

Abstract
Inclusions of Tar DNA- binding protein 43 (TDP-43) are a pathological hallmark of amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration with TDP-43-positive inclusions (FTLD-TDP). Pathological TDP-43 exhibits the disease-specific biochemical signatures, which include its ubiquitination, phosphorylation and truncation. Recently, we demonstrated that the extreme N-terminus of TDP-43 regulates formation of abnormal cytoplasmic TDP-43 aggregation in cultured cells and primary neurons. However, it remained unclear whether this N-terminal domain mediates TDP-43 aggregation and the associated toxicity in vivo. To investigate this, we expressed a GFP-tagged TDP-43 with a nuclear localization signal mutation (GFP-TDP-43NLSm) and a truncated form without the extreme N-terminus (GFP-TDP-4310-414-NLSm) by adeno-associated viral (AAV) vectors in mouse primary cortical neurons and murine central nervous system. Compared to neurons containing GFP alone, expression of GFP-TDP-43NLSm resulted in the formation of ubiquitin-positive cytoplasmic inclusions and activation of caspase-3, an indicator of cell death. Moreover, mice expressing GFP-TDP-43NLSm proteins show reactive gliosis and develop neurological abnormalities. However, by deletion of TDP-43’s extreme N-terminus, these pathological alterations can be abrogated. Together, our study provides further evidence confirming the critical role of the extreme N-terminus of TDP-43 in regulating protein structure as well as mediating toxicity associated with its aggregation.
Keywords: TDP-43, aggregation, the extreme N-terminus, toxicity, in vivo, AAV
1. Introduction
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disease that affects upper and lower motor neurons in the brain and spinal cord. Degeneration of neurons in these affected areas causes muscle weakness and atrophy throughout the body and often leads to death from respiratory failure usually within 3-5 years of onset (Robberecht and Philips, 2013). Frontotemporal dementia (FTD) is a clinical syndrome, caused by the atrophy of frontal and temporal lobes, associated with progressive changes in behavior, personality, language, and/or motor function (Dormont and Seidenwurm, 2008). FTD accounts for 5-15% of all cases of dementia, and is the second most common form of early-onset dementia after Alzheimer’s disease (Rademakers et al., 2012). Frontotemporal lobar degeneration with TDP-43-positive inclusions (FTLD-TDP) is the most common pathological subgroup of FTD (Arai et al., 2006; Neumann et al., 2006); the characteristic formation of ubiquitin-positive inclusions in affected neurons led to its previous description as FTLD-U. ALS and FTLD-TDP are referred to as TDP-43 proteinopathies due to the hallmark presence of TDP-43 cytoplasmic inclusions (Chen-Plotkin et al., 2010; Janssens and Van Broeckhoven, 2013). Other common pathological features include TDP-43 phosphorylation and ubiquitination as well as cleaved fragments of TDP-43 (Arai et al., 2006; Neumann et al., 2006). Unlike in disease, TDP-43 normally localizes to cell nuclei under a physiological steady state and is reported to play roles in regulating transcription, splicing of pre-mRNA, translation, stabilizing mRNA, and processing of microRNAs (Ayala et al., 2011; Baralle et al., 2013; Chen-Plotkin et al., 2010; Fiesel et al., 2010; Fiesel et al., 2012; Strong, 2010; Volkening et al., 2009). TDP-43, a heterogeneous nuclear ribonucleoprotein (hnRNP), contains an N-terminal domain consisting of a nuclear localization signal (NLS) and a nuclear export signal (NES), two RNA-binding domains (RBDs) and a C-terminal glycine-rich domain. Of interest, TDP-43 monomers bind to each other to form homodimers in vitro (Kuo et al., 2009; Shiina et al., 2010; Zhang et al., 2009). Recently, we and others have reported that the N-terminus of TDP-43 is the crucial element in regulating its self-interaction (Chang et al., 2012; Wang et al., 2013; Zhang et al., 2013). Moreover, we demonstrated that TDP-43 homodimerization also plays an important role in modulating the normal biological activity of TDP-43 (Zhang et al., 2013).
TDP-43 pathology is found in the majority of sporadic and familial ALS, and the most common subtype of FTD, FTLD-TDP. Mutations in Tardbp, the gene encoding TDP-43, are associated with familial as well as some cases of sporadic ALS. Considering these factors, TDP-43 is believed to be one of the most important factors in ALS and FTLD-TDP pathogenesis (Cohen et al., 2011). Similar to the roles of the aggregated proteins found in other neurodegenerative diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and polyglutamine diseases (Forman et al., 2004; Taylor et al., 2002), these cytoplasmic TDP-43 inclusions may contribute to disease pathogenesis through a toxic gain of function mechanism (Lee et al., 2012). Indeed, previous studies have shown that the expression of full length TDP-43 with mutations in the NLS sequence (TDP-43NLSm) in mouse or rat brain resulted in formation of cytoplasmic TDP-43 aggregates and neurodegeneration, as well as disease-relevant motor deficits (Dayton et al., 2013; Igaz et al., 2011; Walker et al., 2015). Of importance, suppression of TDP-43NLSm expression rescues above pathological phenotypes, indicating that clearance of cytoplasmic TDP-43 accumulation and aggregation provides protection (Igaz et al., 2011; Walker et al., 2015).
Recently, we demonstrated that the extreme N-terminus of TDP-43 is required for the cytoplasmic aggregation of TDP-43NLSm and the associated toxicity in primary cultured neurons (Zhang et al., 2013). Consistent with our finding, Budini et al. further discovered that the N-terminal region of TDP-43 is required for sequestration of full length TDP-43 into cytoplasmic TDP-43 inclusions and subsequent TDP-43 loss-of-function in cultured cells (Budini et al., 2015). Considering the important roles of the N-terminal region of TDP-43 in regulation of TDP-43 aggregation, we sought to further determine whether this N-terminal domain regulates TDP-43 aggregation and the associated toxicity in vivo. In the present study, we used adeno-associated viral vectors (AAV) to express TDP-43NLSm or TDP-4310-414-NLSm in the murine central nervous system. We found that the deletion of the first 10 amino acids significantly mitigates TDP-43 aggregation and rescues neurodegeneration in vivo. This data confirm the critical role of the extreme N-terminus of TDP-43 in regulating protein structure and aggregation. Therefore, the identification of agents targeting this region might be a promising therapeutic strategy to treat TDP-43 proteinopathies.
2. Results
2.1 Deletion of the TDP-43 N-terminus attenuates aggregate formation and the associated toxicity in mouse primary neurons
We previously showed that the overexpression of TDP-43NLSm, but not TDP-4310-414-NLSm, resulted in the formation of TDP-43 aggregates in the cytoplasm and impairment of neuronal outgrowth in mouse primary neuronal cultures (Zhang et al., 2013). To further investigate the link between N-terminus of TDP-43 and neurotoxicity in vitro, we transduced mouse cortical primary cultures with recombinant AAV2/1 containing GFP, GFP-TDP-43NLSm or GFP-TDP-4310-414-NLSm for 5 days. Consistent with our previous report (Zhang et al., 2013), confocal analysis revealed that expression of GFP-TDP-43NLSm results in formation of cytoplasmic aggregates (Fig. 1A). In contrast, expression of GFP-TDP-4310-414-NLSm and GFP alone in primary neurons showed diffuse distribution (Fig. 1A). Compared to GFP alone, Western blot analysis revealed that expression of GFP-TDP-43NLSm and GFP-TDP-4310-414-NLSm respectively leads to a 3.2 and 1.6-fold increase in levels of active caspase-3 (Fig. 1B, C). Of note, while the levels of TDP-43 protein were comparable between GFP-TDP-43NLSm and GFP-TDP-4310-414-NLSm (Fig. 1B, C), expression of GFP-TDP-43NLSm induced much greater activation of caspase-3 than GFP-TDP-4310-414-NLSm (Fig. 1B, C).
Figure 1. Deletion of the TDP-43 N-terminus attenuates aggregate formation and the associated toxicity in mouse primary neurons.
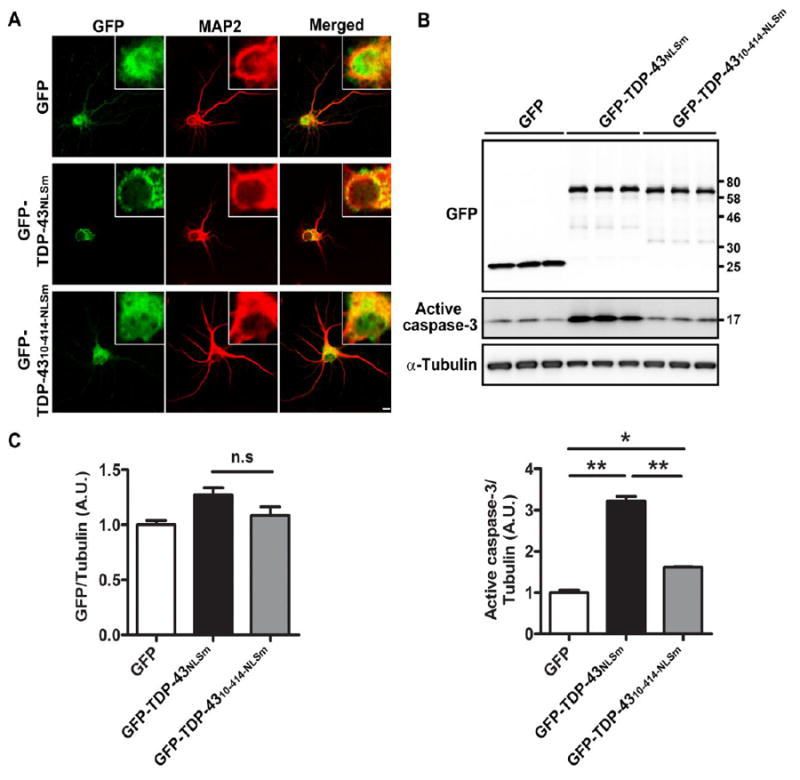
(A) Expression of GFP-TDP-43NLSm resulted information of cytoplasmic aggregates, whereas GFP-TDP-43NLSm-10-414 and GFP alone showed diffuse distribution throughout mouse primary neurons. Scale bar represents 10 μm. Western blot (B) and densitometric analysis of blots (C) revealed that, compared to GFP, expression of GFP-TDP-43NLSm significantly induces the levels of activated caspase-3. In contrast, deletion of the first 10 residues dramatically abrogates caspase-3 activation. Of note, expression levels of TDP-43 protein were comparable between GFP-TDP-43NLSm and GFP-TDP-43NLSm-10-414 (B, C). Data represents mean ± SEM of three separate experiments. * P<0.05, ** P<0.01, n.s. not significant, as analyzed by one-way ANOVA with Tukey’s post-hoc analysis.
2.2 Intracerebroventricular (ICV) injection of AAV2/9-TDP-43 results in expression of TDP-43 proteins throughout mouse brain
Since AAV2/9 capsid serotype has been shown to mediate a more widespread distribution of a target gene than AAV2/1 in mouse brain (Chakrabarty et al., 2013), we injected AAV2/9 containing either GFP, GFP-TDP-43NLSm or GFP-TDP-4310-414-NLSm into cerebroventricles of P0 neonatal mice at low dose (1.25×1010 particles) or high dose (5.0×1010 particles) to further evaluate the neurotoxicity of cytoplasmic TDP-43 aggregates in vivo. Twenty-eight days following ICV delivery, the mice were harvested to measure the expression of GFP-TDP-43 in the brain. We observed that although the protein levels of GFP-TDP-43NLSm were higher than that of GFP-TDP-4310-414-NLSm when the low dose of virus was used (Fig. 2A, B), a high dose of GFP-TDP-4310-414-NLSm produced comparable protein levels as that of GFP-TDP-43NLSm (Fig. 2A, B). In contrast to protein expression, the mRNA levels of GFP-TDP-4310-414-NLSm assessed with primer sets targeting either GFP (Fig. 2C) or human TDP-43 (Fig. 2C) at both low and high doses were significantly higher than that of GFP-TDP-43NLSm at a low dose, suggesting the TDP-43NLSm protein may be more stable than TDP-4310-414-NLSm. To verify that the expression of GFP-TDP-43NLSm or GFP-TDP-4310-414-NLSm did not reduce the protein and mRNA levels of endogenous mouse TDP-43 (Fig. 2A, B, D), the levels of mSort1_17b (mouse sortilin 1 including exon 17b), a splicing variant of mSort1 that is significantly increased during downregulation of mouse TDP-43, were measured (Prudencio et al., 2012; Xu et al., 2013). A decrease in mSort1 RNA was not detected in the brains of GFP-TDP-43NLSm mice (Fig. 2D), further confirming a lack of reduction of mouse TDP-43 and its activity.
Figure 2. ICV injection of AAV2/9-TDP-43 leads to expression of TDP-43 protein throughout mouse brain.
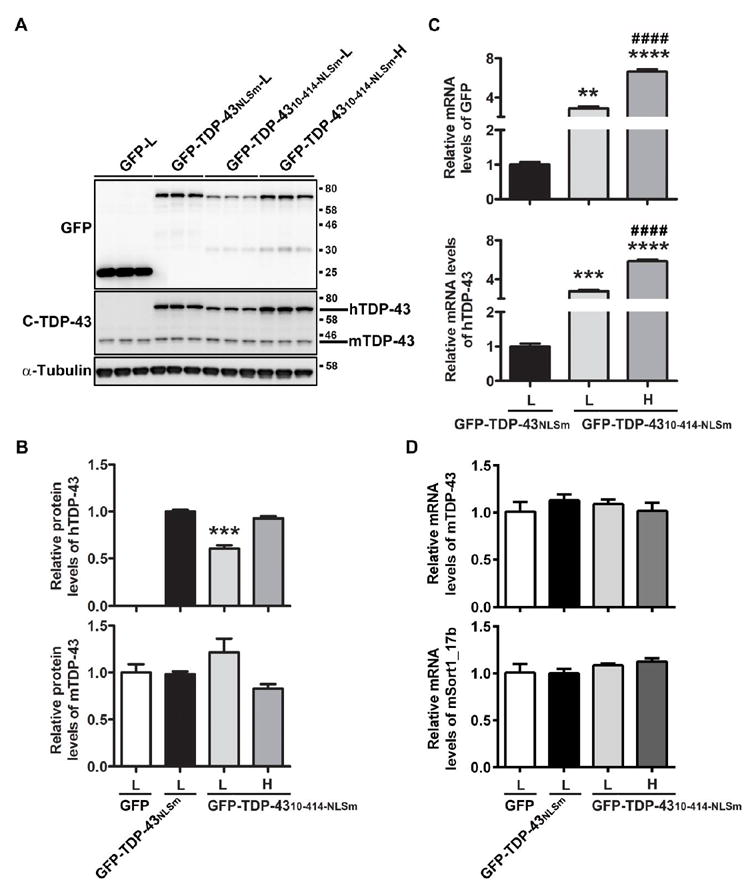
Western blot (A) and densitometric quantification analysis (B) revealed that the levels of GFP-TDP-4310-414-NLSm protein at high dose were comparable to that of GFP-TDP-43NLSm at low dose. Expression of GFP-TDP-43NLSm and GFP-TDP-4310-414-NLSm did not reduce the protein levels of endogenous mouse TDP-43 (B). (C) Quantitative RT-PCR (qRT-PCR) using specific primers against GFP or human TDP-43 showed that the mRNA levels of GFP-TDP-4310-414-NLS were dramatically higher than that of GFP-TDP-43NLS. (D) qRT-PCR using specific primers for mouse TDP-43 or mouse sortin1_17b showed that expression of GFP-TDP-43NLS or GFP-TDP-4310-414-NLS did not reduce mRNA levels of mouse TDP-43 nor induce aberrant splicing of mouse sortilin 1. Data shown are the means ± SEM of 3 mice per group. ** P<0.01, *** P<0.001, **** P<0.0001 versus GFP-TDP-43NLSm (L), #### P<0.0001 versus GFP-TDP-4310-414-NLSm (L), as assessed by one-way ANOVA with Tukey’s post-hoc analysis.
2.3 Deletion of the TDP-43 N-terminus attenuates cell death, reactive gliosis, and motor deficits associated with TDP-43 aggregates
Considering TDP-43 protein levels were comparable in mice injected with low-doses of GFP-TDP-43NLSm and high-doses of GFP-TDP-4310-414-NLSm, we sought to evaluate the resulting behavioral abnormalities of these mice. Twenty-eight days after ICV injection, all the GFP-TDP-43NLSm-expressing mice exhibited a hind limb clasping phenotype when suspended by the tail (Fig. 3A, Supplementary Video 1). In contrast, GFP or GFP-TDP-4310-414-NLSm-injected mice showed a normal escape response by splaying their hind limbs (Fig. 3A, Supplementary Video 2, 3). Pathological analysis of the brains with GFP antibody revealed that TDP-43-positive aggregates were observed in a subset of neurons expressing GFP-TDP-43NLSm in cortical regions (arrow, Fig. 3B). Of note, these TDP-43 aggregates were also ubiquitin-positive (Fig. 3C). In contrast, TDP-43 aggregates were not observed in cells expressing GFP alone or GFP-TDP-4310-414-NLSm (Fig. 3B). Moreover, a small percentage of the cells with TDP-43-positive cytoplasmic aggregates also showed caspase-3 activation (Fig. 3B), which was absent in brains of mice expressing GFP or GFP-TDP-4310-414-NLSm (Fig. 3B). In addition, an astrocytic marker, glial fibrillary acidic protein (GFAP), was activated, but not a microglia marker, ionized calcium-binding adaptor molecule 1 (Iba1), as indicated by mRNA and proteins levels (Fig. 3D, E) in GFP-TDP-43NLSm mice, but not in mice expressing GFP-TDP-4310-414-NLSm or GFP alone.
Figure 3. The extreme N-terminus regulates the aggregation of TDP-43 and neurotoxicity in vivo.
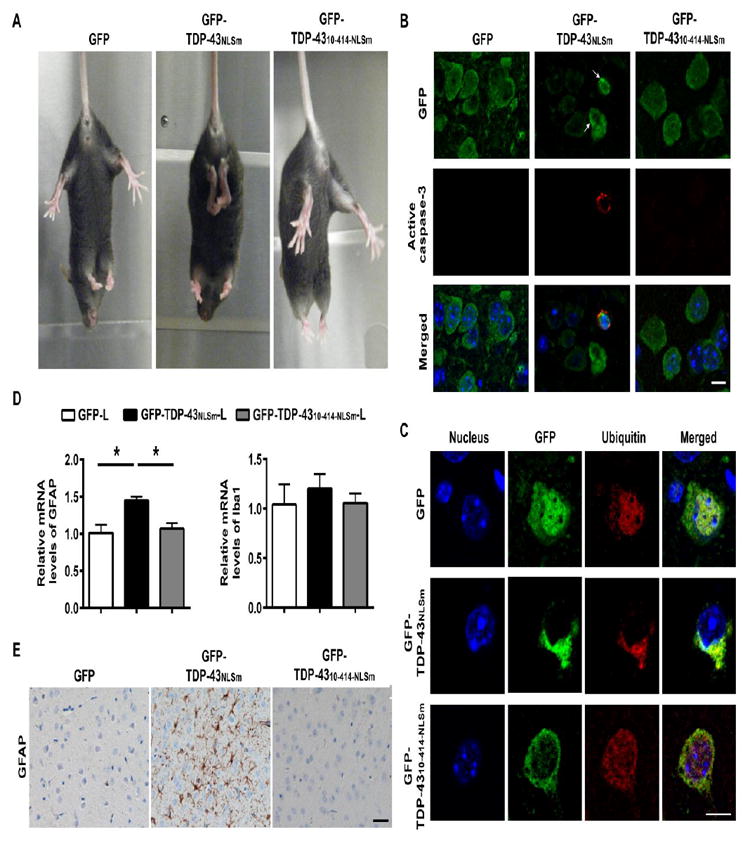
(A) Mice expressing GFP alone or GFP-TDP-4310-414-NLSm splayed their hind legs as normal escape response. In contrast, mice expressing GFP-TDP-43NLSm held their hind limbs close to their body, indicating neurological abnormalities. (B) Immunofluorescence staining with the GFP antibody revealed that some neurons of mice injected with GFP-TDP-43NLSm had small, dense cytoplasmic aggregates (arrows), whereas the distribution pattern of GFP-TDP-4310-414-NLSm was diffuse. Of importance, caspase-3 activation was observed in the subset of neurons with TDP-43 aggregates in cerebral cortex of mice injected with GFP-TDP-43NLSm. The staining of active caspase-3 was not observed in neurons expressing GFP alone or GFP-TDP-4310-414-NLSm. Scale bar represents 10 μm. (C) Double immunofluorescence staining with GFP and ubiquitin antibodies in cerebral cortex of GFP-TDP-43NLSm mice revealed that the TDP-43 cytoplasmic inclusions were ubiquitin-positive. The ubiquitin-positive TDP-43 inclusions were not observed in brains of GFP or GFP-TDP-4310-414-NLSm mice. Scale bar represents 10 μm. (D) Quantitative PCR (qPCR) analysis of the relative mRNA levels of the astrocyte marker, GFAP, but not microglia marker, Iba1, were significantly increased in brains of GFP-TDP-43NLSm mice compared to GFP or GFP-TDP-4310-414-NLSm mice controls. (E) Cerebral cortex and hippocampal sections stained with anti-GFAP antibody showed the activation of astrogliosis in mice expressing GFP-TDP-43NLSm but not GFP alone or GFP-TDP-4310-414-NLSm mice. Scale bar represents 100 μm. Data shown are the means ± SEM of 3 mice per group. * P<0.05, as assessed by one-way ANOVA with Tukey’s post-hoc analysis.
3. Discussion
TDP-43 is the major protein component of ubiquitinated inclusions in FTLD-TDP and ALS (Arai et al., 2006; Neumann et al., 2006). Under physiological conditions, TDP-43 predominantly localizes to the nucleus. However, a substantial loss of nuclear TDP-43 is observed in neurons bearing aberrant cytoplasmic TDP-43 inclusions. Although it has remained unclear how aberrant TDP-43 protein causes neurodegeneration, mounting evidence points to a combination of loss of nuclear TDP-43 function and toxic cytoplasmic TDP-43 aggregates contributing to disease pathogenesis (Lee et al., 2012). Recently, we and others demonstrated that expression of full length TDP-43 containing NLS mutations resulted in cytoplasmic TDP-43 accumulation and aggregation in vitro and in vivo (Barmada et al., 2010; Dayton et al., 2013; Elden et al., 2010; Igaz et al., 2011; Walker et al., 2015; Winton et al., 2008; Zhang et al., 2013). Moreover, the cytoplasmic accumulation and aggregation of TDP-43 induced cellular toxicity in primary neurons (Barmada et al., 2010; Zhang et al., 2013), and neurodegeneration in rat and mouse models (Dayton et al., 2013; Igaz et al., 2011; Walker et al., 2015). Of particular importance, we provided evidence that showed deletion of the first 10 N-terminal amino acids (TDP-4310-414-NLSm) prevented TDP-43 aggregation and rescued neurite outgrowth in primary neurons (Zhang et al., 2013). In the present study, we further confirmed that the GFP-TDP-43NLSm has a much stronger effect than GFP-TDP-4310-414-NLSm to induce activation of caspase-3 in mouse primary neuronal cultures, indicating that this region is critical for TDP-43-mediated neurodegeneration.
Next, we took advantage of the AAV2/9, which efficiently mediates expression of transgenes in neuronal cells when administered to murine neonates through ICV injections (Chakrabarty et al., 2013), to express GFP-TDP-43NLSm or GFP-TDP-4310-414-NLSm in mouse brain. Consistent with previous findings (Dayton et al., 2013; Igaz et al., 2011; Walker et al., 2015), we observed that mice expressing TDP-43NLSm developed behavioral abnormalities, ubiquitin-positive TDP-43 aggregates formation, and astrogliosis. In contrast, while acute cytoplasmic accumulation GFP-TDP-4310-414-NLSm in primary neurons causes a mild toxicity possible through induction of cellular stress, chronic expression of TDP-4310-414-NLSm in mouse brain did not lead to the pathological abnormalities nor cause the behavioral deficits. Of relevance, most TDP-43NLSm detected in neurons was diffusely accumulated in the cytosol and only a small portion had formed aggregates. Considering the proportion of the neurons with TDP-43 aggregates and activated caspase-3 was relatively low, cell death associated with TDP-43 aggregates may only be partially responsible for the observed neuropathological phenotypes in TDP-43NLSm mice. Under normal conditions TDP-43 mainly resides in the nucleus and participates in RNA metabolism, pre-mRNA splicing, and transcription; although small amounts of cytoplasmic TDP-43 also play a critical role in microRNA biogenesis (Buratti et al., 2010; Kawahara and Mieda-Sato, 2012) and mRNA transport, as well as protein translation (Colombrita et al., 2012; Liu-Yesucevitz et al., 2014; Wang et al., 2008). Therefore, excessive accumulation of diffuse full length TDP-43 in the cytosol may result in dysfunctional RNA processing as reported previously (Igaz et al., 2011), which may contribute to the observed neuropathological phenotypes of TDP-43NLSm mice. In contrast, our previous study has reported that deletion of the first 10 amino acids led to loss of TDP-43 biological activity (Zhang et al., 2013), thus the cytoplasmic accumulation of TDP-4310-414-NLSm may not be able perturb the mRNA metabolism and cause toxicity. Although several studies have reported that reduction of mouse TDP-43 in transgenic mice expressing TDP-43NLSm contributes to neurodegeneration (Igaz et al., 2011; Walker et al., 2015), we did not observed this decrease of endogenous mouse TDP-43 protein in our TDP-43NLSm mice, excluding this possibility. Lastly, we have reported that the accumulation of nuclear TDP-43 is required to reach a certain threshold to affect mouse endogenous TDP-43 levels (Xu et al., 2010); the expression level of TDP-43NLSm in our mouse model may not reach the threshold to reduce mouse TDP-43.
4. Conclusions
Our results indicate that the extreme N-terminus of TDP-43 plays critical roles in regulating TDP-43 protein aggregation and toxicity in vivo. As such, targeting TDP-43’s extreme N-terminus to inhibit its activity might be a potential therapeutic strategy to treat ALS and FTD-TDP.
5. Experimental procedures
5.1 Generation of various AAV2/1- and AAV2/9-TDP-43 vectors
GFP-tagged full-length human TDP-43NLSm and TDP-4310-414-NLSm plasmids were generated as described previously (Zhang et al., 2013). AAV2/1 and 2/AAV9-TDP-43 were prepared by standard methods (Zhang et al., 2013). Briefly, AAV vectors expressing GFP, GFP-TDP-43NLSm and GFP-TDP-4310-414-NLSm were transfected with helper plasmids in HEK293 cells. Forty-eight hours after transfection, the cells were harvested and lysed in the presence of 0.5 % sodium deoxycholate and 50 U/ml Benzonase (Sigma) by freeze thawing, and the virus was isolated using a discontinuous iodixanol gradient centrifugation. The genomic titer of each virus was determined by qRT-PCR, and the titer of the virus of GFP, GFP-TDP-43NLSm, or GFP-TDP-4310-414-NLSm was 1.0-3.0×1013 viral genomes per mL. When performed intracerebroventricular injections, the viruses were diluted with sterile phosphate-buffered saline (PBS) and adjusted to the titer indicated in the text.
5.2 Mouse primary neuronal culture and virus treatment
Primary cortical neuronal cultures were prepared according to protocol as we previously described (Zhang et al., 2013). In brief, neurons that were isolated from the cortex from embryonic day 18 (E18) C57BL/6J pups were seeded at a density of 1×105 cells/coverslip in 24-well plates or 8×105 cells/well in 6-well plates. At day in vitro 4, neurons were transduced to express GFP, GFP-TDP-43NLSm, or GFP-TDP-4310-414-NLSm. For 24-well plate, 4×109 genome AAV2/1 virus of GFP, GFP-TDP-43NLSm or GFP-TDP-4310-414-NLSm was added to the cell medium. For 6-well plate, since we observed that more virus of GFP-TDP-4310-414-NLSm have to be used to get comparable protein expression as GFPTDP-43NLSm, 1×1010 of particles of AAV2/1-GFP, GFP-TDP-43NLSm were used and 1.5×1010 particles were used for GFP-TDP-4310-414-NLSm. Five days after addition of AAV2/1 viral particles to the media, the cells grown in 24-well plate were fixed with 4% paraformaldehyde (PFA) and immunostained with a mouse monoclonal anti-MAP2 antibody (Sigma, 1:1,000) for immunofluorescence study. The cell pellets collected from a 6-well plate were lysed using buffer (50 mM Tris-HCl, pH 7.4, 300 mM NaCl, 1% Triton X-100, 5 mM EDTA, 2% SDS, PMSF, and protease and phosphatase inhibitors). Lysates were sonicated and centrifuged at 16,000 g for 20 min. Supernatants were saved and the protein concentration was determined by BCA assay. Samples were then analyzed by Western blot.
5.3 Intracerebroventricular injections of various AAV2/9-TDP-43
Intracerebroventricular (ICV) injections of AAV2/9 were performed as previously described with minor modifications (Chakrabarty et al., 2013; Kim et al., 2013). Briefly, a 32-gauge Hamilton needle attached to a 10 μl Hamilton syringe is inserted at approximately two-fifth of the distance between the lambda and eye of P0 C57BL/6J pups after they underwent cryoanesthesia on ice for 3 minutes. The needle was inserted perpendicular to the surface of the head and held at a depth of approximately 2 mm. Two μl of AAV2/9 solution was manually injected to each cerebroventricle. After injections, pups were placed on a heat pad until they completely recovered from anesthesia and put back to the cages. Motor phenotype was assessed by clasping phenotype of hind limbs while holding the tails. All procedures in this study were performed in accordance with the National Institutes of Health Guide for Care and Use of Experimental Animals and approved by the Mayo Clinic Institutional Animal Care and Use Committee (Protocol number A37811). Of note, both high and low dose of GFP-TDP-4310-414-NLSm viral particles were used for ICV injection for the reason mentioned above.
5.4 Protein and RNA extraction from mouse brains
To extract both protein and RNA from the same brain sample, brains were first homogenized in ice-cold Tris-EDTA (10 mM Tris-HCl, 0.1 mM EDTA, pH 8.0) buffer with 2X PMSF, protease and phosphatase inhibitors, and RNase inhibitors (Ambion, AM2694) by sonication. After all the visible chunks were broken, 2X lysis buffer was added to one volume of the homogenates, and further sonication was performed to extract proteins. To extract RNA, 3 volumes of TRIzol-LS (Invitrogen) were added to 1 volume of the homogenates, and RNA was purified according to manufacturer’s instruction.
5.5 Western blotting
The samples from mouse primary neurons or mouse brains were subjected to SDS-PAGE. After transfer, membranes were incubated with rabbit polyclonal TDP-43 antibody (1:2000, Proteintech), rabbit polyclonal GFP antibody (1:1000, Invitrogen), rabbit polyclonal cleaved-caspase-3 antibody (1:1000, Cell Signaling Technology) or mouse monoclonal α-tubulin antibody (1:10000, Sigma-Aldrich). After incubating with an appropriate secondary antibody, immunoreactivity was visualized by ECL, and signals were recorded by FUJI film imager.
5.6 Quantitative real-time PCR (qRT-PCR)
A total of 2 μg of RNA were used to synthesize cDNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). The qRT-PCR was performed using an HT7900 analyzer (Applied Biosystems). Each 5 µl reaction contained: 2 µl of cDNA diluted 1:10, 0.5 μl primer mix (1 µM for each primer), and 2.5 µl SYBR green (Invitrogen). The qRT-PCR program was as follows: 50°C 2min, 95°C 2min, 40 cycles of 95°C 15s and 60°C 1min. For dissociation curves, a dissociation stage of 95°C 15s, 60°C 15s was added at the end of the program. Tardbp and GFP mRNA values were normalized to Gapdh values, an endogenous transcript control. The data was analyzed by using Software RQ Manager 1.2 (Applied Biosystems). The primers used were: Gapdh: 5’-CATGGCCTTCCGTGTTCCTA-3’ and 5’-CCTGCTTCACCACCTTCTTGAT-3’; Tardbp: 5’-GGGGCAATCTGGTATATGTTG-3’ and 5’-TGGACTGCTCTTTTCACTTTCA-3’; GFP: 5’-GAAGCGCGATCACATGGT-3’ and 5’-CCATGCCGAGAGTGATCC-3’; mSort1_17b: 5’-AAATCCCAGGAGACAAATGC-3’ and 5’-GAGCTGGATTCTGGGACAAG-3’; GFAP: 5’-GGAGAGGGACAACTTTGCAC-3’ and 5’-AGCCTCAGGTTGGTTTCATC-3’; Iba1: 5’-GGATTTGCAGGGAGGAAAAG-3’ and 5’-TGGGATCATCGAGGAATTG-3’.
5.7 Immunohistochemistry and immunofluorescence
Tissues were embedded in paraffin, sectioned (5 μm thick) and mounted on glass slides. Sections were deparaffinized in xylene and rehydrated in a graded series of alcohol, followed by dH2O. Antigen retrieval was performed in a dH2O steam bath for 30 min. Tissues were immunostained with human-specific TDP-43 (1:6,000, Novus Biologicals), glial fibrillary acidic protein (GFAP) (1:2,500, Biogenex) and ionized calcium-binding adaptor molecule 1 (Iba-1) (1:2,000, Wako Chemicals) antibodies, using the DAKO Autostainer (Dako Auto Machine Corporation) and the DAKO EnVision+ HRP system. DAKO Liquid DAB Substrate Chromogen system was the chromogen. After immunostaining, sections were briefly counterstained with hematoxylin to visualize cell nuclei and coverslipped. Paraffin-embedded sections were also stained with hematoxylin and eosin. To examine the colocalization of TDP-43 inclusions and active caspase-3, deparaffinized brain sections were blocked with blocking solution (Dako), and incubated with mouse monoclonal GFP antibody (1:1000, Invitrogen) and rabbit polyclonal cleaved-caspase 3 antibodies (1:250, Cell Signaling Technology). To determine whether TDP-43 inclusions are ubiquitin-positive, the sections were incubated with a rabbit polyclonal anti-GFP antibody (1:1000, Life Technologies) and a mouse monoclonal anti-ubiquitin antibody (1:250, EMD Millipore). Following incubation with secondary antibody, the sections were mounted with Hoechst 33258 (1 μg/ml, Invitrogen). Images were obtained on a Zeiss LSM 510 META confocal microscope.
5.8 Statistics
One-way ANOVA with Tukey’s posthoc analysis were used to compare measures among 3 groups. Two-tailed student’s t-test was used to compare measures among 2 groups. For data presentation, normalized values were averaged and presented as mean ± standard error of mean (SEM). Values of P< 0.05 were considered statistically significant.
Supplementary Material
Video 1: GFP-TDP-43-NLSm 2.5e10 P28
Video 2: GFP 2.5e10 P28
Video 3: GFP-TDP-43-10-414-NLSm 5e10 P28
Highlights.
Deletion of the extreme N-terminus of TDP-43 mitigates aggregation and associated toxicity in vitro.
Expression of TDP-4310-414-NLSm in vivo attenuates neuropathological and behavioral deficits associated with TDP-43 aggregates.
Extreme N-terminus of TDP-43 regulates protein aggregation and toxicity.
Acknowledgments
This study was supported by grants from the National Institutes of Health/National Institute of Neurological Disorders and Stroke [(R21 NS079807 (Y.Z.)]; Amyotrophic Lateral Sclerosis Association (T.F.G, Y.Z.); Alzheimer Association [NIRP-14-304425 (Y.Z.)] and Mayo Graduate School (J.C.). We thank Dr. Leonard Petrucelli for valuable discussions and suggestions during manuscript preparation. We also acknowledge expert technical assistance of Monica Castanedes-Casey, Linda Rousseau, and Virginia Phillips for histology.
Footnotes
Author Contributions
HS, JC and YZ conceived and designed the experiments, HS, JC, YX, TFG, WCL, KJ, POB, EP, JT performed the experiments, HS, JC and YZ analyzed the data, HS, TFG, CS and YZ wrote the paper.
Conflict of Interest
The authors declare no actual or potential conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–11. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- Ayala YM, De Conti L, Avendano-Vazquez SE, Dhir A, Romano M, D’Ambrogio A, Tollervey J, Ule J, Baralle M, Buratti E, Baralle FE. TDP-43 regulates its mRNA levels through a negative feedback loop. Embo Journal. 2011;30:277–288. doi: 10.1038/emboj.2010.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baralle M, Buratti E, Baralle FE. The role of TDP-43 in the pathogenesis of ALS and FTLD. Biochemical Society transactions. 2013;41:1536–40. doi: 10.1042/BST20130186. [DOI] [PubMed] [Google Scholar]
- Barmada SJ, Skibinski G, Korb E, Rao EJ, Wu JY, Finkbeiner S. Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J Neurosci. 2010;30:639–49. doi: 10.1523/JNEUROSCI.4988-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budini M, Romano V, Quadri Z, Buratti E, Baralle FE. TDP-43 loss of cellular function through aggregation requires additional structural determinants beyond its C-terminal Q/N prion-like domain. Human molecular genetics. 2015;24:9–20. doi: 10.1093/hmg/ddu415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratti E, De Conti L, Stuani C, Romano M, Baralle M, Baralle F. Nuclear factor TDP-43 can affect selected microRNA levels. Febs Journal. 2010;277:2268–2281. doi: 10.1111/j.1742-4658.2010.07643.x. [DOI] [PubMed] [Google Scholar]
- Chakrabarty P, Rosario A, Cruz P, Siemienski Z, Ceballos-Diaz C, Crosby K, Jansen K, Borchelt DR, Kim JY, Jankowsky JL, Golde TE, Levites Y. Capsid serotype and timing of injection determines AAV transduction in the neonatal mice brain. PLoS One. 2013;8:e67680. doi: 10.1371/journal.pone.0067680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CK, Wu TH, Wu CY, Chiang MH, Toh EK, Hsu YC, Lin KF, Liao YH, Huang TH, Huang JJ. The N-terminus of TDP-43 promotes its oligomerization and enhances DNA binding affinity. Biochem Biophys Res Commun. 2012 doi: 10.1016/j.bbrc.2012.07.071. [DOI] [PubMed] [Google Scholar]
- Chen-Plotkin AS, Lee VM, Trojanowski JQ. TAR DNA-binding protein 43 in neurodegenerative disease. Nature reviews Neurology. 2010;6:211–20. doi: 10.1038/nrneurol.2010.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen TJ, Lee VM, Trojanowski JQ. TDP-43 functions and pathogenic mechanisms implicated in TDP-43 proteinopathies. Trends Mol Med. 2011;17:659–67. doi: 10.1016/j.molmed.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombrita C, Onesto E, Megiorni F, Pizzuti A, Baralle FE, Buratti E, Silani V, Ratti A. TDP-43 and FUS RNA-binding Proteins Bind Distinct Sets of Cytoplasmic Messenger RNAs and Differently Regulate Their Post-transcriptional Fate in Motoneuron-like Cells. Journal of Biological Chemistry. 2012;287:15635–15647. doi: 10.1074/jbc.M111.333450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayton RD, Gitcho MA, Orchard EA, Wilson JD, Wang DB, Cain CD, Johnson JA, Zhang YJ, Petrucelli L, Mathis JM, Klein RL. Selective forelimb impairment in rats expressing a pathological TDP-43 25 kDa C-terminal fragment to mimic amyotrophic lateral sclerosis. Molecular therapy : the journal of the American Society of Gene Therapy. 2013;21:1324–34. doi: 10.1038/mt.2013.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dormont D, Seidenwurm DJ. Dementia and movement disorders. AJNR. American journal of neuroradiology. 2008;29:204–6. [PMC free article] [PubMed] [Google Scholar]
- Elden AC, Kim HJ, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, Armakola M, Geser F, Greene R, Lu MM, Padmanabhan A, Clay-Falcone D, McCluskey L, Elman L, Juhr D, Gruber PJ, Rub U, Auburger G, Trojanowski JQ, Lee VM, Van Deerlin VM, Bonini NM, Gitler AD. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466:1069–75. doi: 10.1038/nature09320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiesel FC, Voigt A, Weber SS, Van den Haute C, Waldenmaier A, Gorner K, Walter M, Anderson ML, Kern JV, Rasse TM, Schmidt T, Springer W, Kirchner R, Bonin M, Neumann M, Baekelandt V, Alunni-Fabbroni M, Schulz JB, Kahle PJ. Knockdown of transactive response DNA-binding protein (TDP-43) downregulates histone deacetylase 6. EMBO J. 2010;29:209–21. doi: 10.1038/emboj.2009.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiesel FC, Weber SS, Supper J, Zell A, Kahle PJ. TDP-43 regulates global translational yield by splicing of exon junction complex component SKAR. Nucleic acids research. 2012;40:2668–82. doi: 10.1093/nar/gkr1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman MS, Trojanowski JQ, Lee VM. Neurodegenerative diseases: a decade of discoveries paves the way for therapeutic breakthroughs. Nature medicine. 2004;10:1055–63. doi: 10.1038/nm1113. [DOI] [PubMed] [Google Scholar]
- Igaz LM, Kwong LK, Lee EB, Chen-Plotkin A, Swanson E, Unger T, Malunda J, Xu Y, Winton MJ, Trojanowski JQ, Lee VM. Dysregulation of the ALS-associated gene TDP-43 leads to neuronal death and degeneration in mice. The Journal of clinical investigation. 2011;121:726–38. doi: 10.1172/JCI44867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens J, Van Broeckhoven C. Pathological mechanisms underlying TDP-43 driven neurodegeneration in FTLD-ALS spectrum disorders. Human molecular genetics. 2013;22:R77–87. doi: 10.1093/hmg/ddt349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara Y, Mieda-Sato A. TDP-43 promotes microRNA biogenesis as a component of the Drosha and Dicer complexes. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:3347–3352. doi: 10.1073/pnas.1112427109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Ash RT, Ceballos-Diaz C, Levites Y, Golde TE, Smirnakis SM, Jankowsky JL. Viral transduction of the neonatal brain delivers controllable genetic mosaicism for visualising and manipulating neuronal circuits in vivo. The European journal of neuroscience. 2013;37:1203–20. doi: 10.1111/ejn.12126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo PH, Doudeva LG, Wang YT, Shen CK, Yuan HS. Structural insights into TDP-43 in nucleic-acid binding and domain interactions. Nucleic Acids Res. 2009;37:1799–808. doi: 10.1093/nar/gkp013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EB, Lee VM, Trojanowski JQ. Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nature reviews Neuroscience. 2012;13:38–50. doi: 10.1038/nrn3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu-Yesucevitz L, Lin AY, Ebata A, Boon JY, Reid W, Xu YF, Kobrin K, Murphy GJ, Petrucelli L, Wolozin B. ALS-linked mutations enlarge TDP-43-enriched neuronal RNA granules in the dendritic arbor. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2014;34:4167–74. doi: 10.1523/JNEUROSCI.2350-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–3. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Prudencio M, Jansen-West KR, Lee WC, Gendron TF, Zhang YJ, Xu YF, Gass J, Stuani C, Stetler C, Rademakers R, Dickson DW, Buratti E, Petrucelli L. Misregulation of human sortilin splicing leads to the generation of a nonfunctional progranulin receptor. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:21510–5. doi: 10.1073/pnas.1211577110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rademakers R, Neumann M, Mackenzie IR. Advances in understanding the molecular basis of frontotemporal dementia. Nature reviews Neurology. 2012;8:423–34. doi: 10.1038/nrneurol.2012.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nature reviews. Neuroscience. 2013;14:248–64. doi: 10.1038/nrn3430. [DOI] [PubMed] [Google Scholar]
- Shiina Y, Arima K, Tabunoki H, Satoh J. TDP-43 dimerizes in human cells in culture. Cellular and molecular neurobiology. 2010;30:641–52. doi: 10.1007/s10571-009-9489-9. [DOI] [PubMed] [Google Scholar]
- Strong MJ. The evidence for altered RNA metabolism in amyotrophic lateral sclerosis (ALS) Journal of the neurological sciences. 2010;288:1–12. doi: 10.1016/j.jns.2009.09.029. [DOI] [PubMed] [Google Scholar]
- Taylor JP, Hardy J, Fischbeck KH. Toxic proteins in neurodegenerative disease. Science. 2002;296:1991–5. doi: 10.1126/science.1067122. [DOI] [PubMed] [Google Scholar]
- Volkening K, Leystra-Lantz C, Yang W, Jaffee H, Strong MJ. Tar DNA binding protein of 43 kDa (TDP-43), 14-3-3 proteins and copper/zinc superoxide dismutase (SOD1) interact to modulate NFL mRNA stability. Implications for altered RNA processing in amyotrophic lateral sclerosis (ALS) Brain research. 2009;1305:168–82. doi: 10.1016/j.brainres.2009.09.105. [DOI] [PubMed] [Google Scholar]
- Walker AK, Spiller KJ, Ge G, Zheng A, Xu Y, Zhou M, Tripathy K, Kwong LK, Trojanowski JQ, Lee VM. Functional recovery in new mouse models of ALS/FTLD after clearance of pathological cytoplasmic TDP-43. Acta neuropathologica. 2015;130:643–60. doi: 10.1007/s00401-015-1460-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang IF, Wu LS, Chang HY, Shen CKJ. TDP-43, the signature protein of FTLD-U, is a neuronal activity-responsive factor. Journal of Neurochemistry. 2008;105:797–806. doi: 10.1111/j.1471-4159.2007.05190.x. [DOI] [PubMed] [Google Scholar]
- Wang YT, Kuo PH, Chiang CH, Liang JR, Chen YR, Wang S, Shen JC, Yuan HS. The truncated C-terminal RNA recognition motif of TDP-43 protein plays a key role in forming proteinaceous aggregates. The Journal of biological chemistry. 2013;288:9049–57. doi: 10.1074/jbc.M112.438564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winton MJ, Igaz LM, Wong MM, Kwong LK, Trojanowski JQ, Lee VM. Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J Biol Chem. 2008;283:13302–9. doi: 10.1074/jbc.M800342200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu YF, Gendron TF, Zhang YJ, Lin WL, D’Alton S, Sheng H, Casey MC, Tong J, Knight J, Yu X, Rademakers R, Boylan K, Hutton M, McGowan E, Dickson DW, Lewis J, Petrucelli L. Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010;30:10851–9. doi: 10.1523/JNEUROSCI.1630-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu YF, Prudencio M, Hubbard JM, Tong J, Whitelaw EC, Jansen-West K, Stetler C, Cao X, Song J, Zhang YJ. The pathological phenotypes of human TDP-43 transgenic mouse models are independent of downregulation of mouse Tdp-43. PLoS One. 2013;8:e69864. doi: 10.1371/journal.pone.0069864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YJ, Xu YF, Cook C, Gendron TF, Roettges P, Link CD, Lin WL, Tong J, Castanedes-Casey M, Ash P, Gass J, Rangachari V, Buratti E, Baralle F, Golde TE, Dickson DW, Petrucelli L. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc Natl Acad Sci U S A. 2009;106:7607–12. doi: 10.1073/pnas.0900688106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YJ, Caulfield T, Xu YF, Gendron TF, Hubbard J, Stetler C, Sasaguri H, Whitelaw EC, Cai S, Lee WC, Petrucelli L. The dual functions of the extreme N-terminus of TDP-43 in regulating its biological activity and inclusion formation. Human molecular genetics. 2013;22:3112–22. doi: 10.1093/hmg/ddt166. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video 1: GFP-TDP-43-NLSm 2.5e10 P28
Video 2: GFP 2.5e10 P28
Video 3: GFP-TDP-43-10-414-NLSm 5e10 P28