

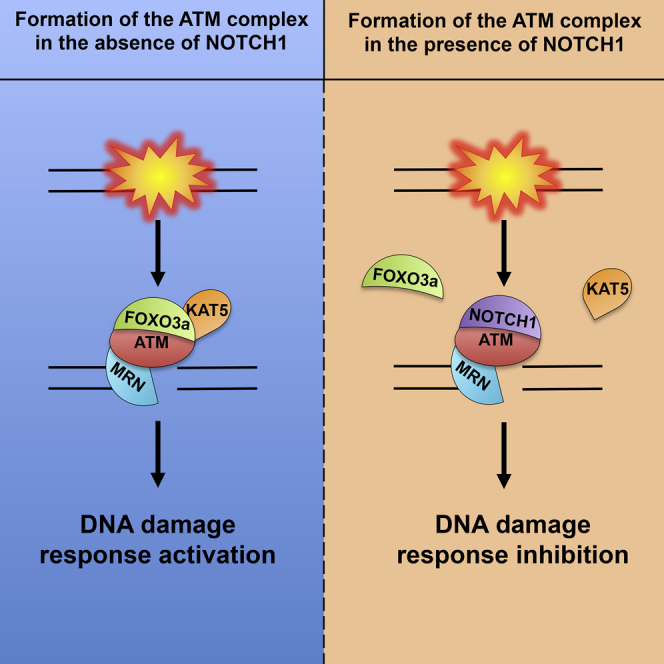
Summary
The DNA damage response (DDR) signal transduction pathway is responsible for sensing DNA damage and further relaying this signal into the cell. ATM is an apical DDR kinase that orchestrates the activation and the recruitment of downstream DDR factors to induce cell-cycle arrest and repair. We have previously shown that NOTCH1 inhibits ATM activation upon DNA damage, but the underlying mechanism remains unclear. Here, we show that NOTCH1 does not impair ATM recruitment to DNA double-strand breaks (DSBs). Rather, NOTCH1 prevents binding of FOXO3a and KAT5/Tip60 to ATM through a mechanism in which NOTCH1 competes with FOXO3a for ATM binding. Lack of FOXO3a binding to ATM leads to the loss of KAT5/Tip60 association with ATM. Moreover, expression of NOTCH1 or depletion of ATM impairs the formation of the FOXO3a-KAT5/Tip60 protein complex. Finally, we show that pharmacological induction of FOXO3a nuclear localization sensitizes NOTCH1-driven cancers to DNA-damage-induced cell death.
Graphical Abstract
Highlights
-
•
NOTCH1 does not inhibit recruitment of ATM to DSBs
-
•
NOTCH1 competes with FOXO3a for binding to the FATC domain of ATM
-
•
FOXO3a is necessary for KAT5/Tip60 binding to ATM
-
•
Induction of FOXO3a nuclear localization sensitizes TALL-1 cancer cells to DNA damage
Adamowicz et al. show that NOTCH1 inhibits ATM not by impairing ATM recruitment to the DSBs but by blocking formation of a three-protein complex composed of ATM, FOXO3a, and KAT5/Tip60. NOTCH1 competes with FOXO3a for binding to the FATC domain of ATM, resulting in NOTCH1-mediated ATM kinase inhibition.
Introduction
Ataxia-telengiectasia mutated (ATM) is a phosphatidylinositol-3-like protein kinase discovered over 20 years ago (Savitsky et al., 1995). Although several reports describe the role of ATM in the DNA damage response (DDR), the underlying molecular mechanisms of ATM activation still awaits complete elucidation. It has been shown that upon DNA damage, ATM is recruited to the double-strand breaks (DSBs) (Andegeko et al., 2001) through its interaction with NBS1 (Falck et al., 2005, Nakada et al., 2003). MRE11, RAD50, and NBS1 form a protein complex known as MRN, which is one of the first to localize to DSBs (Polo and Jackson, 2011). Upon MRN-mediated ATM recruitment, a lysine acetyl-transferase 5 (also known as a Tip60, hereinafter referred to as KAT5), which binds to ATM indirectly (Sun et al., 2005, Sun et al., 2010), interacts with histone H3 trimethylated at lysine 9 (H3K9m3). This interaction induces acetyl-transferase activity of KAT5, which acetylates ATM (Sun et al., 2007, Sun et al., 2009). ATM acetylation has been proposed to be an early step in ATM activation, preceding autophosphorylation and activation (Sun et al., 2007). In addition, c-Abl-mediated phosphorylation of KAT5 was shown to be necessary for KAT5 activation in response to DNA damage (Kaidi and Jackson, 2013).
FOXO3a is a mammalian transcription factor that contains a unique DNA binding forkhead domain and belongs to the forkhead box-O family of transcription factors (Calnan and Brunet, 2008). FOXO3a is involved in many cellular processes such as cell-cycle control, apoptosis, and more recently, DDR. FOXO3a has been proposed to bind to ATM upon DNA damage and to be necessary for its activation. Lack of FOXO3a impairs both ATM autophosphorylation and phosphorylation of its substrates, although the exact mechanism of FOXO3a-mediated ATM activation remains unclear (Chung et al., 2012, Tsai et al., 2008).
NOTCH1 is a transmembrane receptor which, upon interaction with one of its ligands, is processed by gamma secretase protease (Andersen et al., 2012). The cleaved intracellular part of NOTCH1 (N1IC) released in such processes translocates to the nucleus and initiates the transcription of NOTCH1 target genes involved in cell proliferation, differentiation, and survival (Andersen et al., 2012).
We have recently discovered and reported that NOTCH1 is a direct inhibitor of ATM, independent from its transcriptional activity (Vermezovic et al., 2015). Here, we demonstrate that NOTCH1 inactivates ATM by preventing FOXO3a binding to the FRAP-ATM-TRRAP-C-terminal (FATC) domain of ATM. We show that FOXO3a is necessary for KAT5 binding to ATM and the formation of an ATM, FOXO3a, and KAT5 protein complex, hereinafter referred to as the ATM activation complex (AAC). NOTCH1-mediated FOXO3a displacement results in the impairment of KAT5-ATM interaction and ATM inactivation. Additionally, we provide evidence that the expression of NOTCH1 or lack of ATM impairs the formation of the FOXO3a-KAT5 protein complex, suggesting that the interaction between these two proteins takes place only in the context of the AAC. Finally, we show that pharmacological induction of FOXO3a nuclear localization sensitizes NOTCH1-driven cancers to DNA damage-induced cell death.
Results
NOTCH1 Binding to ATM Does Not Impair Recruitment to DSBs
It has been shown that ATM interacts with NBS1 and that this allows its recruitment to DSBs (Nakada et al., 2003, Falck et al., 2005). Therefore, we tested whether NOTCH1 expression interferes with ATM-NBS1 interaction. We immunoprecipitated NBS1 in HEK293T cell lysates expressing, or not expressing, a constitutively active form of NOTCH1 (N1ΔE-Flag) (Rustighi et al., 2009). We observed that NBS1 remains in a complex with ATM both in NOTCH1-expressing and mock-transfected cells and regardless of ionizing radiation (IR) treatment (Figure 1A). Next, we studied whether NOTCH1 expression affects ATM recruitment to chromatin. To that end, we analyzed, by immunoblotting, the chromatin fraction of lysates from cells expressing a nuclear form of NOTCH1 fused to GFP (N1IC-GFP) and treated with IR. Though ATM was inactivated, as indicated by the decreased ATM autophosphorylation on serine 1981 (pATM), we could not observe any statistical difference in ATM recruitment to chromatin upon DNA damage in N1IC-GFP-expressing cells, as compared to control cells (Figures 1B and 1C). Thus, although NOTCH1 inactivates ATM, it does not do so by inhibiting ATM recruitment to the DSBs.
Figure 1.

NOTCH1 Does Not Inhibit ATM Recruitment to DSBs
(A) CoIP of the ATM with NBS1 in cells expressing, or not expressing, NOTCH1. IGG, immunoglobulin G; WCE, whole-cell extract.
(B) Immunoblot analysis of the HEK293T cells transfected with N1IC-GFP or EGFP constructs followed by fluorescence-activated cell sorting (FACS).
(C) Quantification of the signals shown in (B). pATM signal was normalized to ATM for “ATM activation” analysis, and ATM signal was normalized to H3 signal for “ATM recruitment” analysis. In both cases, signal of NO IR samples was subtracted from IR signal (mean ± SEM; n = 3; two-tailed Student’s t test; ∗p ≤ 0.05; ns, not significant).
FOXO3a Binds Directly to the FATC Domain of ATM
FOXO3a has been described as one of the critical protein factors necessary for ATM activation (Chung et al., 2012, Tsai et al., 2008), by virtue of its ability to directly interact with ATM (Tsai et al., 2008). To probe this interaction more in depth, we mapped the domain of ATM involved in FOXO3a binding. We performed immunoprecipitation (IP) of Myc-tagged ATM fragments, spanning the entire length of the ATM protein (Figure S1A). We observed a strong interaction between FOXO3a and a C-terminal ATM fragment (amino acids [aa] 1,965–3,056). Notably, this fragment contains the 33-aa FATC domain (aa 3,023–3,056), previously proposed as a critical domain in the regulation of ATM kinase activity (Jiang et al., 2006). To map with an increased resolution the interaction domain of ATM with FOXO3a, we incubated a lysate of cells overexpressing Myc-FOXO3a with beads bound to purified glutathione S-transferase (GST) fused to 12 individual fragments encompassing the whole ATM. We observed a specific interaction of FOXO3a with the ATM fragment spanning aa 2,842–3,056 but not with that spanning aa 2,682–3,012 (Figure 2A). The same specific interaction could also be observed when the GST-ATM fragment spanning 2,842–3,056 aa (hereinafter referred to as GST-ATM) was incubated with endogenous or recombinant purified FOXO3a, indicating that such interaction is direct (Figures S1B and S1C). To prove the essential role of the FATC domain of ATM in FOXO3a binding, we expressed a full-length ATM (wild-type [WT] ATM) or ATM lacking the FATC domain (1–2992 aa) (ATMΔFATC) in HEK293T cells. IP experiments demonstrated that the FATC domain of ATM is essential for its interaction with FOXO3a (Figure S1D).
Figure 2.

NOTCH1 Competes with FOXO3a for ATM Binding
(A) Immunoblot analysis of the pull-down experiments with GST or GST-ATM fragments (as indicated) incubated with lysate of HEK293T cells transfected with Myc-FOXO3a construct.
(B) Immunoblot analysis of the coIP of ATM with FOXO3a.
(C) Immunoblot analysis of the pull-down experiment with GST or GST-ATM (2,842–3,056 aa) incubated with the lysate of HEK293T (asterisk indicates correct band).
(D) Immunoblot analysis of the coIP of ATM with FOXO3a or NOTCH1 in cells overexpressing FOXO3a or expressing NOTCH1.
(E) Immunofluorescence analysis of the pATM staining in HeLa cells expressing, or not expressing, NOTCH1 or overexpressing FOXO3a (scale bars, 10 μm).
(F) Analysis of the images shown in (E). Percentage of pATM-positive cells (more than three foci) (mean ± SEM; n = 3; two-tailed Student’s t test; p ≤ 0.05).
We decided to further investigate the nature of the endogenous ATM-FOXO3a complex. We immunoprecipitated endogenous FOXO3a in lysates of cells exposed, or not exposed, to IR. We observed that FOXO3a was in a complex with ATM independently of the induction of DNA damage (Figure 2B). Next, to determine whether the entire pool of ATM protein is in a complex with FOXO3a, we performed sequential immunodepletions of either ATM or FOXO3a, followed by reciprocal immunodepletion. We observed that, despite the complete immunodepletion of ATM, the levels of FOXO3a are not dramatically affected. Differently, immunodepletion of FOXO3a led to significant ATM protein depletion (Figure S1E). This result indicates that all of ATM is in a complex with FOXO3a, while not all of FOXO3a is associated with ATM. We conclude that nuclear ATM is quantitatively bound to FOXO3a directly through its FATC domain.
NOTCH1 Competes with FOXO3a for ATM Binding
We have previously shown that NOTCH1 binds directly to the FATC domain of ATM and inactivates ATM in response to IR (Vermezovic et al., 2015). Therefore, we tested whether NOTCH1 competes with FOXO3a in binding to the FATC domain of ATM. Thus, we incubated GST-ATM bound to beads with lysates from cells transfected with N1ΔE-Flag or empty vector. We observed that FOXO3a binding to FATC domain of ATM was diminished in the presence of NOTCH1 (Figure 2C). A similar conclusion was reached by studying the interaction between endogenous ATM and FOXO3a proteins by proximity ligation assay (PLA) (Figures S1F and S1G). Moreover, ATM-FOXO3a interaction was rescued in the presence of a NOTCH1 inhibitor (DAPT) (Figures S1F and S1G), which, at the same time, abolished interaction between ATM and NOTCH1 (Figures S1H and S1I). Reduced interaction between FOXO3a and ATM did not result from NOTCH1-mediated sequestration of FOXO3a, since no interaction between NOTCH1 and FOXO3a could be observed (Figure S1J). Consistently, in living cells, the overexpression of FOXO3a leads to a detectably decreased NOTCH1 and ATM interaction (Figures S1K and S1L).
Next, we compared the strength of the interactions of NOTCH1 and FOXO3a to ATM. To do that, we transfected either Myc-FOXO3a or N1ΔE-Myc and performed an IP of the Myc-tagged proteins. We observed that NOTCH1 was binding more efficiently to ATM than FOXO3a (Figure 2D). This was not due to the differential distribution of expressed tagged proteins in distinct cellular compartments (Figure S1M). Moreover, IP of NOTCH1 or FOXO3a from the nuclear fraction confirmed the results obtained in whole-cell extracts (Figures S1N and S1O).
Next, we asked whether overexpression of FOXO3a could rescue NOTCH1-driven inhibition of ATM activation upon IR. We observed an inhibition of ATM activation, as judged by the inhibition of pATM foci formation, in cells expressing NOTCH1 but not FOXO3a. Furthermore the overexpression of FOXO3 together with NOTCH1 rescues NOTCH1-mediated ATM inactivation (Figures 2E and 2F).
We concluded that NOTCH1 competes with FOXO3a for the FATC domain of ATM and binds to it with higher strength.
KAT5 Binds to ATM through FOXO3a
The role of FOXO3a in the activation of ATM remains unclear. Our results indicate that FOXO3a binds directly to the FATC domain of ATM and that all of ATM is in a complex with it (Figures 2 and S1). Next, we asked whether FOXO3a can form a protein complex with KAT5, which has been reported to bind to the FATC domain of ATM, although indirectly and independently of DNA damage (Sun et al., 2010) (Figure S2A). We immunoprecipitated FOXO3a from cell lysates, and we detected KAT5 in a complex with FOXO3a in cells exposed, or not exposed, to IR (Figure 3A). In order to exclude the possibility of DNA potentially bridging the two proteins, we performed the same IP in the presence or absence of benzonase nuclease, and this did not affect the interaction between KAT5 and FOXO3a (Figure S2B). To test whether such interaction was direct, we performed a pull-down experiment with recombinant purified proteins GST-KAT5 and FOXO3a: we observed that KAT5 and FOXO3a can, indeed, bind directly to each other (Figure S2C).
Figure 3.

FOXO3a Bridges KAT5 Binding to ATM
(A) Immunoblot analysis of the coIP of KAT5 with FOXO3a.
(B) Immunoblot analysis of the coIP of ATM and FOXO3a with KAT5 in cells treated, or not treated, with siRNA against FOXO3a.
(C) Immunoblot analysis of the pull-down experiment with GST-ATM (2,842–3,056 aa) incubated as indicated with the recombinant human KAT5 and recombinant human FOXO3a proteins (asterisk indicates correct band).
(D) Immunoblot analysis of the coIP of ATM and FOXO3a with Myc-KAT5 fragment (450–513 aa) in the presence or absence of NOTCH1.
(E) Three schemes showing the potential interactions among AAC components: (1) AAC complex; (2) AAC complex in the presence of NOTCH1; (3) two possibilities of FOXO3a-KAT5 interaction in the absence of ATM.
(F) Immunoblot analysis of the coIP of FOXO3a and ATM with KAT5.
Next, we aimed at mapping the protein domains of KAT5 and FOXO3a involved in such interaction. We expressed three sequential Myc-tagged fragments of FOXO3a spanning its whole length (Figure S2D; fragments 1–3) in HEK293T cells, followed by IP with anti-Myc antibodies. We observed a specific binding of KAT5 to the C-terminal part of FOXO3a (500–673 aa) (Figure S2E). To define more precisely the domain in the C-terminal region of FOXO3a involved in KAT5 binding, we generated two different C-terminal fragments of FOXO3a (Figure S2D; fragments 4–5). IP of FOXO3a (fragments 3–5) showed that KAT5 binds to FOXO3a in the C-terminal region, between 620 and 650 aa (Figure S2F).
Next, by using four different constructs spanning the entire length of KAT5 (Figure S2G; fragments 1–4), we aimed at mapping the interaction domains between KAT5 and FOXO3a and between KAT5 and ATM. We performed IPs in cells expressing Flag-tagged fragments of KAT5 (1–4). We observed that both FOXO3a and ATM were binding specifically to the C-terminal part of KAT5 (285–513 aa) (Figures S2H and S2I). In addition, we expressed in cells the very C-terminal part of KAT5 (450–513 aa), and we were able to observe both ATM as well as FOXO3a binding to it (Figure S2J).
Given that KAT5 was reported to bind to ATM indirectly, we tested whether FOXO3a was necessary for the binding of KAT5 to ATM. We transfected cells with small interfering RNA (siRNA) against FOXO3a (siFOXO3a) or siRNA targeting LUCIFERASE (control). We observed that, as previously reported (Tsai et al., 2008), cells knocked down for FOXO3a displayed an impaired activation of ATM, as shown by reduced pATM signal (Figure S2K). Next, to assess the interaction between ATM and KAT5, we immunoprecipitated KAT5. Surprisingly, in the absence of FOXO3a also, the interaction between KAT5 and ATM was impaired as compared to mock-transfected cells (Figures 3B and S2L).
Unexpectedly, we observed that reduction of ATM-KAT5 interaction in the cells treated with siRNA against FOXO3a was partial. Therefore, we asked whether other FOXO family members, such as FOXO1 and FOXO4, could contribute to the binding of ATM and KAT5 in the absence of FOXO3a considering high conservation of C-terminal region (Figure S2M). Therefore, we immunoprecipitated FOXO1 and FOXO4, and we observed that ATM was able to form a protein complex with both of them (Figures S2N and S2O). Moreover, the IP of KAT5 showed that FOXO1 and FOXO4 can bind to KAT5 (Figure S2P). Finally, we also observed that knockdown of FOXO3a results in increased levels of endogenous FOXO4 (Figure S2Q). To overcome a problem of potential redundancy between FOXO3a, FOXO1, and FOXO4, we performed an in vitro binding assay. We incubated GST-ATM with recombinant KAT5 and recombinant FOXO3a. As previously shown (Sun et al., 2010), we could not detect any direct interaction between GST-ATM and recombinant KAT5, but in the presence of recombinant FOXO3a, the binding between ATM and KAT5 became evident, demonstrating that FOXO3a can mediate the interaction between ATM and KAT5 (Figure 3C).
Next, we checked whether the formation of the AAC is affected by the presence of DNA damage and whether its formation changes with the cell-cycle progression. Therefore, we performed PLA between the three components of the AAC in cells exposed, or not exposed, to IR. We could not observe any changes in the interaction between AAC components in the presence or absence of DNA damage (Figures S2R–S2W). Next, to check whether the formation of the AAC is differentially modulated during the cell cycle, we performed PLA in HeLa Fucci cells (Sakaue-Sawano et al., 2008). We observed a modest but reproducible increase in the interactions of all AAC components in the S/G2 phase compared to the G1 phase of the cell cycle (Figures S2X–S2AC). Similar to the observed changes in the interactions between AAC components, we observed an increase of ATM autophosphorylation in cells in the S/G2 phase of the cell cycle (Figures S2AD and S2AE). Results consistent with ours were reported by Pandita et al. (2000), who observed increased ATM activation upon IR in S-phase cells as compared to G1 and G2.
Our results demonstrate that NOTCH1 perturbs the binding of FOXO3a to ATM. Since KAT5 binds to FOXO3a, we asked whether NOTCH1 has an impact on KAT5 association with ATM. We immunoprecipitated Myc-tagged KAT5 in cells expressing NOTCH1 or mock-transfected cells. We observed that NOTCH1 expression perturbed binding of KAT5 to ATM, as compared to the control (Figure 3D). Surprisingly, we observed that expression of NOTCH1 impaired the binding of KAT5 not only to ATM but also to FOXO3a (Figure 3D). As we have already excluded the possibility that NOTCH1 could sequester FOXO3a (Figure S1J), we hypothesized that the interaction between FOXO3a and KAT5 may take place only in the context of the formation of the AAC (Figure 3E). To test this hypothesis, we performed an IP of Flag-tagged KAT5 in either WT or Atm knockout mouse embryonic stem cells (mESCs). Indeed, in the absence of ATM, we could not detect any interaction between KAT5 and FOXO3a (Figure 3F).
We propose that KAT5, FOXO3a, and ATM form the AAC, which is perturbed by NOTCH1 binding to ATM and that this interaction between FOXO3a and KAT5 is restricted to the formation of the AAC.
Induction of FOXO3a Nuclear Localization Sensitizes TALL Cells to DNA-Damage-Mediated Apoptosis
T cell acute lymphoblasic leukemia (TALL) cancer cells carry mutations in the NOTCH1 gene that result in increased expression and activity of NOTCH1 (Ferrando, 2009). We have previously shown that treatment of TALL-1 cells with gamma-secretase inhibitor (GSI; a widely used NOTCH1 inhibitor that prevents its nuclear translocation) leads to increased activation of DDR signaling and ensuing DDR-induced cell death (Vermezovic et al., 2015). Consequently, we decided to test whether GSI treatment of TALL cells has an impact on the formation of the AAC. To that end, we used CUTLL1 cells that express high levels of NOTCH1. GSI treatment of CUTLL1 cells caused a decrease of the nuclear form of NOTCH1 as well as of the interaction between ATM and NOTCH1 (Figures S3A–S3C). Next, we performed PLA between ATM and FOXO3a as well as FOXO3a and KAT5 in GSI-treated cells. We observed that inhibition of NOTCH1 resulted in the increase of the interaction between ATM and FOXO3a as well as that between FOXO3a and KAT5 (Figures S3D–S3G).
On the basis of the results showing that NOTCH1 expression in TALL cells leads to the impairment of the AAC formation, we asked whether we can rescue NOTCH1-mediated ATM inactivation in the TALL cells by increasing levels of FOXO3a in the nucleus. Therefore, we tested whether the induced translocation of FOXO3a into the nuclei of TALL-1 cells would have an impact on ATM activation as well as their survival after exposure to IR. We pretreated TALL-1 cells with metformin (MET), which, by acting through AMPK, induces FOXO3a nuclear import (Hu et al., 2014). To confirm the effect of MET, we analyzed the expression levels of FOXO3a target gene (GADD45α). We observed that MET treatment resulted in FOXO3a nuclear accumulation in TALL-1 cells (Figures S3H and S3I). Next, we analyzed the effect of MET on TALL-1 cells subjected to IR. Pretreatment with MET resulted in an increase of ATM activation (pATM) upon IR as compared to mock-treated cells (Figure 4A; Figure S3J). Moreover, we observed an increase of DDR-induced cell death in MET-treated cells (Figure 4B). As FOXO3a is not a direct target of MET, we sought to modulate an independent pathway that has an impact on FOXO3a nuclear localization. For this purpose, we tested the impact of pharmacological inhibition of p38 on TALL-1 cells by the use of a p38 kinase inhibitor (SB203580; hereinafter referred to as SB) previously shown to induce FOXO3a nuclear localization in treated cells (Clavel et al., 2010). We pretreated TALL-1 cells with SB and exposed them to IR. We observed an increase of FOXO3a nuclear import as judged by augmented transcription of a FOXO3a target gene (Figure S3K) and FOXO3a nuclear localization (Figure S3L). Furthermore, we observed that SB pretreatment increased ATM activation (pATM), as well as IR-induced cell death, when compared to mock-treated cells (Figures 4C, S3M, and S3N) and that cell death was dependent on ATM activation (Figure S3O).
Figure 4.

Induction of FOXO3a Nuclear Localization Sensitizes TALL Cells to DNA-Damage-Induced Cell Death
(A) Immunoblot analysis of the ATM activation (pATM) in TALL-1 cells pretreated with MET followed by IR.
(B) Immunoblot analysis of the DNA-damage-induced cell death (cleaved caspase-3) in TALL-1 cells pretreated with MET, followed by IR.
(C) Immunoblot analysis of the DNA-damage-induced cell death (cleaved caspase-3) in TALL-1 cells pretreated with SB, followed by IR.
(D) Model of the NOTCH1-mediated inactivation of ATM upon DNA damage. (1) ATM is recruited to DSB by its interaction with the MRN complex. Upon recruitment of ATM to DSB, KAT5, by interacting with ATM through FOXO3a, binds to H3K9m3 generated by SUV39h1 methyl-transferase, becomes activated, and acetylates ATM. This leads to ATM activation by autophosphorylation. (2) In the presence of NOTCH1, although ATM is still recruited to DSBs, NOTCH1-mediated disruption of the KAT5-FOXO3a-ATM complex results in the lack of KAT5-mediated acetylation of ATM and consequent activation.
NI, not irradiated.
We conclude that induction of FOXO3a nuclear localization sensitizes TALL-1 cells to DNA damage-induced cell death.
Discussion
DDR is one of the most important signaling pathways in the cell, and it is responsible for the maintenance of genome integrity. Here we show that NOTCH1 inhibits DDR by impairing the formation of the AAC composed from ATM, FOXO3a, and KAT5. Our results reveal a hitherto-unknown complex modulation of ATM kinase activity between two factors interacting with ATM.
It has been shown that NBS1-ATM interaction is crucial for the MRN-mediated ATM recruitment to DSBs (Falck et al., 2005, Nakada et al., 2003). Although NBS1 interacts with N-terminal part of ATM, it has been reported that point mutations in the FATC domain of ATM can affect NBS1 binding to ATM and, subsequently, ATM localization at DSBs (You et al., 2005, Ogi et al., 2015). We found that NOTCH1 interacts with ATM through the FATC domain (Vermezovic et al., 2015), but we did not observe any impairment in NBS1 binding to ATM or in its recruitment to DSBs. We show that ATM in the presence of NOTCH1 can localize at the DSB site but that it does not undergo autophosporylation; this is consistent with some recent published data (Hartlerode et al., 2015, Yamamoto et al., 2012). Differently, others reported that autophosphorylation is necessary for ATM retention at DSBs (So et al., 2009), which underlies our still-incomplete understanding of the full impact of ATM autophosphorylation on its activation and recruitment to DSBs.
FOXO3a has been shown to interact with ATM upon DNA damage (Tsai et al., 2008). We demonstrate that FOXO3a interacts with ATM independently of the presence of DNA damage. It has been shown that FOXO3a binds to ATM in a domain spanning 1,764–2,841 aa. Although we did observe FOXO3a binding to this domain, we detected a binding of higher affinity with the 2,842–3,056 aa GST-ATM fragment, which has not been tested in the previously published study (Tsai et al., 2008).
The exact function of FOXO3a in ATM activation was unclear thus far. Here, we propose that FOXO3a participates in the process of ATM activation by bridging KAT5 to ATM. Previous studies have shown that KAT5-ATM interaction is mediated by the FATC domain of ATM and that such binding is indirect, suggesting the existence of one or more bridging proteins (Sun et al., 2005, Sun et al., 2010). Consistent with this observation, we show that FOXO3a, like KAT5, binds specifically to the FATC domain of ATM, but unlike with KAT5, the observed binding is direct. Unexpectedly, we discovered that FOXO3a interacts directly with KAT5, and, in the absence of FOXO3a, KAT5 loses its ability to bind to ATM. It is noteworthy that we observed that the FOXO3a paralogues FOXO1 and FOXO4 could play a similar role in the absence of FOXO3a. Consistent with this, it has been shown before that another FOX family member, FOXP3, can interact with KAT5 (Li et al., 2007).
The interaction between FOXO3a and KAT5 seems to be restricted to the formation of the AAC, since, both in the absence of ATM and upon expression of NOTCH1, an interaction between FOXO3a and KAT5 cannot be detected. Based on that, we would like to propose a model in which ATM activation is dependent on the formation of the AAC, which consists of KAT5, FOXO3a, and ATM proteins. Formation of this three-protein complex allows proper ATM activation upon DNA damage (Figure 4E).
We have already shown that IR can be combined with GSI to sensitize TALL cells to IR-mediated cell death (Vermezovic et al., 2015). Here, we demonstrate the opportunity to use MET and SB to induce FOXO3a nuclear localization and sensitize TALL cells to genotoxic treatments. Our results are consistent with the previous observation that MET stimulates ATM activation in a FOXO3a-dependent manner (Vazquez-Martin et al., 2011, Hu et al., 2014).
Experimental Procedures
Cell Culture and Treatments
HEK293T cells (Interlab Cell Line Collection) were grown in DMEM supplemented with 10% fetal bovine serum (FBS), penicillin/streptomycin, and L-glutamine. HeLa Fucci cells (RIKEN BioResource Center cell bank) (Sakaue-Sawano et al., 2008) were grown in DMEM supplemented with 10% FBS, penicillin/streptomycin, and L-glutamine. HeLa cells (ATCC) were grown in DMEM with GlutaMAX supplemented with 10% FBS, penicillin/streptomycin, and nonessential aa. TALL-1 cells (DSMZ) were grown in RPMI 1640 supplemented with 15% FBS, penicillin/streptomycin, and L-glutamine. CUTLL1 cells (a kind gift from A. Ferrando) (Palomero et al., 2006) were grown in RPMI 1640 and supplemented with 10% FBS, penicillin/streptomycin, and L-glutamine. WT mESCs and ATM KO cells (Xu and Baltimore, 1996) were grown in DMEM with Glutamax (supplemented with 15% FBS, penicillin/streptomycin, nonessential aa, sodium pyruvate, leukemia inhibitory factor, and β-mercaptoethanol). HeLa cells were transfected with the use of lipofectamine (Invitrogen), according to the manufacturer’s protocol. HEK293T cells were transfected with the use of calcium phosphate. The GSI DAPT (N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester) (Sigma) was used at a 10-μM concentration for 16 hr. CUTLL1 cells were treated with 1 μM Compound E (Calbiochem) for 24 hr before fixation. KU60019 (SelleckChem) was used at the concentration of 10 μM for 16 hr before IR. SB203580 (Jena Bioscience) was used at the concentration of 10 μM for 24 hr before the IR. MET (Tocris) was used at the concentration of 500 μM 24 hr before IR. Cells were irradiated with an X-ray machine (Faxitron): 2 Gy, 1 hr, for immunofluorescence experiments; 5 Gy, 1 hr, for immunoblot analysis of DDR activation; and 10 Gy for DDR-induced cell-death analysis. For the knockdown experiments, 20 nM siRNA (GE Dharmacon) against human FOXO3a was transfected with the use of RNAiMAX lipofectamine (Invitrogen) 48 hr before the experiment.
For the purpose of cell sorting, HEK293T cells were transfected with N1IC-GFP or EGFP construct and FACS sorted 1 day after for GFP. Next, cells were plated and, 8 hr later, subjected to IR (10G for recruitment experiments).
Statistical Analysis
All data are represented as means ± SEM. Statistical analysis was performed with the use of a two-tailed Student’s t test. An asterisk indicates p < 0.05.
For more details, please see the Supplemental Experimental Procedures.
Author Contributions
M.A., J.V., and F.d’A.d.F. designed the study. M.A. performed all experiments. M.A., J.V., and F.d’A.d.F. analyzed the data and wrote the manuscript.
Acknowledgments
We would like to thank: A. Sarin (National Centre for Biological Sciences), P.P. Di Fiore (IFOM), G. Del Sal (University of Trieste), S.P. Jackson (Gurdon Institute), K. Yamamoto (Nagasaki University), H.S. Park (Chonnam National University), M. Kastan (Duke Cancer Institute), S.J. Kim (CHA University), B. Amati (Italian Institute of Technology), Yang Xu (University of California, San Diego), Mario Cinquanta (Cogentech), and A. Behrens (Francis Crick Institute) for reagents; F. Kobia (IFOM) and V. Costanzo (IFOM) for advice; and M. Mapelli and S. Pasqualato (Istituto Europeo di Oncologia) for their help and advice regarding protein purification. F.d’A.d.F. is supported by the Fondazione Italiana per la Ricerca sul Cancro, AIRC (application 12971), Human Frontier Science Program (contract RGP 0014/2012), Cariplo Foundation (grant 2010.0818), Association for International Cancer Research (14-1331), Progetti di Ricerca di Interesse Nazionale (PRIN) 2010–2011, the Italian Ministry of Education Universities and Research EPIGEN Project, and a European Research Council advanced grant (322726).
Published: August 11, 2016
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, three figures, and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2016.07.038.
Supplemental Information
References
- Andegeko Y., Moyal L., Mittelman L., Tsarfaty I., Shiloh Y., Rotman G. Nuclear retention of ATM at sites of DNA double strand breaks. J. Biol. Chem. 2001;276:38224–38230. doi: 10.1074/jbc.M102986200. [DOI] [PubMed] [Google Scholar]
- Andersen P., Uosaki H., Shenje L.T., Kwon C. Non-canonical Notch signaling: emerging role and mechanism. Trends Cell Biol. 2012;22:257–265. doi: 10.1016/j.tcb.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calnan D.R., Brunet A. The FoxO code. Oncogene. 2008;27:2276–2288. doi: 10.1038/onc.2008.21. [DOI] [PubMed] [Google Scholar]
- Chung Y.M., Park S.H., Tsai W.B., Wang S.Y., Ikeda M.A., Berek J.S., Chen D.J., Hu M.C. FOXO3 signalling links ATM to the p53 apoptotic pathway following DNA damage. Nat. Commun. 2012;3:1000. doi: 10.1038/ncomms2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavel S., Siffroi-Fernandez S., Coldefy A.S., Boulukos K., Pisani D.F., Dérijard B. Regulation of the intracellular localization of Foxo3a by stress-activated protein kinase signaling pathways in skeletal muscle cells. Mol. Cell. Biol. 2010;30:470–480. doi: 10.1128/MCB.00666-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falck J., Coates J., Jackson S.P. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005;434:605–611. doi: 10.1038/nature03442. [DOI] [PubMed] [Google Scholar]
- Ferrando A.A. The role of NOTCH1 signaling in T-ALL. Hematology Am. Soc. Hematol. Educ. Program. 2009;2009:353–361. doi: 10.1182/asheducation-2009.1.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartlerode A.J., Morgan M.J., Wu Y., Buis J., Ferguson D.O. Recruitment and activation of the ATM kinase in the absence of DNA-damage sensors. Nat. Struct. Mol. Biol. 2015;22:736–743. doi: 10.1038/nsmb.3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu T., Chung Y.M., Guan M., Ma M., Ma J., Berek J.S., Hu M.C. Reprogramming ovarian and breast cancer cells into non-cancerous cells by low-dose metformin or SN-38 through FOXO3 activation. Sci. Rep. 2014;4:5810. doi: 10.1038/srep05810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X., Sun Y., Chen S., Roy K., Price B.D. The FATC domains of PIKK proteins are functionally equivalent and participate in the Tip60-dependent activation of DNA-PKcs and ATM. J. Biol. Chem. 2006;281:15741–15746. doi: 10.1074/jbc.M513172200. [DOI] [PubMed] [Google Scholar]
- Kaidi A., Jackson S.P. KAT5 tyrosine phosphorylation couples chromatin sensing to ATM signalling. Nature. 2013;498:70–74. doi: 10.1038/nature12201. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Li B., Samanta A., Song X., Iacono K.T., Bembas K., Tao R., Basu S., Riley J.L., Hancock W.W., Shen Y. FOXP3 interactions with histone acetyltransferase and class II histone deacetylases are required for repression. Proc. Natl. Acad. Sci. USA. 2007;104:4571–4576. doi: 10.1073/pnas.0700298104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakada D., Matsumoto K., Sugimoto K. ATM-related Tel1 associates with double-strand breaks through an Xrs2-dependent mechanism. Genes Dev. 2003;17:1957–1962. doi: 10.1101/gad.1099003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogi H., Goto G.H., Ghosh A., Zencir S., Henry E., Sugimoto K. Requirement of the FATC domain of protein kinase Tel1 for localization to DNA ends and target protein recognition. Mol. Biol. Cell. 2015;26:3480–3488. doi: 10.1091/mbc.E15-05-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palomero T., Barnes K.C., Real P.J., Glade Bender J.L., Sulis M.L., Murty V.V., Colovai A.I., Balbin M., Ferrando A.A. CUTLL1, a novel human T-cell lymphoma cell line with t(7;9) rearrangement, aberrant NOTCH1 activation and high sensitivity to gamma-secretase inhibitors. Leukemia. 2006;20:1279–1287. doi: 10.1038/sj.leu.2404258. [DOI] [PubMed] [Google Scholar]
- Pandita T.K., Lieberman H.B., Lim D.S., Dhar S., Zheng W., Taya Y., Kastan M.B. Ionizing radiation activates the ATM kinase throughout the cell cycle. Oncogene. 2000;19:1386–1391. doi: 10.1038/sj.onc.1203444. [DOI] [PubMed] [Google Scholar]
- Polo S.E., Jackson S.P. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 2011;25:409–433. doi: 10.1101/gad.2021311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustighi A., Tiberi L., Soldano A., Napoli M., Nuciforo P., Rosato A., Kaplan F., Capobianco A., Pece S., Di Fiore P.P., Del Sal G. The prolyl-isomerase Pin1 is a Notch1 target that enhances Notch1 activation in cancer. Nat. Cell Biol. 2009;11:133–142. doi: 10.1038/ncb1822. [DOI] [PubMed] [Google Scholar]
- Sakaue-Sawano A., Kurokawa H., Morimura T., Hanyu A., Hama H., Osawa H., Kashiwagi S., Fukami K., Miyata T., Miyoshi H. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 2008;132:487–498. doi: 10.1016/j.cell.2007.12.033. [DOI] [PubMed] [Google Scholar]
- Savitsky K., Bar-Shira A., Gilad S., Rotman G., Ziv Y., Vanagaite L., Tagle D.A., Smith S., Uziel T., Sfez S. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1749–1753. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- So S., Davis A.J., Chen D.J. Autophosphorylation at serine 1981 stabilizes ATM at DNA damage sites. J. Cell Biol. 2009;187:977–990. doi: 10.1083/jcb.200906064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y., Jiang X., Chen S., Fernandes N., Price B.D. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc. Natl. Acad. Sci. USA. 2005;102:13182–13187. doi: 10.1073/pnas.0504211102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y., Xu Y., Roy K., Price B.D. DNA damage-induced acetylation of lysine 3016 of ATM activates ATM kinase activity. Mol. Cell. Biol. 2007;27:8502–8509. doi: 10.1128/MCB.01382-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y., Jiang X., Xu Y., Ayrapetov M.K., Moreau L.A., Whetstine J.R., Price B.D. Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat. Cell Biol. 2009;11:1376–1382. doi: 10.1038/ncb1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y., Jiang X., Price B.D. Tip60: connecting chromatin to DNA damage signaling. Cell Cycle. 2010;9:930–936. doi: 10.4161/cc.9.5.10931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai W.B., Chung Y.M., Takahashi Y., Xu Z., Hu M.C. Functional interaction between FOXO3a and ATM regulates DNA damage response. Nat. Cell Biol. 2008;10:460–467. doi: 10.1038/ncb1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez-Martin A., Oliveras-Ferraros C., Cufí S., Martin-Castillo B., Menendez J.A. Metformin activates an ataxia telangiectasia mutated (ATM)/Chk2-regulated DNA damage-like response. Cell Cycle. 2011;10:1499–1501. doi: 10.4161/cc.10.9.15423. [DOI] [PubMed] [Google Scholar]
- Vermezovic J., Adamowicz M., Santarpia L., Rustighi A., Forcato M., Lucano C., Massimiliano L., Costanzo V., Bicciato S., Del Sal G., d’Adda di Fagagna F. Notch is a direct negative regulator of the DNA-damage response. Nat. Struct. Mol. Biol. 2015;22:417–424. doi: 10.1038/nsmb.3013. [DOI] [PubMed] [Google Scholar]
- Xu Y., Baltimore D. Dual roles of ATM in the cellular response to radiation and in cell growth control. Genes Dev. 1996;10:2401–2410. doi: 10.1101/gad.10.19.2401. [DOI] [PubMed] [Google Scholar]
- Yamamoto K., Wang Y., Jiang W., Liu X., Dubois R.L., Lin C.S., Ludwig T., Bakkenist C.J., Zha S. Kinase-dead ATM protein causes genomic instability and early embryonic lethality in mice. J. Cell Biol. 2012;198:305–313. doi: 10.1083/jcb.201204098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Z., Chahwan C., Bailis J., Hunter T., Russell P. ATM activation and its recruitment to damaged DNA require binding to the C terminus of Nbs1. Mol. Cell. Biol. 2005;25:5363–5379. doi: 10.1128/MCB.25.13.5363-5379.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.