

Abstract
Prostate cancer is the most common malignancy in Western men and hormone refractory cancer (HRPC) kills most of the patients. Chemo-resistance is a major obstacle for the treatment of prostate cancer. Platinum-complexes have been used to treat a number of malignancies including prostate cancer. However, it has limited effect to prostate cancer and with significant toxicity at higher doses. In recent years, increasing numbers of new agents targeting cancer specific pathways have become available and with low toxic side-effects. Rapamycin (Sirolimus) is an mTORC1 inhibitor, which inhibits the PI3K/Akt/mTOR signaling pathway, which is commonly altered in prostate cancer. We determined the expression of cyclin D1 and phosphorylated-mTOR proteins in association with the response to rapamycin in two androgen sensitive (22RV1 and LNCaP) and two androgen independent (DU145 and PC3) prostate cancer cell lines and found that the base-line and changes of cyclin D1 level, but not the expression level of p-mTOR, correlated with rapamycin sensitivity. We evaluated the cell killing effect of combined rapamycin and cisplatin treatment and showed that the combination had a more than additive effect in both androgen dependent and independent prostate cancer cells, which may be partially explained by the reduction of cyclin D1 expression by rapamycin. We also evaluated a range of combined treatment schedules, simultaneously or sequentially and found that continuous rapamycin treatment after a short cisplatin exposure was effective. The clinical application of these findings for prostate cancer treatment should be further investigated.
Keywords: Prostate cancer, rapamycin, mTOR, cisplatin, cyclin D1 expression
Introduction
Prostate cancer is the most common cancer and second leading cause of cancer related mortality in Western males, accounting for 903,500 new diagnoses and 258,400 deaths in 2008 [1]. While the majority of prostate cancer cases are indolent and the prognosis for localized disease is good, once the disease progresses into the metastatic stage, it is virtually incurable [2]. Androgen deprivation therapy through either chemical or surgical castration initially works well to control metastatic prostate cancer, but unavoidably patients eventually progress to castration resistant prostate cancer, for which no efficient therapy is currently available [3-5]. Chemotherapy can only prolong the patient survival by a few months in castration-resistant disease [3,5].
Understanding molecular mechanisms/pathways involved in cancer development and progression has helped to identify cancer-specific targets to develop novel effective therapies. The phosphatidylinositol 3-kinase PI3K/AKT/mTOR pathway, which regulates key cellular processes, including survival, angiogenesis and invasion [6], has recently been under extensive investigation of targeted therapy [3,7]. mTOR, a serine/threonine-specific kinase located downstream of PI3K/AKT of this pathway, functions through its effectors to mediate protein synthesis and cell-cycle progression [7,8]. Many human tumors have been noted to have an elevated mTOR activity [8]. Therefore, mTOR is the first of the genes in this pathway, which have been targeted for the treatment of various types of cancers [3]. Rapamycin (now well known as sirolimus), the first anticancer agent targeting mTOR, was initially isolated as an antifungal drug in 1972 and its anticancer effect was demonstrated in 1990s [7]. Through inhibiting mTOR, it blocks cell growth, proliferation and survival. Since then various formulations have been developed (rapalogs), including RAD001 (everolimus, Novartis), CCI-779 (temrapamycin, Wyeth/Pfizer, Inc.) and AP23573 (deferolimus, Merck/Ariad) [7,8]. Although the pre-clinical studies are encouraging, the anticancer efficacy of rapamycin and rapalogs as a single agent is less promising in the clinic than expected. Rapalogs as a single agent only succeeded in a few cancers, including breast cancer, renal cell carcinoma, lymphoma, sarcoma, hepatocellular carcinoma and endometrial cancer [3,8-11]. The PI3K/AKT/mTOR pathway is commonly activated in both primary and metastatic prostate cancer, frequently through the loss of PTEN function, but can also through PI3K and AKT amplification or mutations [2,3,12]. PTEN loss and activation of PI3K/Akt/mTOR pathway has also been associated with castration-resistance and resistance to radiation and chemotherapy [3]. However, single-agent rapamycin and rapalogs treatment for prostate cancer has limited effect [13].
Due to the disappointing efficacy of rapamycin as single agent for cancer therapy, the combination of rapamycin/rapalogs with other therapeutic approaches has been investigated, including the combination of rapamycin/rapalogs with various chemotherapy drugs [3,8]. Rapamycin has been reported to exert its anti-cancer effects in part by inhibiting cell proliferation through reduced synthesis of proteins involved in cell cycle progression and consequent G1 cell cycle arrest [14]. The rapamycin-induced decrease of cyclin D1 expression inhibits the cell cycle progression [8]. Our previous study showed that cyclin D1 overexpression contributed to cisplatin-resistance in testicular germ cell tumors (TGCTs) and that prostate cancer cells have much higher level of cyclin D1 expression than TGCT cells, which are generally sensitive to cisplatin [15]. While platinum-based therapy currently has limited utility in the treatment of prostate cancer [16,17], the combination of an mTOR inhibitor with cisplatin may improve the efficiency of the treatment of prostate cancer, where PI3K/mTOR pathway is commonly altered. In this study, we showed that the base-line cyclin D1 level determined the sensitivity to rapamycin and rapamycin treatment decreased cyclin D1 proteins in androgen sensitive and insensitive prostate cancer cell lines. We evaluated the efficacy of the combined treatment with rapamycin and cisplatin in killing prostate cancer cells using in vitro models of these cell lines and found that rapamycin accentuated the response of prostate cancer cells to cisplatin.
Materials and methods
Cell lines
Two androgen receptor (AR)-positive prostate cancer cell lines, LNCaP and 22RV1 and two AR-negative prostate cancer cell lines, PC3 and DU145 (ATCC, Manassas, VA, USA), were cultured in DMEM medium, supplemented with 10% foetal bovine serum and 100 units/mL penicillin/streptomycin. Cell lines were verified by STR profiling using the ABI AmpF/STR Identifiler kit (Applied Biosystems, Foster City, CA, USA).
Cisplatin and rapamycin dose response
PC3, DU145, LNCaP and 22RV1 cells were seeded in 96-well plate at a concentration of 4000 cells/well. Cells were treated by increasing concentrations of cisplatin or rapamycin for 72 hours. Each treatment for each cell line was performed in six replicates. MTS assay was used to measure the cell viability.
Combined rapamycin and cisplatin treatment schedules
To determine the effect of combined rapamycin and cisplatin treatment, three sets of experiments were done: 1. single agent comparing to combined treatment, 2. short-term (three days or less) combined treatment comparing simultaneous to sequential drug exposure and 3. long-term (12 days) treatment using rapamycin following cisplatin. Predetermined doses of rapamycin and cisplatin based on the dose response data were used for all these experiments. To compare the efficiency of combined treatment with single agents, 4,000 cells were seeded in 96-well plates 24-hour pretreatment and then they were exposed to these drugs together or individually with cell viability determined by MTS assay at 72 hours. For the comparison between short-term simultaneous and sequential combination treatment, 4,000 cells were seeded in 96-well plates 24 hour pretreatment and then cells were either simultaneously treated by adding cisplatin and rapamycin to the culture medium at the same time and left for 24 hours or sequentially treated by cisplatin or rapamycin for 24 hours and then rapamycin or cisplatin for another 24 hours respectively, leaving a 24-hour drug free period in between. Their viability was determined at 72 and 120 hours by MTS assay. For the long-term continuous treatment, 40,000 cells were seeded in 6-well plates and treated by cisplatin and rapamycin together for 24 hours, followed by rapamycin treatment for continuous 11 days with fresh drug added every three days. Various controls with or without short-term cisplatin and/or rapamycin 24 hour exposure were shown in Figure 1. Cells were cultured with refresh DMEM every three days and cell viability was monitored at different time points until day 12 by MTS assay.
Figure 1.
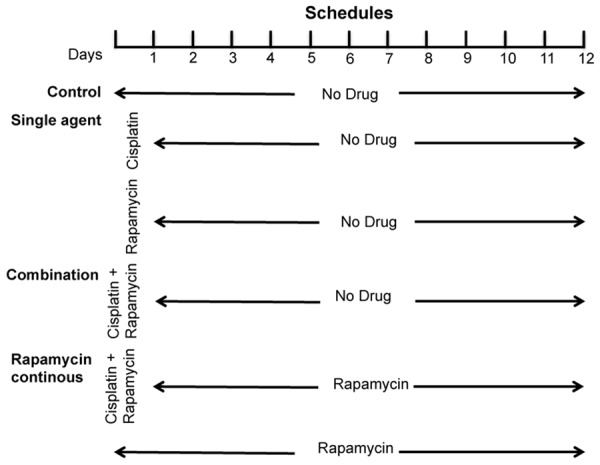
The diagram of long-term rapamycin treatment schedule for different treatment groups. At the beginning of the treatment, cisplatin and rapamycin were added either separately or together and incubated for 24 hours, which were then removed by changing the media and continuing with or without the rapamycin for the remaining period of the treatment. MTS cell viability assays were carried out on day 3, 6, 9 and day 12.
MTS cell viability assay
Cell viability were assessed using the CellTiter 96® AQueous assay (Promega) following the manufacturer’s instructions. Inhibition curves were drawn by means of values obtained by OD percentages versus untreated control for each drug treatment. Cell viability was calculated using the formula: (treatment OD)/(untreated wells OD)×100.
Colony formation assay
500 cells were plated per well of six-well plates. Drug treatment was carried out as shown in Figure 1. After total 12 days of culture, the media was removed and the colonies were stained with crystal violet solution for colony counting. Only the clearly visible colonies, larger than 0.5 mm in diameter, were counted.
Western blot
Whole cell extracts of protein were obtained by lysing cells using PBS-1% Triton X-100 (T8532, Sigma) and 20 µg of protein preparation was loaded onto 10% acrylamide gels and were separated by SDS polyacrylamide gel electrophoresis. Proteins were then transferred to polyvinylidene difluoride membrane (Immobilon-P, Milllipore, Billerica, MA) and incubated with the monoclonal antibodies against cyclin D1 (sc-20044, Santa Cruz Biotechnology, Santa Cruz, CA), phospho-mTOR (Ser2448) (5536, Cell Signalling, Danvers, MA) and β-actin (A5441, Sigma, St Louis, MO). Bands were detected using horseradish peroxidase chemiluminescence based detection kit (Millipore).
Statistical analysis
Data points are given as the standard deviation (SD) of at least three experiments. Statistical analyses were performed using the Prism 5.0b (GraphPad, La Jolla, CA, USA) statistical software package. P values lower than 0.05 were considered significant.
Results
The effect of rapamycin and cisplatin on prostate cancer cell growth
Firstly, we examined the effect of rapamycin and cisplatin on cell growth of the two AR positive (22RV1 and LNCaP) and two AR negative androgen independent (DU145 and PC3) cell lines. After a 72-hour drug treatment, the drug response curve from MTS assay showed that rapamycin had limited toxicity to prostate cancer cells that it cannot kill all cells by increasing dosage, although the responses of individual cell lines to rapamycin varied (Figure 2). The maximum inhibitory effect of rapamycin treatment was reached at around 10 nM in all cell lines that further increase of drug concentration to a maximum of 1 µM did not significantly increased cell killing effect. 22RV1 cells were more sensitive to rapamycin than other cell lines and the least effect was found in DU145 cells, rapamycin treatment was never able to induce 50% cell death. Cisplatin killed cancer cells in a dosage dependent manner and complete cell death can be induced by cisplatin treatment in all cell lines. DU145 and 22RV1 cells were more sensitive to cisplatin than PC3 and LNCaP cells (Figure 2). According to dose-response curve, the drug dosage at EC20-30 for each agent in each cell line was selected in the experiments comparing the efficiency of different treatment schedules (Figure 2 and Table 1).
Figure 2.
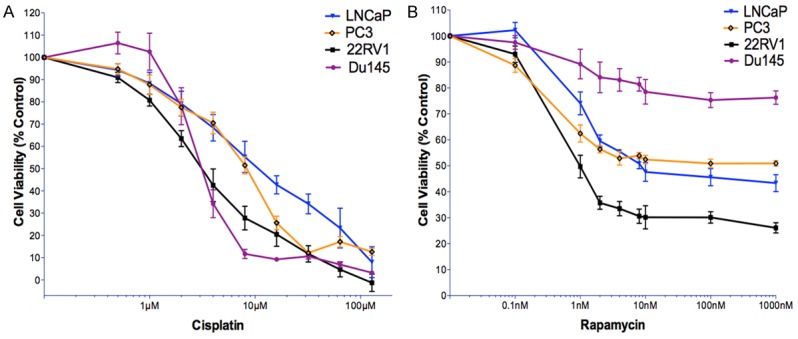
The dose response curves of cisplatin and rapamycin in 22RV1, DU145, PC3 and LNCaP cell lines. A. Cisplatin. B. Rapamycin. Cells were seeded in full media in 96-well plates at concentrations of 4000 cells/well and allowed 24 h to adhere. Cells were then treated with increasing concentrations of rapamycin and cisplatin. After 72 hours, the cell viability was measured via MTS assay. The percentage of cell viability for each drug concentration was normalised to untreated control cells. Each experiment has six replicates and each data point represents at least three independent biological repeats. Bars represent standard deviation.
Table 1.
Cisplatin and rapamycin concentrations used for each cell line in the combination experiments
| Drugs | 22RV1 | LNCaP | PC3 | DU145 |
|---|---|---|---|---|
| Cisplatin (µM) | 1.5 | 2 | 2 | 1.6 |
| Rapamycin (nM) | 0.5 | 0.7 | 0.7 | 10 |
µM: micro molar; nM: nano molar.
Cyclin D1 expression levels in prostate cancer cells correlated to rapamycin sensitivity and were reduced by the treatment
We investigated the effect of rapamycin treatment on cyclin D1 expression. The highest base-line cyclin D1 expression level was found in 22RV1 cells, followed by PC3 and LNCaP, and the lowest in Du145 cells (Figure 3A). This correlated to rapamycin sensitivity, the higher the cyclin D1 expression, the more sensitive to rapamycin. However, cyclin D1 expression level did not correlate to cisplatin sensitivity of those cell lines. After rapamycin treatment at 20 nM and 20 µM, cyclin D1 protein levels were reduced in all cell lines in a dose dependent manner, although the dose effect in PC3 was not apparent (Figure 3B). We also determined the phosphorylated-mTOR (p-mTOR) protein expression in those four cell lines and found that the expression levels did not correlate to rapamycin sensitivity (Figure 3C).
Figure 3.
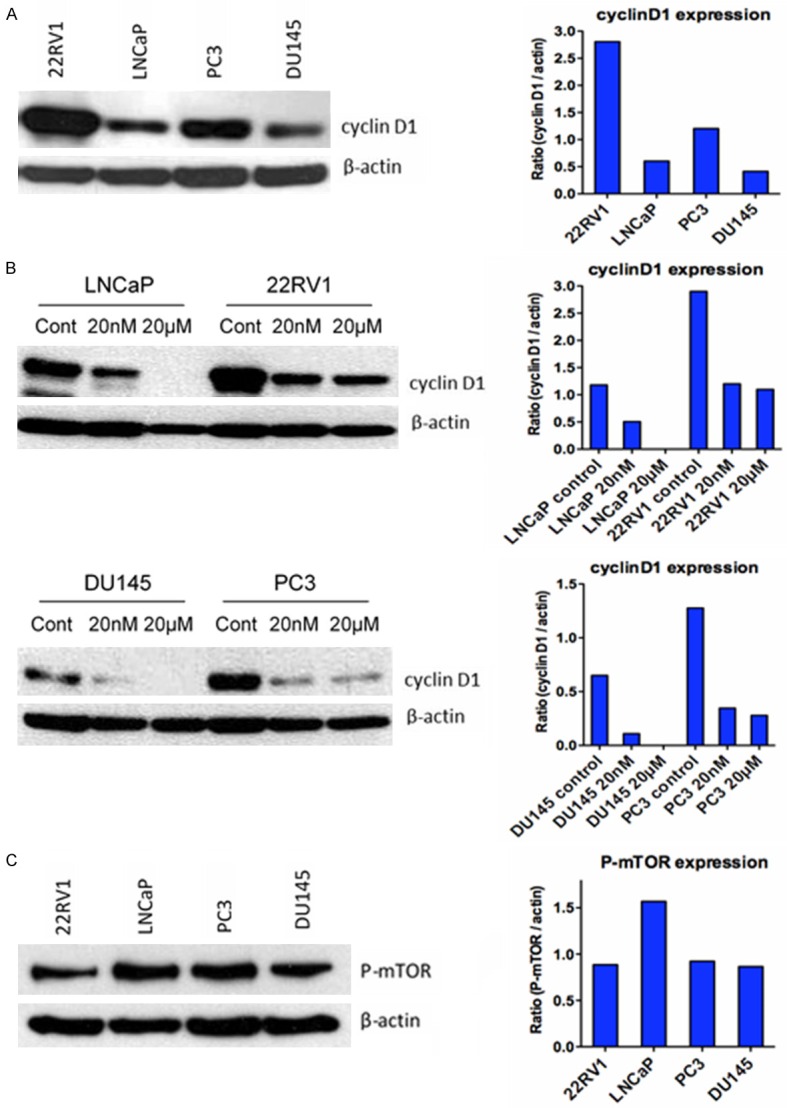
Western blot analysis of cyclin D1 and p-mTOR protein levels in prostate cancer cells pre- and/or post-rapamycin treatment. A. Base-line expression levels of cyclin D1 in prostate cancer cell lines; B. Cyclin D1 protein levels after rapamycin treatment; and C. Base-line expression levels of p-mTOR in those prostate cancer cell lines.
Cisplatin and rapamycin combination treatment has a more than additive effect compared to single treatments
We determined the efficiency of the combination treatment in killing prostate cancer cells. Generally, the cell viability was higher in single agent than combined two agents treatment in all four cell lines. The addition of rapamycin to low-dose cisplatin was associated with a significant reduction in the number of cells compared with treatment with either of the agents alone in all cell lines (Figure 4A).
Figure 4.
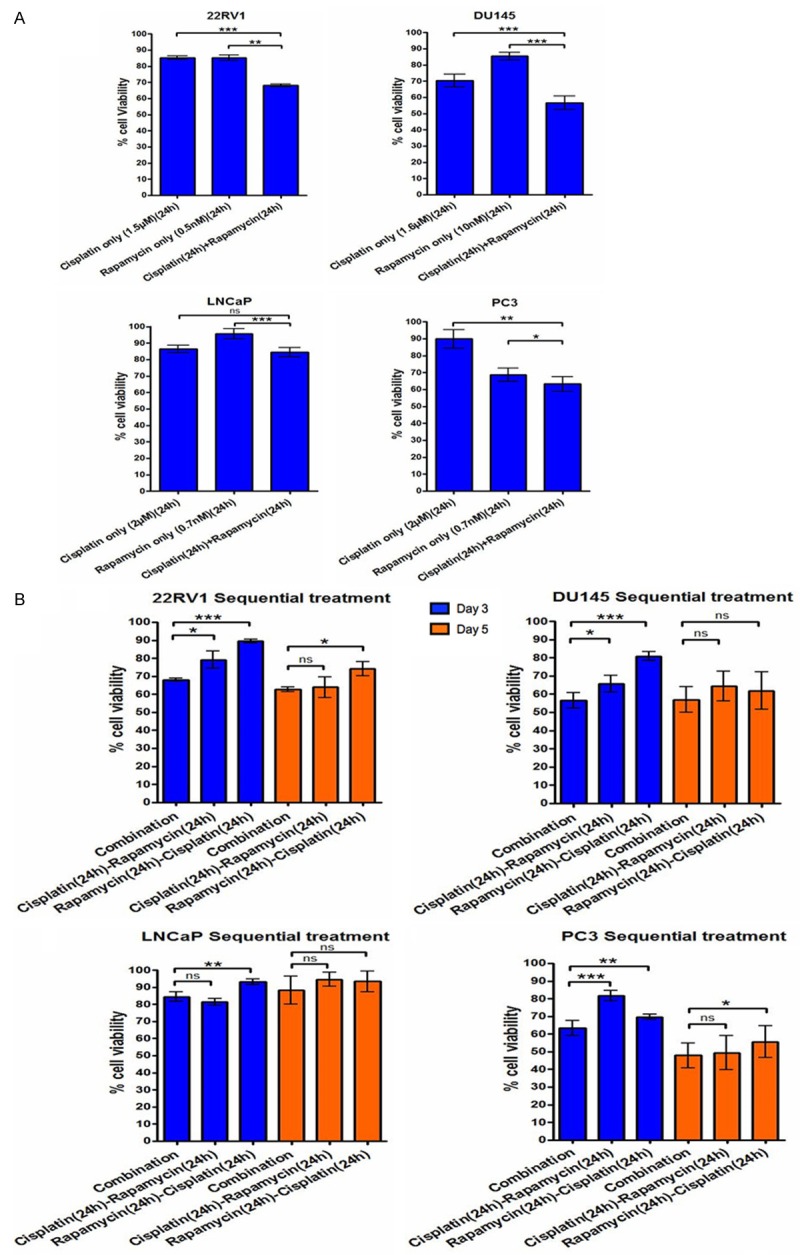
The effect of short-term combined cisplatin and rapamycin treatment. A. Cell viability after treatment by cisplatin and rapamycin alone and in combination of 22RV1, DU145, LNCaP and PC3 cells at day 3. B. Cell viability after treatment by cisplatin and rapamycin simultaneously or one after another of 22RV1, DU145, LNCaP and PC3 cells at day 3 and day 5. Each data point represents three independent experiments with standard deviation bars. ns: not significant, *p<0.05, **p<0.01, ***p<0.001.
In the comparison between sequential and simultaneous combination treatment, simultaneous expression of cisplatin and rapamycin was more efficient (p<0.05 for all) than sequential treatments to inhibit cell growth at day 3, except simultaneous compared to cisplatin followed by rapamycin in LNCaP cells. However, the difference was reduced at day 5 that there was no significant difference between all the treatments in any cell lines except simultaneous vs cisplatin following rapamycin in 22RV1 and PC3 cells (Figure 4B). Cisplatin followed by rapamycin was more efficient than cisplatin following rapamycin treatment to inhibit cell growth in 22RV1, DU145 and LNCaP cells at day 3 (Figure 4B).
To determine the prostate cancer cell inhibition effect of a long-term low dosage/toxicity exposure of rapamycin following initial short-term combined cisplatin and rapamycin treatment, we treated the four prostate cancer cell lines with combination of cisplatin and rapamycin for 24 hours followed with low dosage of rapamycin for a long-term (12 days) together with different control treatment groups as shown in Figure 1. This experiment could not be continued beyond day 12 as the untreated control cells were confluent. We found that cell growth was consistently inhibited during this period. Compared to the different control groups, this combination significantly reduced cell viability at day 12 in all four cell lines (p<0.01) (Figure 5). The greater cell growth inhibition effect of this combined treatment than the control groups become statistically significant at day 6 (p<0.05 for all) and maintained at day 9 before it became more significant at day 12 (Figure 5).
Figure 5.
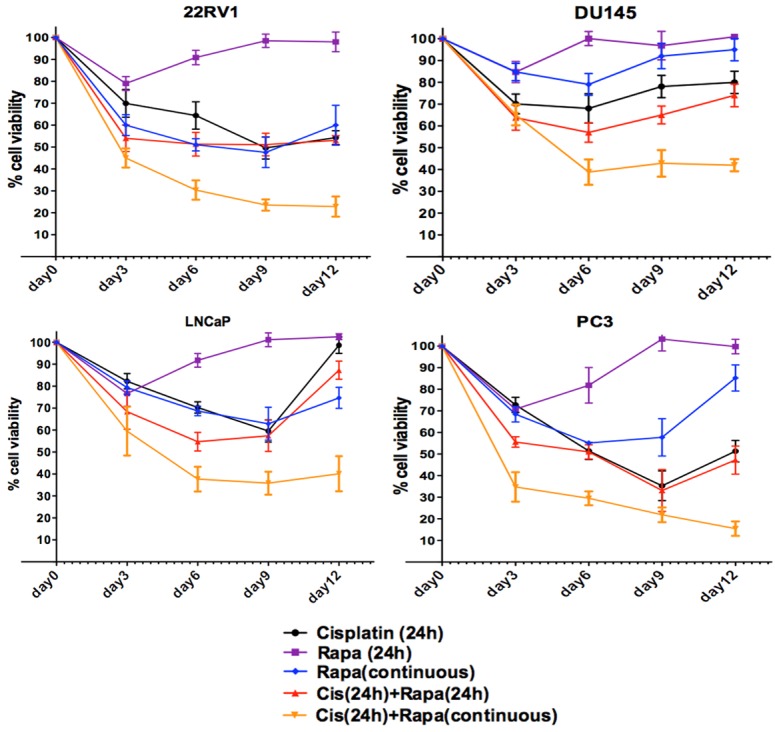
The long-term effect of rapamycin in combination with cisplatin short-term exposure on prostate cancer cell growth. Cells were cultured under various treatments in 6-well plates. Rapamycin was refreshed at every 72 hour for rapamycin continuous treatment and cells under the other treatments only had media refreshed every 72 hours without drugs.
In order to investigate the effect of the combined long-term treatment on the ability of colony formation, we performed a colony formation assay using 22RV1, DU145 and PC3 cell lines. Cisplatin followed by continuous rapamycin treatment significantly decreased the ability of colony formation compared to all the other treatments (Figure 6). Cisplatin only and combination of cisplatin and rapamycin treatment for 24 hours showed similar reduced number of colonies. While 24-hour rapamycin only treatment did not obviously reduce the number of colonies formed in all the cell lines tested compared to the untreated controls, longer period (12 days) rapamycin treatment not only decreased the number of colonies, but also resulted in much smaller colonies compared to the untreated or rapamycin short-term treated cells.
Figure 6.
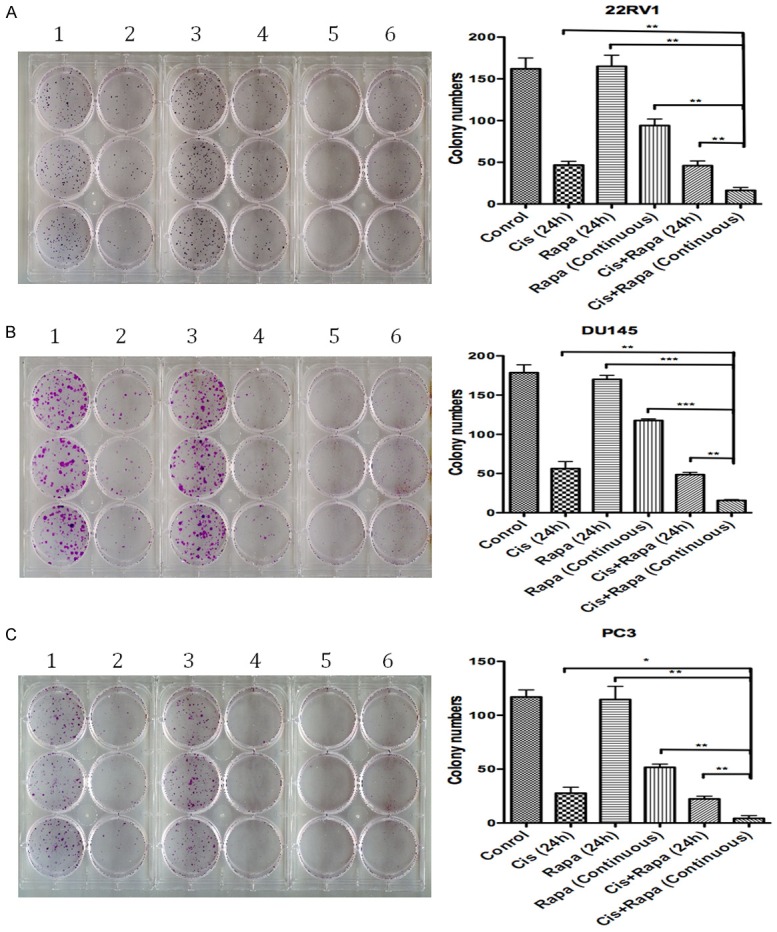
The long-term effect of rapamycin in combination with cisplatin short-term exposure on prostate cancer cell colony formation ability. 500 cells were seeded per well of 6-well plate and treated with different drug schedules. In the rapamycin continuous treatment cells the rapamycin was refreshed every 72 hours. Cells under the other treatment conditions only had their media refreshed at the same time. 1: Control, 2: Cisplatin only (24 h), 3: Rapamycin only (24 h), 4: Cisplatin + rapamycin (24 h), 5: Cisplatin (24 h) + rapamycin (continuous), 6: Rapamycin only (continuous).
Discussion
While rapamycin is supposed to inhibit the PI3K/AKT pathway activity through inhibiting mTOR, our rapamycin sensitivity data on the four prostate cancer cell lines did not correlate with the PI3K/AKT pathway activity status. Tumor suppressor PTEN is a negative regulator of PI3K signaling. Cancer cells with decreased PTEN function are supposed to be especially sensitive to rapamycin treatment. While previous studies have shown that mTOR inhibition reduces neoplastic proliferation and tumor size in PTEN knockout mice [18] and that isogenic PTEN-/-mouse cells and human PTEN deficient cell lines were more preferentially inhibited by mTOR inhibition (CCI-779) than PTEN+/+cells [19], in our study, PTEN mutation status, which influences p-mTOR expression level, did not correlate to rapamycin sensitivity. Although PTEN deficient LNCaP and PC3 cells were more sensitive to rapamycin than DU145 cells, which carry wild type PTEN, the PTEN wild type 22RV1 cells were much more sensitive to rapamycin than LNCaP and PC3 cells. Our p-mTOR data also demonstrated that, although rapamycinin acts through inhibiting mTOR to suppress cell growth, the sensitivity of cells to rapamycin does not depend on baseline mTOR activity.
In this study, we observed a striking correlation between cyclin D1 expression and rapamycin sensitivity of prostate cancer cells. The most sensitive cells to rapamycin, 22RV1, expressed the highest level of base-line cyclin D1 protein and the rapamycin least sensitive DU145 cells had the lowest cyclin D1 expression level. The rapamycin sensitivity of PC3 and LNCaP cells also correlated with cyclin D1 expression level. This correlation between the level of base-line cyclin D1 protein and the level of sensitivity of those cells to rapamycin indicated that cyclin D1 might be an important mediator of cellular response to rapamycin, at least for prostate cancer cells. Our data suggest that cells with higher level of cyclin D1 expression may rely more on cyclin D1 function. When rapamycin reduced cyclin D1 level, potentially through inhibiting mTOR, the growth of cells with high-level cyclin D1 will be affected more than those with low cyclin D1. The correlation between cyclin D1 and sensitivity to rapamycin could also be a potential venue to use cyclin D1 protein expression level as a predictive biomarker to select cancers likely to respond to rapamycin treatment. Further investigation using a large series of samples will be required.
In a previous study using LNCaP cells, AR was found to stimulate prostate cancer cell proliferation through PI3K/Akt-independent activation of mTOR and led to subsequent posttranscriptional increases in cyclin D1 protein expression [20]. The mTOR inhibition with rapamycin blocked androgen stimulated-increase in cyclin D1 protein. This may explain the high cyclin D1 expression and the reduction in cyclin D1 protein expression levels under rapamycin treatment in androgen sensitive 22RV1 and LNCaP cells and may also explains why the reduction in cyclin D1 protein was dramatic in 22RV1, the most sensitive cells to rapamycin. However, this AR-induced cyclin D1 expression cannot explain the reduction of cyclin D1 protein expression levels in AR negative PC3 and DU145 cells under rapamycin treatment. It has been reported that PTEN inhibits cyclin D1 expression by suppressing AKT/mTOR signal pathway [21] and rapamycin treatment induced reduction of cyclin D1 expression has been observed in rapamycin sensitive cancer cells both in vivo and in vitro [22]. Furthermore, a reduction in cyclin D1 has been shown to play an important role in rapamycin induced growth inhibition [23]. However, it has also been reported that rapamycin treatment did not change cyclin D1 levels in neither rapamycin resistant nor sensitive cells [24,25]. Our study clearly supports that cyclin D1 expression level is influenced by rapamycin treatment.
Although PI3K/AKT/mTOR pathway are altered in many human cancers, for most of those cancer types, mTORC1 inhibitors alone have predominantly led to disease stabilization rather than tumor regression, including prostate cancer [3,8,26], which may potentially be explained by our observation that the sensitivity of cells to rapamycin treatment is not correlated to mTOR activity. Therefore, mTOR targeted therapies are now commonly investigated in combination with other drugs or cancer treatment methods and improved therapeutic benefit has been observed in a number of cancers [3,8,27-29]. In prostate cancer, mTOR inhibition has been found to reverse doxorubicin resistance [30]. Our in vitro study of combination of cisplatin and rapamycin also demonstrated that the effect of combining two agents was better than single agent alone in both androgen sensitive and resistant human prostate cancer cells. In consistent with our data, mTOR inhibitors have been shown to additively or synergistically kill cancer cells with carboplatin and cisplatin [31,32]. Rapamycin has also been found to enhance the effect of cisplatin in inhibiting ovarian and bladder cancer cells in vitro and in animal models [33,34]. In a lung cancer study, mTORC1 inhibition restored cisplatin sensitivity of cancer cells [35].
Cisplatin is one of the most effective chemotherapeutic drugs against many types of human cancers, including testicular tumors, bladder, head and neck, ovarian and lung cancers and certain hematological malignancies [36]. It has also been used to efficiently treat BRCA1/2 mutation positive breast cancer [37,38]. Despite the abundant use of cisplatin in oncology, it is associated with severe toxic side effects, which limit its therapeutic dose and consequently therapeutic efficacy [39]. A possible strategy to reduce such toxicity of cisplatin, whist maintaining its therapeutic effect on tumor, would be enhancing the effect of cisplatin at low dose through combination with other drug of limited toxicity. Rapamycin may thus be an appropriate drug to combine with cisplatin.
Platinum-based treatment has rarely been utilized in prostate cancer treatment, but the application of cisplatin in prostate cancer treatment has shown considerable effect in control cancer cell growth [40]. In breast cancer, a type of cancer that platinum-based regimen was not routinely used for chemotherapy (apart from BRCA mutated cases), a phase II clinical trial showed benefit of combined cisplatin and everolimus (mTORC1 inhibitor) treatment in patients with triple-negative metastatic breast cancer [41]. Thus under the premise of equivalent efficacy, addition of rapamycin to cisplatin regimen, with decreased dose of cisplatin and reduced toxicity, would be interesting to be tested in prostate cancer treatment. We showed both by cell viability and colony formation analyses a great effect of prostate cancer cell growth inhibition when cells were initially treated with cisplatin and rapamycin combination and followed by rapamycin alone for a long period continuous treatment. Rapamycin was found mainly acting as an mTORC1 inhibition. However, it has been reported that prolonged rapamycin exposure reduced mTORC2 levels and inhibited Akt activity [42,43]. Hence, the efficient inhibition of prostate cancer cells by long-term rapamycin exposure followed by short-term combined treatment with cisplatin could be explained by inhibiting of both mTOR1 and mTOR2 complexes by a long-term treatment with rapamycin. We found previously that cyclin D1 over-expression contributed to cisplatin resistance in testicular germ cell tumours and PC3 prostate cancer cells and that knockdown of cyclin D1 sensitized cells to cisplatin treatment [15]. Although in this study with prostate cancer cell lines, a correlation between cyclin D1 expression level and cell sensitivity to cisplatin treatment was not observed, rapamycin may preserve the cell growth inhibiting effect of cisplatin by decreasing cyclin D1 expression level, that cells cannot recover from the damage caused by cisplatin. This finding suggests that a long cisplatin treatment gap filled with low toxic rapamycin worth to be explored in clinical trials to determine its benefit for the management of patients with prostate cancer and also potentially other cancers.
In conclusion, we showed that cell sensitivity to rapamycin was correlated to their cyclin D1 but not p-mTOR protein expression levels. We also demonstrated that combined treatment of prostate cancer cells with cisplatin and rapamycin, in particular in the setting of short-term exposure of both followed by long period continuous rapamycin treatment efficiently inhibited prostate cancer cell growth. While further preclinical studies using more cell lines and in vivo models are required to confirm these findings, this combined therapy may be a useful approach for treating patients with prostate cancer and cyclin D1 expression level may be used as a biomarker to predict rapamycin response.
Disclosure of conflict of interest
None.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Barbieri CE, Bangma CH, Bjartell A, Catto JW, Culig Z, Gronberg H, Luo J, Visakorpi T, Rubin MA. The mutational landscape of prostate cancer. Eur Urol. 2013;64:567–576. doi: 10.1016/j.eururo.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bitting RL, Armstrong AJ. Targeting the PI3K/Akt/mTOR pathway in castration-resistant prostate cancer. Endocr Relat Cancer. 2013;20:R83–99. doi: 10.1530/ERC-12-0394. [DOI] [PubMed] [Google Scholar]
- 4.Edlind MP, Hsieh AC. PI3K-AKT-mTOR signaling in prostate cancer progression and androgen deprivation therapy resistance. Asian J Androl. 2014;16:378–386. doi: 10.4103/1008-682X.122876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Katzenwadel A, Wolf P. Androgen deprivation of prostate cancer: Leading to a therapeutic dead end. Cancer Lett. 2015;367:12–17. doi: 10.1016/j.canlet.2015.06.021. [DOI] [PubMed] [Google Scholar]
- 6.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 7.Vignot S, Faivre S, Aguirre D, Raymond E. mTOR-targeted therapy of cancer with rapamycin derivatives. Ann Oncol. 2005;16:525–537. doi: 10.1093/annonc/mdi113. [DOI] [PubMed] [Google Scholar]
- 8.Meric-Bernstam F, Gonzalez-Angulo AM. Targeting the mTOR signaling network for cancer therapy. J. Clin. Oncol. 2009;27:2278–2287. doi: 10.1200/JCO.2008.20.0766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ashworth RE, Wu J. Mammalian target of rapamycin inhibition in hepatocellular carcinoma. World J Hepatol. 2014;6:776–782. doi: 10.4254/wjh.v6.i11.776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beck JT. Potential role for mammalian target of rapamycin inhibitors as first-line therapy in hormone receptor-positive advanced breast cancer. Onco Targets Ther. 2015;8:3629–3638. doi: 10.2147/OTT.S88037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hortobagyi GN, Chen D, Piccart M, Rugo HS, Burris HA 3rd, Pritchard KI, Campone M, Noguchi S, Perez AT, Deleu I, Shtivelband M, Masuda N, Dakhil S, Anderson I, Robinson DM, He W, Garg A, McDonald ER 3rd, Bitter H, Huang A, Taran T, Bachelot T, Lebrun F, Lebwohl D, Baselga J. Correlative Analysis of Genetic Alterations and Everolimus Benefit in Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer: Results From BOLERO-2. J. Clin. Oncol. 2016;34:419–26. doi: 10.1200/JCO.2014.60.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, Antipin Y, Mitsiades N, Landers T, Dolgalev I, Major JE, Wilson M, Socci ND, Lash AE, Heguy A, Eastham JA, Scher HI, Reuter VE, Scardino PT, Sander C, Sawyers CL, Gerald WL. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Templeton AJ, Dutoit V, Cathomas R, Rothermundt C, Bärtschi D, Dröge C, Gautschi O, Borner M, Fechter E, Stenner F, Winterhalder R, Müller B, Schiess R, Wild PJ, Rüschoff JH, Thalmann G, Dietrich PY, Aebersold R, Klingbiel D, Gillessen S Swiss Group for Clinical Cancer Research (SAKK) Phase 2 trial of single-agent everolimus in chemotherapy-naive patients with castration-resistant prostate cancer (SAKK 08/08) Eur Urol. 2013;64:150–158. doi: 10.1016/j.eururo.2013.03.040. [DOI] [PubMed] [Google Scholar]
- 14.Easton JB, Houghton PJ. mTOR and cancer therapy. Oncogene. 2006;25:6436–6446. doi: 10.1038/sj.onc.1209886. [DOI] [PubMed] [Google Scholar]
- 15.Noel EE, Yeste-Velasco M, Mao X, Perry J, Kudahetti SC, Li NF, Sharp S, Chaplin T, Xue L, McIntyre A, Shan L, Powles T, Oliver RT, Young BD, Shipley J, Berney DM, Joel SP, Lu YJ. The association of CCND1 overexpression and cisplatin resistance in testicular germ cell tumors and other cancers. Am J Pathol. 2010;176:2607–2615. doi: 10.2353/ajpath.2010.090780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matos CS, de Carvalho AL, Lopes RP, Marques MP. New strategies against prostate cancer--Pt(II)-based chemotherapy. Curr Med Chem. 2012;19:4678–4687. doi: 10.2174/092986712803306394. [DOI] [PubMed] [Google Scholar]
- 17.Wang Y, Nangia-Makker P, Balan V, Hogan V, Raz A. Calpain activation through galectin-3 inhibition sensitizes prostate cancer cells to cisplatin treatment. Cell Death Dis. 2010;1:e101. doi: 10.1038/cddis.2010.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Podsypanina K, Lee RT, Politis C, Hennessy I, Crane A, Puc J, Neshat M, Wang H, Yang L, Gibbons J, Frost P, Dreisbach V, Blenis J, Gaciong Z, Fisher P, Sawyers C, Hedrick-Ellenson L, Parsons R. An inhibitor of mTOR reduces neoplasia and normalizes p70/S6 kinase activity in Pten+/-mice. Proc Natl Acad Sci U S A. 2001;98:10320–10325. doi: 10.1073/pnas.171060098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Neshat MS, Mellinghoff IK, Tran C, Stiles B, Thomas G, Petersen R, Frost P, Gibbons JJ, Wu H, Sawyers CL. Enhanced sensitivity of PTEN-deficient tumors to inhibition of FRAP/mTOR. Proc Natl Acad Sci U S A. 2001;98:10314–10319. doi: 10.1073/pnas.171076798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu Y, Chen SY, Ross KN, Balk SP. Androgens induce prostate cancer cell proliferation through mammalian target of rapamycin activation and post-transcriptional increases in cyclin D proteins. Cancer Res. 2006;66:7783–7792. doi: 10.1158/0008-5472.CAN-05-4472. [DOI] [PubMed] [Google Scholar]
- 21.Radu A, Neubauer V, Akagi T, Hanafusa H, Georgescu MM. PTEN induces cell cycle arrest by decreasing the level and nuclear localization of cyclin D1. Mol Cell Biol. 2003;23:6139–6149. doi: 10.1128/MCB.23.17.6139-6149.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gera JF, Mellinghoff IK, Shi Y, Rettig MB, Tran C, Hsu JH, Sawyers CL, Lichtenstein AK. AKT activity determines sensitivity to mammalian target of rapamycin (mTOR) inhibitors by regulating cyclin D1 and c-myc expression. J Biol Chem. 2004;279:2737–2746. doi: 10.1074/jbc.M309999200. [DOI] [PubMed] [Google Scholar]
- 23.Law M, Forrester E, Chytil A, Corsino P, Green G, Davis B, Rowe T, Law B. Rapamycin disrupts cyclin/cyclin-dependent kinase/p21/proliferating cell nuclear antigen complexes and cyclin D1 reverses rapamycin action by stabilizing these complexes. Cancer Res. 2006;66:1070–1080. doi: 10.1158/0008-5472.CAN-05-1672. [DOI] [PubMed] [Google Scholar]
- 24.Noh WC, Mondesire WH, Peng J, Jian W, Zhang H, Dong J, Mills GB, Hung MC, Meric-Bernstam F. Determinants of rapamycin sensitivity in breast cancer cells. Clin Cancer Res. 2004;10:1013–1023. doi: 10.1158/1078-0432.ccr-03-0043. [DOI] [PubMed] [Google Scholar]
- 25.Shi Y, Gera J, Hu L, Hsu JH, Bookstein R, Li W, Lichtenstein A. Enhanced sensitivity of multiple myeloma cells containing PTEN mutations to CCI-779. Cancer Res. 2002;62:5027–5034. [PubMed] [Google Scholar]
- 26.Amato RJ, Wilding G, Bubley G, Loewy J, Haluska F, Gross ME. Safety and preliminary efficacy analysis of the mTOR inhibitor ridaforolimus in patients with taxane-treated, castration-resistant prostate cancer. Clin Genitourin Cancer. 2012;10:232–238. doi: 10.1016/j.clgc.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seto B. Rapamycin and mTOR: a serendipitous discovery and implications for breast cancer. Clin Transl Med. 2012;1:29. doi: 10.1186/2001-1326-1-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yao JC, Phan AT, Chang DZ, Wolff RA, Hess K, Gupta S, Jacobs C, Mares JE, Landgraf AN, Rashid A, Meric-Bernstam F. Efficacy of RAD001 (everolimus) and octreotide LAR in advanced low- to intermediate-grade neuroendocrine tumors: results of a phase II study. J. Clin. Oncol. 2008;26:4311–4318. doi: 10.1200/JCO.2008.16.7858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Husseinzadeh N, Husseinzadeh HD. mTOR inhibitors and their clinical application in cervical, endometrial and ovarian cancers: a critical review. Gynecologic Oncology. 2014;133:375–381. doi: 10.1016/j.ygyno.2014.02.017. [DOI] [PubMed] [Google Scholar]
- 30.Grunwald V, DeGraffenried L, Russel D, Friedrichs WE, Ray RB, Hidalgo M. Inhibitors of mTOR reverse doxorubicin resistance conferred by PTEN status in prostate cancer cells. Cancer Res. 2002;62:6141–6145. [PubMed] [Google Scholar]
- 31.Mondesire WH, Jian W, Zhang H, Ensor J, Hung MC, Mills GB, Meric-Bernstam F. Targeting mammalian target of rapamycin synergistically enhances chemotherapy-induced cytotoxicity in breast cancer cells. Clin Cancer Res. 2004;10:7031–7042. doi: 10.1158/1078-0432.CCR-04-0361. [DOI] [PubMed] [Google Scholar]
- 32.Geoerger B, Kerr K, Tang CB, Fung KM, Powell B, Sutton LN, Phillips PC, Janss AJ. Antitumor activity of the rapamycin analog CCI-779 in human primitive neuroectodermal tumor/medulloblastoma models as single agent and in combination chemotherapy. Cancer Res. 2001;61:1527–1532. [PubMed] [Google Scholar]
- 33.Mabuchi S, Altomare DA, Cheung M, Zhang L, Poulikakos PI, Hensley HH, Schilder RJ, Ozols RF, Testa JR. RAD001 inhibits human ovarian cancer cell proliferation, enhances cisplatin-induced apoptosis, and prolongs survival in an ovarian cancer model. Clin Cancer Res. 2007;13:4261–4270. doi: 10.1158/1078-0432.CCR-06-2770. [DOI] [PubMed] [Google Scholar]
- 34.Makhlin I, Zhang J, Long CJ, Devarajan K, Zhou Y, Klein-Szanto AJ, Huang M, Chernoff J, Boorjian SA. The mTOR pathway affects proliferation and chemosensitivity of urothelial carcinoma cells and is upregulated in a subset of human bladder cancers. BJU Int. 2011;108:E84–90. doi: 10.1111/j.1464-410X.2010.09844.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu C, Wangpaichitr M, Feun L, Kuo MT, Robles C, Lampidis T, Savaraj N. Overcoming cisplatin resistance by mTOR inhibitor in lung cancer. Mol Cancer. 2005;4:25. doi: 10.1186/1476-4598-4-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dasari S, Tchounwou PB. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol. 2014;740:364–378. doi: 10.1016/j.ejphar.2014.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Narod SA. BRCA mutations in the management of breast cancer: the state of the art. Nat Rev Clin Oncol. 2010;7:702–707. doi: 10.1038/nrclinonc.2010.166. [DOI] [PubMed] [Google Scholar]
- 38.Shamseddine AI, Farhat FS. Platinum-based compounds for the treatment of metastatic breast cancer. Chemotherapy. 2011;57:468–487. doi: 10.1159/000334093. [DOI] [PubMed] [Google Scholar]
- 39.Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov. 2005;4:307–320. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- 40.Dhar S, Kolishetti N, Lippard SJ, Farokhzad OC. Targeted delivery of a cisplatin prodrug for safer and more effective prostate cancer therapy in vivo. Proc Natl Acad Sci U S A. 2011;108:1850–1855. doi: 10.1073/pnas.1011379108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Singh J, Novik Y, Stein S, Volm M, Meyers M, Smith J, Omene C, Speyer J, Schneider R, Jhaveri K, Formenti S, Kyriakou V, Joseph B, Goldberg JD, Li X, Adams S, Tiersten A. Phase 2 trial of everolimus and carboplatin combination in patients with triple negative metastatic breast cancer. Breast Cancer Res. 2014;16:R32. doi: 10.1186/bcr3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 43.Zeng Z, Sarbassov dos D, Samudio IJ, Yee KW, Munsell MF, Ellen Jackson C, Giles FJ, Sabatini DM, Andreeff M, Konopleva M. Rapamycin derivatives reduce mTORC2 signaling and inhibit AKT activation in AML. Blood. 2007;109:3509–3512. doi: 10.1182/blood-2006-06-030833. [DOI] [PMC free article] [PubMed] [Google Scholar]