

Abstract
The IgG4 subclass of antibodies exhibits unique characteristics that suggest it may function in an immunoregulatory capacity. The inhibitory function of IgG4 has been well documented in allergic disease by the demonstration of IgG4 blocking antibodies, but similar functions have not been explored in autoimmune disease. Bullous pemphigoid (BP) is a subepidermal autoimmune blistering disease characterized by autoantibodies directed against BP180 and an inflammatory infiltrate including eosinophils and neutrophils. Animal models have revealed that the NC16A region within BP180 harbors the critical epitopes necessary for autoantibody mediated disease induction. BP180 NC16A-specific IgG belong to the IgG1, IgG3, and IgG4 subclasses. The purpose of this study was to determine effector functions of different IgG subclasses of NC16A-specific autoantibodies in BP. We find that IgG4 anti-NC16A autoantibodies inhibit the binding of IgG1 and IgG3 autoantibodies to the NC16A region. Moreover, IgG4 anti-NC16A blocks IgG1 and IgG3 induced complement fixation, neutrophil infiltration, and blister formation clinically and histologically in a dose-dependent manner following passive transfer to humanized BP180-NC16A mice. These findings highlight the inhibitory role of IgG4 in autoimmune disease and have important implications for the treatment of BP as well as other antibody mediated inflammatory and autoimmune diseases.
Keywords: IgG4, bullous pemphigoid, inhibitory, autoimmune
1. INTRODUCTION
IgG4 is unique among IgG subclasses as it lacks the ability to fix complement and does not effectively bind Fcγ receptors II or III [1–3]. Additionally, due to specific amino acid differences in the Fc region, IgG4 is also able to participate in half molecule (or Fab arm) exchange, which results in asymmetric and functionally monovalent antibodies [4–6]. These features have led to the premise that IgG4 acts in an immunoregulatory role. Indeed, while IgG4 is present in relatively low amounts in the sera of healthy individuals, IgG4 levels increase upon repeat antigen exposure [7]. In fact, increases in IgG4 levels can be used as a marker of successful immunotherapy in patients with allergies, where IgG4 antibodies are felt to dampen the IgE response by acting as blocking antibodies [8–10].
Despite the accumulating evidence that IgG4 functions in a regulatory capacity, there are many diseases in which IgG4 is pathogenic. It is well accepted that the pathogenic autoantibodies in the intraepidermal blistering diseases pemphigus vulgaris and pemphigus foliaceus are predominantly of the IgG4 subclass [11, 12]. IgG4 antibodies have also been reported to play a role in the pathogenesis of membranous glomerulonephritis and thrombotic thrombocytopenic purpura [13–15]. In addition, diseases in which IgG4 plasma cells are involved are becoming more frequently recognized and reported [16–18]. These diseases, collectively termed IgG4-related diseases, can manifest in multiple organ systems exhibiting a wide range of clinical presentations, but are generally characterized by fibroinflammatory changes, IgG4 plasma cell infiltrate, and mild tissue eosinophilia with variable elevation in serum IgG4 levels [19, 20]. Whereas in pemphigus, the IgG4 anti-desmoglein 1 and 3 autoantibodies are directly pathogenic to the epidermis, impairing the cell adhesion mechanism without the intervention of complement or inflammatory cells [11, 12], the exact role of IgG4 antibodies in other IgG4 mediated and IgG4-related diseases is not well understood. Moreover, while the inhibitory capacity of IgG4 has been well demonstrated in the setting of allergy, the potential regulatory role of IgG4 antibodies in inflammatory and antibody mediated autoimmune diseases had not been investigated.
Bullous pemphigoid (BP) is an autoantibody mediated autoimmune blistering disease of the skin characterized clinically by tense bullae formation and histologically by subepidermal clefting and dermal infiltration of eosinophils and neutrophils [21]. Direct immunofluorescence studies show IgG and complement deposition along the dermal epidermal junction [22, 23]. The disease is caused by autoantibodies directed against BP180 (also termed type XVII collagen), a transmembrane glycoprotein that is located in the hemidesmosome and critical for adhesion of the basal keratinocytes to the dermis [24–26]. Previous studies have shown that the noncollagenous 16A (NC16A) region of the BP180 ectodomain contains the major epitopes recognized by autoantibodies from BP sera [27, 28]. As the NC16A domain is poorly conserved between humans and mice, the development of the humanized NC16A mice containing the human NC16A domain has allowed for a more precise understanding of how autoantibodies from BP patients induce disease [29, 30]. Passive transfer of BP IgG is pathogenic in NC16A mice, but not wildtype mice, confirming the importance of the NC16A domain in pathogenicity of disease [30, 31].
IgG anti-NC16A autoantibodies are predominantly of the IgG1 and IgG4 subclasses [32, 33]. While many aspects of BP immunopathology are well understood, the role of different IgG subclasses remains unclear. The aim of this study was to determine the role of IgG anti-NC16A subclasses in the pathogenesis of BP using the humanized NC16A mouse model. We show that IgG4 anti-NC16A autoantibodies inhibit the binding of anti-NC16A IgG1,3 to the cutaneous basement membrane zone (BMZ), block complement fixation and neutrophil infiltration, and prevent blister formation, clinically and histologically, in a dose dependent manner following passive transfer to NC16A mice. These findings have important implications for the treatment of BP as well as other antibody mediated inflammatory and autoimmune diseases as well.
2. MATERIALS AND METHODS
2.1 Patients, sera, and antibody purification
Serum samples were collected from three patients with active BP (BP1, BP2, and BP3). NC16A-specific total IgG were purified from BP patient sera using a protein G column, followed by an NC16A-specific glutathione sepharose column as described [30]. NC16A-specific IgG1,3 and 4 were purified from NC16A-specific total IgG using human IgG4-specific columns (Amersham Biosciences). The concentration and purity of NC16A-specific IgG4 (eluate) and NC16A-specific IgG1 and 3 (flow through) were determined by human IgG subclass-specific ELISA (Southern Biotechology). The purity of NC16A-specific IgG1,3 and IgG4 were 95% and 98%, respectively. Purified IgG and IgG subclass fractions were concentrated by ultrafiltration (Millipore) and used for in vitro and in vivo experiments.
2.2 In vitro inhibition of IgG binding and C′ activation at the basement membrane zone
Anti-NC16A IgG1,3 (1 μg/ml) were incubated with normal human IgG4 or NC16A-specific IgG4 (0.25 μg/ml) and then incubated with skin sections of NC16A mice. Binding of anti-NC16A IgG1 and 3 to the BMZ was detected by FITC-conjugated anti-human IgG1 and IgG3. For the in vitro C′ fixation assay, anti-NC16A IgG1,3 were incubated with normal human IgG4 or NC16A-specific IgG4 and then incubated with skin sections of NC16A mice in the presence of freshly prepared human serum or commercial rabbit serum as a complement source. The complement C3 deposition at the BMZ was detected by FITC-conjugated anti-human or anti-rabbit C3 (Cappel Laboratories).
2.3 In vitro complement fixation assay
Antibody-sensitized sheep erythrocytes (EA), human serum complement, and GVB++ buffer were purchased from Complement Technology (Tyler, TX). The ability of NC16A-specific IgG1,3 and IgG4 to fix complement was determined by following the protocol by the company and as described [34, 35] with some modifications. Briefly, EA (1×108 cells/ml), complement (1:400 dilution), and NC16A-specific IgG (100 μg/ml) were incubated in the presence of GST-NC16A fusion protein (0–100 μg/ml) at 37°C for 1 h. After centrifugation, the absorbance of the supernatant was determined at 540 nm by a microtiter plate reader. The complement fixation activity was expressed as percent of sheep erythrocyte lysis inhibition.
2.4 Flow cytometry
Mouse neutrophils were isolated as described [36]. 1×106 neutrophils were incubated with anti-NC16A IgG (0–10 μg/ml) and goat anti-human IgG-FITC conjugate. FITC positive cells were quantified by flow cytrometry.
2.5 Mice and antibody passive transfer
The humanized NC16A mice were generated as described [30]. A 50 μl aliquot of sterile IgG (control human IgG or affinity-purified NC16A-specific IgG) in PBS was administered to neonatal mice by intradermal or intraperitoneal injection [30]. The mouse skin was examined 24 h after injection. The extent of cutaneous disease (clinical disease activity) was scored as follows: “(−)”, no detectable skin disease; “1+”, mild erythematous reaction with no evidence of the “epidermal detachment” sign; “2+”, intense erythema and “epidermal detachment” sign involving 10–50% of the epidermis in localized areas; and “3+”, intense erythema with frank “epidermal detachment” sign involving more than 50% of the epidermis. After clinical examination, the animals were sacrificed, and skin and serum specimens were obtained. The skin sections were used for routine histological examination by light microscopy (H/E staining) to localize the lesional site and neutrophil infiltration. Direct immunofluorescence assays were done to detect deposition of human IgG and mouse C3 at the basement membrane zone using commercially available FITC-conjugated goat anti-human and monospecific goat anti-mouse C3 IgG (Cappel Laboratories). Myeloperoxidase (MPO) enzymatic assay was performed to quantify the neutrophil accumulation at the skin injection site as described below.
2.6 In vivo inhibition of NC16A-specific IgG1,3-induced BP
Neonatal NC16A mice were injected intradermally with anti-NC16A IgG1,3 in the presence of normal human IgG4 control or different ratios of anti-NC16A IgG4. The mice were examined clinically for disease activity 24 hours post injection.
2.7 Quantification of infiltrating neutrophils
Infiltrating neutrophils in the antibody-injected skin were quantified by measuring tissue MPO activity as described [30, 37, 38] using purified MPO as standard. MPO content was expressed as relative MPO activity (OD460nm reading/mg protein of the mouse skin injected with pathogenic anti-mBP180 IgG minus OD460nm reading/mg protein of the mouse skin injected with control IgG). Protein concentrations were determined by the Bio-Rad dye-binding assay using BSA as a standard.
2.8 Quantification of C5a levels
Skin from the IgG injection site of the diseased and control mice was homogenized in PBS to extract proteins. The level of mouse C5a (mC5a) in the skin was measured by ELISA (R&D Systems, Minneapolis, MN) [39]. Microtiter plates were coated with a rat anti-mouse C5a antibody, incubated with skin protein extracts, followed by goat anti-mC5a detection antibody, and then developed and read at OD492nm. The C5a level was expressed as OD492 reading/mg protein.
2.9 Statistics
The data are expressed as mean ± SEM and were analyzed using the Student’s t-test. A p value less than 0.05 is considered significant.
2.10 Study approval
Animal care and animal experiments were in accordance with the Animal Care Committee at the University of North Carolina-Chapel Hill. Written informed consent was received from participants prior to inclusion in the study.
3. RESULTS
3.1 IgG1, IgG3, and IgG4 are the dominant IgG subclasses among NC16A-specific autoantibodies
To determine the concentration of IgG subclasses among NC16A-specific autoantibodies, anti-NC16A specific total IgG was purified from the sera of three BP patients by NC16A affinity purification. Levels of anti-NC16A IgG1, IgG2, IgG3, and IgG4 were determined by quantitative human subclass ELISA on the purified NC16A specific fraction. As shown in Figure 1, IgG1 is the dominant IgG subclass among NC16A-specific autoantibodies ranging from 0.55–1.41 mg/ml serum. Lower levels of IgG3 and IgG4 anti-NC16A autoantibodies are detected in all three patients as well. IgG2 is detected at very low levels in BP1 and is below the level of detection in BP2 and BP3.
Figure 1. Levels of anti-NC16A IgG subclasses.

NC16A-specific total IgG from three BP sera were purified using protein A/G column, followed by NC16A-specific affinity column. Levels of anti-NC16A IgG1, G2, G3, and G4 were quantified by human IgG1-, G2-, G3- and G4-specific ELISA. IgG1, 3, and 4 are dominant IgG subclasses in 3 BP sera and IgG2 were below detection limit in BP2 and BP3. Data are expressed as mg IgG/ml serum.
3.2 IgG anti-NC16A induced clinical blistering in NC16A mice is dose dependent
Our group has previously shown that IgG anti-NC16A autoantibodies are pathogenic upon passive transfer to NC16A mice [30]. In an effort to determine the optimal dose necessary to induce blister formation, we conducted a dose titration study using anti-NC16A total IgG derived from each of three BP sera (BP1, BP2, and BP3). Neonatal NC16A mice were injected with different amounts of purified anti-NC16A total IgG ranging from 100 to 300 ug/g of body weight. The mice were examined for clinical signs of disease at 24 hours post injection. Mice injected with 300 (Figure 2A, panels a, b) and 200 (Figure 2A, panels c, d) but not 100 ug/g of body weight (Figure 2A, panels e, f) anti-NC16A IgG develop clinical and histological subepidermal blisters. Anti-NC16A IgG induces clinical blistering in a dose-dependent fashion as shown in Figure 2B with optimal dosing of 300 ug anti-NC16A IgG/g body weight for BP1 and 3 and 200 ug anti-NC16A IgG/g body weight for BP2. Optimal dosing is defined as the dose necessary to reach a clinical disease score of approximately 2.5.
Figure 2. Anti-NC16A IgG induced dose-dependent BP disease in NC16A mice.

Neonatal NC16A mice were injected i.d. with different amounts of anti-NC16A total IgG (100–300 μg/g body weight) and examined clinically for disease activity 24 hours post injection. A. Mice injected with 300 μg/g body weight (panels a, b) and 200 μg/g body weight (panels c, d) but not 100 μg/g body weight (panels e, f) developed clinical and histological subepidermal blister. D, dermis; E, epidermis; V, vesicle. Arrow, basal keratinocytes. B. Anti-NC16A IgG induced BP in a dose-dependent fashion. p<0.05, n=6 for each group.
3.3 IgG4 anti-NC16A inhibits binding and complement fixation of anti-NC16A IgG1,3 at the basement membrane zone in vitro
While previous studies have shown that anti-NC16A total IgG can bind the basement membrane zone (BMZ) and fix complement on NC16A murine skin in vitro [30], we wanted to investigate possible differences between the anti-NC16A subclasses in terms of BMZ binding and complement fixation. IgG4 anti-NC16A autoantibodies were purified from anti-NC16A total IgG using a human IgG4 affinity column, resulting in an anti-NC16A IgG1,3 flow through fraction and an anti-NC16A IgG4 eluate fraction. Skin sections of NC16A mice were incubated with anti-NC16A IgG1,3, IgG4, or both IgG1,3 and IgG4. As shown in Figure 3A, IgG1,3 and IgG4 are both independently able to bind the BMZ in NC16A murine skin (Figure 3A, panels a and b). Pre- or co-incubation of IgG4 anti-NC16A, but not control IgG4 reduces binding of anti-NC16A IgG1,3 at the BMZ as detected by FITC-conjugated IgG1 specific secondary antibody (Figure 3A, panels c and d) or FITC-conjugated IgG3 specific secondary antibody (data not shown).
Figure 3. Anti-NC16A IgG4 inhibit binding and C.
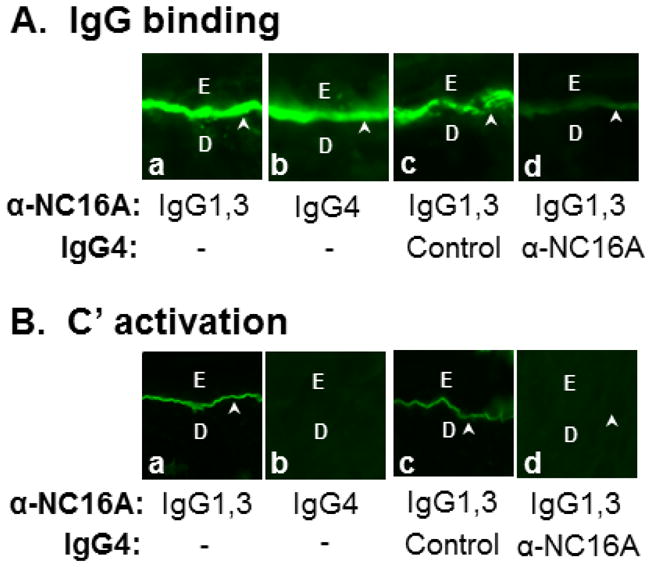
′ fixation of anti-NC16A IgG1,3 at the BMZ in vitro.
Skin sections of NC16A mice were incubated with anti-NC16A IgG1,3 (1 μg/ml), IgG4 (0.25 μg/ml), or both IgG1,3 and IgG4 in the absence or presence of freshly prepared human serum (providing complete complement system). A. IgG binding. Anti-NC16A igG1,3, and 4 stained the BMZ. Pre- or co-incubation of anti-NC16A IgG4 but not control IgG4 with anti-NC16A IgG1,3 drastically reduced binding of anti-NC16A IgG1,3 at the BMZ as detected by FITC-conjugated IgG1-spefici secondary antibody (panel d). B. C′ fixation. In the presence of human serum, anti-NC16A IgG1,3 and not IgG4 fixed complement as determined by C3 deposition at the BMZ using FITC-conjugated human C3-specific secondary antibody. Pre- or co-incubation of anti-NC16A IgG4 but not control IgG4 with anti-NC16A IgG1,3 significantly inhibited C′ fixation by anti-NC16A IgG1,3 (panel d).
Anti-NC16A IgG1,3 is able to fix complement as determined by C3 deposition at the BMZ, while IgG4 anti-NC16A is not (Figure 3B, panels a and b). Pre- or co-incubation of IgG4 anti-NC16A, but not control IgG4 significantly inhibits complement fixation by anti-NC16A IgG1,3 (Figure 3B, panels c and d).
3.4 IgG4 anti-NC16A inhibits anti-NC16A IgG1,3 induced BP in vivo in a dose dependent manner
Knowing that IgG4 anti-NC16A inhibits binding and C′ fixation of anti-NC16A IgG1,3 in vitro, we then tested whether anti-NC16A IgG4 were inhibitory in vivo. Neonatal NC16A mice were injected intradermally with BP1-derived anti-NC16A IgG1,3 (300ug/g body weight) in the presence of control IgG4 or BP1-derived anti-NC16A IgG4 at a ratio of 10:2.5 (anti-NC16A IgG1,3:IgG4). The neonates were examined 24 hours post injection. As shown in Figure 4A anti-NC16A IgG1,3 binds to the BMZ (Figure 4A, panel b and c), fixes complement (Figure 4A, panel d), and induces clinical and histologic blistering in vivo (Figure 4A, panels a and e) in the presence of control IgG4. In contrast, the presence of anti-NC16A IgG4 inhibits IgG1 binding (Figure 4A, panel h) and complement fixation (Figure 4A, panel i) and blocks clinical and histologic blister formation in vivo (Figure 4A, panels f and j) although mouse skin showed IgG (due to IgG4) deposition at the BMZ (Figure 4A, panel g).
Figure 4. Anti-NC16A IgG4 inhibit BP in vivo.

Neonatal NC16A mice were injected i.d. with BP1-derived anti-NC16A IgG1,3 (300 μg/g body weight) in the presence of normal human IgG4 control or anti-NC16A IgG4 (10:0 – 10:5 ratio IgG1,3 vs. IgG4) and examined 24 h post injection. A. IgG passive transfer. Anti-NC16A IgG1,3 bind to the BMZ (panels b, c), activate C′ as evidenced by C3 deposition at the BMZ (panel d) and induced BP blistering clinically (panel a) and histologically (panel e). In contrast, anti-NC16A IgG4 bind to the BMZ (panel g) and blocked anti-NC16A IgG1 binding (panel h), C′ activation (panel i) and BP blistering (panel j). IgG1,3 vs. IgG4 ratio was 10:2.5. B. Inhibition of anti-NC16A IgG1,3-induced disease activity (clinical disease score), C′ activation (skin C5a levels) and neutrophil infiltration (skin tissue MPO enzymatic activity) by anti-NC16A IgG4 were dose dependent. The inhibition was expressed as % change. n=8.
Using different ratios of BP1-derived anti-NC16A IgG1,3 to IgG4 (10:0 – 10:5), we further demonstrated that anti-NC16A IgG4 inhibits anti-NC16A IgG1,3-induced C′ activation (as measured by levels of C5a in the skin), neutrophil infiltration (as measured by skin MPO activity) and clinical disease severity (clinical disease score) in a dose dependent fashion (Figure 4B).
3.5 Anti-NC16A IgG4 inhibits anti-NC16A IgG1,3 induced BP from multiple BP sera in vivo
Although BP1 derived anti-NC16A IgG4 was able to block BP1-derived anti-NC16A IgG1,3 induced BP in vivo, we wanted to determine if anti-NC16A IgG4 could block anti-NC16A IgG1,3 induced BP using anti-NC16A IgG1,3 derived from multiple BP sera. Neonatal NC16A mice were injected intradermally with anti-NC16A IgG1,3 (300ug/g body weight) from BP1, BP2, and BP3 in the presence of control IgG4 or anti-NC16A IgG4 from BP1 at a ratio of 10:5 and examined at 24 hours post injection. The presence of BP1 anti-NC16A IgG4 inhibits clinical blister formation (Figure 5A and 5B), complement fixation and activation (Figure 5C and 5D), and neutrophil infiltration (Figure 5E) induced by anti-NC16A IgG1,3 from BP1, BP2, and BP3.
Figure 5. Anti-NC16A IgG4 inhibit BP in vivo.

Neonatal NC16A mice were injected i.d. with anti-NC16A IgG1,3 (300 μg/g body weight) from BP1, BP2, and BP3 in the presence of normal human IgG4 control or anti-NC16A IgG4 from BP1 (10:5 ratio IgG1,3 vs. IgG4) and examined 24 h post injection. A. IgG passive transfer. Anti-NC16A IgG4 and not control IgG (panel a) abolished BP clinical blistering induced by anti-NC16A IgG1,3 (panels b–d). B. Anti-NC16A IgG4 and not control IgG significantly inhibited BP disease activity (disease score) induced by anti-NC16A IgG1,3. C. Direct IF. Anti-NC16A IgG4 and not control IgG (panel a) blocked anti-NC16A IgG1,3-induced C3 deposition at the BMZ (panels b–d). Anti-NC16A IgG4 and not control IgG also significantly inhibited activation (skin C5a level, D) and neutrophil infiltration (skin MPO activity, E) caused by anti-NC16A IgG1,3 from three BP patients. *p<0.05, n=8 for BP1, n=6 for BP2 and BP3.
3.6 Anti-NC16A IgG4 bind to Fcγ receptors and induce BP in C5a reconstituted NC16A mice
Anti-NC16A IgG4 inhibit complement fixation activity of anti-NC16A IgG1,3 in the skin tissue assay (Figure 3) and anti-NC16A IgG1,3-mediated complement fixation in vivo (Figures 4, 5). This inhibition activity is most likely due to competition for IgG binding (occupying) site(s) on the NC16A domain between anti-NC16A IgG1,3 and IgG4. To further explore this possibility, we determined whether anti-NC16A IgG4 are capable of blocking anti-NC16A IgG1,3 complement fixing activity in vitro using a liquid phase sheep red blood cell lysis assay. As expected, immune complexes of NC16A and NC16A IgG1,3 but not IgG4 fix complement (Figure 6A). Further, when mixing NC16A, anti-NC16A IgG1,3 and IgG4 together, the complement fixation activity of anti-NC16A IgG1,3 was drastically reduced in a IgG4 dose dependent fashion (Figure 6B). In experimental BP, binding of anti-BP180 IgG Fc to IgG FcγRs (especially FcγRIII) is required for activating infiltrating neutrophils to release tissue injury proteolytic enzymes [40]. To rule out the possibility that anti-NC16A IgG1,3 and IgG4 are different in their ability to bind FcγRs, we incubated anti-NC16A IgG1,3 and IgG4 with purified mouse neutrophils and compared their FcγR binding by flow cytometry. Anti-NC16A IgG1,3 and IgG4 at 1, 2.5 and 10 μg/ml concentrations show comparable binding to neutrophil FcγRs (Figure 7A, 7B).
Figure 6. In vitro complement fixation of anti-NC16A IgG1,3 and IgG4.
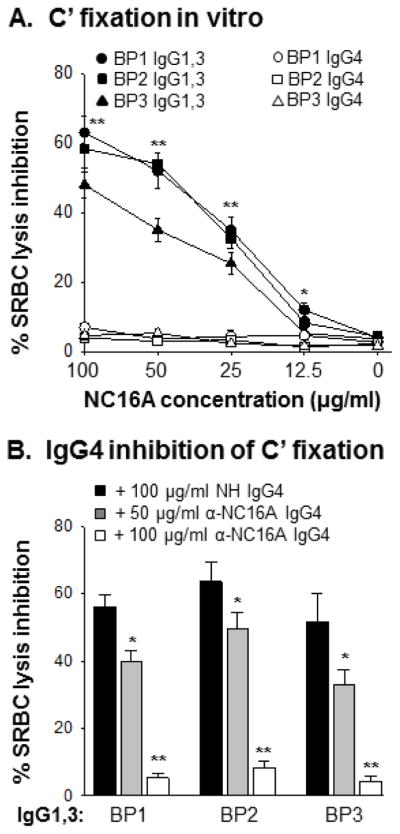
Antibody-sensitized sheep erythrocytes were incubated with complement, GST-NC16A fusion protein (0–100 μg/ml), anti-NC16A IgG1,3 or IgG4 (100 μg/ml) or both anti-NC16A IgG1,3 and IgG4. After 1 h incubation, lysis of sheep erythrocytes was quantified by reading OD540nm of the supernatant. Complement-fixing activity of anti-NC16A IgG was expressed as percent of inhibition of cell lysis. A. Complement fixation. Anti-NC16A IgG1,3 and not IgG4 fixed complement. B. Inhibition of C′ fixing activity of anti-NC16A IgG1,3 by IgG4. Anti-NC16A IgG4 inhibited C′ fixing activity of anti-NC16A IgG1,3 in a dose-dependent fashion. n=3, *p<0.05, **p<0.01, IgG1,3 vs. IgG4.
Figure 7. Anti-NC16A IgG4 bind to FcγRs on neutrophils and induce BP in C5a reconstituted NC16A mice.

Mouse neutrophils (1×106) were stained with anti-NC16A IgG1,3 or IgG4 from BP1. IgG bound neutrophils were quantified by flow cytometry using goat-anti-human IgG FITC conjugates. Anti-NC16A IgG1,3 and IgG4 exhibited comparable binding to FcγRs on mouse neutrophils (A) at 1–10 μg/ml concentrations (B). In vivo, anti-NC16A IgG4 induced BP blister in mC5a locally reconstituted NC16A mice (C). Clinical disease activity (D) and skin neutrophil infiltration (E) in the mC5a reconstituted NC16A mice were mC5a-dependent. Similar results were seen using BP2 and BP3 anti-NC16A IgG1,3 and IgG4 (data not shown). n=6, p<0.01, mC5a vs. BSA groups.
The finding that anti-NC16A IgG4 lack complement fixing activity, but are capable of binding to FcγRs lead us to speculate that non-pathogenic anti-NC16A IgG4 should induce BP in mice when C5a is provided locally. As shown in Figure 7, NC16A mice were injected locally with anti-NC16A IgG4 plus mC5a or BSA control. Both groups of mice show IgG4, but not C3 deposition at the BMZ by direct IF; however, only mice reconstituted with mC5a developed typical BP skin lesions (Figure 7C). Clinical disease activity (Figure 7D) and neutrophil infiltration (Figure 7E) in the diseased mice were mC5a dose-dependent. Thus, once neutrophils are recruited by exogenous mC5a, IgG4 anti-NC16A antibodies are able to activate neutrophils via FcR binding initiating the disease cascade.
4. DISCUSSION
IgG4 and its contribution to the pathogenesis of disease have gained significant attention over the past several years across a number of fields including inflammatory disease, allergy, and autoimmunity. The unique properties of IgG4 suggest that it may function to regulate immune responses mediated by other IgG subclasses. While the inhibitory capacity of IgG4 has been well demonstrated in the field of allergy, the potential regulatory role of IgG4 in inflammatory and antibody mediated autoimmune diseases had not been investigated. Using BP as a model of autoantibody mediated autoimmune disease, we clearly demonstrate that IgG4 can function in an inhibitory capacity in autoantibody mediated autoimmune disease. To our knowledge, this study is the first to show inhibition as a major effector function of IgG4 in autoimmunity.
BP provides an excellent model for exploring the inhibitory role of IgG4 in autoantibody mediated autoimmune disease. Studies using animal models of BP have demonstrated that the pathogenic mechanisms of autoantibody mediated blister formation in BP are complex and involve a multi-step process that includes autoantibody binding, complement fixation and activation, inflammatory cell infiltration, BMZ degradation, and ultimately subepidermal clefting that results in clinical disease. Earlier studies using total IgG from BP patients have shown that IgG mediated blister formation is complement dependent. Subsequent studies using monoclonal antibodies have raised the possibility that some autoantibodies are capable of inducing blister formation in the absence of complement [41, 42]. A recent report describes two cases of BP with dominant IgG4 and without C3 deposition at the BMZ, indicating that blister formation could be complement-independent in some BP cases [43]. However, it is well documented that the majority of human BP skin (>90%) have complement (C3 deposition), and less than 10% of BP patients are C3-negative at the basement membrane zone by direct IF [44, 45]. Here, we show using highly purified IgG subclass fractions derived from BP patients with more classic or typical BP that NC16A specific IgG1,3 are able to bind the BMZ, fix and activate complement, and induce blister formation in vivo. While anti-NC16A IgG4 is able to bind the BMZ, it is unable to fix complement and does not induce blister formation. Furthermore, co- or pre-incubation of IgG1,3 with IgG4 inhibits complement fixation, activation, neutrophil infiltration and blister formation.
There are several possible mechanisms by which IgG4 anti-NC16A antibodies inhibit BP. The most obvious explanation is that IgG4 anti-NC16A binds NC16A, thereby blocking IgG1,3 NC16A specific antibodies from binding the same region. This mechanism is strongly supported by our studies as the pre- or co-incubation of IgG4 anti-NC16A blocks IgG1,3 anti-NC16A binding in vitro and in vivo by immunofluorescence (Figure 3A panel d and Figure 4A panel h).
However, the more fundamental and underlying question is why IgG4 anti-NC16A antibodies bind the pathogenic NC16A domain, but do not induce disease. It is known that IgG4 lacks the ability to fix complement [2, 3] and our current studies corroborate that this finding extends to IgG4 anti-NC16A antibodies. Another function of IgG4 that may play a role in BP pathogenesis or lack thereof is the ability of IgG4 to bind FcγRs. While older studies have suggested that IgG4 is not able to bind FcγRs, more recent studies suggest that human IgG4 indeed bind FcγRs [1–3, 46]. In this study we demonstrate that IgG1,3 and IgG4 anti-NC16A antibodies show comparable binding to neutrophil FcγRs (Figure 7A, 7B). These features may explain why IgG4 anti-NC16A antibodies are not directly pathogenic as both complement activation for recruiting neutrophils and FcR binding for activating infiltrating neutrophils are pivitol in experimental BP [30, 39, 40]. Supporting this theory, IgG4 anti-NC16A IgG4 induce BP in mice when C5a is provided locally to induce neutrophil infiltration (Figure 7C). Clinical disease activity (Figure 7D) and neutrophil infiltration (Figure 7E) in the diseased mice were mC5a dose-dependent. Taken together, these results clearly demonstrate that anti-NC16A IgG4 bind to NC16A domain and block or reduce pathogenic anti-NC16A IgG1,3 binding to the same or nearby sites. This blockade results in reduced anti-NC16A IgG1,3-mediated complement activation and subsequent neutrophil infiltration and BP blistering.
While this study did not set out to address the requirement of complement in the pathophysiology of BP, the results suggest that complement is a critical part of the pathogenesis of blister formation. There are many factors that may account for the seemingly disparate findings between this and studies from other groups. First of all, differences in autoantibody source and composition may have a significant impact on both complement and pathogenesis studies. The clinical relevance of our results are strengthened by the use of highly purified, yet oligoclonal anti-NC16A IgG1,3 and IgG4 fractions, which are specific to the critical NC16A domain, but still inclusive of anti-NC16A autoantibodies to multiple epitopes within the NC16A region. This approach eliminates the possible misinterpretation of results that can occur when using monoclonal antibodies against a single epitope on NC16A, which may not be fully representative of the true oligoclonal anti-NC16A response. It is possible that patient sera have varying ratios of IgG1,3:IgG4. When total IgG is used, it is conceivable that some sera would be predominantly IgG1,3 and therefore able to fix complement and induce blisters, whereas other sera would have more significant levels of IgG4 that would block complement fixation and blister formation. For example, anti-NC16A IgG4 inhibition of anti-NC16A IgG1,3 induced disease is most significant when it is present at ratios of 10:5 or 10:2.5 (anti-NC16A IgG1,3:IgG4). As determined in Figure 1, the naturally occurring ratio of anti-NC16A IgG1:IgG4 in BP patient sera is approximately 10:1. At this ratio, the inhibitory capacity of anti-NC16A IgG4 is less remarkable (see Figure 4B). Secondly, differences in autoantibody dosing may influence results. In this study, dose finding studies were performed to avoid possible confounding results that can be seen with supraphysiologic dosing that may not be relevant clinically. Finally, another possible explanation that cannot be ruled out is the role of epitope specificity in pathogenesis. It is feasible that while the majority of the NC16A specific IgG4 binds an epitope that enables it to function in an inhibitory manner, a small pool of the IgG4 fraction may bind an epitope capable of inducing disease. Further studies are needed to explore this intriguing possibility.
In conclusion, our findings clearly show that anti-NC16A subclasses differ in their ability to induce disease upon passive transfer, and, moreover, that IgG4 anti-NC16A has a primary function of blocking the pathogenic effects of anti-NC16A IgG1,3 in vivo. These results highlight the potential role for IgG4 anti-NC16A blocking autoantibodies as a useful strategy for both the monitoring and treatment of BP. For example, it is possible that an increase in IgG4 anti-NC16A antibodies may serve as a biomarker for patients that are entering into long term remission. As such, the presence of IgG4 anti-NC16A antibodies may predict better outcomes upon reduction of immunosuppressive medications. On the other hand, administration of IgG4 anti-NC16A antibodies may represent a new treatment strategy in patients with suboptimal ratios of IgG1,3:1gG4 NC16A specific antibodies. These possibilities need to be more thoroughly explored, but highlight the translational aspect of our findings. More importantly, these findings may ultimately have implications for the monitoring and treatment of other autoantibody mediated diseases as well.
HIGHLIGHTS.
Autoantibodies in bullous pemphigoid belong to the IgG1, IgG3, and IgG4 subclasses
Patient derived antigen specific IgG1/3 induce disease in vivo
Patient derived antigen specific IgG4 inhibits IgG1/3 induced disease in vivo
Inhibitory IgG4 may be useful for treating bullous pemphigoid
Acknowledgments
The authors acknowledge Dr. Daniel Zedek for his assistance with histology. This study was supported by NIH grants AI07924 and AI40768 (ZL), AR06372 (NL), and AR32599 (LAD) and the Dermatology Foundation (DAC).
Abbreviations
- BP
bullous pemphigoid
- BMZ
basement membrane zone
- NC16A
noncollagenous 16A
- MPO
myeloperoxidase
- C′
complement
Footnotes
Conflict of interest statement: The authors have declared that no conflict of interest exists.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Huizinga TW, Roos D, von dem Borne AE. Neutrophil Fc-gamma receptors: a two-way bridge in the immune system. Blood. 1990;75:1211–4. [PubMed] [Google Scholar]
- 2.Jefferis R, Pound J, Lund J, Goodall M. Effector mechanisms activated by human IgG subclass antibodies: clinical and molecular aspects. Review article Ann Biol Clin (Paris) 1994;52:57–65. [PubMed] [Google Scholar]
- 3.Nirula A, Glaser SM, Kalled SL, Taylor FR. What is IgG4? A review of the biology of a unique immunoglobulin subtype. Curr Opin Rheumatol. 2011;23:119–24. doi: 10.1097/BOR.0b013e3283412fd4. [DOI] [PubMed] [Google Scholar]
- 4.Aalberse RC, Schuurman J. IgG4 breaking the rules. Immunology. 2002;105:9–19. doi: 10.1046/j.0019-2805.2001.01341.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Labrijn AF, Rispens T, Meesters J, Rose RJ, den Bleker TH, Loverix S, et al. Species-specific determinants in the IgG CH3 domain enable Fab-arm exchange by affecting the noncovalent CH3-CH3 interaction strength. J Immunol. 2011;187:3238–46. doi: 10.4049/jimmunol.1003336. [DOI] [PubMed] [Google Scholar]
- 6.Rispens T, Ooijevaar-de Heer P, Bende O, Aalberse RC. Mechanism of immunoglobulin G4 Fab-arm exchange. J Am Chem Soc. 2011;133:10302–11. doi: 10.1021/ja203638y. [DOI] [PubMed] [Google Scholar]
- 7.Lee SI, Heiner DC, Wara D. Development of serum IgG subclass levels in children. Monogr Allergy. 1986;19:108–21. [PubMed] [Google Scholar]
- 8.Nouri-Aria KT, Wachholz PA, Francis JN, Jacobson MR, Walker SM, Wilcock LK, et al. Grass pollen immunotherapy induces mucosal and peripheral IL-10 responses and blocking IgG activity. J Immunol. 2004;172:3252–9. doi: 10.4049/jimmunol.172.5.3252. [DOI] [PubMed] [Google Scholar]
- 9.Akdis CA, Akdis M. Mechanisms of allergen-specific immunotherapy and immune tolerance to allergens. World Allergy Organ J. 2015;8:17. doi: 10.1186/s40413-015-0063-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Subbarayal B, Schiller D, Mobs C, de Jong NW, Ebner C, Reider N, et al. Kinetics, cross-reactivity, and specificity of Bet v 1-specific IgG4 antibodies induced by immunotherapy with birch pollen. Allergy. 2013;68:1377–86. doi: 10.1111/all.12236. [DOI] [PubMed] [Google Scholar]
- 11.Rock B, Martins CR, Theofilopoulos AN, Balderas RS, Anhalt GJ, Labib RS, et al. The pathogenic effect of IgG4 autoantibodies in endemic pemphigus foliaceus (fogo selvagem) N Engl J Med. 1989;320:1463–9. doi: 10.1056/NEJM198906013202206. [DOI] [PubMed] [Google Scholar]
- 12.Warren SJ, Arteaga LA, Rivitti EA, Aoki V, Hans-Filho G, Qaqish BF, et al. The role of subclass switching in the pathogenesis of endemic pemphigus foliaceus. J Invest Dermatol. 2003;120:104–8. doi: 10.1046/j.1523-1747.2003.12017.x. [DOI] [PubMed] [Google Scholar]
- 13.Ferrari S, Mudde GC, Rieger M, Veyradier A, Kremer Hovinga JA, Scheiflinger F. IgG subclass distribution of anti-ADAMTS13 antibodies in patients with acquired thrombotic thrombocytopenic purpura. J Thromb Haemost. 2009;7:1703–10. doi: 10.1111/j.1538-7836.2009.03568.x. [DOI] [PubMed] [Google Scholar]
- 14.Omokawa A, Komatsuda A, Hirokawa M, Wakui H. Membranous nephropathy with monoclonal IgG4 deposits and associated IgG4-related lung disease. Clin Kidney J. 2014;7:475–8. doi: 10.1093/ckj/sfu077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saeki T, Ito T, Youkou A, Ishiguro H, Sato N, Yamazaki H, et al. Thrombotic thrombocytopenic purpura in IgG4-related disease with severe deficiency of ADAMTS-13 activity and IgG4 autoantibody against ADAMTS-13. Arthritis Care Res (Hoboken) 2011;63:1209–12. doi: 10.1002/acr.20484. [DOI] [PubMed] [Google Scholar]
- 16.Stone JH. IgG4-related disease: nomenclature, clinical features, and treatment. Semin Diagn Pathol. 2012;29:177–90. doi: 10.1053/j.semdp.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 17.Stone JH, Khosroshahi A, Deshpande V, Chan JK, Heathcote JG, Aalberse R, et al. Recommendations for the nomenclature of IgG4-related disease and its individual organ system manifestations. Arthritis Rheum. 2012;64:3061–7. doi: 10.1002/art.34593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med. 2012;366:539–51. doi: 10.1056/NEJMra1104650. [DOI] [PubMed] [Google Scholar]
- 19.Sthoeger Z, Asher I, Rosenberg-Bezalel S, Mahlab-Guri K. Immunoglobulin G4 and related diseases. Isr Med Assoc J. 2012;14:642–5. [PubMed] [Google Scholar]
- 20.Stone JH, Brito-Zeron P, Bosch X, Ramos-Casals M. Diagnostic Approach to the Complexity of IgG4-Related Disease. Mayo Clin Proc. 2015;90:927–39. doi: 10.1016/j.mayocp.2015.03.020. [DOI] [PubMed] [Google Scholar]
- 21.Lever W. In: Pemphigus and Pemphigoid. Thomas CC, editor. Springfield; Illinois: 1965. [Google Scholar]
- 22.Jordon RE, Nordby JM, Milstein H. The complement system in bullous pemphigoid. III. Fixation of C1q and C4 by pemphigoid antibody. J Lab Clin Med. 1975;86:733–40. [PubMed] [Google Scholar]
- 23.Jordon RE, Schroeter AL, Good RA, Day NK. The complement system in bullous pemphigoid. II. Immunofluorescent evidence for both classical and alternate-pathway activation. Clin Immunol Immunopathol. 1975;3:307–14. doi: 10.1016/0090-1229(75)90017-3. [DOI] [PubMed] [Google Scholar]
- 24.Diaz LA, Ratrie H, 3rd, Saunders WS, Futamura S, Squiquera HL, Anhalt GJ, et al. Isolation of a human epidermal cDNA corresponding to the 180-kD autoantigen recognized by bullous pemphigoid and herpes gestationis sera. Immunolocalization of this protein to the hemidesmosome. J Clin Invest. 1990;86:1088–94. doi: 10.1172/JCI114812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giudice GJ, Emery DJ, Diaz LA. Cloning and primary structural analysis of the bullous pemphigoid autoantigen BP180. J Invest Dermatol. 1992;99:243–50. doi: 10.1111/1523-1747.ep12616580. [DOI] [PubMed] [Google Scholar]
- 26.Nishizawa Y, Uematsu J, Owaribe K. HD4, a 180 kDa bullous pemphigoid antigen, is a major transmembrane glycoprotein of the hemidesmosome. J Biochem. 1993;113:493–501. doi: 10.1093/oxfordjournals.jbchem.a124072. [DOI] [PubMed] [Google Scholar]
- 27.Giudice GJ, Emery DJ, Zelickson BD, Anhalt GJ, Liu Z, Diaz LA. Bullous pemphigoid and herpes gestationis autoantibodies recognize a common non-collagenous site on the BP180 ectodomain. J Immunol. 1993;151:5742–50. [PubMed] [Google Scholar]
- 28.Zillikens D, Rose PA, Balding SD, Liu Z, Olague-Marchan M, Diaz LA, et al. Tight clustering of extracellular BP180 epitopes recognized by bullous pemphigoid autoantibodies. J Invest Dermatol. 1997;109:573–9. doi: 10.1111/1523-1747.ep12337492. [DOI] [PubMed] [Google Scholar]
- 29.Liu Z, Diaz LA, Troy JL, Taylor AF, Emery DJ, Fairley JA, et al. A passive transfer model of the organ-specific autoimmune disease, bullous pemphigoid, using antibodies generated against the hemidesmosomal antigen, BP180. J Clin Invest. 1993;92:2480–8. doi: 10.1172/JCI116856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu Z, Sui W, Zhao M, Li Z, Li N, Thresher R, et al. Subepidermal blistering induced by human autoantibodies to BP180 requires innate immune players in a humanized bullous pemphigoid mouse model. J Autoimmun. 2008;31:331–8. doi: 10.1016/j.jaut.2008.08.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nishie W, Sawamura D, Goto M, Ito K, Shibaki A, McMillan JR, et al. Humanization of autoantigen. Nat Med. 2007;13:378–83. doi: 10.1038/nm1496. [DOI] [PubMed] [Google Scholar]
- 32.Dopp R, Schmidt E, Chimanovitch I, Leverkus M, Brocker EB, Zillikens D. IgG4 and IgE are the major immunoglobulins targeting the NC16A domain of BP180 in Bullous pemphigoid: serum levels of these immunoglobulins reflect disease activity. J Am Acad Dermatol. 2000;42:577–83. [PubMed] [Google Scholar]
- 33.Hofmann S, Thoma-Uszynski S, Hunziker T, Bernard P, Koebnick C, Stauber A, et al. Severity and phenotype of bullous pemphigoid relate to autoantibody profile against the NH2- and COOH-terminal regions of the BP180 ectodomain. J Invest Dermatol. 2002;119:1065–73. doi: 10.1046/j.1523-1747.2002.19529.x. [DOI] [PubMed] [Google Scholar]
- 34.Hennessey PJ, Black CT, Andrassy RJ. Nonenzymatic glycosylation of immunoglobulin G impairs complement fixation. JPEN J Parenter Enteral Nutr. 1991;15:60–4. doi: 10.1177/014860719101500160. [DOI] [PubMed] [Google Scholar]
- 35.Giclas PC. Complement. In: Coligan JE, Kruisbeek AM, Margulies DH, Shevach EM, Strober W, editors. Current Protocols in Immunology. John Wiley & Sons; 1994. pp. 1–26. [Google Scholar]
- 36.Liu Z, Zhou X, Shapiro SD, Shipley JM, Twining SS, Diaz LA, et al. The serpin alpha1-proteinase inhibitor is a critical substrate for gelatinase B/MMP-9 in vivo. Cell. 2000;102:647–55. doi: 10.1016/s0092-8674(00)00087-8. [DOI] [PubMed] [Google Scholar]
- 37.Bradley PP, Priebat DA, Christensen RD, Rothstein G. Measurement of cutaneous inflammation: estimation of neutrophil content with an enzyme marker. J Invest Dermatol. 1982;78:206–9. doi: 10.1111/1523-1747.ep12506462. [DOI] [PubMed] [Google Scholar]
- 38.Liu Z, Giudice GJ, Zhou X, Swartz SJ, Troy JL, Fairley JA, et al. A major role for neutrophils in experimental bullous pemphigoid. J Clin Invest. 1997;100:1256–63. doi: 10.1172/JCI119639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heimbach L, Li Z, Berkowitz P, Zhao M, Li N, Rubenstein DS, et al. The C5a receptor on mast cells is critical for the autoimmune skin-blistering disease bullous pemphigoid. J Biol Chem. 2011;286:15003–9. doi: 10.1074/jbc.M111.221036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao M, Trimbeger ME, Li N, Diaz LA, Shapiro SD, Liu Z. Role of FcRs in animal model of autoimmune bullous pemphigoid. J Immunol. 2006;177:3398–405. doi: 10.4049/jimmunol.177.5.3398. [DOI] [PubMed] [Google Scholar]
- 41.Natsuga K, Nishie W, Shinkuma S, Ujiie H, Nishimura M, Sawamura D, et al. Antibodies to pathogenic epitopes on type XVII collagen cause skin fragility in a complement-dependent and - independent manner. J Immunol. 2012;188:5792–9. doi: 10.4049/jimmunol.1003402. [DOI] [PubMed] [Google Scholar]
- 42.Ujiie H, Sasaoka T, Izumi K, Nishie W, Shinkuma S, Natsuga K, et al. Bullous pemphigoid autoantibodies directly induce blister formation without complement activation. J Immunol. 2014;193:4415–28. doi: 10.4049/jimmunol.1400095. [DOI] [PubMed] [Google Scholar]
- 43.Dainichi T, Nishie W, Yamagami Y, Sonobe H, Ujiie H, Kaku Y, et al. Bullous pemphigoid suggestive of complement-independent blister formation with anti-BP180 IgG4 autoantibodies. Br J Dermatol. 2016 doi: 10.1111/bjd.14411. [DOI] [PubMed] [Google Scholar]
- 44.Bird P, Friedmann PS, Ling N, Bird AG, Thompson RA. Subclass distribution of IgG autoantibodies in bullous pemphigoid. J Invest Dermatol. 1986;86:21–5. doi: 10.1111/1523-1747.ep12283737. [DOI] [PubMed] [Google Scholar]
- 45.Brooks WS, Lee YY, Abell E, Deng JS. Comparison of IgG subclasses and complement binding activity of autoantibodies from patients with bullous pemphigoid and pemphigus. J Clin Lab Anal. 1989;3:307–11. doi: 10.1002/jcla.1860030509. [DOI] [PubMed] [Google Scholar]
- 46.Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, et al. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood. 2009;113:3716–25. doi: 10.1182/blood-2008-09-179754. [DOI] [PubMed] [Google Scholar]