

Abstract
Objective
To explore the transcriptome of epicardial adipose tissue (EAT) as compared to subcutaneous adipose tissue (SAT) and its modifications in a small number of patients with coronary artery disease (CAD) versus valvulopathy.
Design and Methods
SAT and EAT samples were obtained during elective cardiothoracic surgeries. The transcriptome of EAT was evaluated using an unbiased, whole-genome approach as compared to SAT in subjects with CAD (n=6) and without CAD (n=5), where the patients without CAD had cardiac valvulopathy.
Results
Relative to SAT, EAT is a highly inflammatory tissue enriched with genes involved in endothelial function, coagulation, immune signaling, potassium transport and apoptosis. EAT is lacking in expression of genes involved in protein metabolism, TGF-beta signaling, and oxidative stress. Although underpowered, in subjects with severe CAD, there is an expression trend suggesting widespread downregulation of EAT encompassing a diverse group of gene sets related to intracellular trafficking, proliferation/transcription regulation, protein catabolism, innate immunity/lectin pathway and ER stress.
Conclusions
The EAT transcriptome is unique when compared to SAT. In the setting of CAD versus valvulopathy, there is possible alteration of the EAT transcriptome with gene suppression. This pilot study explores the transcriptome of EAT in CAD and valvulopathy, providing new insight into its physiologic and pathophysiologic roles.
Keywords: Adipose Tissue, Genetics, Coronary artery disease, Adiposity, Subcutaneous Adipose Tissue, epicardial fat, microarray, transcriptome
Introduction
The adipose organ plays an active role in the development and progression of cardiometabolic syndromes. It is well accepted that anatomically distinct fat depots differ in their biomolecular properties and contribution to disease; considerable data suggests metabolically detrimental effects of increased visceral adipose tissue (VAT). Epicardial adipose tissue (EAT) is the VAT of the heart (1) and is present in humans and other large mammals, but not in rats or mice (2). EAT has unique embryologic, anatomic, and histologic features (3), including smaller adipocytes (4) and higher prevalence of immune cells when compared to subcutaneous adipose tissue (SAT) (5). EAT lies in direct contiguity with the myocardium without fascial barrier (Figure 1) such that some EAT adipocytes even appear to invaginate the epicardial surface. The myocardium and EAT are supplied by branches of the coronary arteries whereas other paracardiac adipose depots are located external to the parietal pericardium (Figure 1) and are supplied by branches of noncoronary arteries (2). Hence, the physical proximity, lack of fascial barrier and shared microcirculation potentially allows for a bi-directional crosstalk through paracrine and vasocrine pathways (6, 7).
Figure 1. Epicardial adipose tissue (EAT) is an anatomically distinct adipose depot.
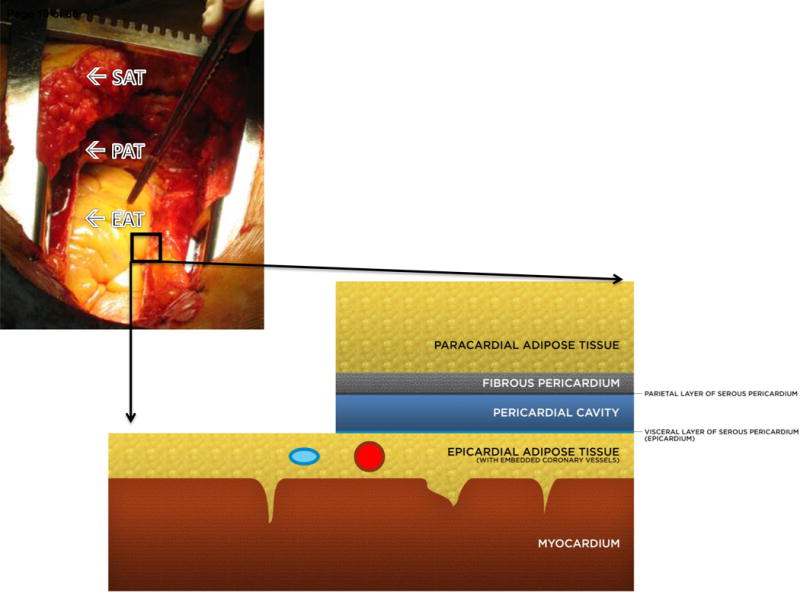
EAT lies on the myocardial surface without fascial separation such that epicardial adipocytes are in direct contact with the myocardium, even appearing to invaginate the myocardial surface on histology. In healthy adult subjects, EAT lies in the atrioventricular grooves and follows the branches of the major coronary arteries such that the coronary vessels are embedded within the EAT, although some patients may present with a massive epicardial fat pad as shown. Superficial to the epicardial fat are the visceral and parietal layers of the pericardial sac. Paracardial adipose tissue (PAT) is superficial to the pericardium and thus has no direct anatomical contact with the EAT or myocardium; it also has a separate microcirculation from EAT, as EAT shares the microcirculation of the heart. Paracardial fat is alternatively termed mediastinal fat. Although EAT and PAT are anatomically distinct, they are sometimes discussed under the umbrella term pericardial fat which refers to epicardial plus paracardial fat (6).
Its unique features and the clinical evidence that EAT thickness might serve as a marker of visceral adiposity (1), have sparked interest in defining its biomolecular properties. EAT is enriched with saturated fatty acids, has high protein content, and has greater capacity for lipogenesis and fatty acid metabolism (2, 8, 9). Most studies have evaluated the EAT in subjects with CAD and thus the nature of the EAT-myocardial relationship in normal conditions is not clear. It has been hypothesized in CAD that EAT could negatively affect the heart via macrophage activation, oxidative stress, innate inflammatory response and plaque destabilization (7, 10, 11, 12). Clinically, EAT as measured by echocardiography or CT has been associated with increased cardiovascular risk, CAD (13) and metabolic syndrome (14) independent of traditional risk factors.
Microarray offers an unbiased approach to the study of genomewide expression analysis; the relevance is strengthened when gene expression is considered in the context of mechanistically related gene sets (15). Few analyses of EAT’s transcriptome have been performed. In one study, a microarray of EAT versus SAT in CAD patients found that upregulated genes in EAT were associated with the inflammatory and immune responses, whereas downregulated genes were adipocyte-related (5). In other patients, a microarray of EAT compared to SAT in both CAD and nonCAD subjects found the highest ranked gene encoding for a secreted protein to be secretory type II phospholipase A2, the rate limiting enzyme in the synthesis of a proinflammatory lipid mediator (16). Lastly, another study evaluated the genome-wide mRNA profile of EAT versus mediastinal adipose tissue (MAT, or paracardial adipose tissue) and SAT in men with CAD and found enrichment in genes involved in cardiovascular disease including ADORA1 (17). In the present study, microarrays were performed and the data evaluated to i) comprehensively define the EAT transcriptome compared to SAT and ii) determine the transcriptional profile of EAT of CAD subjects compared to the EAT of subjects without CAD but with cardiac valve disease.
Methods and Procedures
Subjects
Once weekly from March, 2012 through April, 2013, consecutive patients undergoing elective cardiothoracic surgery (CABG or cardiac valve replacement) who provided informed consent were enrolled to participate; this enrollment approach resulted in 23 sequentially enrolled patients. The study was conducted in accordance with The Declaration of Helsinki and the protocol approved by the University of Miami IRB. Participants were analyzed in two groups based on clinical findings indicating the lack or presence of CAD on preoperative testing. Of the 23 participants enrolled, 18 patients (14M:4F) aged 45–79 (57.8 ± 8.8 years) were found to have severe CAD (triple-vessel involvement meeting criteria for CABG) on preoperative cardiac angiography; this was termed the CAD group. The other five patients (3M:2F), aged 33–58 (45.0 ± 9.3 years) in the valvulopathy group had no clinical signs of CAD and normal coronary arteries on preoperative angiography; these patients were undergoing cardiac valve replacement (4 mitral: 1 tricuspid). Clinical characteristics are shown (Table 1). Laboratory analyses and anthropometrics were performed during routine preoperative evaluation and fasting blood samples were obtained. In this cross-sectional study, clinical data was obtained at perioperative evaluation (adipose samples obtained during surgery). Clinical outcomes were not followed beyond surgery.
Table 1.
Anthropometric and Clinical Characteristics of Participants
| CAD–All BMI (n = 18) | CAD—microarray subgroup (n = 6) | Valvulopathy (n=5) | p value | |
|---|---|---|---|---|
| Age (years) | 57.8 (8.8) | 60.0 (10.8) | 45.0 (9.33) | 0.0498 |
| F:M | 4:14 | 2:4 | 2:3 | >0.999 |
| BMI (kg/m2) | 30.5 (6.2) | 25.7 (2.6) | 23.4 (2.5) | 0.1255 |
| Waist (cm) | 41.9 (6.5) (n=15) | 36.8 (3.6) (n =5) | 34.5 (4.1) (n=4) | 0.3730 |
| SBP (mmHg) | 129.2 (14.8) | 133.2 (17.7) | 110.8 (17.1) | 0.0476 |
| DBP (mmHg) | 68.9 (11.2) | 66.8 (7.1) | 73.4 (8.8) | 0.2424 |
| Glucose (mg/dl) | 133.6 (47.9) | 140.0 (49.7) | 85.0 (9.6) | 0.0173 |
| Total cholesterol (mg/dl) | 213.0 (60.6) (n=17) | 198.3 (30.5) | 117.7 (22.0)(n=3) | 0.0238 |
| HDL (mg/dl) | 48.2 (17.1) (n=17) | 46.8 (18.4) | 28.0 (16.5) (n = 3) | 0.3333 |
| Triglycerides (mg/dl) | 145.4 (77.5) (n = 17) | 121.8 (40.0) | 94.3 (31.0) (n=3) | 0.5952 |
| LDL (mg/dl) | 135.7 (58.8) (n = 17) | 127.2 (34.8) | 71.0 (22.5) (n = 3) | 0.0238 |
| EATt (mm) | 7.14 (2.41) (n = 16) | 8.87 (2.60) | 7.34 (3.19) | 0.5108 |
| Risk factors: | ||||
| Dyslipidemia | 6/18 | 3/6 | 1/5 | 0.5455 |
| Diabetes | 13/18 | 5/6 | 0/5 | 0.0152 |
| HTN | 15/18 | 6/6 | 2/5 | 0.0606 |
| Medications: | ||||
| ACE-Inhibitors | 11/18 | 4/6 | 1/5 | 0.2424 |
| ARBs | 3/18 | 1/6 | 0/5 | >0.999 |
| Beta-Blockers | 14/18 | 5/6 | 3/5 | 0.5455 |
| Statins | 12/18 | 3/6 | 0/5 | 0.1818 |
| Fibrates | 1/18 | 0/6 | 1/5 | 0.4545 |
| Nitroderivates | 5/18 | 3/6 | 1/5 | 0.5455 |
| Metformin | 3/18 | 1/6 | 0/5 | >0.999 |
| Insulin | 9/18 | 3/6 | 1/5 | 0.5455 |
| Antiplatelet | 15/18 | 4/6 | 2/5 | 0.5671 |
Values presented as mean (SD); n is indicated at the column header unless otherwise stated (in cases where the value was not obtained from all participants). The ‘CAD—all BMI’ column represents the characteristics of all of the enrolled participants within the CAD group, whereas the ‘CAD-microarray subgroup’ column represents these data from the patients selected for microarray analysis based on BMI-matching to valve patients; p values correspond to the comparison of ‘CAD-microarray subgroup’ and valvulopathy subjects. The data were analyzed using a Mann-Whitney test; Fisher’s exact test was used for categorical data. Blood samples were drawn pre-operatively from fasting patients. CAD, coronary artery disease; F:M, female:male; BMI, body mass index; mmHg, millimeters of mercury; SBP, systolic blood pressure; DBP, diastolic blood pressure; HDL, high-density lipoprotein; LDL, low-density lipoprotein; EATt, epicardial adipose tissue thickness; ACE, angiotensin converting enzyme; ARB, angiotensin II receptor antagonist; antiplatelet: aspirin and/or clopidogrel.
Epicardial Fat Thickness
Subjects had preoperative echocardiographic measurements of EAT thickness, by the previously described method (18). Briefly, EAT was identified as the echo-free space between the outer wall of the myocardium and the visceral layer of pericardium. Maximum EAT thickness was measured perpendicularly on the free wall of the right ventricle at end-systole.
Adipose tissue collection
Subcutaneous and epicardial adipose samples were collected during cardiothoracic surgeries of all 23 patients. SAT samples were obtained from the median sternotomy site and EAT samples (~ 1 gram) were obtained near the proximal segment of the right coronary artery, deep to the visceral layer of the pericardium (Figure 1) prior to the patients being placed on-pump. Each tissue sample was immediately frozen over dry ice and then stored at −80°C until processing.
RNA extraction
RNA was extracted from frozen samples; samples were homogenized on ice with TRIzol Reagent (Invitrogen, Carlsbad, CA). RNA was extracted using a kit for lipid-containing tissues (Qiagen RNeasy Lipid Tissue Mini Kit, Germantown, MD) following the manufacturer’s protocol.
Microarray
From the 5 valvulopathy patients, all SAT and EAT samples were analyzed by microarray. Only a subset of samples from the CAD group were analyzed by microarray; from the 18 CAD patients, 6 were chosen by BMI-matching compared to the valvulopathy group (these were the 6 with the lowest BMIs). From these 6 CAD patients, SAT and EAT samples were analyzed by microarray. Table 1 shows clinical characteristics of the subset of patients analyzed by microarray. Gene expression was evaluated using Genechip Human Gene 2.0 ST arrays (Affymetrix) which utilizes a whole-transcript design to assess >30,000 coding genes. Microarray was performed at the Joslin Diabetes Center Genomics Core Laboratory (Boston, MA).
Microarray analysis
Gene expression data was preprocessed using GenePattern (Broad Institute, Cambridge, MA). Differential analysis was performed in GenePattern to identify individual genes demonstrating enrichment in two separate comparisons: EAT versus SAT and EATCAD versus EATVal. The Student’s t-test yielded p-values for individual genes. Select genes were highlighted in Table 2. Then, to discern functional patterns among enriched genes, gene ontology analysis was used to determine enrichment of gene sets (Gene Set Enrichment Analysis (GSEA), Broad Institute)(15). No filter was applied to eliminate genes with low expression. GSEA included calculation of enrichment scores (ES), estimation of significance level of ES (nominal p-value), and adjustment for multiple hypothesis testing including the normalized enrichment score (NES) and FDR (15). Gene sets with an FDR ≤25% were limited and the goal was to generate hypotheses, so a nominal p-value of <1% was chosen to indicate significance for gene sets (Figure 2). Core enrichment of individual genes was defined as those genes contributing to the leading-edge subset within the gene set (Tables S1, S2). All individual genes demonstrating core enrichment within enriched gene sets were further considered by investigators (investigator analysis) consisting of searches in publically available databases (PubMed, UniProt) to determine the context in which each gene had been previously evaluated. Gene sets enriched in EAT versus SAT are listed (Table 3) and for EATCAD versus EATVal (Table 4).
Table 2.
Expression of select individual genes by differential analysis.
| EAT versus SAT | ||||||
|---|---|---|---|---|---|---|
| Gene name | Fold Change | P value | FDR | Mean Expression | ||
| EAT (n = 11) |
SAT (n = 11) |
|||||
| Enriched in EAT | ITLN1 | 34.8 | <0.001 | 0.011 | 1726 ± 1559 | 50 ± 11 |
| CCL21 | 11.1 | <0.001 | 0.007 | 5633 ± 3157 | 506 ± 346 | |
| KCNN3 | 2.47 | <0.001 | 0.007 | 180 ± 47 | 73 ± 10 | |
| PLA2G2A | 2.23 | 0.001 | 0.020 | 855 ± 341 | 384 ± 214 | |
| PLAT | 1.84 | <0.001 | 0.011 | 1037 ± 302 | 563 ± 184 | |
| KCNA3 | 1.65 | 0.004 | 0.049 | 265 ± 87 | 161 ± 53 | |
| KCNK17 | 1.57 | <0.001 | 0.007 | 124 ± 22 | 79 ± 7 | |
| CXCR5 | 1.31 | 0.001 | 0.017 | 42 ± 9 | 32 ± 3 | |
| CCR2 | 1.43 | 0.002 | 0.030 | 127 ± 25 | 89 ± 20 | |
| ADORA1 | 1.16 | 0.044 | 0.238* | 96 ± 16 | 82 ± 13 | |
| Enriched in SAT | MAOB | 2.73 | <0.001 | 0.007 | 318 ± 102 | 870 ± 158 |
| LOXL2 | 2.02 | <0.001 | 0.007 | 230 ± 52 | 467 ± 124 | |
| LOXL4 | 1.63 | <0.001 | 0.007 | 138 ± 20 | 226 ± 43 | |
| SMAD7 | 1.51 | 0.003 | 0.038 | 206 ± 64 | 312 ± 78 | |
| USP30 | 1.43 | <0.001 | 0.007 | 168 ± 27 | 240 ± 33 | |
| USP14 | 1.31 | <0.001 | 0.007 | 839 ± 98 | 1099 ± 166 | |
| SOD1 | 1.31 | <0.001 | 0.007 | 1337 ± 198 | 1747 ± 206 | |
| SMAD2 | 1.23 | <0.001 | 0.011 | 727 ± 70 | 897 ± 94 | |
| TGFBR3 | 1.19 | 0.003 | 0.042 | 1775 ± 221 | 2120 ± 247 | |
| SMURF1 | 1.17 | 0.003 | 0.038 | 487 ± 69 | 572 ±39 | |
| EATCAD versus EATVal | ||||||
| EATCAD (n = 6) |
EATVal (n = 5) |
|||||
| Enriched in EATCAD | TECRL | 14.6 | 0.022 | 0.999* | 147 ± 331 | 10 ± 1 |
| CTSE | 2.51 | 0.034 | 0.999* | 247 ± 144 | 98 ± 44 | |
| FLRT3 | 2.48 | 0.005 | 0.999* | 182 ± 111 | 74 ± 30 | |
| TRDN | 2.43 | 0.028 | 0.999* | 139 ± 92 | 58 ± 23 | |
| HSPB7 | 2.25 | 0.005 | 0.925* | 722 ± 183 | 321 ± 160 | |
| Enriched in EAT-Val | O3FAR1 | 1.73 | <0.001 | 0.114* | 159 ± 25 | 274 ± 58 |
| CD33 | 1.73 | <0.001 | 0.114* | 233 ± 49 | 402 ± 75 | |
| ENG | 1.35 | <0.001 | 0.114* | 1431 ± 204 | 1927 ± 312 | |
| CLEC9A | 1.28 | <0.001 | 0.114* | 22 ± 1 | 28 ± 6 | |
| MAP3K1 | 1.22 | <0.001 | 0.114* | 834 ± 76 | 1019 ± 123 | |
The signal intensity for each individual gene was measured by microarray and analyzed using GenePattern differential analysis. Expression data are presented as mean ± SD. A 2-sided student’s t-test of paired samples was used to calculate p-values in GenePattern as 2signal(EAT-SAT) or 2signal(EATCAD-EATVal). Select genes from this differential analysis are listed with their p-values, FDR and fold changes.
indicates genes with an FDR ≥5% that were included to facilitate discussion.
Figure 2. Microarray analysis protocol.
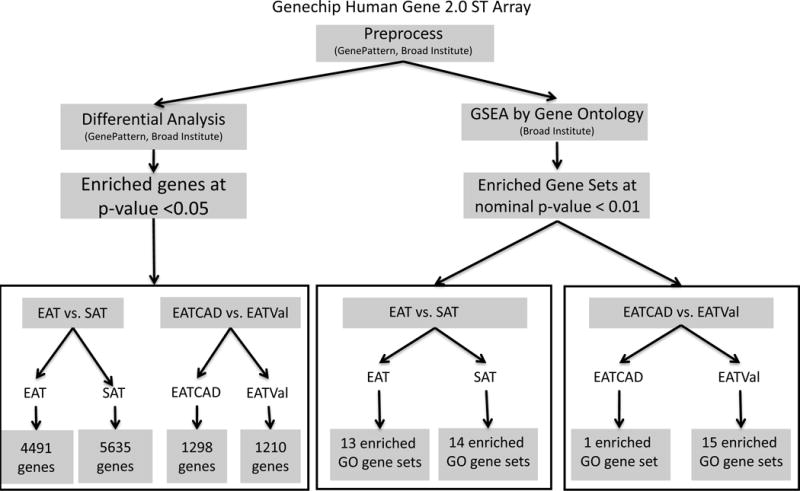
Gene expression was evaluated using Genechip Human Gene 2.0 ST arrays (Affymetrix, Inc., Santa Clara, CA) which utilizes a whole-transcript design to assess >30,000 coding genes. The resulting data files were preprocessed with GenePattern (www.broadinstitute.org, Cambridge, MA). Files were then analyzed by two methods. Differential analysis identified individual genes with enrichment and GSEA identified enriched gene sets by gene ontology. Individual genes were considered statistically significant with an FDR <0.05 in the EAT versus SAT analysis, and in the EATCAD versus EATVal analysis at an FDR <0.15. Gene sets were considered enriched at statistical significance with nominal p-value <1%. Two comparisons were made: EAT versus SAT to define the transcriptome of EAT and EATCAD versus EATVal to clarify how the transcriptome of EAT is modified in CAD. GO: gene ontology; EAT: epicardial adipose tissue; SAT: subcutaneous adipose tissue; CAD: coronary artery disease; Val: valvulopathy.
Table 3.
Significantly enriched gene sets in EAT versus SAT.
| Enriched in EAT compared to SAT at nominal p value < 1% | |||||||
|---|---|---|---|---|---|---|---|
| GO ID | GO term | Genes with core enrichment | Total # genes in set | ES | NES | NOM p-val | FDR q-val |
| GO:0007498 | Mesoderm development | 10 | 22 | 0.64 | 1.64 | 0.002 | 0.619 |
| GO:0004620 | Phospholipase activity | 12 | 41 | 0.51 | 1.62 | 0.004 | 0.656 |
| GO:0005267 | Potassium channel activity | 18 | 49 | 0.47 | 1.56 | 0.006 | 0.740 |
| GO:0007599 | Hemostasis | 17 | 46 | 0.58 | 1.66 | 0.010 | 0.537 |
| GO:0050817 | Coagulation | 16 | 42 | 0.61 | 1.71 | 0.008 | 0.512 |
| GO:0007596 | Blood coagulation | 16 | 41 | 0.61 | 1.69 | 0.008 | 0.558 |
| GO:0003704 | Specific RNA polymerase II transcription factor activity | 6 | 34 | 0.54 | 1.81 | 0.004 | 0.708 |
| GO:0050878 | Regulation of body fluid levels | 18 | 55 | 0.54 | 1.67 | 0.008 | 0.561 |
| GO:0042060 | Wound healing | 18 | 51 | 0.59 | 1.74 | <0.001 | 0.870 |
| GO:0016860 | Intramolecular oxidoreductase activity | 10 | 19 | 0.7 | 1.74 | 0.008 | 0.607 |
| GO:0008624 | Induction of apoptosis by extracellular signals | 8 | 26 | 0.57 | 1.73 | 0.006 | 0.539 |
| GO:0016337 | Cell cell adhesion | 15 | 82 | 0.36 | 1.46 | 0.004 | 0.500 |
| GO 0007267 | Cell cell signaling | 94 | 389 | 0.33 | 1.35 | 0.008 | 0.447 |
| Enriched in SAT compared to EAT at nominal p value < 1% | |||||||
| GO:0044257 | Cellular protein catabolic process | 34 | 55 | −0.62 | −1.87 | 0.002 | 0.157 |
| GO:0030163 | Protein catabolic process | 36 | 63 | −0.59 | −1.88 | 0.002 | 0.262 |
| GO:0007179 | Transforming growth factor beta receptor Signaling pathway | 15 | 35 | −0.63 | −1.75 | 0.002 | 0.241 |
| GO:0015457 | Auxiliary Transport Protein Activity | 7 | 24 | −0.6 | −1.79 | <0.001 | 0.227 |
| GO:0007584 | Response to nutrient | 5 | 17 | −0.60 | −1.67 | 0.004 | 0.361 |
| GO:0048037 | Cofactor binding | 8 | 20 | −0.73 | −1.73 | 0.008 | 0.251 |
| GO:0016247 | Channel regulator activity | 6 | 22 | −0.53 | −1.58 | 0.008 | 0.613 |
| GO:0043406 | Positive regulation of MAP kinase activity | 8 | 46 | −0.45 | −1.52 | 0.008 | 0.491 |
| GO:0008639 | Small protein conjugating enzyme activity | 25 | 50 | −0.49 | −1.71 | 0.010 | 0.297 |
| GO:0051402 | Neuron apoptosis | 4 | 17 | −0.49 | −1.58 | 0.010 | 0.583 |
| GO:0016879 | Ligase activity forming carbon nitrogen bonds | 31 | 67 | −0.47 | −1.69 | 0.010 | 0.333 |
| GO:0007178 | Transmembrane receptor protein serine threonine kinase signaling pathway | 23 | 46 | −0.57 | −1.68 | 0.010 | 0.339 |
| GO:0016881 | Acid amino acid ligase activity | 26 | 56 | −0.52 | −1.80 | 0.006 | 0.263 |
| GO:0009653 | Anatomical structure morphogenesis | 73 | 360 | −0.30 | −1.37 | 0.004 | 0.444 |
GO, gene ontology; ES, enrichment score; NES, normalized enrichment score; FDR q-val, false discovery rate q-value; NOM p-val, nominal p value.
Table 4.
Significantly enriched gene sets in EATCAD versus EATVal.
| Enriched in EAT of CAD patients compared to EAT of valve patients at nominal p value < 1% | |||||||
|---|---|---|---|---|---|---|---|
| GO ID | GO term | Genes with core enrichment | Total # genes in set | ES | NES | NOM p-val | FDR q-val |
| GO:0050906 | Detection of stimulus involved in sensory perception | 7 | 21 | 0.57 | 1.79 | 0.008 | 0.306 |
| Enriched in EAT of valve patients compared to EAT of CAD patients at nominal p value < 1% | |||||||
| GO:0030166 | Proteoglycan biosynthetic process | 4 | 15 | −0.71 | −1.85 | <0.001 | 0.265 |
| GO:0008080 | N acyltransferase activity | 12 | 21 | −0.59 | −1.74 | <0.001 | 0.226 |
| GO:0008270 | Zinc ion binding | 22 | 85 | −0.43 | −1.65 | <0.001 | 0.399 |
| GO:0016044 | Membrane organization and biogenesis | 50 | 130 | −0.40 | −1.59 | <0.001 | 0.504 |
| GO:0046914 | Transition metal ion binding | 23 | 105 | −0.35 | −1.47 | 0.002 | 0.53 |
| GO:0051128 | Regulation of cellular component organization and biogenesis | 41 | 116 | −0.38 | −1.58 | 0.004 | 0.473 |
| GO:0051347 | Positive regulation of transferase activity | 29 | 84 | −0.4 | −1.43 | 0.004 | 0.585 |
| GO:0006984 | ER nuclear signaling pathway | 8 | 16 | −0.6 | −1.78 | 0.006 | 0.273 |
| GO:0008234 | Cysteine type peptidase activity | 20 | 53 | −0.52 | −1.77 | 0.006 | 0.238 |
| GO:0008144 | Drug binding | 7 | 15 | −0.51 | −1.7 | 0.006 | 0.314 |
| GO:0006508 | Proteolysis | 62 | 181 | −0.39 | −1.61 | 0.006 | 0.466 |
| GO:0004197 | Cysteine type endopeptidase activity | 15 | 39 | −0.53 | −1.76 | 0.008 | 0.223 |
| GO:0006944 | Membrane fusion | 14 | 28 | −0.61 | −1.7 | 0.008 | 0.3 |
| GO:0016540 | Protein autoprocessing | 11 | 31 | −0.55 | −1.6 | 0.008 | 0.465 |
| GO:0008233 | Peptidase activity | 51 | 168 | −0.36 | −1.51 | 0.008 | 0.59 |
GO, gene ontology; ES, enrichment score; NES, normalized enrichment score; FDR q-val, false discovery rate q value; NOM p-val, nominal p value.
Statistical analysis
Clinical data were analyzed using PRISM software (GraphPad, San Diego, CA). Comparisons between groups were performed using the Mann-Whitney test or Fisher’s exact test where differences with p<0.05 were considered statistically significant for clinical data. Microarray statistical analysis was performed as above.
For statistical power analysis, the raw microarray data was log2-transformed and baseline-transformed to median of all samples using Agilent Genespring 12.6. Welch’s t-test was performed for each individual gene, then the p-values were corrected using Benjamini-Hochberg FDR. For gene sets, the gene values for each sample were averaged. Cohen’s d was computed using group means and pooled standard deviation for the averaged or individual gene values. For individual genes, statistical power was computed from Cohen’s d and an FDR-corrected significance level of 0.05. For gene sets, statistical power was computed from an FDR-corrected significance level of 0.05. The pwr package of R was used for power calculations.
Results
CAD study participants exhibit features of metabolic syndrome
Over the course of about one year 18 patients with CAD (14M:4F) aged 45–79 (57.8 ± 8.8 years) and 5 patients with valve disease (3M:2F) aged 33–58 (45.0 ± 9.3 years) were studied. Patients were defined as either CAD or valvulopathy based on the presence or lack of CAD/valvulopathy on preoperative coronary angiography. CAD patients had ~25% higher BMI, ~20% larger waist circumference, ~50% higher fasting glucose, ~80% higher total cholesterol, and ~90% higher low-density lipoprotein. None of the valve patients had a diagnosis of diabetes whereas > 70% of the CAD patients were diabetics. Further studies were performed in a subset of 6 CAD patients and all of the 5 valvulopathy patients (Table 1). These groups of patients were matched for BMI but the subset of CAD patients was older (~33%), had higher systolic blood pressure (~15%), fasting glucose (~60%), total serum cholesterol (~65%), and LDL (~75%) compared to valve patients. Most of the CAD patients carried diagnoses of diabetes but none of the valve patients were known to be diabetic. EAT thickness did not differ between these groups.
EAT and SAT samples were evaluated using Genechip Human Gene 2.0 ST arrays. Differential analysis was used to identify enrichment of individual genes at a p-value <0.05 and GSEA was used to determine enrichment of gene ontology sets where a nominal p-value <1% was chosen to indicate significance. Determination of core enrichment of individual genes within these sets was according to GSEA criteria, where an enriched gene contributed to the leading-edge subset within the gene set. All individual genes with core enrichment were further considered investigators. This consisted of searches in public databases to determine the context in which each gene had been previously evaluated. The samples are referred to as follows: EAT of CAD patients (EATCAD), EAT of valvulopathy patients (EATVal), SAT of CAD patients (SATCAD) and SAT of valvulopathy patients (SATVal). Two different differential analysis and GSEA comparisons were made: EAT versus SAT and EATCAD versus EATVal (Figure 2).
EAT exhibits a unique transcriptome
The first approach was to compare the EAT and the SAT samples from the 11 (6 CAD and 5 valve) patients. Differential analysis revealed enrichment at a p-value <0.05 of 4491 genes in EAT and 5635 genes in SAT; select individual genes are highlighted for discussion and shown in Table 2. Only 1420 of these genes had a fold change of expression that was detectable with significance <5% and 80% power. GSEA revealed enrichment at nominal p-value <1% for 13 gene sets in EAT and 14 gene sets in SAT (Table 3) at adequate power; three of these sets (GO:0015457/auxiliary transport protein activity, GO:0044257/cellular protein catabolic process, and GO:0007179/transforming growth factor beta (TGF-beta) receptor signaling pathway) were additionally enriched at an FDR q-value <25% in SAT (Table 3). The individual genes demonstrating core enrichment by GSEA within the 13 enriched gene sets in EAT are listed (Table S1) and within the 14 enriched gene sets in SAT (Table S2). The 50 most enriched individual genes are displayed by heat map (Figure 3).
Figure 3. Heat map of the 50 most enriched genes in EAT samples compared with SAT samples.

Heat mapping demonstrates the unique features of EAT where expression values are represented as colors (red: increased expression; blue: decreased expression) with the degree of color saturation indicating the degree of expression. Heat map generated by Broad Institute’s Gene Set Enrichment Analysis. EAT: epicardial adipose tissue; SAT: subcutaneous adipose tissue.
Gene expression in the EAT of CAD patients may be downregulated
Within EAT samples, a comparison of CAD and valvulopathy subjects was performed (EATCAD versus EATVal) to generate hypotheses on the modifications in gene expression associated with CAD. To provide a comparison between the transcriptome of EATCAD and EATVal, the 6 samples from EATCAD were compared against the 5 EATVal samples as described (Figure 2). Although our study was underpowered for this comparison, the results provide a foundation for hypothesis generation and can be considered exploratory.
In the EATCAD versus EATVal, there were 1298 individual genes enriched in EATCAD and 1210 genes in EATVal (Table 2). GSEA revealed enrichment for 1 gene set in EATCAD and 15 gene sets in EATVal (Table 3); although differential analysis and gene set analysis in these comparison were underpowered. The individual genes displaying core enrichment within the 1 enriched gene set in EATCAD, just 7 genes, are listed in Table S3, the individual genes with core enrichment within the 15 enriched gene sets in EATVal are listed in Table S4. A heat map of the 50 most enriched genes is shown in Figure 4.
Figure 4. Heat map of the 50 most enriched genes in EATCAD samples compared with EATVal samples.

Heat mapping demonstrates the unique features of EAT where expression values are represented as colors (red: increased expression; blue: decreased expression) with the degree of color saturation indicating the degree of expression. Heat map generated by Broad Institute’s Gene Set Enrichment Analysis. EAT: epicardial adipose tissue; CAD: coronary artery disease; Val: valvulopathy.
Discussion
The comparison of EAT to SAT samples (from CAD and valvulopathy patients) yields the transcriptional identity of EAT in these patients. Although the majority of enriched genes are related to endothelial function, coagulation, and inflammation, the enrichment of many genes is unexpected such as those involved in potassium transport (Table 3). The genes less represented in EAT, and thus demonstrating core enrichment in SAT, were related to protein metabolism including ubiquitin and proteasome components, oxidative stress, and TGF-beta signaling. In addition to defining the EAT transcriptome, this study served to explore the transcriptional profile of the EAT in subjects with severe CAD versus subjects without CAD but with valvulopathy. This analysis, although underpowered for definitive conclusions, found a trend toward suppression of diverse genes and gene sets involved in housekeeping cellular functions in the EAT of CAD patients, suggesting a global downregulation of the tissue (Table 4).
Inflammatory markers in the EAT transcriptome include T-cell and macrophage markers as well as B-cell-associated factors like TGFbeta2 (Table S1). Multiple chemokine ligands and chemokine receptors including CCL21, CXCR5 and CCR2 (Table 2) were expressed in EAT; chemokine expression in fat is associated with obesity and increased systemic inflammation (19). This supports data from previous studies demonstrating inflammatory infiltrates in the EAT (5, 12, 20). The gene with the highest fold change (34.8, FDR 0.01) in EAT in comparison to SAT was ITLN1, omentin, an adipokine expressed in stomal vascular cells of omental tissue (21).
Endothelial function and coagulation enrichment further demonstrates the inflammatory properties of EAT. This likely reflects a difference in gene expression between peripheral versus the cardiac vasculature, as the EAT is supplied by branches of the coronary arteries (3). There were many enriched genes related to coagulation in the EAT; most were classified into the hemostasis (GO:0007599) and coagulation (GO:0050817) gene sets. These included tissue plasminogen activator (PLAT, Table 2) an inhibitor of endogenous fibrinolysis that links fibrinolysis and inflammation in fat (22) and impairs fat development (23).
Unexpected gene sets included the potassium channel activity set (GO:0005267). Of these enriched genes, several are of particular interest (Table 2). KCNK17 has been noted to be overexpressed in human BAT compared to WAT (24) but is a nonspecific marker (25); this gene has also been associated with cerebrovascular events in a genome-wide study (26). KCNA3 plays a role in immunomodulation of effector memory T cells (27); this gene is also involved in glucose tolerance and insulin sensitivity (28). Another gene in this group that may suggest a link to inflammation is KCNN3; it is involved in endothelial function and is downregulated in hypoxia (29).
In agreement with previous studies we found that EAT was enriched in secretory type II phospholipase A2 (sPLA2-IIA or PLA2G2A) (16) at more than twice its mean expression in SAT (Table 2). ADORA1 as highlighted in previous microarray of EAT(17) did not reach significance at the FDR <0.05 level as an individual gene (Table 2); this receptor is implicated in cardioprotection in the face of ischemia (30, 31).
The genes downregulated in EAT, thus enriched in SAT, were involved in processes including protein metabolism, TGF-beta signaling and oxidative stress (Table 2, Table S2). The cellular protein catabolic process gene set (GO:0044257) was enriched not only at a nominal p-value of <1% but also at an FDR of <25%; the individual genes demonstrating core enrichment in this set are involved in ubiquitin pathways and proteasome-mediated degradation. The ubiquitin/proteasome pathway has been implicated in obesity and insulin resistance (32). Furthermore, ubiquitin removal is related to the TGF-beta pathway and SMAD; all of these pathways are downregulated in EAT (33).
Markers of oxidative stress are implicated in insulin resistance (34) and increased in obesity and type 2 diabetes. Several genes related to oxidation/detoxification were enriched in the SAT, particularly represented in the cofactor binding gene set (GO:0048037). AOC3, MAOB and LOXL2 and -4 were also enriched (Table 2). SOD1 was of particular interest as it has been shown to prevent oxidation of LDL in the arterial wall (Table 2) (35).
The EATCAD versus EATVal transcriptome analyses were limited due to small sample size but the results may lay the foundation for future studies and hypothesis generation. Differential analysis in the EATCAD versus EATVal revealed enrichment of fewer individual genes (Figure 2). GSEA was able to identify a diverse array of genes that are downregulated in EATCAD (Table 3); these are related to a broad spectrum of cellular processes including intracellular trafficking, proliferation/transcription regulation, protein catabolism, innate immunity/lectin pathway and ER stress. This is in contrast to the very limited upregulated gene enrichment profile of EATCAD, consisting of just one gene set, GO:0050906/Detection of stimulus involved in sensory perception. Individual genes demonstrating core enrichment in this set were related to taste and photo perception (Table S3); this has not been previously described in EAT. Because previous microarray studies of EATCAD used SATCAD as the control rather than EATVal, gene enrichment in EATCAD could have been masked by the inherent differences between EAT and SAT (5, 16, 17). Future studies in which EATCAD is compared with EAT from healthy subjects (as opposed to valvulopathy subjects as in the present study) would help to characterize the relationship between CAD and EAT.
The high diversity of downregulated genes in EATCAD may suggest relative downregulation of robust cellular activities in this tissue in the setting of severe CAD resulting from adaptive, or even maladaptive, mechanisms of CAD progression. Evaluation of EAT taken from individuals with variable stages of CAD, or in which samples are collected from the same individuals as disease progresses, would aid in determining causation.
The inflammatory components of the EATVal were numerous (Table S4); inflammation in the EAT of these patients could be a consequence of the valvulopathy itself. It will be interesting to further evaluate EAT inflammatory activity in vivo using non-invasive, imaging techniques such as positron emission tomography, in different clinical settings (36, 37). There were also markers of cellular stress. Elements of the MAP kinase stress response were enriched including MAP3K1 (Table 2). GSEA also identified gene sets related to ER stress and proteases that play roles in lysosomal degradation and apoptosis, including cathepsin E (CTSE, Table 2).
This study has several notable limitations. The CAD and valvulopathy patient groups differed significantly in terms of their cardiovascular risk factor profiles where the CAD patients were older, exhibited higher risk lipid profiles and were mostly diabetics. This could introduce confounding in the EATCAD versus EATVal analysis; future studies could be considered in which the CAD and valvulopathy groups are more similar in terms of their cardiovascular risk. However the lack of difference between the two groups in BMI or EAT thickness makes it unlikely that the gene expression pattern we observed was due to a difference in amount of general or organ-specific adiposity. Sample size was also a limitation of the study as the EATCAD vs. EATVal comparisons were underpowered. The analysis in the EAT versus SAT, however, was sufficient to determine enrichment of many individual genes and all of the gene ontology sets. Specifically, it would be important to repeat these analyses in a larger sample of nondiabetic patients to eliminate this source of confounding and to improve the power to detect gene enrichment while accounting for multiple hypotheses.
This is the first study providing an exploration of the transcriptome of EAT in both CAD and nonCAD, valvulopathy subjects. In addition to being an anatomically unique adipose depot, EAT demonstrates a transcriptome different from that of SAT in the same subjects. The EAT transcriptome is primarily characterized by markers of inflammation and is modified in subjects with CAD versus valve disease. These data may provide the basis for new hypothesis generation in the understanding of the physiologic role of EAT and its pathophysiologic involvement in CAD and valve disease.
Supplementary Material
What is already known about this subject?
Epicardial adipose tissue (EAT) is an anatomically unique adipose depot; it is the visceral adipose depot of the heart.
EAT is enriched with saturated fatty acids, has high protein content, and has greater capacity for lipogenesis and fatty acid metabolism.
EAT thickness as measured by echocardiography or CT has been associated with increased cardiovascular risk, coronary artery disease and metabolic syndrome independent of traditional risk factors.
What this study adds.
The epicardial adipose transcriptome is distinct compared to subcutaneous fat.
Epicardial fat is an inflammatory visceral fat depot.
Epicardial adipose tissue is altered in coronary artery disease versus valvulopathy.
Acknowledgments
Thanks to Katelyn Hughes and Dr. Mary-Elizabeth Patti (Joslin Diabetes Center, microarray performance), the Broad Institute (GSEA/GenePattern softwares) and Cezar Bianchi (graphic design).
Footnotes
Conflicts of Interest Statement
The authors declare no conflicts of interest.
Author Contributions: EM consented and enrolled patients, retrieved samples from surgeries, processed samples, analyzed microarray and composed manuscript. TF assisted in sample processing. RP drafted the IRB protocol. AP and TS performed surgeries including harvest of samples and provided clinical care and follow-up. AB proposed methods for tissue processing/analysis, assisted with microarray analysis, provided funding support and assisted with manuscript composition. GI is the PI for this study and is the guarantor; designed study protocol, oversaw IRB submission, and assisted with manuscript composition.
References
- 1.Iacobellis G, Assael F, Ribaudo MC, Zappaterreno A, Alessi G, Di Mario U, et al. Epicardial fat from echocardiography: a new method for visceral adipose tissue prediction. Obes Res. 2003;11:304–310. doi: 10.1038/oby.2003.45. [DOI] [PubMed] [Google Scholar]
- 2.Marchington JM, Mattacks CA, Pond CM. Adipose tissue in the mammalian heart and pericardium: structure, foetal development and biochemical properties. Comp Biochem Physiol B. 1989;94:225–232. doi: 10.1016/0305-0491(89)90337-4. [DOI] [PubMed] [Google Scholar]
- 3.Iacobellis G, Corradi D, Sharma AM. Epicardial adipose tissue: anatomic, biomolecular and clinical relationships with the heart. Nat Clin Pract Cardiovasc Med. 2005;2:536–543. doi: 10.1038/ncpcardio0319. [DOI] [PubMed] [Google Scholar]
- 4.Bambace C, Telesca M, Zoico E, Sepe A, Olioso D, Rossi A, et al. Adiponectin gene expression and adipocyte diameter: a comparison between epicardial and subcutaneous adipose tissue in men. Cardiovasc Pathol. 2011;20:e153–156. doi: 10.1016/j.carpath.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 5.Mazurek T, Zhang L, Zalewski A, Mannion JD, Diehl JT, Arafat H, et al. Human epicardial adipose tissue is a source of inflammatory mediators. Circulation. 2003;108:2460–2466. doi: 10.1161/01.CIR.0000099542.57313.C5. [DOI] [PubMed] [Google Scholar]
- 6.Sacks HS, Fain JN. Human epicardial adipose tissue: a review. Am Heart J. 2007;153:907–917. doi: 10.1016/j.ahj.2007.03.019. [DOI] [PubMed] [Google Scholar]
- 7.Iacobellis G, Bianco AC. Epicardial adipose tissue: emerging physiological, pathophysiological and clinical features. Trends Endocrinol Metab. 2011;22:450–457. doi: 10.1016/j.tem.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marchington JM, Pond CM. Site-specific properties of pericardial and epicardial adipose tissue: the effects of insulin and high-fat feeding on lipogenesis and the incorporation of fatty acids in vitro. Int J Obes. 1990;14:1013–1022. [PubMed] [Google Scholar]
- 9.Pezeshkian M, Noori M, Najjarpour-Jabbari H, Abolfathi A, Darabi M, Darabi M, et al. Fatty acid composition of epicardial and subcutaneous human adipose tissue. Metab Syndr Relat Disord. 2009;7:125–131. doi: 10.1089/met.2008.0056. [DOI] [PubMed] [Google Scholar]
- 10.Salgado-Somoza A, Teijeira-Fernandez E, Fernandez AL, Gonzalez-Juanatey JR, Eiras S. Proteomic analysis of epicardial and subcutaneous adipose tissue reveals differences in proteins involved in oxidative stress. Am J Physiol Heart Circ Physiol. 2010;299:H202–209. doi: 10.1152/ajpheart.00120.2010. [DOI] [PubMed] [Google Scholar]
- 11.Fain JN, Sacks HS, Bahouth SW, Tichansky DS, Madan AK, Cheema PS. Human epicardial adipokine messenger RNAs: comparisons of their expression in substernal, subcutaneous, and omental fat. Metabolism: clinical and experimental. 2010;59:1379–1386. doi: 10.1016/j.metabol.2009.12.027. [DOI] [PubMed] [Google Scholar]
- 12.Baker AR, Silva NF, Quinn DW, Harte AL, Pagano D, Bonser RS, et al. Human epicardial adipose tissue expresses a pathogenic profile of adipocytokines in patients with cardiovascular disease. Cardiovasc Diabetol. 2006;5:1. doi: 10.1186/1475-2840-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mahabadi AA, Berg MH, Lehmann N, Kalsch H, Bauer M, Kara K, et al. Association of epicardial fat with cardiovascular risk factors and incident myocardial infarction in the general population: the Heinz Nixdorf Recall Study. J Am Coll Cardiol. 2013;61:1388–1395. doi: 10.1016/j.jacc.2012.11.062. [DOI] [PubMed] [Google Scholar]
- 14.Pierdomenico SD, Pierdomenico AM, Cuccurullo F, Iacobellis G. Meta-analysis of the relation of echocardiographic epicardial adipose tissue thickness and the metabolic syndrome. The American journal of cardiology. 2013;111:73–78. doi: 10.1016/j.amjcard.2012.08.044. [DOI] [PubMed] [Google Scholar]
- 15.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dutour A, Achard V, Sell H, Naour N, Collart F, Gaborit B, et al. Secretory type II phospholipase A2 is produced and secreted by epicardial adipose tissue and overexpressed in patients with coronary artery disease. The Journal of clinical endocrinology and metabolism. 2010;95:963–967. doi: 10.1210/jc.2009-1222. [DOI] [PubMed] [Google Scholar]
- 17.Guauque-Olarte S, Gaudreault N, Piche ME, Fournier D, Mauriege P, Mathieu P, et al. The transcriptome of human epicardial, mediastinal and subcutaneous adipose tissues in men with coronary artery disease. PloS one. 2011;6:e19908. doi: 10.1371/journal.pone.0019908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iacobellis G, Willens HJ. Echocardiographic epicardial fat: a review of research and clinical applications. J Am Soc Echocardiogr. 2009;22:1311–1319. doi: 10.1016/j.echo.2009.10.013. quiz 1417–1318. [DOI] [PubMed] [Google Scholar]
- 19.Huber J, Kiefer FW, Zeyda M, Ludvik B, Silberhumer GR, Prager G, et al. CC chemokine and CC chemokine receptor profiles in visceral and subcutaneous adipose tissue are altered in human obesity. The Journal of clinical endocrinology and metabolism. 2008;93:3215–3221. doi: 10.1210/jc.2007-2630. [DOI] [PubMed] [Google Scholar]
- 20.Cheng KH, Chu CS, Lee KT, Lin TH, Hsieh CC, Chiu CC, et al. Adipocytokines and proinflammatory mediators from abdominal and epicardial adipose tissue in patients with coronary artery disease. Int J Obes (Lond) 2008;32:268–274. doi: 10.1038/sj.ijo.0803726. [DOI] [PubMed] [Google Scholar]
- 21.Yang RZ, Lee MJ, Hu H, Pray J, Wu HB, Hansen BC, et al. Identification of omentin as a novel depot-specific adipokine in human adipose tissue: possible role in modulating insulin action. American journal of physiology Endocrinology and metabolism. 2006;290:E1253–1261. doi: 10.1152/ajpendo.00572.2004. [DOI] [PubMed] [Google Scholar]
- 22.Ekstrom M, Liska J, Eriksson P, Sverremark-Ekstrom E, Tornvall P. Stimulated in vivo synthesis of plasminogen activator inhibitor-1 in human adipose tissue. Thromb Haemost. 2012;108:485–492. doi: 10.1160/TH11-11-0822. [DOI] [PubMed] [Google Scholar]
- 23.Lijnen HR. Functional role of the fibrinolytic system in development of adipose tissue. Verh K Acad Geneeskd Belg. 2009;71:101–113. [PubMed] [Google Scholar]
- 24.Svensson PA, Jernas M, Sjoholm K, Hoffmann JM, Nilsson BE, Hansson M, et al. Gene expression in human brown adipose tissue. Int J Mol Med. 2011;27:227–232. doi: 10.3892/ijmm.2010.566. [DOI] [PubMed] [Google Scholar]
- 25.Decher N, Maier M, Dittrich W, Gassenhuber J, Bruggemann A, Busch AE, et al. Characterization of TASK-4, a novel member of the pH-sensitive, two-pore domain potassium channel family. FEBS Lett. 2001;492:84–89. doi: 10.1016/s0014-5793(01)02222-0. [DOI] [PubMed] [Google Scholar]
- 26.Matarin M, Brown WM, Scholz S, Simon-Sanchez J, Fung HC, Hernandez D, et al. A genome-wide genotyping study in patients with ischaemic stroke: initial analysis and data release. Lancet Neurol. 2007;6:414–420. doi: 10.1016/S1474-4422(07)70081-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koo GC, Blake JT, Talento A, Nguyen M, Lin S, Sirotina A, et al. Blockade of the voltage-gated potassium channel Kv1.3 inhibits immune responses in vivo. J Immunol. 1997;158:5120–5128. [PubMed] [Google Scholar]
- 28.Tschritter O, Machicao F, Stefan N, Schafer S, Weigert C, Staiger H, et al. A new variant in the human Kv1.3 gene is associated with low insulin sensitivity and impaired glucose tolerance. The Journal of clinical endocrinology and metabolism. 2006;91:654–658. doi: 10.1210/jc.2005-0725. [DOI] [PubMed] [Google Scholar]
- 29.Yang Q, Huang JH, Man YB, Yao XQ, He GW. Use of intermediate/small conductance calcium-activated potassium-channel activator for endothelial protection. J Thorac Cardiovasc Surg. 2011;141:501–510. 510 e501. doi: 10.1016/j.jtcvs.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 30.Regan SE, Broad M, Byford AM, Lankford AR, Cerniway RJ, Mayo MW, et al. A1 adenosine receptor overexpression attenuates ischemia-reperfusion-induced apoptosis and caspase 3 activity. Am J Physiol Heart Circ Physiol. 2003;284:H859–866. doi: 10.1152/ajpheart.00251.2002. [DOI] [PubMed] [Google Scholar]
- 31.Matherne GP, Linden J, Byford AM, Gauthier NS, Headrick JP. Transgenic A1 adenosine receptor overexpression increases myocardial resistance to ischemia. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:6541–6546. doi: 10.1073/pnas.94.12.6541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wing SS. The UPS in diabetes and obesity. BMC Biochem. 2008;9(Suppl 1):S6. doi: 10.1186/1471-2091-9-S1-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aggarwal K, Massague J. Ubiquitin removal in the TGF-beta pathway. Nat Cell Biol. 2012;14:656–657. doi: 10.1038/ncb2534. [DOI] [PubMed] [Google Scholar]
- 34.Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006;440:944–948. doi: 10.1038/nature04634. [DOI] [PubMed] [Google Scholar]
- 35.Holvoet P, De Keyzer D, Jacobs DR., Jr Oxidized LDL and the metabolic syndrome. Future Lipidol. 2008;3:637–649. doi: 10.2217/17460875.3.6.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mazurek T, Kiliszek M, Kobylecka M, Skubisz-Gluchowska J, Kochman J, Filipiak K, et al. Relation of proinflammatory activity of epicardial adipose tissue to the occurrence of atrial fibrillation. The American journal of cardiology. 2014;113:1505–1508. doi: 10.1016/j.amjcard.2014.02.005. [DOI] [PubMed] [Google Scholar]
- 37.Mazurek T, Kochman J, Kobylecka M, Wilimski R, Filipiak KJ, Krolicki L, et al. Inflammatory activity of pericoronary adipose tissue may affect plaque composition in patients with acute coronary syndrome without persistent ST-segment elevation: preliminary results. Kardiologia polska. 2014;72:410–416. doi: 10.5603/KP.a2013.0320. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.