associated proteins FUS and TDP-43 function together to affect Drosophila locomotion and life span. J Clin Invest. 2011;121:4118–4126. doi: 10.1172/JCI57883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WY, Pan L, Su SC, Quinn EJ, Sasaki M, Jimenez JC, Mackenzie IR, Huang EJ, Tsai LH. Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat Neurosci. 2013;16:1383–1391. doi: 10.1038/nn.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Arai S, Song X, Reichart D, Du K, Pascual G, Tempst P, Rosenfeld MG, Glass CK, Kurokawa R. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature. 2008;454:126–130. doi: 10.1038/nature06992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Schwartz JC, Cech TR. Nucleic acid-binding specificity of human FUS protein. Nucleic Acids Res. 2015;43:7535–7543. doi: 10.1093/nar/gkv679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki T, Chen S, Yu Y, Yan B, Haertlein TC, Carrasco MA, Tapia JC, Zhai B, Das R, Lalancette-Hebert M, et al. FUS-SMN protein interactions link the motor neuron diseases ALS and SMA. Cell reports. 2012;2:799–806. doi: 10.1016/j.celrep.2012.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Deng HX, Siddique N, Fecto F, Chen W, Yang Y, Liu E, Donkervoort S, Zheng JG, Shi Y, et al. Frameshift and novel mutations in FUS in familial amyotrophic lateral sclerosis and ALS/dementia. Neurology. 2010;75:807–814. doi: 10.1212/WNL.0b013e3181f07e0c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Embree LJ, Tsai S, Hickstein DD. Oncoprotein TLS interacts with serine-arginine proteins involved in RNA splicing. J Biol Chem. 1998;273:27761–27764. doi: 10.1074/jbc.273.43.27761. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Liu S, Ozturk A, Hicks GG. FUS-regulated RNA metabolism and DNA damage repair: Implications for amyotrophic lateral sclerosis and frontotemporal dementia pathogenesis. Rare diseases. 2014;2:e29515. doi: 10.4161/rdis.29515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinszner H, Albalat R, Ron D. A novel effector domain from the RNA-binding protein TLS or EWS is required for oncogenic transformation by CHOP. Genes Dev. 1994;8:2513–2526. doi: 10.1101/gad.8.21.2513. [DOI] [PubMed] [Google Scholar]
- Zinszner H, Sok J, Immanuel D, Yin Y, Ron D. TLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling. J Cell Sci. 1997;110(Pt 15):1741–1750. doi: 10.1242/jcs.110.15.1741. [DOI] [PubMed] [Google Scholar]