

Abstract
Constitutive activation of extracellular signal regulated kinase (ERK)-Jun NH2-terminal kinase (JNK) signaling commonly occurs in tumors. The activation of ERK promotes cell proliferation, whereas that of JNK induces cell apoptosis. However, the apoptotic mechanism of ERK-JNK signaling in cancer is not well understood. Recently, we identified that apoptosis and activation of the JNK signaling pathway were induced after cordycepin treatment in human renal cancer, suggesting that JNK signaling might contribute to TK-10 cell apoptosis. We investigated the apoptotic effects of cordycepin by evaluating the activation of the ERK-JNK signaling pathway in renal cancer TK-10 cells. We found that cordycepin downregulated ERK and DUSP5, upregulated phosphorylated-JNK (p-JNK), and induced apoptosis. Moreover, we showed that siRNA-mediated inhibition of ERK downregulated DUSP5, whereas ERK overexpression upregulated DUSP5, and that DUSP5 knockdown by siRNA upregulated p-JNK. The JNK-specific inhibitor SP600125 upregulated nuclear translocation of β-catenin, and downregulated Dickkopf-1 (Dkk1), which has been shown to be a potent inhibitor of Wnt signaling. Dkk1 knockdown by siRNA upregulated nuclear β-catenin, suggesting the involvement of the Wnt/β-catenin signaling pathway. DUSP5 overexpression in TK-10 cells decreased p-JNK and increased nuclear β-catenin. The decreased Bax activation markedly protected against cordycepin-induced apoptosis. Bax subfamily proteins induced apoptosis through caspase-3. Taken together, we show that JNK signaling activation by cordycepin mediated ERK inhibition, which might have induced Bax translocation and caspase-3 activation via regulation of DUSP5 in TK-10 cells, thereby promoting the apoptosis of TK-10 cells. Targeting ERK-JNK signaling via the apoptotic effects of cordycepin could be a potential therapeutic strategy to treat renal cancer.
Keywords: Cordycepin, ERK, DUSP5, JNK, apoptosis, TK-10
Introduction
Cordycepin has been reported to have numerous biological activities, including antiproliferative [1,2], apoptotic [3-5], inhibition of platelet aggregation [6], inhibition of cell migration and invasiveness, and anti-inflammatory activities. In mice, cordycepin can reduce tumor formation in a model of metastasis and has therefore been proposed as an anticancer drug [7]. Cordycepin induces cell apoptosis through three signaling pathways: the PKA/TdT signaling pathway, caspase signaling pathway, and p38/JNK signaling pathway. The caspase signaling pathway is the most prominent pathway by which cell apoptosis is induced by cordycepin. Following cordycepin binding to DR3 receptor, DR3 activates caspase-8 via Fas-associated protein with death domain (FADD). Subsequently, caspase-8 may directly activate caspase-3 or induce mitochondria to release cytochrome c, which also activates caspase-3 with the help of Apaf1 and caspase-9 [8-10].
The extracellular signal-regulated kinase 1/2 (ERK1/2) cascade is a central signaling pathway that regulates a wide variety of cellular processes, including mainly proliferation, differentiation, transformation, and survival, but apoptosis and stress response as well. ERK1 and ERK2 isoforms are both phosphorylated at the conserved T-X-Y motif in the activation loop of the kinase. Generally, the ERK signaling is activated by growth factors and plays a role in cell proliferation [11]. Conversely, the JNK signaling pathway is stimulated by genotoxic agents and cytokines mediating growth arrest and apoptosis [12-16], indicating that more complex roles of these mitogen activated protein kinase (MAPK) pathways exist to transmit other ultimately distinct cellular effects in various subcellular organelles [17,18].
ERK is negatively regulated by specific protein phosphatases. Dual-specificity MAPK phosphatases (DUSPs) are a form of phosphatase that can act on tyrosine or serine/threonine residues. Among them, DUSP5 and DUSP6, localized in the nucleus [19] and cytoplasm, respectively [20], specifically dephosphorylate ERK [21]. These phosphatases belong to the large family of DUSPs, since they dephosphorylate both tyrosine and serine/threonine residues [22]. Since DUSP5 possesses a functional nuclear localization signal and has been proposed to act as a nuclear anchor for ERK, its substrate selectivity is only determined by the specific interaction with nuclear ERK [23]. ERK signaling is required for the induction of DUSP5 and DUSP6 [24-27], which are involved in a negative feedback loop that tightly controls the levels of phosphorylated ERK (p-ERK). The role of DUSPs in both cancer progression and cancer resistance is obvious, making them rational targets for new therapeutics [24]. DUSP5 negatively regulates members of the MAPK superfamily (MAPK/ERK, SAPK/JNK, p38), which are associated with cellular proliferation and differentiation. In this study, we analyzed the effects of cordycepin on renal cancer cell apoptosis and studied the relationship between ERK and JNK. We attempted to identify the apoptotic pathway by which cordycepin-mediated inhibition of ERK promoted JNK signaling by regulating DUSP5, resulting in inhibition of nuclear β-catenin translocation, thus inducing apoptosis in human renal-cancer cells. The data presented herein clearly show that cordycepin is involved in the ERK-JNK signaling pathway by modulating DUSP5 signaling, and that the consequent activation of the Bax/caspase-3-mediated pathway causes cancer cell death.
Materials and methods
Cell lines and cell viability assay
The human renal adenocarcinoma cell line TK-10 was obtained from the National Cancer Center (Ilsan, Korea). The cells were grown in RPMI1640 medium supplemented with 10% (v/v) FBS and 1% (w/v) penicillin-streptomycin at 37°C under 5% (v/v) CO2. Cells (5 × 103/well) were seeded into a 96-well plate. After 24-h incubation, the cells were treated with 0, 20, 40, 60, 80, 100, and 120 μM cordycepin for 48 h. Cell viability assays were conducted as reported previously [28]. At the end of the treatment, 10 μl of CCK-8 solution was added to the cell solution and incubated at 37°C for 1 h. Cell viability was determined by measuring the absorbance at 450 nm using a microplate reader (Sunrise, Tecan, Switzerland). The assays were performed in triplicate. The appropriate dose was determined by evaluating the cytotoxicity of cordycepin after 48 h.
Reagents and chemicals
Fetal bovine serum (FBS), 1% (w/v) penicillin-streptomycin stock solution, phosphate-buffered saline (PBS), and Roswell Park Memorial Institute (RPMI) 1640 medium were obtained from Thermo (Paisley, Scotland, UK). Cordycepin (3’-deoxyadenosine, from Cordyceps militaris) was purchased from Sigma-Aldrich (St. Louis, MO, USA, C3394). An Annexin-V-FLUOS staining kit was purchased from Roche Diagnostics GmbH (Mannheim, Germany). Whole cell lysis buffer was purchased from Intron (Seoul, Korea), and transfection reagent Hilymax and cell-counting kit-8 (CCK-8) from Dojindo (Dojindo, Japan). DAPI (4’,6-Diamidino-2-Phenylindole, Dihydrochloride) and TRIZOL reagent were purchased from Thermo. Antibodies against β-catenin, p-JNK, caspase-3, p-Akt, and β-actin were purchased from Cell Signaling (Beverly, MA, USA). Antibodies against ERK, JNK, Akt, DUSP5, Dkk1, and Bax, and the JNK inhibitor SP600125 were from Santa Cruz (Dallas, TX, USA).
Cell cycle analysis by propidium iodide (PI)-Annexin V staining
To detect the effect of cordycepin on apoptosis, we analyzed the PI-Annexin V staining pattern by using the Annexin V-FLUOS staining kit (Roche Diagnostics). Cells were treated with 0, 20, 40, 60, 80, and 100 μM of cordycepin for 48 h, collected, and washed twice with PBS. The cell suspension was centrifuged at 2000 × g for 2 min and incubated at 24°C with 0.2 mg/mL Annexin V FLUOS and 1.4 mg/mL PI and RNase solution for 15 min under dark conditions. Measurements were conducted using an Image Cytometer (NUCLEOCOUNTER® NC-3000TM; Chemometec, Copenhagen, Denmark) with an excitation wavelength of 488 nm and a 530/30-nm band-pass filter to detect Annexin V and a 670 nm high-pass filter to detect PI.
Microarray analysis
Total RNA was extracted from vehicle- or 100 µM cordycepin-treated TK-10 renal call adenocarcinima. The total RNA from each sample was extracted using TRIZOL reagent according to the manufacturer’s instructions. For microarray analysis of the cordycepin-treated renal cancer cells, the Human Twin ChipTM Human 44 K (Genocheck, Seoul, Korea) was used for the transcription profiling analysis. In brief, the Cy3- (vehicle) and Cy5-labeled (cordycepin-treated) cRNAs were hybridized with the Human 44 K microarray, and the hybridization images were analyzed with an Agilent DNA Microarray Scanner. All data normalization and selection of up-regulated and down-regulated genes were performed using GeneSpring GX 7.3 (Agilent Technology). The microarray data have been submitted to the Gene Expression Omnibus database (GEO accession number: GSE81718).
Gene ontology-based network analysis
To study the biological functions of the regulated genes through their interaction network, we conducted a network analysis by using ingenuity pathway analysis (IPA, http://www.ingenuity.com) to examine the biological functions of the differentially regulated genes and proteins according to ontology-related interaction networks, including apoptosis signaling. Network generation was optimized from the obtained expression profiles when possible and was aimed at producing highly connected networks.
Gene overexpression and silencing
For overexpression of ERK, we used lentivirus carrying RFP-conjugated full-length ERK1 and DUSP5 (Lenti H1.4-ERK1, DUSP5/RFP; Bioneer Corp., Daejeon, Korea). Small interfering RNAs (siRNAs) were purchased from Cell signaling and ST Pharm (Seoul, Korea). ERK1/2 siRNA was purchased from Cell signaling (SignalSilence® p44/42 MAPK (Erk1/2) siRNA #6560). The nucleotide sequences of the siRNAs used in this study were as follows: for DUSP5, 5’-GGC CUU CGA UUA CAU CAA G-3’; for β-catenin, 5’-GCU UGG AAU GAG ACU GCU GAU-3’ and for Dkk1 siRNA, 5’-AAG AAC GGA AGU GUG AUA UGU-3’. Scrambled control siRNA (Silencer Negative Control 5) was provided by Ambion (Waltham, MA, USA). Lentiviral infection was performed according to the manufacturer’s method. Briefly, TK-10 cells were seeded (2 × 105 cells/well) into a 6-well plate and infected with 1 mL of lentivirus for 8 h. After incubation, the cells were supplied with growth medium containing 10% FBS and were harvested 48 h later for further assays. Transfection of siRNA into the TK-10 cells was performed using Lipofectamine RNAiMAX reagent (Invitrogen, Carlsbad, CA, USA) in accordance with the manufacturer’s instructions. Cells were then treated with 100 mM cordycepin for 48 h.
Fractionation and protein extraction
TK-10 cells were incubated with cordycepin for 2 days. The cells were collected with 2 mL of homogenization buffer A (25 mM Tris (pH 7.5), 2 mM EDTA, 0.5 mM EGTA, 1 mM DTT, protease inhibitor cocktail, 1 mM PMSF, and 0.02% Triton X-100) per culture dish, homogenized 15 times using a 15-mL Dounce homogenizer with pestle A, and centrifuged at 100,000 × g for 30 min. The supernatant cytosolic fraction was transferred to a new tube and 500 μL of homogenization buffer B (homogenization buffer A containing 1% Triton X-100) was added to the pellet. The pellet was resuspended by sonication, incubated for 30 min at 4°C by shaking, and centrifuged at 100,000 × g for 30 min. The supernatant nuclear fraction was transferred to a fresh tube. The protein contents of the cytosolic and nuclear fractions were determined by using a bicinchoninic acid (BCA) assay kit (Thermo Scientific, Rockford, IL, USA) and analyzed by western blotting using anti-β-catenin antibody.
Western blotting
Cell lysates were mixed with loading buffer and separated by SDS-PAGE. Proteins were then transferred to nitrocellulose membranes. Non-specific binding sites on the membranes were blocked using 5% non-fat dry milk for 90 min at room temperature. Membranes were incubated with primary antibodies against ERK (1:1000), JNK (1:200), p-JNK (1:200), β-catenin (1:500), p-β-catenine (1:100), caspase-3 (1:500), Bax (1:1000), Akt (1:1000), p-Akt (1:1000), DUSP5 (1:1000), Dkk1 (1:1000), and β-actin (1:2000) at 4°C overnight, followed by secondary antibodies for 1 h at room temperature. Protein bands on the membranes were visualized using an ECL Plus Blotting Detection System from Santa Cruz Biotechnology Inc. and ChemiDoc MP system (Bio-Rad, Hercules, CA, USA). Densitometric measurements of bands were made using the ImageJ software. The expression levels of proteins were quantitatively analyzed through comparison with actin as an internal control.
Immunofluorescence microscopy
Untreated control and over-DUSP5 transfected TK-10 cells were seeded at a density of 4 × 104 cells/well on a coverslip in a 12-well plate. The cells were pretreated with cordycepin at 100 mM for 48 h. The cells were washed twice with 250 μM (1 ×) PBS (pH 7.5) and fixed with 4% (v/v) formaldehyde for 15 min. They were permeabilized with 0.1% (v/v) Triton X-100 for 15 min and then incubated with 3% (w/v) bovine serum albumin (BSA) for 1 h to prevent nonspecific binding. The cells were incubated with β-catenin monoclonal purified mouse IgG1 as a primary antibody (diluted to 1:100 in 3% (w/v) BSA) overnight at 4°C followed by 1-h incubation in the dark with fluorescein isothiocyanate-anti-mouse antibody (diluted to 1:200 in 3% (w/v) BSA; Invitrogen Life Technologies, Carlsbad, CA, USA) as a secondary antibody. Fixed cells were washed with PBS and stained with 4,6-diamidino-2-phenylindole (DAPI) at room temperature. The cells were again washed twice with PBS and mounted with mounting solution for observation. Images were acquired using an LSM 710 laser-scanning confocal microscope (Carl Zeiss, Jena, Germany) equipped with a C-Apochromat 40 ×/1.2 water immersion lens (488 nm Ar laser/505-550 nm detection range). Image data were analyzed with the ZEN 2009 Light Edition software (Carl Zeiss).
Statistical analysis
GraphPad Prism software (GraphPad, San Diego, CA, USA) was used for the statistical analyses. Student’s t-test was used to assess differences between the control and the cordycepin-treated groups. P-values less than 0.05 were considered statistically significant.
Results
Cordycepin inhibits lung cancer cell growth
To investigate the effects of cordycepin on renal cancer cell proliferation, TK-10 cells were treated directly with 0, 10, 20, 40, 60, 80, 100, or 120 μM cordycepin for 48 h. As shown in Figure 1A, cordycepin inhibited the growth of the cells during the 48-h incubation period in a dose-dependent manner. At 100 μM, cordycepin inhibited approximately half of the TK-10 cell population. Thus, the half-maximal inhibitory concentration (IC50) was determined as 100 μM (Figure 1A). The apoptotic effect of cordycepin on TK-10 renal cancer cells was analyzed with Annexin V- and PI-staining using flow cytometry after 48-h treatment with 0 to 100 μM cordycepin. The relative proportion of non-viable cells was quantitatively measured as cells at the early stage of apoptosis (Annexin V-stained, non-disrupted cells) or as cells entering the late stage of apoptosis (disrupted or lysed cells). In 20 to 40 μM cordycepin-treated cells, no drastic change in the Annexin V-stained viable fraction was observed (91% to 89% and 88%) (Figure 1B). However, in cells treated with 100 μM cordycepin, a marked shift from the normal state to the early apoptotic stage (4% to 26%), was observed whereas the viable fraction decreased from 91% to 65%. Thus, cordycepin at 100 μM induces apoptosis in renal cancer cells.
Figure 1.
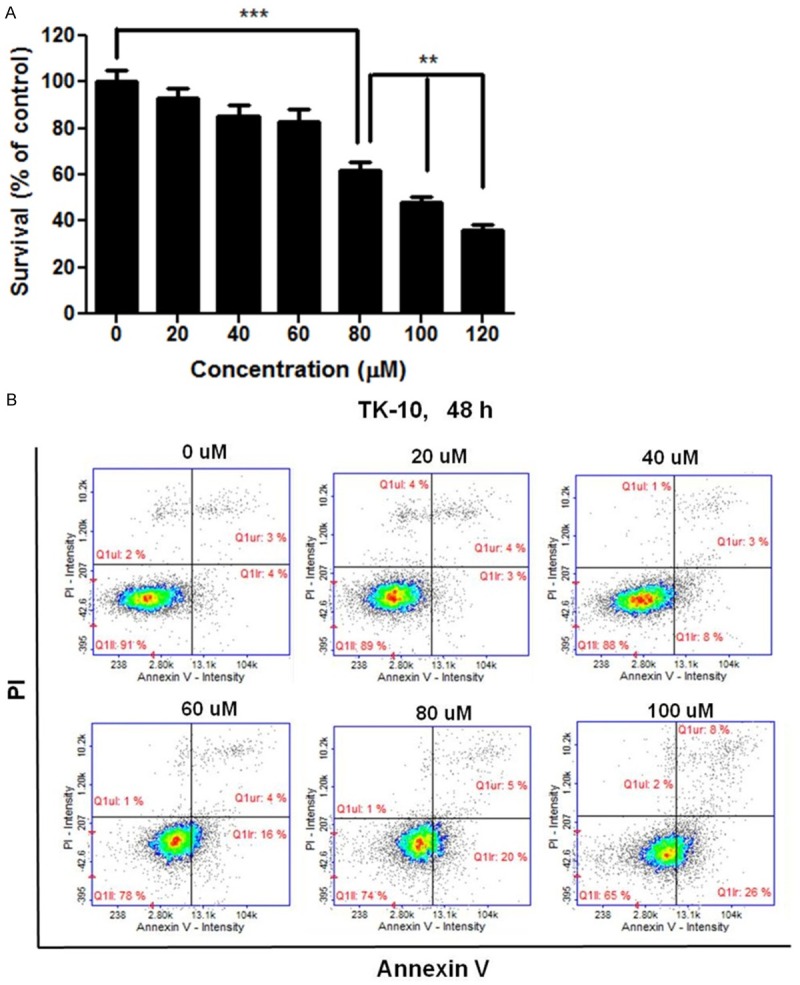
Cordycepin induces apoptosis in renal cancer cells. A: Inhibition of the growth of renal cancer cells by cordycepin. TK-10 cells were exposed to cordycepin at 0, 20, 40, 60, 80, 100, or 120 μM for 48 h. Data are presented as the mean ± standard deviation from triplicate experiments. *P<0.05, **P<0.01, and ***P<0.001 vs. untreated control. B: Apoptosis analysis of TK-10 cells exposed to cordycepin. The cells were treated with cordycepin at 0, 20, 40, 60, 80, or 100 μM for 48 h. The cells were doubly strained with PI and Annexin V and analyzed by flow cytometry. The data are representative of three independent experiments.
Cordycepin alters gene expression
To identify the potential genes involved in the anti-cancer activity of cordycepin, we conducted microarray analysis of TK-10 cancer cells after treatment with 100 μM cordycepin. Among the 43,142 unique genes tested, 30,165 genes were expressed in the cordycepin-treated cells. Among these 30,165 genes, 2,243 and 1,442 genes were up- and downregulated, respectively, after 48-h treatment with cordycepin. Genes that were significantly up- or downregulated by more than 2-fold were subjected to GO enrichment analysis using the Database for Annotation, Visualization, and Integrated Discovery (DAVID) tools (http://david.abcc.ncifcrf.gov/). The upregulated genes were mainly involved in developmental process, positive regulation of apoptosis, immunity and defense, central nervous system development, sensory perception, endocytosis, cell cycle control, and negative regulation of cell development (Figure 2A). The downregulated genes were related to intracellular signaling cascade, apoptosis, regulation of cellular protein, and cell adhesion-mediated signaling. To identify genes potentially involved in apoptosis among the cordycepin-induced genes, we used the GeneCards database (http://www.genecards.org/) (Figure 2B, 2C). Quantitative alterations in gene expression were observed in cordycepin-treated renal cancer cells as compared to that in control cells. The signal network of apoptotic genes regulated in response to cordycepin is shown in Figure 2D. Among these, DUSP5 and ERK1 were identified as central hubs of the apoptosis-related interactome network in the cordycepin-treated lung cancer cells.
Figure 2.
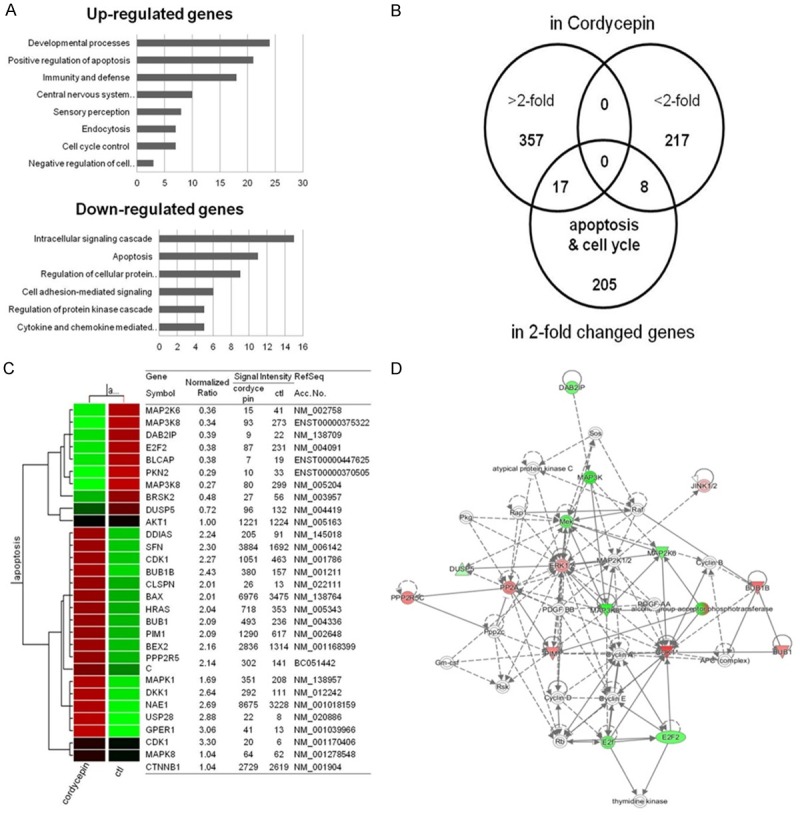
Gene expression analysis and signal network of apoptotic genes. A: Results of microarray analysis of gene expression in response to 100 μM cordycepin for 48 h. B: Venn diagram of expression of genes that were altered more than 2-fold and apoptosis-related genes in response to cordycepin. C: List of apoptosis-related genes that were altered more than 2-fold in response to cordycepin. D: Signal network of the apoptotic genes generated using a Qiagen IPA (red: upregulated genes, green: downregulated genes).
Cordycepin downregulates p-ERK and DUSP5 and upregulates p-JNK and Bax
Because the expression of p-ERK decreases after cordycepin treatment, we examined the cell signaling modulated by ERK in TK-10 cells treated with cordycepin. The expression of p-ERK (active ERK), DUSP5, p-JNK (active JNK), and total JNK at 24 and 48 h after cordycepin treatment was analyzed using western blot analysis (Figure 3A). The expression of total JNK protein was not obviously altered at different time points when compared to the control (0 h). However, p-ERK and DUSP5 protein expression decreased after cordycepin treatment in a time-dependent manner. A 2-fold increase in p-JNK expression was observed at 48 h after cordycepin treatment as compared with the control. The cell lysates were probed with various antibodies to examine the cordycepin-induced apoptotic cell signaling. Western blot analysis showed that Bax, caspase-3, and cleaved caspase-3 expression increased after cordycepin treatment.
Figure 3.
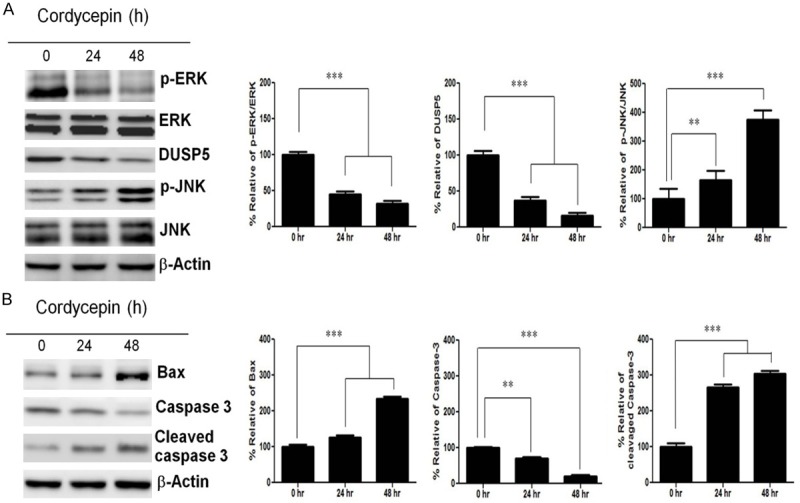
Expression of p-ERK, ERK, DUSP5, p-JNK, JNK, and Bax proteins after cordycepin treatment. A: Western blots showing expression of p-ERK, ERK, DUSP5, p-JNK, and JNK at 0, 24, and 48 h in TK-10 cells after cordycepin treatment. B: Western blots showing expression of Bax, caspase-3, and cleaved caspase-3 at 0, 24, and 48 h in TK-10 cells after cordycepin treatment. Bar graphs represent densitometric analysis of at least three separate experiments (means ± SD). *P<0.05, **P<0.01, and ***P<0.001.
Cordycepin-mediated ERK inhibition results in JNK phosphorylation and β-catenin nuclear translocation
We assessed whether ERK was involved in the negative regulation of p-JNK after cordycepin treatment by siRNA-mediated silencing of ERK. Cordycepin decreased the expression of ERK and increased that of p-JNK, whereas siRNA-mediated inhibition of ERK effectively lowered the DUSP5 protein level and abrogated p-JNK expression, indicating that ERK-DUSP5 signaling is involved in the regulation of JNK phosphorylation (Figure 4A). To confirm whether JNK is involved in β-catenin regulation after cordycepin treatment, we used SP600125, a highly specific JNK inhibitor, which blocks JNK phosphorylation by binding to its ATP-binding site. In TK-10 cells treated with 10 mM SP600125, the level of phosphorylated JNK increased at 48 h after cordycepin treatment as compared to the controls (Figure 4B), which indicated that cordycepin induced the upregulation of JNK activity. Next, we investigated whether SP600125 affected the nuclear translocation of β-catenin. We found that SP600125 treatment significantly increased the nuclear translocation of β-catenin at 48 h after cordycepin treatment as compared to the control (Figure 4B). However, cytosol β-catenin was markedly decreased at 48 h by SP600125 treatment. We then examined the expression of DUSP5 and JNK. Cordycepin increased the level of Bax protein (Figure 4B). These results suggest that the downregulation of ERK and DUSP5 contributes to the expression of Bax after cordycepin treatment. We next performed a loss-of-function analysis using DUSP5 knockdown by siRNA. si-DUSP5 enhanced p-JNK expression, whereas DUSP5 overexpression suppressed the p-JNK protein-mediated increase in Bax (Figure 4B). Taken together, these results indicate that cordycepin-mediated ERK inhibition downregulates DUSP5, which phosphorylates JNK and regulates β-catenin nuclear translocation. In addition, si-β-catenin significantly downregulated nuclear β-catenin and upregulated Bax, whereas SP600125 significantly upregulated nuclear β-catenin and downregulated Bax (Figure 5A, 5B), indicating that β-catenin signaling promotes apoptosis by upregulating Bax in TK-10 cells. Taken together, these results indicate that cordycepin-induced DUSP5 downregulation upregulated the p-JNK-induced decrease in nuclear β-catenin translocation, leading to the upregulation of Bax.
Figure 4.
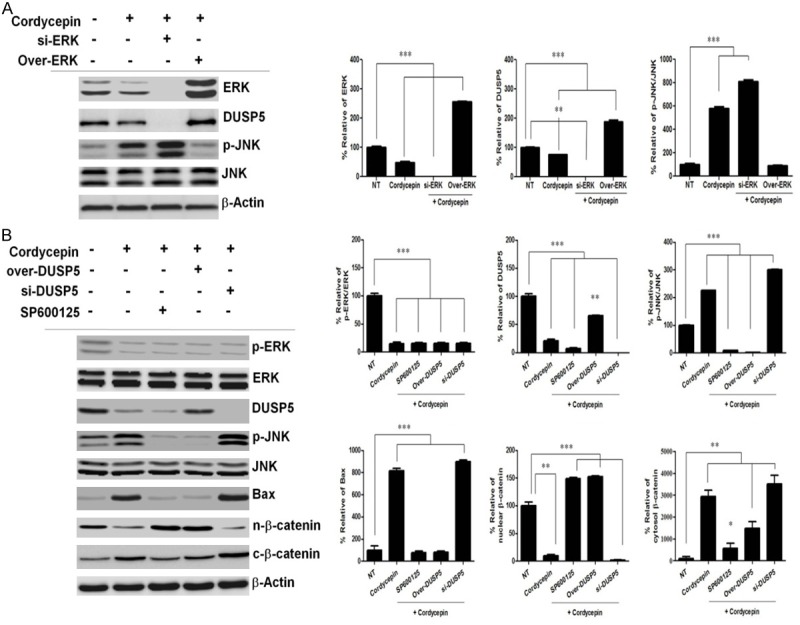
Cordycepin-mediated ERK inhibition downregulates DUSP5 and phosphorylates JNK. A: Representative western blots of TK-10 cells treated with cordycepin showing the expression of p-ERK, ERK, DUSP5, p-JNK, JNK. TK-10 cells were incubated with siRNA directed against ERK (si-ERK) or negative control siRNA for 48 h, and transfected with an ERK-overexpressing construct for 48 h. B: Cordycepin-mediated DUSP5 inhibition phosphorylates JNK and increases Bax. TK-10 cells were incubated with siRNA directed against DUSP5 (si-DUSP5) for 48 h, and transfected with a DUSP5-overexpressing construct for 48 h. TK-10 cells were treated with cordycepin or cordycepin and 10 mM SP600125 for 48 h. Bar graphs represent densitometric analysis of at least three separate experiments (means ± SD). *P<0.05, **P<0.01, and ***P<0.001.
Figure 5.
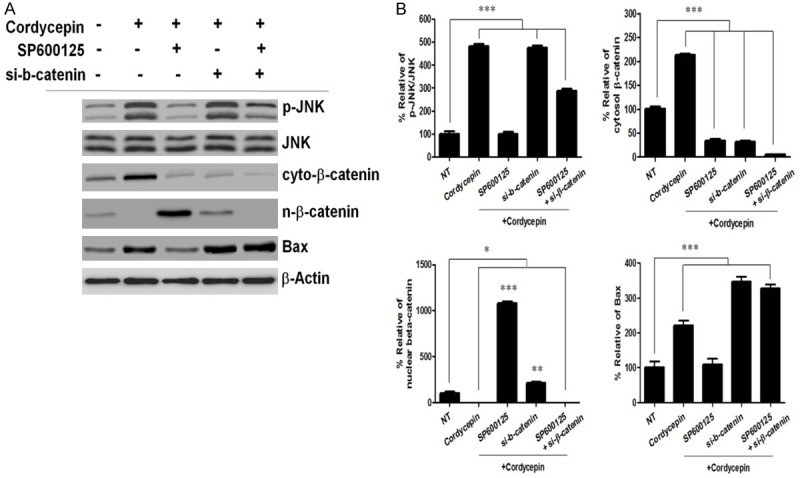
Cordycepin upregulates p-JNK to prevent nuclear β-catenin translocation. A: TK-10 cells were treated with cordycepin or cordycepin and 10 mM SP600125 for 48 h. TK-10 cells were incubated with siRNA directed against β-catenin (si-β-catenin) or negative control siRNA for 48 h. B: Band intensities are represented as percentage of p-JNK, nuclear β-catenin. Data were densitometric analysis of at least three separate experiments (means ± SD). *P<0.05, **P<0.01, and ***P<0.001.
DUSP5-JNK signaling regulates nuclear β-catenin translocation signaling through Dkk1 expression
We next examined the potential role of JNK in the regulation of β-catenin signaling in TK-10 cells. As shown in Figure 6A, Dkk1 was downregulated in JNK inhibitor SP600125-treated TK-10 cells and siRNA-mediated inhibition of Dkk1 upregulated the levels of nuclear β-catenin protein and phosphorylated protein kinase B (p-Akt) after cordycepin treatment (Figure 6B). These findings indicate that JNK regulates nuclear β-catenin translocation signaling through Dkk1 expression. There is a crosstalk between the pro-apoptotic JNK pathway and the pro-survival Akt pathway at multiple levels. To further investigate whether the regulation of β-catenin by JNK after cordycepin treatment is related to Akt, we examined the expression of p-Akt and Akt protein after cordycepin treatment by western blotting. Additionally, we silenced β-catenin using specific siRNA and blocked JNK activation with SP600125. We found that p-Akt was upregulated and total Akt was not affected at 48 h in the SP600125-treated TK-10 cells as compared to that in the untreated control (Figure 6B), suggesting that JNK is dependent on Akt in this study. Cordycepin enhanced p-JNK (Figure 6A). We next performed a loss-of-function analysis using DUSP5 knockdown by siRNA. si-DUSP5 enhanced p-JNK, and the selective JNK inhibitor SP600125 blocked JNK, whereas DUSP5 overexpression suppressed the p-JNK protein-mediated increase in Bax (Figure 6B). Consistent with this finding, β-catenin nuclear translocation was inhibited after cordycepin treatment, whereas both cytosolic and nuclear β-catenin increased in untreated control as confirmed by confocal microscopy (Figure 6C). DUSP5 over-expressing constructs were transfected into TK-10 cells, which were then treated with cordycepin for 24 h; this upregulated nuclear β-catenin translocation and JNK mediated Dkk1, and DUSP5 overexpression enhanced the nuclear translocation of β-catenin (Figure 6C) and markedly protected against cordycepin-induced apoptosis (Figure 6D).
Figure 6.
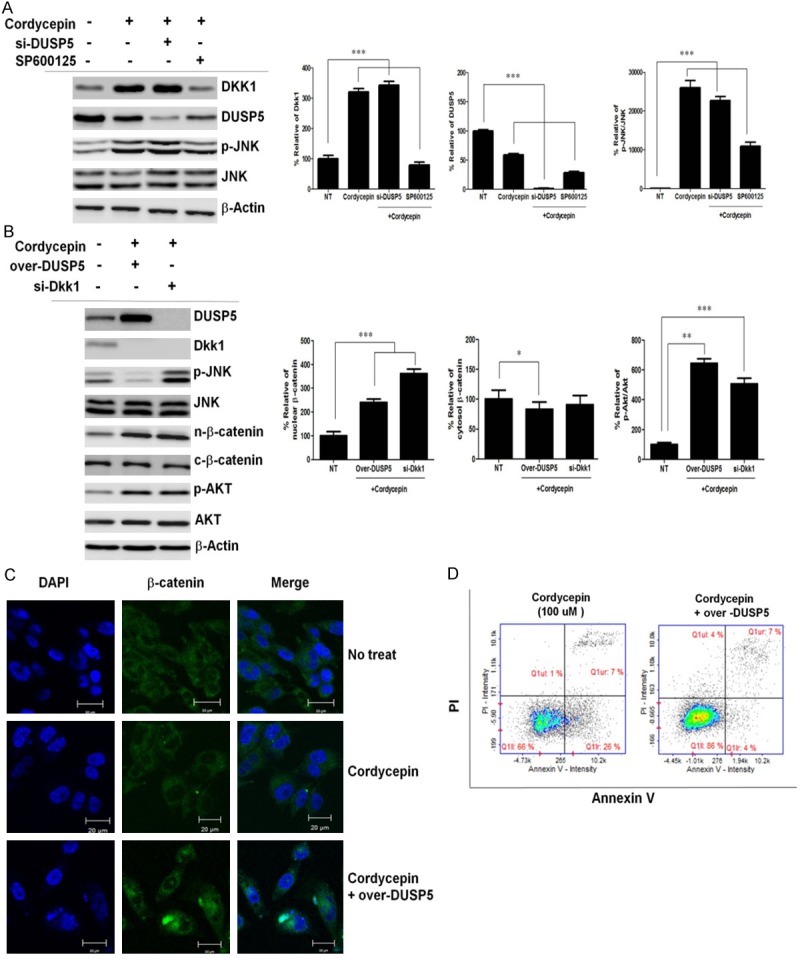
DKK1 signaling through JNK attenuates p-Akt, resulting in inhibition of nuclear β-catenin translocation. A: Cordycepin-mediated p-JNK upregulation enhances Dkk1. TK-10 cells were treated with cordycepin or cordycepin and 10 mM SP600125 for 48 h. TK-10 cells were incubated with siRNA directed against DUSP5 (si-DUSP5) or negative control siRNA for 48 h. B: TK-10 cells were incubated with siRNA directed against Dkk1 (si-Dkk1) or negative control siRNA for 48 h, and transfected with a DUSP5-overexpressing construct for 48 h. Bar graphs represent densitometric analysis of at least three separate experiments (means ± SD). *P<0.05, **P<0.01, and ***P<0.001. C: TK-10 cells were grown on glass coverslips and treated with cordycepin or cordycepin with DUSP5 overexpression for 48 h. Samples were analyzed by indirect immunofluorescence with confocal microscopy. The cellular localization of β-catenin (cytosol, green; nucleus, blue) is shown in the middle panels. The right panels are merged images. The bar indicates 20 μm. D: Analysis of apoptotic protection by DUSP5 overexpression using double-labeled flow cytometry in TK-10 cells treated with cordycepin. Control (left panel) and DUSP5 over-expressing (right panel) constructs were transfected into TK-10 cells, which were then treated with cordycepin for 24 h.
Discussion
Cordycepin has been reported to have various biological activities, including inhibition of protein synthesis and cell adhesion, inflammation, platelet aggregation, and mRNA polyadenylation [29-31]. In addition, it has remarkable anticancer potential, such as inhibition of cell proliferation [32,33], induction of apoptosis, and inhibition of cell growth, migration and invasiveness [34,35]. These inhibitory effects on tumor have been observed in oral, lung, prostate, and colorectal carcinoma, and mainly involve the induction of apoptosis via the targeting of specific molecules and pathways [36-39]. However, the roles of ERK and JNK signaling in the induction of apoptosis are not clearly understood. This study showed that treatment with 100 μM cordycepin reduced viability and strongly inhibited the growth of renal cancer TK-10 cells (Figure 1A). Moreover, 100 μM cordycepin had an effect on the morphology of TK-10 cells as compared to the untreated control and 20-80 μM cordycepin (Figure 1B, 1C). The results of Annexin V/PI staining using FACS demonstrated that cordycepin could induce pro-apoptosis. Cordycepin at 100 μM induced the transformation of cells from the normal state (untreated group: 91.0% normal, 4.0% early apoptotic, and 3% late apoptotic) to the apoptotic state (65.0% normal, 26.0% early apoptotic, and 8.0% late apoptotic) (Figure 1D). These results suggest that cordycepin exhibits anti-lung cancer activity by promoting proapoptosis.
To analyze cordycepin-related gene expression in renal cancer cells, we used a cDNA microarray approach. Clustering of the microarray data identified groups of genes that were differentially regulated upon treatment of TK-10 cells with 100 μM cordycepin. The GO categories of genes whose expression was altered by at least 2-fold are shown in Figure 2A. Among these, 20 genes whose expression increased and 9 with decreased expression were related to apoptosis (Figure 2B, 2C). To explore the major cordycepin-regulated proteins identified using GO analysis, we used IPA to query 29 proteins that were up- or downregulated by cordycepin, yielding a distinct interconnected network of 33 proteins (Figure 2D). Among these, ERK/JNK (MAPK8) were the center of the apoptosis-related protein network. In present study, we showed that the ERK/DUSP5/JNK pathway was involved in TK-10 cell apoptosis after cordycepin treatment. First, we found that cordycepin-mediated ERK inhibition upregulated p-JNK and the reduction of nuclear β-catenin preceded TK-10 renal cancer cell apoptosis, suggesting that JNK phosphorylation and nuclear β-catenin were involved in mediating cordycepin-induced TK-10 cell apoptosis. Second, cordycepin upregulated JNK phosphorylation through ERK-mediated DUSP5 regulation. Third, treatment with the JNK inhibitor SP600125 significantly increased nuclear β-catenin levels, leading to a decrease in the protein levels of Bax and cleaved caspase-3. Finally, siRNA-mediated inhibition of β-catenin attenuated Bax levels after cordycepin treatment. Previous studies on the expression and function of the ERK and JNK signaling pathways in cancer have shown controversial results, indicating that the physiological role of ERK varies according to the cancer type. ERK increases the level of c-Jun expression by affecting its transcription and stability [40] whereas ERK inhibition increased JNK activity [41]. We found that ERK decreased, whereas p-JNK increased at 24 h and 48 h after cordycepin treatment (Figure 3A). JNK plays a pivotal role in death receptor-initiated extrinsic as well as mitochondrial intrinsic apoptotic pathways [42]. After phosphorylation, JNK can activate its downstream transcriptional factors, inducing cell apoptosis by triggering the expression of the pro-apoptotic target gene Bax [43].
Thus, our results suggest that cordycepin-mediated p-JNK upregulation by downregulating ERK induces TK-10 cell apoptosis. Because DUSP5 is an inducible, nuclear, dual-specificity phosphatase, which specifically interacts with and inactivates the ERK1/2 MAP kinases in mammalian cells [25], we assessed whether DUSP5 was affected at 24 h and 48 h after cordycepin treatment. DUSP5 levels decreased at both time points (Figure 3A). We found that phosphorylation of ERK reduced along with the upregulation in the expression of p-JNK at 24 h and 48 h after cordycepin treatment (Figure 3B). This suggests that cordycepin-mediated ERK inhibition promotes p-JNK, inducing the expression of the pro-apoptotic proteins Bax and cleaved caspase-3, leading to TK10 cell apoptosis after cordycepin treatment.
These findings are consistent with those of previous studies [44,45]. The JNK specific inhibitor SP600125 significantly suppressed cordycepin-induced Bax (Figure 4B), strongly indicating that JNK regulates the activity of Bax in TK-10 cells. Next, we investigated whether ERK was involved in the negative regulation of p-JNK after cordycepin treatment (Figure 5A). We found that ERK overexpression decreased the expression of p-JNK, whereas siRNA-mediated inhibition of ERK increased p-JNK, indicating that ERK indeed mediated JNK dephosphorylation. ERK-silenced TK-10 cells treated with cordycepin also showed decreased DUSP5 and increased p-JNK levels (Figure 4A), while siRNA-mediated inhibition of DUSP5 increased p-JNK levels (Figure 4B). These findings indicated that cordycepin-mediated ERK inhibition upregulated p-JNK through decreased function of the JNK inhibitor DUSP5, inducing TK-10 cell apoptosis. The mechanism by which JNK regulates β-catenin in an Akt-dependent manner after cordycepin is not clear. JNK-regulated phosphatase activities may be involved in the nuclear translocation of β-catenin [46]. We investigated whether SP600125 affected nuclear β-catenin accumulation. We found that JNK inhibition by SP600125 increased the nuclear translocation of β-catenin at 48 h after cordycepin treatment, suppressing β-catenin cytosol translocation (Figure 4B). The transcription factor β-catenin can downregulate Bax gene expression and prevents caspase-dependent apoptosis [47]. In addition, the direct effect of JNK on Bax and consequent Bax translocation to mitochondria are essential to initiating mitochondrial apoptosis [48]. We further studied whether the inhibition of β-catenin by siRNA could enhance the expression of Bax (Figure 5A, 5B). We found that the inhibition of β-catenin by siRNA increased the expression of Bax, indicating that it also significantly induces TK-10 cell apoptosis after cordycepin treatment. Because β-catenin/Wnt signaling activates Akt, which phosphorylates and inhibits Bax [49], the effect of Akt inactivation on Bax activation was elucidated using phosphatidylinositol 3 kinase (PI3K) inhibitor, LY-294002, an agent that attenuates PI3K-mediated Akt activation [50]. Our results demonstrated that Dkk1, a negative regulator of Wnt signaling, affected β-catenin expression, which in turn affected Akt activation in TK-10 cells after cordycepin treatment (Figure 6B). Nuclear β-catenin translocation decreased after cordycepin treatment, but recovered in si-Dkk1-transfected cells after cordycepin treatment. siRNA-mediated Dkk1 inhibition also activated Akt (Figure 6B). The results of the present study suggest that the inhibition of p-JNK-mediated β-catenin/Wnt signaling downregulates Akt activation, a kinase that phosphorylates and upregulates Bax, and that ERK regulated JNK-mediated β-catenin signaling, which is essential for cordycepin-induced TK-10 cell apoptosis. Our findings are consistent with a recent report showing that ERK downregulation may induce cancer apoptosis and that the ERK/DUSP5/JNK signaling pathway is involved in TK-10 cell apoptosis after cordycepin treatment. These results indicate that ERK-mediated JNK negative regulation of β-catenin may be Akt-dependent. Thus, ERK-mediated negative regulation of β-catenin by JNK should be considered as a potential therapeutic target.
Taken together, the pro-apoptotic function and unique capability of negative regulation by JNK of β-catenin nuclear translocation by modulating ERK-JNK signaling suggest that cordycepin has potential as a new drug that could suppress the growth of renal-cancer cells.
Acknowledgements
This work was supported by institutional funds from the Edinburg Regional Academic Health Center, University of Texas Rio Grande Valley and National Institutes of Health, NIEHS, Grant R01ES022250 (to D. J. Kim) and supported by the Korea Basic Science Institute (D36403).
Disclosure of conflict of interest
None.
References
- 1.Chang W, Lim S, Song H, Song BW, Kim HJ, Cha MJ, Sung JM, Kim TW, Hwang KC. Cordycepin inhibits vascular smooth muscle cell proliferation. Eur J Pharmacol. 2008;597:64–69. doi: 10.1016/j.ejphar.2008.08.030. [DOI] [PubMed] [Google Scholar]
- 2.Shi P, Huang Z, Tan X, Chen G. Proteomic detection of changes in protein expression induced by cordycepin in human hepatocellular carcinoma BEL-7402 cells. Methods Find Exp Clin Pharmacol. 2008;30:347–353. doi: 10.1358/mf.2008.30.5.1186085. [DOI] [PubMed] [Google Scholar]
- 3.Wehbe-Janek H, Shi Q, Kearney CM. Cordycepin/Hydroxyurea synergy allows low dosage efficacy of cordycepin in MOLT-4 leukemia cells. Anticancer Res. 2007;27:3143–3146. [PubMed] [Google Scholar]
- 4.Wu WC, Hsiao JR, Lian YY, Lin CY, Huang BM. The apoptotic effect of cordycepin on human OEC-M1 oral cancer cell line. Cancer Chemother Pharmacol. 2007;60:103–111. doi: 10.1007/s00280-006-0354-y. [DOI] [PubMed] [Google Scholar]
- 5.Thomadaki H, Scorilas A, Tsiapalis CM, Havredaki M. The role of cordycepin in cancer treatment via induction or inhibition of apoptosis: implication of polyadenylation in a cell type specific manner. Cancer Chemother Pharmacol. 2008;61:251–265. doi: 10.1007/s00280-007-0467-y. [DOI] [PubMed] [Google Scholar]
- 6.Cho HJ, Cho JY, Rhee MH, Kim HS, Lee HS, Park HJ. Inhibitory effects of cordycepin (3’-deoxyadenosine), a component of Cordyceps militaris, on human platelet aggregation induced by thapsigargin. J Microbiol Biotechnol. 2007;17:1134–1138. [PubMed] [Google Scholar]
- 7.Nakamura K, Konoha K, Yoshikawa N, Yamaguchi Y, Kagota S, Shinozuka K, Kunitomo M. Effect of cordycepin (3’-deoxyadenosine) on hematogenic lung metastatic model mice. In Vivo. 2005;19:137–141. [PubMed] [Google Scholar]
- 8.Tian X, Li Y, Shen Y, Li Q, Wang Q, Feng L. Apoptosis and inhibition of proliferation of cancer cells induced by cordycepin. Oncol Lett. 2015;10:595–599. doi: 10.3892/ol.2015.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Imesch P, Hornung R, Fink D, Fedier A. Cordycepin (3’-deoxyadenosine), an inhibitor of mRNA polyadenylation, suppresses proliferation and activates apoptosis in human epithelial endometriotic cells in vitro. Gynecol Obstet Invest. 2011;72:43–49. doi: 10.1159/000322395. [DOI] [PubMed] [Google Scholar]
- 10.Choi S, Lim MH, Kim KM, Jeon BH, Song WO, Kim TW. Cordycepin-induced apoptosis and autophagy in breast cancer cells are independent of the estrogen receptor. Toxicol Appl Pharmacol. 2011;257:165–173. doi: 10.1016/j.taap.2011.08.030. [DOI] [PubMed] [Google Scholar]
- 11.Zhang W, Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002;12:9–18. doi: 10.1038/sj.cr.7290105. [DOI] [PubMed] [Google Scholar]
- 12.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 13.Johnson NL, Gardner AM, Diener KM, Lange-Carter CA, Gleavy J, Jarpe MB, Minden A, Karin M, Zon LI, Johnson GL. Signal transduction pathways regulated by mitogen-activated/extracellular response kinase kinase kinase induce cell death. J Biol Chem. 1996;271:3229–3237. doi: 10.1074/jbc.271.6.3229. [DOI] [PubMed] [Google Scholar]
- 14.Zanke BW, Boudreau K, Rubie E, Winnett E, Tibbles LA, Zon L, Kyriakis J, Liu FF, Woodgett JR. The stress-activated protein kinase pathway mediates cell death following injury induced by cis-platinum, UV irradiation or heat. Curr Biol. 1996;6:606–613. doi: 10.1016/s0960-9822(02)00547-x. [DOI] [PubMed] [Google Scholar]
- 15.Goillot E, Raingeaud J, Ranger A, Tepper RI, Davis RJ, Harlow E, Sanchez I. Mitogen-activated protein kinase-mediated Fas apoptotic signaling pathway. Proc Natl Acad Sci U S A. 1997;94:3302–3307. doi: 10.1073/pnas.94.7.3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T, Takagi M, Matsumoto K, Miyazono K, Gotoh Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275:90–94. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- 17.Wortzel I, Seger R. The ERK Cascade: Distinct Functions within Various Subcellular Organelles. Genes Cancer. 2011;2:195–209. doi: 10.1177/1947601911407328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chuang SM, Wang IC, Yang JL. Roles of JNK, p38 and ERK mitogen-activated protein kinases in the growth inhibition and apoptosis induced by cadmium. Carcinogenesis. 2000;21:1423–1432. [PubMed] [Google Scholar]
- 19.Theodosiou A, Ashworth A. MAP kinase phosphatases. Genome Biol. 2002;3:REVIEWS3009. doi: 10.1186/gb-2002-3-7-reviews3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jeffrey KL, Camps M, Rommel C, Mackay CR. Targeting dual-specificity phosphatases: manipulating MAP kinase signalling and immune responses. Nat Rev Drug Discov. 2007;6:391–403. doi: 10.1038/nrd2289. [DOI] [PubMed] [Google Scholar]
- 21.Guan KL, Butch E. Isolation and characterization of a novel dual specific phosphatase, HVH2, which selectively dephosphorylates the mitogen-activated protein kinase. J Biol Chem. 1995;270:7197–7203. doi: 10.1074/jbc.270.13.7197. [DOI] [PubMed] [Google Scholar]
- 22.Vang T, Miletic AV, Arimura Y, Tautz L, Rickert RC, Mustelin T. Protein tyrosine phosphatases in autoimmunity. Annu Rev Immunol. 2008;26:29–55. doi: 10.1146/annurev.immunol.26.021607.090418. [DOI] [PubMed] [Google Scholar]
- 23.Mandl M, Slack DN, Keyse SM. Specific inactivation and nuclear anchoring of extracellular signal-regulated kinase 2 by the inducible dual-specificity protein phosphatase DUSP5. Mol Cell Biol. 2005;25:1830–1845. doi: 10.1128/MCB.25.5.1830-1845.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buffet C, Catelli MG, Hecale-Perlemoine K, Bricaire L, Garcia C, Gallet-Dierick A, Rodriguez S, Cormier F, Groussin L. Dual Specificity Phosphatase 5, a Specific Negative Regulator of ERK Signaling, Is Induced by Serum Response Factor and Elk-1 Transcription Factor. PLoS One. 2015;10:e0145484. doi: 10.1371/journal.pone.0145484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kucharska A, Rushworth LK, Staples C, Morrice NA, Keyse SM. Regulation of the inducible nuclear dual-specificity phosphatase DUSP5 by ERK MAPK. Cell Signal. 2009;21:1794–1805. doi: 10.1016/j.cellsig.2009.07.015. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Z, Kobayashi S, Borczuk AC, Leidner RS, Laframboise T, Levine AD, Halmos B. Dual specificity phosphatase 6 (DUSP6) is an ETS-regulated negative feedback mediator of oncogenic ERK signaling in lung cancer cells. Carcinogenesis. 2010;31:577–586. doi: 10.1093/carcin/bgq020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ekerot M, Stavridis MP, Delavaine L, Mitchell MP, Staples C, Owens DM, Keenan ID, Dickinson RJ, Storey KG, Keyse SM. Negative-feedback regulation of FGF signalling by DUSP6/MKP-3 is driven by ERK1/2 and mediated by Ets factor binding to a conserved site within the DUSP6/MKP-3 gene promoter. Biochem J. 2008;412:287–298. doi: 10.1042/BJ20071512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stockert JC, Blazquez-Castro A, Canete M, Horobin RW, Villanueva A. MTT assay for cell viability: Intracellular localization of the formazan product is in lipid droplets. Acta Histochem. 2012;114:785–796. doi: 10.1016/j.acthis.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 29.Wong YY, Moon A, Duffin R, Barthet-Barateig A, Meijer HA, Clemens MJ, de Moor CH. Cordycepin inhibits protein synthesis and cell adhesion through effects on signal transduction. J Biol Chem. 2010;285:2610–2621. doi: 10.1074/jbc.M109.071159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mehta RG, Murillo G, Naithani R, Peng X. Cancer chemoprevention by natural products: how far have we come? Pharm Res. 2010;27:950–961. doi: 10.1007/s11095-010-0085-y. [DOI] [PubMed] [Google Scholar]
- 31.Yin JQ, Shen JN, Su WW, Wang J, Huang G, Jin S, Guo QC, Zou CY, Li HM, Li FB. Bufalin induces apoptosis in human osteosarcoma U-2OS and U-2OS methotrexate300-resistant cell lines. Acta Pharmacol Sin. 2007;28:712–720. doi: 10.1111/j.1745-7254.2007.00559.x. [DOI] [PubMed] [Google Scholar]
- 32.Liang CZ, Zhang JK, Shi Z, Liu B, Shen CQ, Tao HM. Matrine induces caspase-dependent apoptosis in human osteosarcoma cells in vitro and in vivo through the upregulation of Bax and Fas/FasL and downregulation of Bcl-2. Cancer Chemother Pharmacol. 2012;69:317–331. doi: 10.1007/s00280-011-1699-4. [DOI] [PubMed] [Google Scholar]
- 33.Lin CC, Chuang YJ, Yu CC, Yang JS, Lu CC, Chiang JH, Lin JP, Tang NY, Huang AC, Chung JG. Apigenin induces apoptosis through mitochondrial dysfunction in U-2 OS human osteosarcoma cells and inhibits osteosarcoma xenograft tumor growth in vivo. J Agric Food Chem. 2012;60:11395–11402. doi: 10.1021/jf303446x. [DOI] [PubMed] [Google Scholar]
- 34.Nakamura K, Yoshikawa N, Yamaguchi Y, Kagota S, Shinozuka K, Kunitomo M. Antitumor effect of cordycepin (3’-deoxyadenosine) on mouse melanoma and lung carcinoma cells involves adenosine A3 receptor stimulation. Anticancer Res. 2006;26:43–47. [PubMed] [Google Scholar]
- 35.Zhou X, Luo L, Dressel W, Shadier G, Krumbiegel D, Schmidtke P, Zepp F, Meyer CU. Cordycepin is an immunoregulatory active ingredient of Cordyceps sinensis. Am J Chin Med. 2008;36:967–980. doi: 10.1142/S0192415X08006387. [DOI] [PubMed] [Google Scholar]
- 36.Bender F, Montoya M, Monardes V, Leyton L, Quest AF. Caveolae and caveolae-like membrane domains in cellular signaling and disease: identification of downstream targets for the tumor suppressor protein caveolin-1. Biol Res. 2002;35:151–167. doi: 10.4067/s0716-97602002000200006. [DOI] [PubMed] [Google Scholar]
- 37.Capozza F, Williams TM, Schubert W, McClain S, Bouzahzah B, Sotgia F, Lisanti MP. Absence of caveolin-1 sensitizes mouse skin to carcinogen-induced epidermal hyperplasia and tumor formation. Am J Pathol. 2003;162:2029–2039. doi: 10.1016/S0002-9440(10)64335-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Du ZM, Hu CF, Shao Q, Huang MY, Kou CW, Zhu XF, Zeng YX, Shao JY. Upregulation of caveolin-1 and CD147 expression in nasopharyngeal carcinoma enhanced tumor cell migration and correlated with poor prognosis of the patients. Int J Cancer. 2009;125:1832–1841. doi: 10.1002/ijc.24531. [DOI] [PubMed] [Google Scholar]
- 39.Savage K, Lambros MB, Robertson D, Jones RL, Jones C, Mackay A, James M, Hornick JL, Pereira EM, Milanezi F, Fletcher CD, Schmitt FC, Ashworth A, Reis-Filho JS. Caveolin 1 is overexpressed and amplified in a subset of basal-like and metaplastic breast carcinomas: a morphologic, ultrastructural, immunohistochemical, and in situ hybridization analysis. Clin Cancer Res. 2007;13:90–101. doi: 10.1158/1078-0432.CCR-06-1371. [DOI] [PubMed] [Google Scholar]
- 40.Lopez-Bergami P, Huang C, Goydos JS, Yip D, Bar-Eli M, Herlyn M, Smalley KS, Mahale A, Eroshkin A, Aaronson S, Ronai Z. Rewired ERK-JNK signaling pathways in melanoma. Cancer Cell. 2007;11:447–460. doi: 10.1016/j.ccr.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Monick MM, Powers LS, Gross TJ, Flaherty DM, Barrett CW, Hunninghake GW. Active ERK contributes to protein translation by preventing JNK-dependent inhibition of protein phosphatase 1. J Immunol. 2006;177:1636–1645. doi: 10.4049/jimmunol.177.3.1636. [DOI] [PubMed] [Google Scholar]
- 42.Dhanasekaran DN, Reddy EP. JNK signaling in apoptosis. Oncogene. 2008;27:6245–6251. doi: 10.1038/onc.2008.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Papadakis ES, Finegan KG, Wang X, Robinson AC, Guo C, Kayahara M, Tournier C. The regulation of Bax by c-Jun N-terminal protein kinase (JNK) is a prerequisite to the mitochondrial-induced apoptotic pathway. FEBS Lett. 2006;580:1320–1326. doi: 10.1016/j.febslet.2006.01.053. [DOI] [PubMed] [Google Scholar]
- 44.Wang XA, Xiang SS, Li HF, Wu XS, Li ML, Shu YJ, Zhang F, Cao Y, Ye YY, Bao RF, Weng H, Wu WG, Mu JS, Hu YP, Jiang L, Tan ZJ, Lu W, Wang P, Liu YB. Cordycepin induces S phase arrest and apoptosis in human gallbladder cancer cells. Molecules. 2014;19:11350–11365. doi: 10.3390/molecules190811350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee SY, Debnath T, Kim SK, Lim BO. Anti-cancer effect and apoptosis induction of cordycepin through DR3 pathway in the human colonic cancer cell HT-29. Food Chem Toxicol. 2013;60:439–447. doi: 10.1016/j.fct.2013.07.068. [DOI] [PubMed] [Google Scholar]
- 46.Liao G, Tao Q, Kofron M, Chen JS, Schloemer A, Davis RJ, Hsieh JC, Wylie C, Heasman J, Kuan CY. Jun NH2-terminal kinase (JNK) prevents nuclear beta-catenin accumulation and regulates axis formation in Xenopus embryos. Proc Natl Acad Sci U S A. 2006;103:16313–16318. doi: 10.1073/pnas.0602557103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Z, Havasi A, Gall JM, Mao H, Schwartz JH, Borkan SC. Beta-catenin promotes survival of renal epithelial cells by inhibiting Bax. J Am Soc Nephrol. 2009;20:1919–1928. doi: 10.1681/ASN.2009030253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Desagher S, Martinou JC. Mitochondria as the central control point of apoptosis. Trends Cell Biol. 2000;10:369–377. doi: 10.1016/s0962-8924(00)01803-1. [DOI] [PubMed] [Google Scholar]
- 49.Gardai SJ, Hildeman DA, Frankel SK, Whitlock BB, Frasch SC, Borregaard N, Marrack P, Bratton DL, Henson PM. Phosphorylation of Bax Ser184 by Akt regulates its activity and apoptosis in neutrophils. J Biol Chem. 2004;279:21085–21095. doi: 10.1074/jbc.M400063200. [DOI] [PubMed] [Google Scholar]
- 50.Sinha D, Wang Z, Ruchalski KL, Levine JS, Krishnan S, Lieberthal W, Schwartz JH, Borkan SC. Lithium activates the Wnt and phosphatidylinositol 3-kinase Akt signaling pathways to promote cell survival in the absence of soluble survival factors. Am J Physiol Renal Physiol. 2005;288:F703–713. doi: 10.1152/ajprenal.00189.2004. [DOI] [PubMed] [Google Scholar]