

Abstract
Afatinib is the second generation of irreversible inhibitor of EGFR, HER2 and HER4, which has shown encouraging phase II and III clinical outcomes in the treatment of head and neck squamous cell carcinoma (HNSCC). However, the molecular mechanism of afatinib-induced apoptosis in HNSCC is poorly understood. In the present investigation, we discovered that down-regulation of MCL-1, an anti-apoptotic member of BCL-2 family, was responsible for afatinib-triggered apoptosis. And the inactivation of AKT-mTOR signaling caused by afatinib lead to translational inhibition of MCL-1 expression. As a crucial branch of ER stress, PERK-eIF2α-ATF4 axis was also stimulated in HNSCC cells after afatinib incubation. Silencing either eIF2α or ATF4 by siRNA transfection relieved afatinib-caused suppression of AKT-mTOR activity, attenuating MCL-1 down-regulation as well as subsequent apoptosis. Collectively, the results show that afatinib hampers AKT-mTOR activation by stimulating PERK-eIF2α-ATF4 signaling pathway, giving rise to MCL-1 down-regulation mediated apoptosis in HNSCC cells. Therefore, our findings reveal the elaborate molecular network of afatinib-induced apoptosis in HNSCC, which would provide substantial theoretical underpinnings for afatinib clinical application and highlight its promising prospect in HNSCC treatment.
Keywords: Afatinib, apoptosis, PERK, eIF2α, ATF4, MCL-1
Introduction
Head and neck squamous cell carcinoma (HNSCC) is the sixth most common cancer type with depressing prognosis and high mortality worldwide [1]. A vast majority (~90%) of HNSCC patients over-express epidermal growth factor receptor (EGFR), which is related to the poor clinical outcomes [2]. Thus, EGFR has become the hot therapeutic target for developing novel anti-cancer agents in HNSCC. Besides the monoclonal antibody, tyrosine kinase inhibitor (TKI) is another targeted approach against EGFR [3]. Nevertheless, the first-generation TKI such as gefitinib and erlotinib, generated frustrating results in HNSCC clinical research [4-6].
Afatinib, the second generation of TKI, is an oral and irreversible inhibitor that targets the ErbB family members including EGFR, HER2 and HER4 [7], which has received FDA approval for first-line treatment of EGFR mutation-positive non-small cell lung cancer (NSCLC) [8]. Nowadays, phase II and III clinical results demonstrate that recurrent and/or metastatic HNSCC patients obtain benefits from afatinib treatment [9,10]. In addition to advances in clinical research, mechanistic investigations show that afatinib induces apoptosis through triggering pro-apoptotic autophagy or via Elk-1/CIP2A/PP2A/AKT pathway in NSCLC cells [11,12]. The expression of Ki67 and p53 is also down-regulated in afatinib-caused apoptosis in retinoblastoma [13]. Moreover, Young NR et al. demonstrate that afatinib effectively suppresses the viability of a panel of HNSCC cells [14]. However, the molecular mechanism of afatinib-induced apoptosis in HNSCC is poorly elucidated.
In general, there are two extensively studied signaling pathways contributing to apoptosis: the extrinsic death receptor pathway and the intrinsic mitochondrial pathway [15]. The intrinsic apoptosis pathway is initiated by the release of cytochrome-c from the pores correlated with the oligomerization of Bax and Bak [16]. Once turn into cytosol, cytochrome-c recruits apoptosis protease-activating factor 1 (Apaf1) and pre-caspase9 to form apoptosome, which evokes auto-activation of pro-caspase9, leads to stimulation of caspase3 and succedent caspase cascade, resulting in apoptosis ultimately [17]. Importantly, the process is tightly regulated by the activity coordination between pro- and anti-apoptotic members of BCL-2 family [18]. MCL-1, a crucial anti-apoptotic member of BCL-2 family, negatively modulates apoptosis by interfering polymerization of Bax and Bak, blocking the release of cytochrome-c from mitochondria, hence performs a protective role in cell fate determination [19].
Based on the significant pro-survival function of MCL-1, it is rigorously modulated in multi-levels and promoted as a potential therapeutic target in a variety of malignancies. Notably, mTORC1 has been identified to translationally regulate MCL-1 expression [20]. mTORC1 and mTORC2 are two structurally and functionally distinct complex of serine/threonine kinase mTOR [21]. Initiated mTORC1 modulates protein translation and cell metabolism by phosphorylating two vital downstream effectors: eukaryotic initiation factor 4E binding protein 1 (4EBP1) and p70 ribosomal protein S6-kinase (P70S6K) [22]. Moreover, it is well described that activated AKT inactivates Tsc2 and PRAS40, eliminating the suppression of mTORC1 and leading to mTORC1 stimulation [23].
To cope with amount of misfolded proteins accumulation and aggregation in endoplasmic reticulum (ER), cells evolve with a protective mechanism termed unfolded protein response (UPR) which is tightly regulated by three pivotal ER transmembrane proteins including PERK, ATF6 and IRE1α. Whereas when the insults are prolonged and exceeded cells control, the defense mechanism is overwhelmed, and apoptosis is thereafter occurred via activating PERK signaling pathway [24]. eIF2α serves as the effective downstream of PERK stimulation and the phosphorylation of which gives rise to translational restraint in global protein synthesis, while the expression of activating transcription factor 4 (ATF4) is enhanced during this procedure [25]. Many studies have documented that ATF4 up-regulation participates apoptosis induction by anti-tumor agents. Moreover, it also involves autophagy modulation through disturbing AKT-mTOR stimulation [26,27].
In the present study, we found that afatinib dramatically activates PERK-eIF2α-ATF4 axis in HNSCC cell lines, which further contributes to AKT-mTOR suppression, leading to MCL-1 down-regulation and subsequent apoptosis. Based on the data, we build up an innovative bridge between ER stress and afatinib-triggered intrinsic apoptosis. Furthermore, the elaborate interpretation of MCL-1 regulation is illustrated in HNSCC for the first time. We confirm our results could establish a solid foundation for afatinib clinical application in HNSCC treatment.
Materials and methods
Reagents
The pure powder of afatinib and rapamycin was obtained from Selleckchem (Houston, TX, USA). Caspase-8, Caspase-9, PARP, p-PERK, p-eIF2α, p-P70S6K, p-4EBP1 antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). ATF4 and MCL-1 antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Caspase-3 antibody was purchased from Imgenex (San Diego, CA, USA). AKT (phosphor S473) was purchased from Abcam (Cambridge, MA, USA). β-actin antibody was obtained from Sigma-Aldrich (St. Louis, MO, USA).
Cell lines and cell culture
Human HNSCC cell lines FaDu, Detroit 562, HN6, CAL-27 were purchased from the American Type Culture Collection (Manassas, VA). FaDu, HN6, CAL-27 cell lines were cultured in DMEM/F12 (1:1) medium (Gibco, C11330500BT) containing 10% fetal bovine serum (FBS) (Ausbian, VS500T), while Detroit 562 cell line was cultured in MEM/EBSS medium (Hyclone, SH30024.01B) containing 10% FBS. All cells were grown in monolayer culture at 37°C in a humidified atmosphere consisting of 5% CO2 and 95% air.
Cell survival assay
Human HNSCC cells were seeded in 96-well plates and incubated with the indicated concentrations of afatinib for 24 h. Then cell survival was evaluated by sulforhodamine B (SRB) assay as described previously [28]. Briefly, treatment medium was discarded after 24 h incubation and the cells were immobilized with 100 μl cold trichloroacetic acid (10% (w/v)) at 4°C for 1 h. Then, the plates were washed five times with deionized water and air dried. Afterwards, 50 μl 0.4% SRB solution (dissolved in 1% acetic acid) were added into each well for 5 min at room temperature. The plates were then washed with 1% acetic acid for five times to remove uncombined SRB. The combined SRB was dissolved with 100 μl 10 mM Tris base buffer (pH 10.5) and detected absorbency by a microtiter plate reader at 495 nm. Absorbency was regarded to be positively connected with cell survival.
Western blot analysis
The preparation of whole-cell protein lysates and Western blot assay procedure were depicted earlier [29].
Gene knocking down using small interfering RNA
All small interfering RNAs (siRNAs) were synthesized by GenePharma (Shanghai, China). Control siRNA duplexes target the sequence of 5’-UUCUCCGAACGUGUCACGU-3’. eIF2α siRNA duplexes target the sequence of 5’-GAUUGGCACCCUUAUGGUU-3’. ATF4 siRNA duplexes target the sequence of 5’-GCCTAGGTCTCTTAGATGA-3’. The Polyplus Transfection Reagent (Strasbourg, Alsace, France) was applied to transfect siRNAs into cells according to the manufacture’s protocol.
Plasmid transient transfection
The plasmid of pcDNA3.1-MCL-1 was purchased from Addgene (Cambridge, MA, USA). FaDu and HN6 cells were seeded in 6-well plates and transfected with pcDNA3.1 and pcDNA3.1-MCL-1 plasmids using X-treme GENE HP DNA Transfection Reagent (Roche Molecular Biochemicals, Mannheim, Germany) following the manufacture’s instruction. Then, cells were treated with 2 μM afatinib for 24 h and subjected to Western blot analysis.
Apoptosis assays
Apoptosis was assessed by Annexin V-FITC Apoptosis Detection Kit (Bio-BOX Biotech, China) following the manufacturer’s protocol. Caspase activation was examined by Western blot assay.
Statistical analysis
All assays were carried out in three independent experiments and the results were expressed as the mean value ± standard deviation (SD) by GraphPad Prism software. The statistical significance of differences between two groups was analyzed with two-sided unpaired Student’s t-tests and the data were deemed to be statistically significant when the P value was less than 0.05.
Results
Afatinib triggers caspase-dependent apoptosis in HNSCC cell lines
Considering afatinib exerts cytotoxicity effect on various neoplastic cells, we used SRB assay to detect the inhibitory potential of afatinib in HNSCC cells. After incubated with 0, 0.5, 1, 2 μM afatinib for 24 h, the viability of FaDu, Detroit 562, HN6 and CAL-27 cells was dramatically suppressed in a dose-dependent fashion (Figure 1A). To further determine the mechanism through which afatinib elicited survival inhibition, we conducted western blot assay to inspect whether afatinib induced apoptosis in HNSCC cells and the data revealed that afatinib significantly triggered the cleavage of caspase8, caspase9, caspase3 and PARP in both a concentration- and a time-dependent manner (Figure 1B and 1C). Collectively, these results suggest that afatinib effectively suppresses the growth of HNSCC cells via apoptosis induction.
Figure 1.

Afatinib induces apoptosis in HNSCC cells. A. FaDu, Detroit 562, HN6 and CAL-27 cells were cultured in 96-well plates and treated with 0, 1, 2, 4 μM afatinib for 24 h. Thereafter, the cell survival was evaluated by SRB assay. Points: mean of four replicate determinations; bars: S.D. B. Four human HNSCC cell lines were incubated with 0, 0.5, 1, 2 μM afatinib for 24 h. Then, cells were harvested and subjected to Western blot analysis. C. FaDu, Detroit 562 and HN6 cells were incubated with 1 μM afatinib for 0, 12, 24, 36 or 48 h. Then, cells were harvested and subjected to Western blot analysis. CF: cleaved form.
MCL-1 down-regulation is required for afatinib-caused apoptosis
As a pivotal pro-survival component of BCL-2 family, MCL-1 negatively regulates apoptosis during the process of intrinsic apoptosis. Hence, we wondered whether afatinib had an impact on the expression of MCL-1. As shown in Figure 2A, MCL-1 was down-regulated greatly in HNSCC cells after afatinib treatment. And accompanied with elevated concentration of afatinib, a dose-dependent reduction of MCL-1 levels was occurred in FaDu, Detroit 562, HN6 and CAL-27 cells (Figure 2A). In addition, time course assay demonstrated that afatinib markedly decreased MCL-1 expression in a time-dependent manner (Figure 2B). To confirm whether MCL-1 down-regulation was account for afatinib-involved apoptosis, pcDNA3.1-MCL-1 plasmid was transfected into FaDu and HN6 cells. Western blot analysis showed that MCL-1 over-expression distinctly impaired afatinib-induced cleavage of caspase9, caspase3 and PARP (Figure 2C). Therefore, our data demonstrate that MCL-1 down-regulation caused by afatinib contributes to subsequent apoptosis in HNSCC cells.
Figure 2.
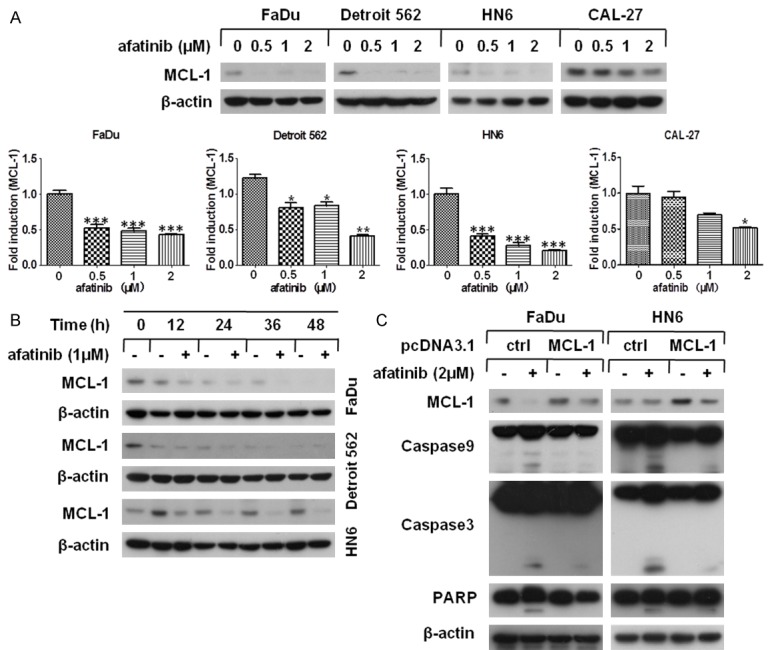
Afatinib down-regulates MCL-1 expression in HNSCC cells. (A) The indicated cell lines were treated with 0, 0.5, 1, 2 μM afatinib for 24 h. Then the expression of MCL-1 was examined by Western blot analysis and quantified using Image J software, bars S.D. *P<0.05, **P<0.01 as compared with control (B) FaDu, Detroit 562 and HN6 cells were treated with 1 μM afatinib for 0, 12, 24, 36 or 48 h. Then, whole-cell lysates were collected and prepared for Western blot analysis. (C) FaDu and HN6 cells were seeded in 6-well plates and transfected with pc-DNA3.1-MCL-1 plasmid on the second day. After 48 h transfection, cells were exposed to 2 μM afatinib for 24 h and carried out Western blot analysis.
Afatinib decreases MCL-1 expression through mTOR suppression
Previous reports have suggested that mTORC1 facilitated cell survival via translational modulation of MCL-1 [30]. Hence, we questioned whether afatinib triggered MCL-1 down-regulation through mTOR inhibition. There are two well characterized downstream substrates of mTORC1: P70S6K and 4EBP1, which actually exert critical protein synthesis function [22]. Thus, we examined the phosphorylation levels of P70S6K and 4EBP1 in four HNSCC cell lines with afatinib treatment for the indicated concentrations or times, and found that the levels of p-P70S6K and p-4EBP1 were both decreased in a concentration- and a time-dependent manner (Figure 3A and 3B). To further justify whether afatinib-induced mTOR inactivation contributes to MCL-1 reduction, rapamycin, an inhibitor of mTOR [31], was used to mimic the inhibitory effect of afatinib. Western blot assay showed that MCL-1 expression was obviously down-regulated in a concentration-dependent manner after rapamycin incubation (Figure 3C). Thus, we come to a conclusion that afatinib reduced MCL-1 expression through inhibiting mTOR activity.
Figure 3.
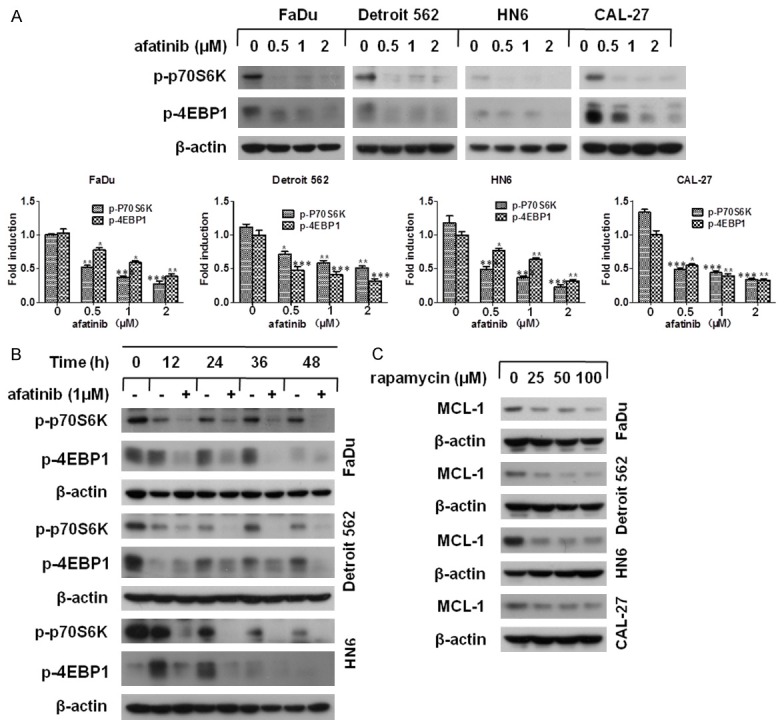
Afatinib inactivates mTOR and contributes to MCL-1 down-regulation. (A) The indicated cells were incubated with 0, 0.5, 1, 2 μM afatinib for 24 h. Then, cells were subjected to Western blot analysis and quantified by Image J software (*P<0.05, **P<0.01 versus control) (B) FaDu, Detroit 562 and HN6 cells were incubated with 1 μM afatinib for the indicated time. Then, cells were collected and subjected to Western blot analysis. (C) Four human HNSCC cell lines were treated with 0, 25, 50, 100 nM rapamycin for 24 h. Subsequently, the levels of MCL-1 were measured by Western blot analysis.
ATF4-AKT-mTOR axis contributes to MCL-1 mediated apoptosis
It has been documented that AKT stimulates mTORC1 by inactivating Tsc2 and PRAS40, which leads to succedent phosphorylation of P70S6K and 4EBP1 [32]. As afatinib works as an irreversible inhibitor of tyrosine kinase, we speculated that afatinib restrained AKT activity and, thereby, suppressed mTOR activity. To confirm our speculation, the phosphorylation level of AKT was measured by western blot analysis in HNSCC cell lines after treatment with the indicated concentration of afatinib for 24 h. And the level of p-AKT was dramatically decreased in a dose-dependent fashion (Figure 4A). Moreover, the time-course assay obtained the consistent results (Figure 4B).
Figure 4.
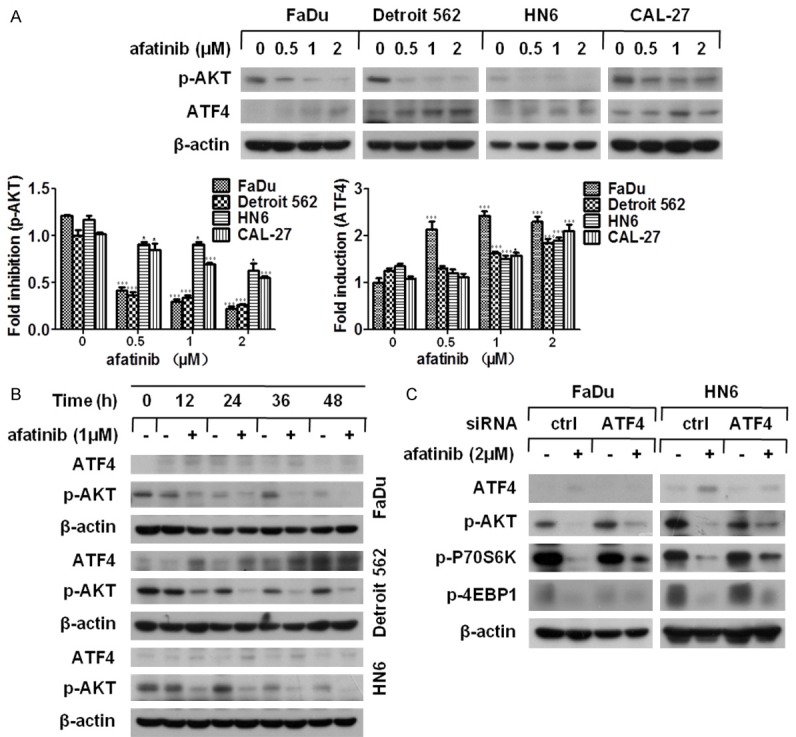
Afatinib suppresses AKT-mTOR via ATF4 up-regulation. (A) The indicated cell lines were treated with afatinib at various concentrations for 24 h. Thereafter, the whole cell lysates were harvested and prepared for Western blot analysis. The given proteins were quantified by Image J software, bars S.D. *P<0.05, **P<0.01 as compared with control (B) FaDu, Detroit 562 and HN6 cells were treated with 1 μM afatinib for various lengths of time. Then, the whole cell lysates were harvested and prepared for Western blot analysis. (C) FaDu and HN6 cells were seeded in 6-well plates and transfected with ATF4 siRNA on the second day. Forty-eight hours later, cells were cultured with 2 μM afatinib for 24 h. Then, cells were collected for Western blot analysis.
Because ATF4 participates in the suppression of AKT-mTOR signaling [27], we detected the level of ATF4 in HNSCC cells after exposure to afatinib. As shown in Figure 4A and 4B, the expression of ATF4 was significantly elevated by afatinib treatment both in a concentration- and a time-dependent fashion. To inspect whether afatinib-caused ATF4 up-regulation resulted in AKT-mTOR inhibition, FaDu and HN6 cells were transfected with ATF4 siRNA and the phosphorylation level of ATK, P70S6K and 4EBP1 was measured by western blot assay following afatinib incubation. We found that the reduction of p-ATK, p-P70S6K and p-4EBP1 caused by afatinib was effectively relieved coupled with ATF4 knockdown (Figure 4C). Hence, the data indicated that ATF4 up-regulation promoted afatinib-triggered AKT-mTOR inhibition.
As ATF4 took part in the process of apoptosis induction as well as AKT-mTOR inactivation [26], we further determined whether afatinib-triggered ATF4 up-regulation influenced MCL-1-mediated apoptosis in HNSCC. FaDu and HN6 cells were treated with ATF4 siRNA to silence ATF4 expression as depicted above, then the levels of MCL-1 and apoptotic-related proteins were examined by western blot analysis. Compared with control cells, both the reduction of MCL-1 and the cleaved form of caspase9, caspase3 and PARP caused by afatinib were attenuated in ATF4 siRNA-transfected cells (Figure 5A). Correspondingly, a dramatic decline of afatinib-induced apoptosis in FaDu and HN6 cells with ATF4 absence was observed in flow cytometery analysis (Figure 5B). In conclusion, our data show that ATF4-AKT-mTOR axis plays a critical role in afatinib-induced MCL-1 down-regulation and subsequent apoptosis in HNSCC cells.
Figure 5.

ATF4 expression is essential for afatinib-caused apoptosis. (A and B) FaDu and HN6 cells were seeded in 6-well plated and transfected with ATF4 siRNA on the next day. After 48 h transfection, cells were exposed to 2 μM afatinib for 24 h. Subsequently, cell lysates were prepared for Western blot analysis (A) or subjected to apoptosis inspection by flow cytometery analysis. *P<0.05, **P<0.01 as compared with control (B).
PERK-eIF2α activation is responsible for afatinib-induced apoptosis
Since ATF4 serves as the vital downstream substrate of PERK-eIF2α signaling pathway [33], we hypothesized that afatinib stimulated PERK-eIF2α signaling which elicits ensuing events. Our research revealed that the phosphorylation level of PERK and eIF2α was greatly enhanced in both a dose- and a time-dependent manner (Figure 6A and 6B). So we then questioned whether eIF2α, the acute responsor of PERK activation, had an effect on afatinib-caused AKT-mTOR inhibition. We used siRNA to silence eIF2α expression and found both afatinib-caused ATF4 induction and AKT-mTOR inhibition were distinctly diminished (Figure 6C). To further explore the function of eIF2α in afatinib-induced apoptosis, western blot assay was performed to detect apoptotic-associated proteins. The results presented that not only did the decline of MCL-1 attenuate, but the cleavage of caspase9, caspase3 and PARP was also weakened when eIF2α inhibited by siRNA treatment (Figure 7A). Moreover, flow cytometery analysis was carried out to evaluate the effect of eIF2α on afatinib-induced apoptosis. And a marked decrease of afatinib-induced apoptosis in FaDu and HN6 cells after eIF2α silence was observed (Figure 7B). Taking together, our data suggest that PERK-eIF2α signaling pathway, the important branch of ER stress, is required for afatinib-induced apoptosis.
Figure 6.
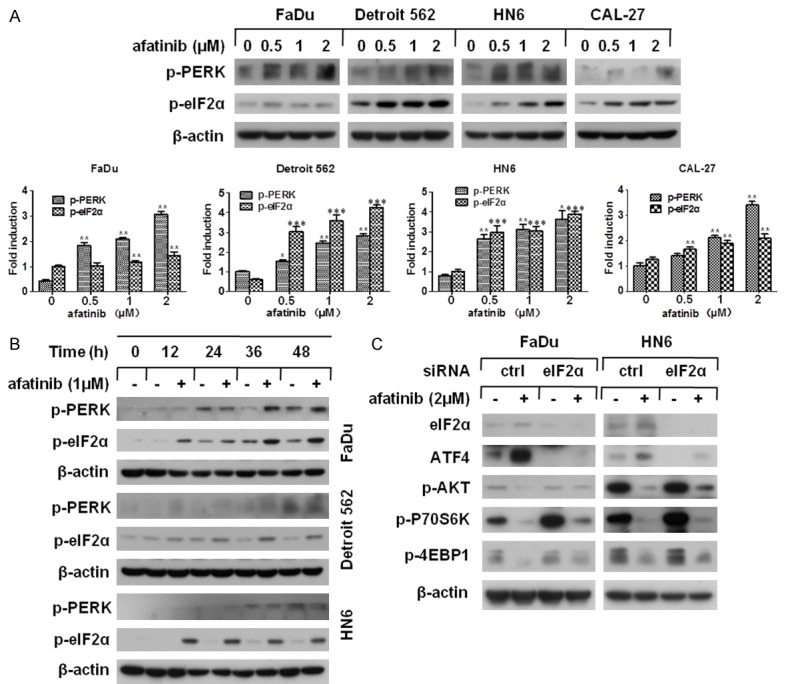
Afatinib activates PERK-eIF2α signaling and conctributes to AKT-mTOR inhibition. (A) The indicated cell lines were treated with 0, 0.5, 1, 2 μM afatinib for 24 h. Hereafter the whole cell lysates were harvested and prepared for Western blot analysis. The detected proteins were quantified using Image J software, bars S.D. *P<0.05, **P<0.01 as compared with control (B) FaDu, Detroit 562 and HN6 cells were treated with 1 μM afatinib for 0, 12, 24, 36 or 48 h. Then, the whole cell lysates were harvested and prepared for Western blot analysis. (C) FaDu and HN6 cells were seeded in 6-well plated and transfected with eIF2α siRNA on the second day. After 48 h transfection, cells were treated with 2 μM afatinib for 24 h. Then cells were harvested and prepared for Western blot analysis.
Figure 7.
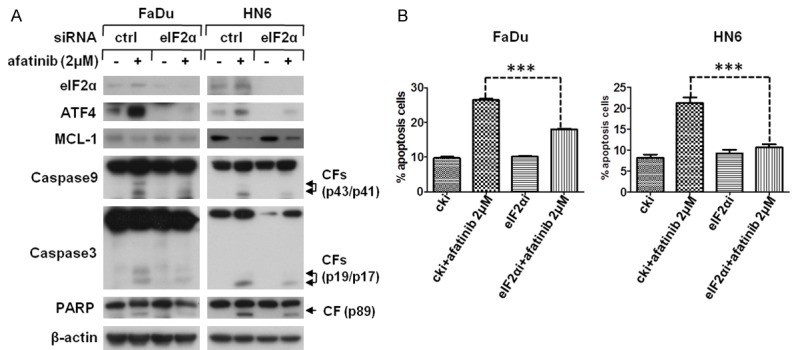
eIF2α activation a is required for MCL-1 mediated apoptosis. (A and B) FaDu and HN6 cells were seeded in 6-well plated and transfected with eIF2α siRNA on the second day. After 48 h transfection, cells were treated with 2 μM afatinib for 24 h. Then cells were havested and prepared for Western blot analysis (A) or subjected to apoptosis inspection by flow cytometery analysis. *P<0.05, **P<0.01 as compared with control (B).
Discussion
Afatinib, a novel irreversible TKI targeting multiple ErbB family members, displays outstanding survival inhibition effects in a broad spectrum of cancer types [11-14,34-36]. Many efforts have been devoted to illuminate the molecular mechanism of afatinib-induced cell death, whereas the mechanism of apoptosis caused by afatinib in HNSCC is still unclear. Here, we exhibit that afatinib stimulates PERK-eIF2α-ATF4 signaling, leading to AKT-mTOR inhibition, which in turn gives rise to MCL-1 down-regulation and subsequent apoptosis in HNSCC cells.
In this study, we found that the cell viability of HNSCC cell lines was apparently decreased after afatinib treatment, indicating afatinib has obvious toxic effects on HNSCC cells. Western blot assay further showed that the cleaved forms of apoptosis-associated proteins (caspase8, caspase9, caspase3 and PARP) were distinctly increased both in a dose- and a time-dependent manner followed by afatinib incubation in FaDu, Detroit 562, HN6 and CAL-27 cells. Thus, the data demonstrate that afatinib induces caspase-dependent apoptosis in HNSCC cells.
MCL-1 is viewed as an anti-apoptotic member belonged to BCL-2 family which is closely connected to mitochondrial apoptosis initiation [37], and blocking MCL-1 makes tumor cells more susceptible to anti-cancer agents [38]. Our work revealed that the level of MCL-1 was dramatically decreased following afatinib treatment and MCL-1 over-expression significantly weakened the cleavage of apoptosis-associated proteins, implying MCL-1 is essential for afatinib-caused apoptosis.
It is well known that MCL-1 is translationally modulated by mTORC1 thereby in parallel to apoptosis regulation [30]. In the present investigation, we identified that the substrates of mTORC1 such as p-P70S6K and p-4EBP1 were dose- and time-dependently declined when MCL-1 was correspondingly reduced after afatinib exposure. To further confirm whether afatinib-caused MCL-1 down-regulation relied on mTOR regulation, rapamycin was utilized to imitate the inhibitory action of afatinib on mTOR, and the expression of MCL-1 was dropped greatly, revealing mTOR guides MCL-1 expression in the treatment of afatinib. Studies have shown that AKT, the vital upstream of mTORC1, inactivates Tsc2 and PRAS40 to relieve the inhibition of mTORC1, thereafter gives rise to the phosphorylation of P70S6K and 4EBP1 [23]. Our results showed that the level of p-AKT was simultaneously decreased by afatinib in HNSCC cells. Therefore, we propose that afatinib down-regulates MCL-1 expression by suppressing AKT-mTOR signaling in HNSCC cells.
Previous research demonstrates that ATF4 up-regulates CHOP-TRB3 axis to inactivate AKT-mTORC1 and plays a key role in both extrinsic and intrinsic apoptosis induction [27]. Here we discovered that ATF4 up-regulation was accompanied with p-AKT reduction after afatinib treatment. And ATF4 silence abolished the decrease of p-AKT, p-P70S6K and p-4EBP1, indicating afatinib-caused AKT-mTOR suppression is dependent on ATF4 elevation. What’s more, ATF4 knockdown not only attenuated afatinib-induced MCL-1 reduction, but also decreased subsequent apoptosis. Collectively, the data suggest that ATF4 is a pivotal medium bridges AKT-mTOR inactivation and MCL-1-mediated apoptosis. Furthermore, to explore how ATF4 influences AKT-mTOR activity, the function of CHOP and TRB3 may need to be put into consideration.
Diverse cell damages that perturb protein synthesizing and folding property of ER bring about ER stress. To fight against this abnormal physiological status, cells develop a defense measure referred to as UPR [39]. Accumulating evidence demonstrated that persistent or violent ER stress initiates apoptosis, the molecular mechanism of which is correlated with IRE1 and PERK signaling [33]. In the PERK signaling, eIF2α is phosphorylated to restrain a majority of mRNA translation, while provides a translational permission for rare proteins such as ATF4, ATF5 and CHOP. Given ATF4 obtained expression advantages from afatinib treatment, we hypothesized that afatinib stimulated PERK signaling pathway of ER stress. The results displayed that afatinib enhanced the phosphorylation of PERK and eIF2α. Further investigation showed that suppressing eIF2α impaired afatinib-caused ATF4 up-regulation and AKT-mTOR inhibition. Moreover, both MCL-1 down-regulation and apoptosis induction by afatinib were concordantly weakened in eIF2α siRNA-transfected cells. Our data suggest that afatinib triggers PERK signaling pathway of ER stress, which renders afatinib to induce apoptotic cell death in HNSCC cells.
In summary, we demonstrate that afatinib stimulates PERK-eIF2α-ATF4 axis which contributes to MCL-1 down-regulation and subsequent apoptosis via suppressing AKT-mTOR signaling. Importantly, we illustrate a novel mechanism of apoptosis triggered by afatinib therapy and this would provide theoretical basis for promoting afatinib as a potent clinical therapeutic strategy in HNSCC treatment.
Acknowledgements
This work was supported by the Key Project of Shandong Provincial Programs for Research and Development (2015GGB14257).
Disclosure of conflict of interest
None.
References
- 1.Rothenberg SM, Ellisen LW. The molecular pathogenesis of head and neck squamous cell carcinoma. J Clin Invest. 2012;122:1951–1957. doi: 10.1172/JCI59889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gold KA, Lee HY, Kim ES. Targeted therapies in squamous cell carcinoma of the head and neck. Cancer. 2009;115:922–935. doi: 10.1002/cncr.24123. [DOI] [PubMed] [Google Scholar]
- 3.Bonner JA, Harari PM, Giralt J, Azarnia N, Shin DM, Cohen RB, Jones CU, Sur R, Raben D, Jassem J, Ove R, Kies MS, Baselga J, Youssoufian H, Amellal N, Rowinsky EK, Ang KK. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006;354:567–578. doi: 10.1056/NEJMoa053422. [DOI] [PubMed] [Google Scholar]
- 4.Cohen EE, Rosen F, Stadler WM, Recant W, Stenson K, Huo D, Vokes EE. Phase II trial of ZD1839 in recurrent or metastatic squamous cell carcinoma of the head and neck. J. Clin. Oncol. 2003;21:1980–1987. doi: 10.1200/JCO.2003.10.051. [DOI] [PubMed] [Google Scholar]
- 5.Soulieres D, Senzer NN, Vokes EE, Hidalgo M, Agarwala SS, Siu LL. Multicenter phase II study of erlotinib, an oral epidermal growth factor receptor tyrosine kinase inhibitor, in patients with recurrent or metastatic squamous cell cancer of the head and neck. J. Clin. Oncol. 2004;22:77–85. doi: 10.1200/JCO.2004.06.075. [DOI] [PubMed] [Google Scholar]
- 6.Cohen EE, Kane MA, List MA, Brockstein BE, Mehrotra B, Huo D, Mauer AM, Pierce C, Dekker A, Vokes EE. Phase II trial of gefitinib 250 mg daily in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck. Clin Cancer Res. 2005;11:8418–8424. doi: 10.1158/1078-0432.CCR-05-1247. [DOI] [PubMed] [Google Scholar]
- 7.Solca F, Dahl G, Zoephel A, Bader G, Sanderson M, Klein C, Kraemer O, Himmelsbach F, Haaksma E, Adolf GR. Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J Pharmacol Exp Ther. 2012;343:342–350. doi: 10.1124/jpet.112.197756. [DOI] [PubMed] [Google Scholar]
- 8.Wang XK, To KK, Huang LY, Xu JH, Yang K, Wang F, Huang ZC, Ye S, Fu LW. Afatinib circumvents multidrug resistance via dually inhibiting ATP binding cassette subfamily G member 2 in vitro and in vivo. Oncotarget. 2014;5:11971–11985. doi: 10.18632/oncotarget.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seiwert TY, Fayette J, Cupissol D, Del Campo JM, Clement PM, Hitt R, Degardin M, Zhang W, Blackman A, Ehrnrooth E, Cohen EE. A randomized, phase II study of afatinib versus cetuximab in metastatic or recurrent squamous cell carcinoma of the head and neck. Ann Oncol. 2014;25:1813–1820. doi: 10.1093/annonc/mdu216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Machiels JP, Haddad RI, Fayette J, Licitra LF, Tahara M, Vermorken JB, Clement PM, Gauler T, Cupissol D, Grau JJ, Guigay J, Caponigro F, de Castro G Jr, de Souza Viana L, Keilholz U, Del Campo JM, Cong XJ, Ehrnrooth E, Cohen EE LUX-H&N 1 investigators. Afatinib versus methotrexate as second-line treatment in patients with recurrent or metastatic squamous-cell carcinoma of the head and neck progressing on or after platinum-based therapy (LUX-Head & Neck 1): an open-label, randomised phase 3 trial. Lancet Oncol. 2015;16:583–594. doi: 10.1016/S1470-2045(15)70124-5. [DOI] [PubMed] [Google Scholar]
- 11.Lee TG, Jeong EH, Kim SY, Kim HR, Kim CH. The combination of irreversible EGFR TKIs and SAHA induces apoptosis and autophagy-mediated cell death to overcome acquired resistance in EGFR T790M-mutated lung cancer. Int J Cancer. 2015;136:2717–2729. doi: 10.1002/ijc.29320. [DOI] [PubMed] [Google Scholar]
- 12.Chao TT, Wang CY, Chen YL, Lai CC, Chang FY, Tsai YT, Chao CH, Shiau CW, Huang YC, Yu CJ, Chen KF. Afatinib induces apoptosis in NSCLC without EGFR mutation through Elk-1-mediated suppression of CIP2A. Oncotarget. 2015;6:2164–2179. doi: 10.18632/oncotarget.2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhan WJ, Zhu JF, Wang LM. Inhibition of proliferation and induction of apoptosis in RB116 retinoblastoma cells by afatinib treatment. Tumour Biol. 2016;37:9249–54. doi: 10.1007/s13277-015-4768-1. [DOI] [PubMed] [Google Scholar]
- 14.Young NR, Soneru C, Liu J, Grushko TA, Hardeman A, Olopade OI, Baum A, Solca F, Cohen EE. Afatinib efficacy against squamous cell carcinoma of the head and neck cell lines in vitro and in vivo. Target Oncol. 2015;10:501–508. doi: 10.1007/s11523-014-0353-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 16.Goldstein JC, Waterhouse NJ, Juin P, Evan GI, Green DR. The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat Cell Biol. 2000;2:156–162. doi: 10.1038/35004029. [DOI] [PubMed] [Google Scholar]
- 17.Kang MH, Reynolds CP. Bcl-2 inhibitors: targeting mitochondrial apoptotic pathways in cancer therapy. Clin Cancer Res. 2009;15:1126–1132. doi: 10.1158/1078-0432.CCR-08-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 19.Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15:49–63. doi: 10.1038/nrm3722. [DOI] [PubMed] [Google Scholar]
- 20.Mojsa B, Lassot I, Desagher S. Mcl-1 ubiquitination: unique regulation of an essential survival protein. Cells. 2014;3:418–437. doi: 10.3390/cells3020418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caron A, Richard D, Laplante M. The Roles of mTOR Complexes in Lipid Metabolism. Annu Rev Nutr. 2015;35:321–348. doi: 10.1146/annurev-nutr-071714-034355. [DOI] [PubMed] [Google Scholar]
- 22.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 23.Lauring J, Park BH, Wolff AC. The phosphoinositide-3-kinase-Akt-mTOR pathway as a therapeutic target in breast cancer. J Natl Compr Canc Netw. 2013;11:670–678. doi: 10.6004/jnccn.2013.0086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7:880–885. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bernales S, Soto MM, McCullagh E. Unfolded protein stress in the endoplasmic reticulum and mitochondria: a role in neurodegeneration. Front Aging Neurosci. 2012;4:5. doi: 10.3389/fnagi.2012.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu L, Su L, Liu X. PKCdelta regulates death receptor 5 expression induced by PS-341 through ATF4-ATF3/CHOP axis in human lung cancer cells. Mol Cancer Ther. 2012;11:2174–2182. doi: 10.1158/1535-7163.MCT-12-0602. [DOI] [PubMed] [Google Scholar]
- 27.Li T, Su L, Zhong N, Hao X, Zhong D, Singhal S, Liu X. Salinomycin induces cell death with autophagy through activation of endoplasmic reticulum stress in human cancer cells. Autophagy. 2013;9:1057–1068. doi: 10.4161/auto.24632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Su L, Liu G, Hao X, Zhong N, Zhong D, Liu X, Singhal S. Death receptor 5 and cellular FLICE-inhibitory protein regulate pemetrexed-induced apoptosis in human lung cancer cells. Eur J Cancer. 2011;47:2471–2478. doi: 10.1016/j.ejca.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 29.Liu X, Yue P, Zhou Z, Khuri FR, Sun SY. Death receptor regulation and celecoxib-induced apoptosis in human lung cancer cells. J Natl Cancer Inst. 2004;96:1769–1780. doi: 10.1093/jnci/djh322. [DOI] [PubMed] [Google Scholar]
- 30.Mills JR, Hippo Y, Robert F, Chen SM, Malina A, Lin CJ, Trojahn U, Wendel HG, Charest A, Bronson RT, Kogan SC, Nadon R, Housman DE, Lowe SW, Pelletier J. mTORC1 promotes survival through translational control of Mcl-1. Proc Natl Acad Sci U S A. 2008;105:10853–10858. doi: 10.1073/pnas.0804821105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.LoPiccolo J, Blumenthal GM, Bernstein WB, Dennis PA. Targeting the PI3K/Akt/mTOR pathway: effective combinations and clinical considerations. Drug Resist Updat. 2008;11:32–50. doi: 10.1016/j.drup.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tasian SK, Teachey DT, Rheingold SR. Targeting the PI3K/mTOR Pathway in Pediatric Hematologic Malignancies. Front Oncol. 2014;4:108. doi: 10.3389/fonc.2014.00108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hiramatsu N, Chiang WC, Kurt TD, Sigurdson CJ, Lin JH. Multiple Mechanisms of Unfolded Protein Response-Induced Cell Death. Am J Pathol. 2015;185:1800–1808. doi: 10.1016/j.ajpath.2015.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Janjigian YY, Viola-Villegas N, Holland JP, Divilov V, Carlin SD, Gomes-DaGama EM, Chiosis G, Carbonetti G, de Stanchina E, Lewis JS. Monitoring afatinib treatment in HER2-positive gastric cancer with 18F-FDG and 89Zr-trastuzumab PET. J Nucl Med. 2013;54:936–943. doi: 10.2967/jnumed.112.110239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang SQ, Liu ST, Zhao BX, Yang FH, Wang YT, Liang QY, Sun YB, Liu Y, Song ZH, Cai Y, Li GF. Afatinib reverses multidrug resistance in ovarian cancer via dually inhibiting ATP binding cassette subfamily B member. Oncotarget. 2015;6:26142–26160. doi: 10.18632/oncotarget.4536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guan SS, Chang J, Cheng CC, Luo TY, Ho AS, Wang CC, Wu CT, Liu SH. Afatinib and its encapsulated polymeric micelles inhibits HER2-overexpressed colorectal tumor cell growth in vitro and in vivo. Oncotarget. 2014;5:4868–4880. doi: 10.18632/oncotarget.2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quinn BA, Dash R, Azab B, Sarkar S, Das SK, Kumar S, Oyesanya RA, Dasgupta S, Dent P, Grant S, Rahmani M, Curiel DT, Dmitriev L, Hedvat M, Wei J, Wu B, Stebbins JL, Reed JC, Pellecchia M, Sarkar D. Targeting Mcl-1 for the therapy of cancer. Expert Opin Investig Drugs. 2011;20:1397–1411. doi: 10.1517/13543784.2011.609167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Akgul C. Mcl-1 is a potential therapeutic target in multiple types of cancer. Cell Mol Life Sci. 2009;66:1326–1336. doi: 10.1007/s00018-008-8637-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yoshida H. ER stress and diseases. FEBS J. 2007;274:630–658. doi: 10.1111/j.1742-4658.2007.05639.x. [DOI] [PubMed] [Google Scholar]