

Abstract
Macrophage stimulating 1 receptor (MST1R) is a C-MET proto-oncogene family receptor tyrosine kinase. Promoter methylation patterns determine transcription of MST1R variants as hypermethylation of a region upstream of transcription start site (TSS) is associated with lack of MST1R long transcript (MST1R long) and expression of a short transcript with oncogenic potential. Thus, we aimed to investigate MST1R variant transcript regulation in renal cell tumors (RCT) and assess their prognostic potential. We found, in a series of 120 RCT comprising the four main subtypes (clear cell, papillary and chromophobe renal cell carcinoma, and oncocytoma), that higher methylation levels close to TSS were associated with total MST1R expression levels (MST1R total) in primary tumors (p=0.049) and renal cancer cell lines. After demethylating treatment, MST1R long/MST1R total ratio increased, as expected, in two renal cell carcinoma cell lines tested. However, in primary tumors with hypermethylation upstream of TSS, a decrease in MST1R long/MST1R total ratio was not detected, although higher expression ratio of nuclear factor-κB was apparent. Furthermore, survival analysis demonstrated that MST1R long/MST1R total ratio was independently associated with shorter disease-specific and disease-free survival, whereas MST1R total expression associated with shorter disease-specific survival. In conclusion, although promoter methylation patterns seem to determine MST1R global transcription regulation in renal cell carcinoma, other mechanisms might contribute to deregulate MST1R variant expression in RCT. Nevertheless, MST1R total expression and MST1R long/MST1R total ratio modulate the biological and clinical aggressiveness of renal cell carcinoma, as depicted by its prognostic significance, a finding that requires validation in a larger independent series.
Keywords: Renal cell tumors, MST1R, RON, MST1R promoter methylation, MST1R expression, epigenetics
Introduction
The macrophage stimulating 1 receptor (MST1R), also known as RON (recepteur d’origine nantais), is a C-MET proto-oncogene family receptor tyrosine kinase [1]. Both MST1R/RON and its ligand, macrophage-stimulating protein (MSP) [2], are mapped at chromosome 3p21 [1,3], and MSP binding triggers MST1R dimerization and subsequent activation [4]. This leads to downstream signaling activation of RAS-MAPK and PI-3K-AKT pathways [4], determining increased proliferation, survival and invasion [5], epithelial to mesenchymal transition (EMT) [6] and chemoresistance [7]. Since the nomenclature used for MST1R/RON varies in different references, we will follow the designation used in the original study whenever we consider that it prevents further confusion, but otherwise we will use MST1R.
MST1R is constitutively transcribed in epithelial cells, macrophages, osteoclasts and hematopoietic cells [8-12], and its signaling was reported to be altered in several human cancers, including those of the breast [13], lung [14], liver [15], ovary [16], colon [17], bladder [18] and nasopharynx [19].
In addition to ligand-induced dimerization, MST1R activation may be accomplished by receptor overexpression, kinase domain activating mutations and generation of constitutively active MST1R variants [4,20]. Most of these variants originate from full-length MST1R (flRON) alternative mRNA splicing (RONΔ170, RONΔ165, RONΔ160, RONΔ155), but may also be generated from protein truncation (RONΔ110, RONΔ75) and alternative transcription start site (short-form RON, sfRON, or RONΔ55) [21]. Some of these variants are constitutively active and thought to be oncogenic, including RONΔ165, RONΔ160, RONΔ155 and RONΔ110 [21].
Concerning alternative transcription start site, two MST1R transcripts are often found in both normal and neoplastic cells, named full-length RON (flRON) and short-form RON (sfRON) [22,23]. Whereas flRON transcription is initiated through a classical promoter upstream transcription start site (TSS) and it is enhanced by hypoxia-inducible factor 1α (HIF-1α) [24], early growth response-1 (Egr-1) [25] and nuclear factor-κB (NF-κB) [25] in cancer cells, sfRON transcription is initiated at the codon that encodes for Met913, using an alternative intragenic promoter located between introns 8 and 10 [12,22]. Scarce data is available on alternative transcription start site regulation, but it has been reported that methylation pattern of MST1R promoter associates with differential flRON and sfRON expression: hypermethylation at an area upstream of MST1R promoter, named ‘island 1’, was associated with absence of flRON and the presence of sfRON expression, whereas ‘island 1’ low or absent methylation was associated with concomitant flRON and sfRON expression [22]. It was also suggested that sfRON endogenous activity might be influenced by flRON expression, since a protein complex that is promptly degraded is formed when both sfRON and flRON are co-expressed [22]. Hence, when ‘island 1’ is hypermethyalted, MST1R homeostasis is shifted towards flRON null or low expression levels, and increased sfRON expression and activity. sfRON protein is constitutively active and its overexpression has been associated with an aggressive tumor phenotype: cancer cells grow faster, lose epithelial morphology, cease to form cell aggregates and become motile [23], features that promote local invasion and metastatic spread.
Despite MST1R signaling was found to be deregulated in several neoplasms [13-19,22,23], few studies have focused on MST1R promoter methylation [22], particularly in renal cell tumors (RCT). We have previously reported that MST1R promoter hypermethylation in renal cell tumors (RCT) was a sensitive and specific biomarker for clear cell renal cell carcinoma [26], and the 307 renal tumors available in the “Catalogue of somatic mutations in cancer” (COSMIC) dataset (cancer.sanger.ac.uk) were reported as highly methylated [27]. RCTs, a clinical, morphological, genetically and epigenetically heterogeneous group of tumors, comprise both benign [e.g., oncocytoma (RO)], and malignant [e.g., clear cell renal cell carcinoma (ccRCC), papillary RCC (pRCC) and chromophobe RCC (chRCC)] neoplasms, among which ccRCC is the most frequent (75%) and aggressive subtype, followed by pRCC (10%), and then chRCC (5%), the least aggressive subtype that rarely metastasizes [28,29]. Although MST1R protein expression has been previously investigated in RCTs, it mainly focused on chRCC and RO [30,31], and, thus, studies on MST1R mRNA expression deregulation through promoter methylation, as well as its biological and clinical impact are lacking. Thus, we aimed to characterize MST1R promoter methylation in RCT to investigate whether altered patterns might associate with different transcript variant expression in RCT primary tumors and cell lines, and how it might impact on tumor aggressiveness.
Material and methods
Patients and sample collection
Fresh-frozen tissue was prospectively collected, after informed consent, from 130 nephrectomy specimens at the Portuguese Oncology Institute - Porto (Portugal) between 2003 and 2007, comprising samples from ccRCC, pRCC, chRCC and oncocytoma (30 of each), and 10 morphologically normal kidney (cortical) tissue (from patients with upper urinary tract neoplasia not invading the renal parenchyma). Tissue samples were snap-frozen immediately after surgery, stored at -80°C and later cut in a cryostat. An H&E slide was performed before and after the sections used for nucleic acid extraction, to ensure at least 70% of neoplastic cells in the tumor samples and negligible inflammation in morphologically normal kidney samples.
Routine assessment of tumor classification (WHO), grading (Fuhrman) and staging (TNM) was performed for all tumor cases in formalin-fixed paraffin-embedded tissue [29,32]. Relevant clinical data was collected from clinical charts.
This study was approved by the Institutional Review Board (Comissão de Ética para a Saúde) of Portuguese Oncology Institute of Porto, Portugal (CES518/2010).
Cancer cell lines
Cell lines representative of ccRCC, two established from primary tumors (769-P, 786-O) and one from a metastatic site (Caki-1) were obtained from the American Type Culture Collection (Manassas, VA). All cell lines were cultured according to the manufacturer’s specifications, with 10% fetal bovine serum (Gibco, Invitrogen, Carlsbad, CA) and antibiotics (100 units/mL penicillin G and 100 μg/mL streptomycin, Gibco), in a humidified atmosphere of 5% CO2 at 37°C.
769-P and 786-O cancer cell lines were subjected to treatment with the demethylating drug 5-aza-2’deoxycytidine (1 μM for 72 h). In parallel, the same cell lines were cultured without treatment for 72 h and harvested before confluence. Demethylating treatment was conducted in triplicate for both cell lines.
Nucleic acid extraction
Genomic DNA from fresh-frozen samples and cell lines was extracted as previously described [33]. In brief, DNA was digested overnight with proteinase K (20 mg/mL) in the presence of 10% SDS at 55°C, and then extracted with phenolchloroform and precipitated with 100% ethanol.
RNA extraction was performed as previously reported [34] both for fresh-frozen tissues and cell lines. Briefly, TRIzol® reagent (InvitrogenTM, Carlsbad, CA, USA) was used to suspend the samples, chloroform (Merk Millipore, Darmstadt, Germany) was added to the lysed cells, and total RNA was then purified using Ambion® PureLink RNA Mini Kit (Invitrogen™, Carlsbad, CA, USA), according to manufacturer’s recommendations. RNA purity ratios and concentration were measured in a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA) and RNA quality was confirmed by electrophoresis.
Bisulfite modification and bisulfite sequencing
Bisulfite conversion of unmethylated cytosine residues to uracil, whereas methylated cytosine residues remain as such, was performed using the EZ DNA Methylation-Gold Kit (Zymo Research, Orange, CA, USA), according to manufacturer’s instructions. The modified DNA was eluted in 60 mL of water and stored at -80°C.
Subsequently, MST1R [GenBank: NM_002447] promoter was subjected to direct bisulfite sequencing in 5 samples: 1 ccRCC, 1 pRCC, 1 chRCC, 1 RO and 1 normal kidney. Primers were specifically designed to amplify fragments containing the MST1R promoter CpG “island 1” [22], using Methyl Primer Express v 1.0 (Applied Biosystems, Foster City, CA, USA). Primer sequences and location, amplicons, and annealing temperatures are listed in Table 1.
Table 1.
Primer sequences, amplicon size, and annealing temperatures for MST1R [GenBank: NM_002447] bisulfite sequencing (BSP), quantitative methylation specific PCR (QMSP) and expression
| Primer set | Sense primer sequence (5’-3’) | Antisense primer sequence (5’-3’) | Amplicon size (bp) | Location [bp upstream (up) or downstream (down) TSS] | Annealing temp (°C) |
|---|---|---|---|---|---|
| BSP | |||||
| MST1R_B_1 | GTTATTGAGGGTGTTGTTATTAAGTG | ACCTAACCCAAACCCTCC | 264 | 612 up to 348 up | 60 |
| MST1R_B_2 | AGGTGAAGGTATAGGAGTTAGG | AAATTCCTATAAAACCCAAATC | 272 | 417 up to 145 up | 60 |
| MST1R_B_3 | GGTAGGGATTTTTTAGGGTTT | CACCATAACCTATACCAAACCTC | 210 | 33 up to 177 down | 60 |
| QMSP | |||||
| MST1R up | TTAAGGGTCGGAAGAGTC | ATACACTAACGCTTAACGCTC | 128 | 540 up to 412 up | 60 |
| MST1R TSS | AGCGTTAGTGTATAGCGGC | TAAACAACGATCCCGACA | 169 | 270 up to 101 up | 60 |
| Expression | |||||
| MST1R total | GGCTGAGGTCAAGGATGTGCT | GCCTTTGCCAATGACTCGGT | 73 | - | 62 |
| MST1R long | CTCTGGGGACCAGGTTTTC | ATGAAATGCCATGCCCTTAGa | 93 | - | 62 |
| NF-κB | GCTTAGGAGGGAGAGCCCT | CTGCCATTCTGAAGCCGGG | 86 | - | 61 |
Primer sequence from [23].
PCR reactions included a 94°C denaturation 10 min. step followed by 40 cycles at 94°C for 30 sec., annealing temperature for 30 sec., and 72°C for 30 sec. PCR products were loaded in a 2% agarose gel, stained with ethidium bromide and visualized under an ultraviolet transilluminator. Excess primer and nucleotides were removed by Illustra GFX PCR DNA and Gel Band Purification kit (GE Healthcare, USB Corporation, Cleveland, OH, USA) following manufacturer’s protocol. The purified products were sequenced using the dGTP BigDye Terminator Cycle Sequencing ReadyReaction kit (Applied Biosystems, Foster City, CA, USA) in an ABI PRISM 310 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA), and data were analyzed by Sequencer Version 4.2.2 software. The peak height of the cytosine signal and the sum of the cytosine and thymine peak height signals were compared to calculate the approximate amount of methylcytosine of each CpG site. CpG sites with ratios 0-0.20, 0.21-0.80, and 0.81-1.0 were considered unmethylated, partially methylated, and fully methylated, respectively, as previously described [33,35].
Quantitative MSP
Quantitative methylation specific real-time polymerase chain reaction (QMSP) was performed in cell lines before and after demethylating treatment, and in all frozen tissue samples, after DNA bisulfite conversion.
Primers were designed to specifically amplify methylated bisulfite converted complementary sequences of MST1R promoter using Methyl Primer Express v 1.0 (Applied Biosystems, Foster City, CA, USA), enclosing the region previously described as MST1R promoter “island 1” [22], located upstream of TSS [26]. Two areas were amplified, one upstream “island 1” but still in the MST1R promoter CpG island, and another downstream, more close to TSS, named MST1R up and MST1R TSS respectively (Figure 1). Primer sequences and location are listed in Table 1. β-actin (ACTB) was used as reference gene to normalize for DNA input in each sample.
Figure 1.
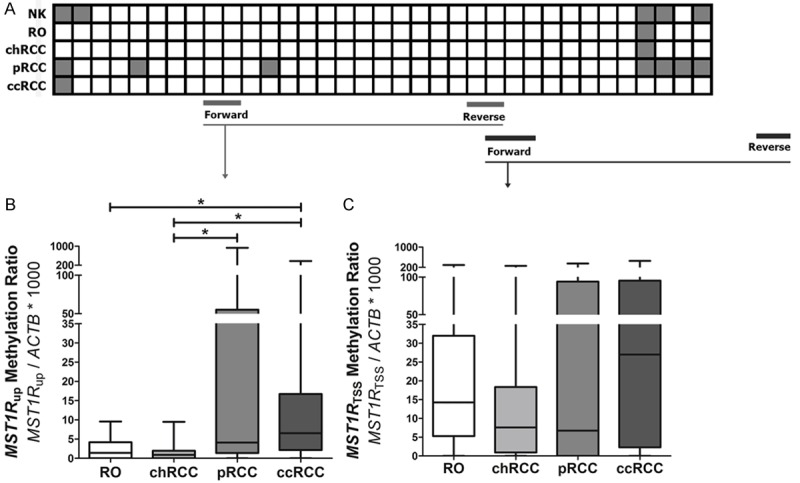
MST1R promoter methylation in renal cell tumors (RCT): bisulfite sequencing of MST1R promoter in 5 cases (A) and QMSP methylation levels in two distinct regions, one upstream TSS (MST1R up) and another closer to TSS (MST1R TSS), in 120 cases. White squares: CpG unmethylated; gray squares: CpG partially methylated. NK: normal kidney; RO: renal oncocytoma; chRCC: chromophobe renal cell carcinoma; pRCC: papillary renal cell carcinoma; ccRCC: clear cell renal cell carcinoma.
For QMSP analysis, a reaction volume of 20 µL consisting of 10 µL of SYBR® Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA), 7 µL of H2O, 0.5 µL of forward primer, 0.5 µL of reverse primer and 2 μL of bisulfate-modified DNA, was run in an 7500 Real-time PCR system (Applied Biosystems, Foster City, CA, USA). Each sample was run in triplicate, a calibration curve was constructed with serial dilutions (1:5) of bisulfite converted universally methylated DNA at all CpGs (CpGenome Universal Methylated DNA; Millipore, Billerica, MA) to quantify the amount of fully methylated alleles in each reaction, and “no template controls” were included as a control for contamination. The amplification reaction was carried out at 95°C for 2 min followed by 45 cycles of 95°C for 15 s, and at annealing temperature (Table 1) for 1 min, followed by a melt curve.
Relative levels of methylated promoter DNA in each sample were determined by the ratio of the mean quantity obtained by QMSP analysis for each gene and the respective value of the internal reference gene (ACTB), multiplied by 1000 for easy tabulation (methylation level = target gene/reference gene × 1000).
Quantitative gene expression analysis
MST1R gene expression was evaluated in ccRCC cell lines before and after treatment when done, and in the 120 RCTs samples. For cell lines, 1 μg of total RNA was reversely transcribed using the High Capacity cDNA Reverse Transcription kit (Applied Biosystems®, Foster City, CA, USA) according to manufacturer instructions. For frozen tissue, 300 ng of total RNA was reversely transcribed and amplified using TransPlex® Whole Transcriptome Amplification Kit (Sigma-Aldrich®, St. Louis, MO, United States) purified with QIAquick PCR Purification Kit (QIAGEN, Germany). Total MST1R expression (MST1R total) and long form MST1R expression (MST1R long) was evaluated using custom primers designed respectively to a region common to all MST1R described transcripts and to a region specific of the long form transcript (Table 1), using a Light Cycler 480 Real-time PRC system (Roche, Basel, Switzerland), in a reaction volume of 10 µL consisting of 5 µL of KAPA SYBR FAST® qPCR Master Mix (Kapa Biosystems, Wilmington, MA, USA), 3.7 µL of H2O, 0.15 µL of forward primer, 0.15 µL of reverse primer and 1 μL of cDNA.
Each sample was run in triplicate and the amount of cDNA was normalized to Glucuronidase beta (GUSβ) reference gene, as the ratio of the mean expression level obtained by QMSP analysis for each transcript and the respective value of the internal reference gene (GUSβ), multiplied by 1000 for easy tabulation. Each plate included multiple non-template controls and serial dilutions (1:5) of a cDNA Human Reference Total RNA (Agilent Technologies, La Jolla, CA, USA) to construct a standard curve.
NF-κB expression was evaluated in the 120 RCTs, as described above, using custom primers (Table 1).
Statistical analysis
Median and interquartile range of promoter methylation and expression levels were determined for cell lines and tumor samples. For tumor samples analysis and for each QMSP primer pair, each RCT sample was classified as “methylated” if the methylation level was higher than the highest value determined in the normal kidney samples (MST1R up: 17.58; MST1R TSS: 2.22), and as “not methylated” if the methylation level was lower than that value. MST1R long/total ratio was computed as the ratio MST1R long/MST1R total x 100, after linear normalization of MST1R total relative expression [(MST1R total value - MST1R total min)/(MST1R total max - MST1R total min)] and MST1R long relative expression [(MST1R long value - MST1R long min)/(MST1R long max - MST1R long min)].
Non-parametric tests were used to ascertain the statistical significance of differences among groups of samples, namely Kruskal-Wallis ANOVA test (KW) for multiple comparisons and Mann-Whitney U test (MW) with Bonferroni’s correction for pair-wise comparisons, as appropriate. Spearman’s test was carried out to ascertain correlations between age and gene expression levels.
Prognostic significance of standard clinicopathological variables (histological subtype, pathological stage, Fuhrman grade, age, gender) and of MST1R up and MST1R TSS methylation level, MST1R long/total ratio, MST1R total and NF-κB expression levels, was assessed by constructing disease-specific and disease-free survival (defined, respectively, as the time between diagnosis and death for renal cell carcinoma, and the time between treatment and the first metastasis or local recurrence) curves using the Kaplan-Meier method, with log-rank test (univariable test). For this purpose, expression levels and ratio were classified as low or high using as cut-off the 75th percentile expression value of each gene/ratio. Multivariable survival analysis was conducted only for ccRCC and pRCC. The exclusion of chRCC from the analysis was due to the paucity of events (one patient presented progression/metastasis during follow-up and none has died from cancer). Age, stage and histological subtype were also included in the final Cox-regression model, both for disease-specific (DSS) and disease-free (DFS) survival.
Statistical significance was set at p < 0.05. Statistical analysis was performed using SPSS software for Windows, version 22.0 (IBM-SPSS Inc., Chicago, IL, USA), and graphs were built using GraphPad Prism 6.0 software for Windows (GraphPad Software Inc., La Jolla, CA, USA).
Results
MST1R promoter methylation is higher near TSS in renal cell tumors
The methylation pattern of MST1R promoter in RCTs was characterized by QMSP using two primer sets, one upstream TSS (MST1R up) and another more close to TSS (MST1R TSS). Globally, MST1R TSS methylation levels [median (range): 14 (0-458)] was higher than those of MST1R up [median (range): 2 (0-933)], and 74% of samples were hypermethylated at MST1R TSS (22 ccRCC, 20 pRCC, 22 chRCC and 25 oncocytomas) whereas only 10% of samples were hypermethylated at MST1R up (4 ccRCC and 8 pRCC). This is in line with overall results of bisulfite sequencing in the 5 samples analyzed, which revealed rare methylated CpG in the MST1R up area, and an increase of methylated CpG dinucleotides near TSS (Figure 1A). Additionally, at MST1R up, significantly higher methylation level were depicted for ccRCC and pRCC compared to chRCC, and for ccRCC compared to oncocytoma (p<0.001 for all) (Figure 1B). There were no statistically significant differences in MST1R TSS methylation levels among RCT subtypes (p=0.291) (Figure 1C).
NF-κB expression associates with MST1Rlong/total ratio in hypermethylated RCTs
RCTs with MST1R TSS hypermethylation showed a significantly lower MST1R total expression ratio (p=0.049), and RCTs with MST1R up hypermethylation displayed a trend for higher expression of MST1R long/total, (p=0.053) (Figure 2A). Interestingly, a significantly higher expression of NF-κB (p=0.013) was also observed in these RCTs (Figure 2B).
Figure 2.
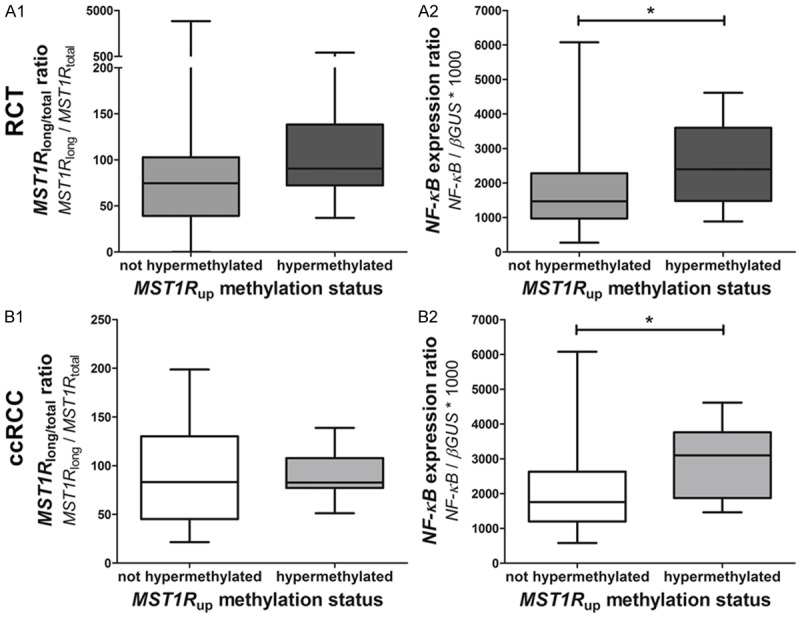
Expression levels in tumors hypermethylated or not at MST1R up (upstream area of MST1R promoter): MST1R long/total ratio in RCTs (n=120) (A1) and in ccRCC (n=30) (B1), and NF-κB expression ratio in RCTs (A2) and ccRCC (B2).
When analyzing ccRCC and pRCC independently (MST1R up hypermethylation was not apparent in chRCC or oncocytomas), there were no differences in MST1R long/total in ccRCC with or without MST1R up hypermethylation (p=0.756) (Figure 2C), but ccRCC with MST1R up hypermethylation displayed a significantly higher NF-κB expression (p=0.036) (Figure 2D). No statistically significant differences were depicted for pRCC.
MST1R expression is regulated by promoter methylation pattern in ccRCC cell lines
MST1R promoter methylation levels more close to TSS (MST1R TSS) and MST1R total expression was evaluated in 769-P, 786-O and Caki-1 ccRCC cell lines. MST1R total expression was lowest in 769-P and 786-O cells (Figure 3A2), which also displayed the highest MST1R TSS methylation levels (Figure 3A1), paralleling the observations in primary tumors. Demethylating treatment in those two cell lines restored MST1R total expression (Figure 3B2), mainly MST1R long (flRON) expression, which was apparent through a higher MST1R long/total ratio (Figure 3C2), and decreased MST1R TSS and MST1R up methylation levels (Figure 3B1 and C1).
Figure 3.

MST1R methylation and expression levels in ccRCC cell lines. A: MST1R TSS methylation level (A1) and MST1R total expression level (A2) in three cell lines. B: MST1R TSS methylation level (B1) and MST1R total expression level (B2) in 769-P and 786-O cell lines before and after demethylating treatment with 1 µM 5-aza-2’deoxycytidine for 72 h. C. MST1R up methylation level (C1) and MST1R long/total ratio (C2) in 769-P and 786-O cell lines before and after demethylating treatment with 1 µM 5-aza-2’deoxycytidine for 72 h.
Clinical-pathological associations and survival analysis
Clinical and pathological features of the 120 patients included in this study are depicted in Table 2. The methylation levels of MST1R up and MST1R TSS, as well as MST1R total expression level, MST1R long/total ratio and NF-κB expression level, were not associated with gender (p=0.563, p=0.263, p=0.561, p=0.159 and p=0.576, respectively), age (p=0.352, p=0.979, p=0.676, p=0.119 and p=0.056, respectively) or pathological stage (p=0.661, p=0.908, p=0.132, p=0.579 and p=0.822, respectively).
Table 2.
Clinical and pathological data of patients included in the present study
| Tumor | Normal | |
|---|---|---|
| Number of patients, n | 120 | 10 |
| Age, median (range) | 60 (29-83) | 67.5 (20-83) |
| Gender, n (%) | ||
| Male | 73 (61) | 7 (70.0) |
| Female | 47 (39) | 3 (30.0) |
| Histological subtype, n (%) | n.a. | |
| Clear cell RCC | 30 (25) | |
| Papillary RCC | 30 (25) | |
| Chromophobe RCC | 30 (25) | |
| Oncocytoma | 30 (25) | |
| Pathological Stage, n (%) | n.a. | |
| Stage I | 47 (39) | |
| Stage II | 19 (16) | |
| Stage III | 21 (17.5) | |
| Stage IV | 3 (2.5) | |
| n.a. (oncocytoma) | 30 (25) | |
| Fuhrman grade, n (%) | n.a. | |
| 1 | 3 (2.5) | |
| 2 | 28 (23) | |
| 3 | 45 (37.5) | |
| 4 | 14 (12) | |
| n.a. | 30 (25) |
n.a.: not applicable.
A significantly lower NF-κB expression level (p<0.001) was observed in oncocytomas compared to RCC, whereas for MST1R up and MST1R TSS methylation levels, MST1R total expression levels and MST1R long/total ratio, no significant differences were found.
During follow-up [median (range): 60 months (2-392 months)], 12 (13%) patients died from RCC and 17 (19%) developed metastatic disease. Among molecular parameters, only MST1R total expression levels associated with development of metastasis during follow-up (p=0.049). Patients with a low RCC MST1R total expression displayed shorter DSS, and those with high MST1R long/total ratio presented shorter DSS and DFS (Figure 4), which was statistically significant in multivariable analysis, controlling for stage, histological subtype and age (Table 3).
Figure 4.

Kaplan-Meier analysis for disease-specific survival in 60 RCC patients, according to MST1R total expression level (A) and MST1R long/total ratio (B), and for disease-free survival in 60 RCC patients, according to MST1R long/total ratio (C). The results presented were categorized using third quartile (75th percentile) value as cutoff.
Table 3.
Prognostic value of pathological stage, histological subtype and MST1R expression in renal cell carcinomas, following multivariable analysis using Cox-regression model
| Prognostic Factor | Multivariable Analysis | ||
|---|---|---|---|
|
| |||
| Hazard Ratio (HR) | 95% CI for HR | Cox regression p value | |
| Disease Specific Survivala | |||
| - Stage III/Stage IV (vs Stage I/Stage II) | 38 | 5.4-269 | < 0.001 |
| - pRCC (vs ccRCC) | 22 | 3.1-157 | 0.002 |
| - High MST1Rtotal expression level (vs low MST1Rtotal expression level) | 10 | 1-96 | 0.046 |
| - Stage III/Stage IV (vs Stage I/Stage II) | 26 | 4.4-153 | < 0.001 |
| - pRCC (vs ccRCC) | 14.6 | 2.2-99 | 0.006 |
| - Low MST1Rlong/MST1Rtotal ratio (vs high MST1Rlong/MST1Rtotal ratio) | 4.9 | 1.2-20 | 0.025 |
| Disease Free Survivalb | |||
| - Stage III/Stage IV (vs Stage I/Stage II) | 14 | 3.5-59 | < 0.001 |
| - Low MST1Rlong/MST1Rtotal ratio (vs high MST1Rlong/MST1Rtotal ratio) | 3.2 | 1.1-9.5 | 0.038 |
Only ccRCC and pRCC were included due to insufficient events in chRCC. CI: Confidence Interval; ccRCC: clear cell renal cell carcinoma; pRCC: papillary renal cell carcinoma.
Adjusted for patient age.
Adjusted for patient age and histological subtype;
MST1Rtotal expression level did not attained statistical significance in multivariable analysis for disease free survival.
Discussion
Gene expression regulation by promoter methylation is a well described epigenetic mechanism and its deregulation is considered an early event in carcinogenesis [36]. Indeed, aberrant promoter hypermethylation is associated with transcriptional repression [36,37] and, thus, gene re-expression after demethylating treatment has been widely used as a strategy for identification of genes regulated by promoter methylation, namely in RCC [38-42]. MST1R promoter had been previously reported as hypermethylated in RCC in an area downstream of TSS [26,27] and in the regions investigated by Angeloni and co-workers [22], but its putative association with altered MST1R expression pattern was not further explored. Our findings suggest that MST1R global expression (MST1R total) is predominantly modulated by promoter methylation near TSS (MST1R TSS), because significantly lower MST1R total expression was found in primary RCT with MST1R TSS hypermethylation, lower MST1R total expression was found in ccRCC cell lines with higher MST1R TSS methylation levels (769-P and 786-O), and MST1R total increased expression was observed in those cell lines after demethylating treatment.
It has been previously suggested that the pattern of promoter methylation was associated with the expression of different MST1R variants, specifically that the methylation of a particular promoter region upstream TSS - ‘island 1’ - was associated with lack of flRON/MST1R long and an increase of sfRON transcription, through an alternative internal promoter, with a consequent decrease in MST1R long/total ratio [22]. By bisulfite sequencing we found that not only the region previously described as ‘island 1’ but also its’ upstream region within the CpG island were not methylated in RCTs, and thus we designed primers slightly upstream ‘island 1’ to further explore this MST1R promoter area. The quantification of sfRON expression could provide additional information concerning the variation of expression of different transcripts, but this was not possible due to the inability to design primers specific for sfRON. Surprisingly, a higher MST1R long/total ratio was found in RCTs with MST1R up hypermethylation (using a QMSP primer set specific to ‘island 1’), although it did not reach statistical significance.
Because MST1R long is under control of the classical MST1R promoter, we hypothesized that transcription factors acting on MST1R in cancer cells might contribute to MST1R long expression, overcoming the methylation inhibitory effect. Since NF-κB has more predicted binding sites in the MST1R promoter than HIF-1α and Egr-1, NF-κB expression was determined in RCTs and, indeed, we found that RCTs with MST1R up hypermethylation displayed a significantly higher level of NF-κB expression, suggesting that promoter methylation is not the sole mechanism regulating MST1R expression.
Nevertheless, aberrant promoter methylation seems to be a relevant cause of MST1R silencing, because in 769-P and 786-O cells the MST1R long/total ratio increased after demethylating treatment. Importantly, increase in flRON expression after demethylating treatment had already been reported for other cell lines, including TF1 (erythroleukemia) and lung cancer cell lines [22].
We have previously reported that promoter methylation in a region downstream MST1R TSS identifies ccRCC with high sensitivity and specificity [26]. Similar diagnostic performance was not demonstrated for methylation of MST1R up or MST1R TSS, neither for MST1R total expression or MST1R long/total ratio. Nevertheless, the present study demonstrated that lower MST1R total expression and higher MST1R long/total ratio independently predict worse prognosis in ccRCC and pRCC. Intriguingly, in urothelial carcinoma of the bladder, MST1R protein expression was associated with a worse prognosis [18] and MST1R overexpression is one of the mechanisms for activation of MST1R signaling, which seems to confer a more aggressive phenotype to cancer cells. However, it should be taken in account that in our series, MST1R overexpression is not a common alteration driving activation of signaling pathways that lead to cancer cell proliferation, invasion and metastization in RCC. On the contrary, we found significantly lower MST1R total expression in association with MST1R TSS hypermethylation in RCTs. Indeed, this contrasts with the more prominent role of MST1R in other cancer models, including nasopharyngeal carcinoma (NPC), in which latent membrane protein 1 (LMP1) stimulates NF-κB binding to MST1R promoter, inducing EMT, a finding that may explain the higher metastatic potential of NPC with LMP1 overexpression [19].
It should, however, be noted that the biological interpretation of MST1R expression in RCT primary tumor is not straightforward. We explored the association of MST1R promoter methylation pattern and MST1R long/total ratio, and given that sfRON is a constitutively active variant with oncogenic potential, it would be expectable that most aggressive tumors should display a lower MST1R long/total ratio. However, some MST1R long splicing variants have oncogenic potential, and even the overexpression of MST1R could lead to the activation of cell signaling pathways related to proliferation and metastization. The presence of such splicing variants, although functionally relevant for the understanding of MST1R role in renal carcinogenesis, was not further explored mainly because all are transcribed from the classical promoter and the MST1R long primer set was unable to discriminate splicing variants. Other MST1R activating mechanisms might also be relevant but their relative contribution might be limited. Indeed, the frequency of activating point mutations for RCC reported in COSMIC dataset (cancer.sanger.ac.uk) is low (3/1474, 0.2%), and the same holds true for the frequency of copy number variations (loss in 8/417, 1.9%) [27].
In conclusion, although promoter methylation patterns seem to determine MST1R global transcription regulation in renal cell carcinoma, other mechanisms might contribute to deregulate MST1R variant expression in RCT. Nevertheless, MST1R total expression and MST1R long/MST1R total ratio modulate the biological and clinical aggressiveness of renal cell carcinoma, as depicted by its prognostic significance, a finding that requires validation in a larger independent series.
Acknowledgements
This study was funded by research grants from Research Center of Portuguese Oncology Institute of Porto (CI-IPOP 4-2012) and from Associação Portuguesa de Urologia (APU). ASP-L, JRC and IG are supported by FCT-Fundação para a Ciência e a Tecnologia fellowships (SFRH/SINTD/94217/2013, SFRH/BD/71293/2010 and CI-IPOP-BPD/UID/DTP/00776/2013, respectively).
Disclosure of conflict of interest
None.
References
- 1.Ronsin C, Muscatelli F, Mattei MG, Breathnach R. A novel putative receptor protein tyrosine kinase of the met family. Oncogene. 1993;8:1195–1202. [PubMed] [Google Scholar]
- 2.Gaudino G, Follenzi A, Naldini L, Collesi C, Santoro M, Gallo KA, Godowski PJ, Comoglio PM. RON is a heterodimeric tyrosine kinase receptor activated by the HGF homologue MSP. EMBO J. 1994;13:3524–3532. doi: 10.1002/j.1460-2075.1994.tb06659.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yoshimura T, Yuhki N, Wang MH, Skeel A, Leonard EJ. Cloning, sequencing, and expression of human macrophage stimulating protein (MSP, MST1) confirms MSP as a member of the family of kringle proteins and locates the MSP gene on chromosome 3. J Biol Chem. 1993;268:15461–15468. [PubMed] [Google Scholar]
- 4.Wang MH, Zhang R, Zhou YQ, Yao HP. Pathogenesis of RON receptor tyrosine kinase in cancer cells: activation mechanism, functional crosstalk, and signaling addiction. J Biomed Res. 2013;27:345–356. doi: 10.7555/JBR.27.20130038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang MH, Dlugosz AA, Sun Y, Suda T, Skeel A, Leonard EJ. Macrophage-stimulating protein induces proliferation and migration of murine keratinocytes. Exp Cell Res. 1996;226:39–46. doi: 10.1006/excr.1996.0200. [DOI] [PubMed] [Google Scholar]
- 6.Cote M, Miller AD, Liu SL. Human RON receptor tyrosine kinase induces complete epithelial-to-mesenchymal transition but causes cellular senescence. Biochem Biophys Res Commun. 2007;360:219–225. doi: 10.1016/j.bbrc.2007.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McClaine RJ, Marshall AM, Wagh PK, Waltz SE. Ron receptor tyrosine kinase activation confers resistance to tamoxifen in breast cancer cell lines. Neoplasia. 2010;12:650–658. doi: 10.1593/neo.10476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gaudino G, Avantaggiato V, Follenzi A, Acampora D, Simeone A, Comoglio PM. The proto-oncogene RON is involved in development of epithelial, bone and neuro-endocrine tissues. Oncogene. 1995;11:2627–2637. [PubMed] [Google Scholar]
- 9.Wang MH, Wang D, Chen YQ. Oncogenic and invasive potentials of human macrophage-stimulating protein receptor, the RON receptor tyrosine kinase. Carcinogenesis. 2003;24:1291–1300. doi: 10.1093/carcin/bgg089. [DOI] [PubMed] [Google Scholar]
- 10.Danilkovitch A, Leonard EJ. Kinases involved in MSP/RON signaling. J Leukoc Biol. 1999;65:345–348. doi: 10.1002/jlb.65.3.345. [DOI] [PubMed] [Google Scholar]
- 11.Danilkovitch A, Donley S, Skeel A, Leonard EJ. Two independent signaling pathways mediate the antiapoptotic action of macrophage-stimulating protein on epithelial cells. Mol Cell Biol. 2000;20:2218–2227. doi: 10.1128/mcb.20.6.2218-2227.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Angeloni D, Danilkovitch-Miagkova A, Ivanov SV, Breathnach R, Johnson BE, Leonard EJ, Lerman MI. Gene structure of the human receptor tyrosine kinase RON and mutation analysis in lung cancer samples. Genes Chromosomes Cancer. 2000;29:147–156. [PubMed] [Google Scholar]
- 13.Maggiora P, Marchio S, Stella MC, Giai M, Belfiore A, De Bortoli M, Di Renzo MF, Costantino A, Sismondi P, Comoglio PM. Overexpression of the RON gene in human breast carcinoma. Oncogene. 1998;16:2927–2933. doi: 10.1038/sj.onc.1201812. [DOI] [PubMed] [Google Scholar]
- 14.Willett CG, Wang MH, Emanuel RL, Graham SA, Smith DI, Shridhar V, Sugarbaker DJ, Sunday ME. Macrophage-stimulating protein and its receptor in non-small-cell lung tumors: induction of receptor tyrosine phosphorylation and cell migration. Am J Respir Cell Mol Biol. 1998;18:489–496. doi: 10.1165/ajrcmb.18.4.2978. [DOI] [PubMed] [Google Scholar]
- 15.Chen Q, Seol DW, Carr B, Zarnegar R. Co-expression and regulation of Met and Ron proto-oncogenes in human hepatocellular carcinoma tissues and cell lines. Hepatology. 1997;26:59–66. doi: 10.1002/hep.510260108. [DOI] [PubMed] [Google Scholar]
- 16.Maggiora P, Lorenzato A, Fracchioli S, Costa B, Castagnaro M, Arisio R, Katsaros D, Massobrio M, Comoglio PM, Flavia Di Renzo M. The RON and MET oncogenes are co-expressed in human ovarian carcinomas and cooperate in activating invasiveness. Exp Cell Res. 2003;288:382–389. doi: 10.1016/s0014-4827(03)00250-7. [DOI] [PubMed] [Google Scholar]
- 17.Chen WS, Kung HJ, Yang WK, Lin W. Comparative tyrosine-kinase profiles in colorectal cancers: enhanced arg expression in carcinoma as compared with adenoma and normal mucosa. Int J Cancer. 1999;83:579–584. doi: 10.1002/(sici)1097-0215(19991126)83:5<579::aid-ijc1>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 18.Cheng HL, Liu HS, Lin YJ, Chen HH, Hsu PY, Chang TY, Ho CL, Tzai TS, Chow NH. Co-expression of RON and MET is a prognostic indicator for patients with transitional-cell carcinoma of the bladder. Br J Cancer. 2005;92:1906–1914. doi: 10.1038/sj.bjc.6602593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chou YC, Chen CL, Yeh TH, Lin SJ, Chen MR, Doong SL, Lu J, Tsai CH. Involvement of recepteur d’origine nantais receptor tyrosine kinase in Epstein-Barr virus-associated nasopharyngeal carcinoma and its metastasis. Am J Pathol. 2012;181:1773–1781. doi: 10.1016/j.ajpath.2012.07.014. [DOI] [PubMed] [Google Scholar]
- 20.Yao HP, Zhou YQ, Zhang R, Wang MH. MSP-RON signalling in cancer: pathogenesis and therapeutic potential. Nat Rev Cancer. 2013;13:466–481. doi: 10.1038/nrc3545. [DOI] [PubMed] [Google Scholar]
- 21.Lu Y, Yao HP, Wang MH. Multiple variants of the RON receptor tyrosine kinase: biochemical properties, tumorigenic activities, and potential drug targets. Cancer Lett. 2007;257:157–164. doi: 10.1016/j.canlet.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 22.Angeloni D, Danilkovitch-Miagkova A, Ivanova T, Braga E, Zabarovsky E, Lerman MI. Hypermethylation of Ron proximal promoter associates with lack of full-length Ron and transcription of oncogenic short-Ron from an internal promoter. Oncogene. 2007;26:4499–4512. doi: 10.1038/sj.onc.1210238. [DOI] [PubMed] [Google Scholar]
- 23.Bardella C, Costa B, Maggiora P, Patane S, Olivero M, Ranzani GN, De Bortoli M, Comoglio PM, Di Renzo MF. Truncated RON tyrosine kinase drives tumor cell progression and abrogates cell-cell adhesion through E-cadherin transcriptional repression. Cancer Res. 2004;64:5154–5161. doi: 10.1158/0008-5472.CAN-04-0600. [DOI] [PubMed] [Google Scholar]
- 24.Thangasamy A, Rogge J, Ammanamanchi S. Recepteur d’origine nantais tyrosine kinase is a direct target of hypoxia-inducible factor-1alpha-mediated invasion of breast carcinoma cells. J Biol Chem. 2009;284:14001–14010. doi: 10.1074/jbc.M809320200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xia Y, Lian S, Khoi PN, Yoon HJ, Han JY, Chay KO, Kim KK, Jung YD. Chrysin inhibits cell invasion by inhibition of Recepteur d’origine Nantais via suppressing early growth response-1 and NF-kappaB transcription factor activities in gastric cancer cells. Int J Oncol. 2015;46:1835–1843. doi: 10.3892/ijo.2015.2847. [DOI] [PubMed] [Google Scholar]
- 26.Pires-Luis ASL F, Vieira-Coimbra M, Costa-Pinheiro P, Antunes L, Oliveira J, Henrique R, Jerónimo C. MST1R methylation as a diagnostic biomarker in renal cell tumors. Acta Urológica Portuguesa. 2015;32:64–70. [Google Scholar]
- 27.Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S, Kok CY, Jia M, De T, Teague JW, Stratton MR, McDermott U, Campbell PJ. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43:D805–811. doi: 10.1093/nar/gku1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moch H. An overview of renal cell cancer: pathology and genetics. Semin Cancer Biol. 2013;23:3–9. doi: 10.1016/j.semcancer.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 29.Eble J SG, Epstein J, Sesterhenn I. Tumours of the kidney. In: J E, editor. WHO classification of tumours. Tumours of the urinary system and male genital organs. Lyon: IARC Press; 2004. [Google Scholar]
- 30.Patton KT, Tretiakova MS, Yao JL, Papavero V, Huo L, Adley BP, Wu G, Huang J, Pins MR, Teh BT, Yang XJ. Expression of RON Proto-oncogene in Renal Oncocytoma and Chromophobe Renal Cell Carcinoma. Am J Surg Pathol. 2004;28:1045–1050. doi: 10.1097/01.pas.0000128661.58697.7d. [DOI] [PubMed] [Google Scholar]
- 31.Wang HY, Mills SE. KIT and RCC are useful in distinguishing chromophobe renal cell carcinoma from the granular variant of clear cell renal cell carcinoma. Am J Surg Pathol. 2005;29:640–646. doi: 10.1097/01.pas.0000157943.33903.92. [DOI] [PubMed] [Google Scholar]
- 32.Edge SB BD, Compton CC, Fritz AG, Greene FL, Trotti A. AJCC Cancer Staging Manual. New York: Springer; 2010. [Google Scholar]
- 33.Patricio P, Ramalho-Carvalho J, Costa-Pinheiro P, Almeida M, Barros-Silva JD, Vieira J, Dias PC, Lobo F, Oliveira J, Teixeira MR, Henrique R, Jeronimo C. Deregulation of PAX2 expression in renal cell tumours: mechanisms and potential use in differential diagnosis. J Cell Mol Med. 2013;17:1048–1058. doi: 10.1111/jcmm.12090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pires-Luis AS, Vieira-Coimbra M, Vieira FQ, Costa-Pinheiro P, Silva-Santos R, Dias PC, Antunes L, Lobo F, Oliveira J, Goncalves CS, Costa BM, Henrique R, Jeronimo C. Expression of histone methyltransferases as novel biomarkers for renal cell tumor diagnosis and prognostication. Epigenetics. 2015;10:1033–43. doi: 10.1080/15592294.2015.1103578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Costa VL, Henrique R, Danielsen SA, Duarte-Pereira S, Eknaes M, Skotheim RI, Rodrigues A, Magalhaes JS, Oliveira J, Lothe RA, Teixeira MR, Jeronimo C, Lind GE. Three epigenetic biomarkers, GDF15, TMEFF2, and VIM, accurately predict bladder cancer from DNA-based analyses of urine samples. Clin Cancer Res. 2010;16:5842–5851. doi: 10.1158/1078-0432.CCR-10-1312. [DOI] [PubMed] [Google Scholar]
- 36.Feinberg AP, Ohlsson R, Henikoff S. The epigenetic progenitor origin of human cancer. Nat Rev Genet. 2006;7:21–33. doi: 10.1038/nrg1748. [DOI] [PubMed] [Google Scholar]
- 37.Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31:27–36. doi: 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ibanez de Caceres I, Dulaimi E, Hoffman AM, Al-Saleem T, Uzzo RG, Cairns P. Identification of novel target genes by an epigenetic reactivation screen of renal cancer. Cancer Res. 2006;66:5021–5028. doi: 10.1158/0008-5472.CAN-05-3365. [DOI] [PubMed] [Google Scholar]
- 39.Kagara I, Enokida H, Kawakami K, Matsuda R, Toki K, Nishimura H, Chiyomaru T, Tatarano S, Itesako T, Kawamoto K, Nishiyama K, Seki N, Nakagawa M. CpG hypermethylation of the UCHL1 gene promoter is associated with pathogenesis and poor prognosis in renal cell carcinoma. J Urol. 2008;180:343–351. doi: 10.1016/j.juro.2008.02.044. [DOI] [PubMed] [Google Scholar]
- 40.Morris MR, Gentle D, Abdulrahman M, Maina EN, Gupta K, Banks RE, Wiesener MS, Kishida T, Yao M, Teh B, Latif F, Maher ER. Tumor suppressor activity and epigenetic inactivation of hepatocyte growth factor activator inhibitor type 2/SPINT2 in papillary and clear cell renal cell carcinoma. Cancer Res. 2005;65:4598–4606. doi: 10.1158/0008-5472.CAN-04-3371. [DOI] [PubMed] [Google Scholar]
- 41.Morris MR, Gentle D, Abdulrahman M, Clarke N, Brown M, Kishida T, Yao M, Teh BT, Latif F, Maher ER. Functional epigenomics approach to identify methylated candidate tumour suppressor genes in renal cell carcinoma. Br J Cancer. 2008;98:496–501. doi: 10.1038/sj.bjc.6604180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morris MR, Ricketts C, Gentle D, Abdulrahman M, Clarke N, Brown M, Kishida T, Yao M, Latif F, Maher ER. Identification of candidate tumour suppressor genes frequently methylated in renal cell carcinoma. Oncogene. 2010;29:2104–2117. doi: 10.1038/onc.2009.493. [DOI] [PMC free article] [PubMed] [Google Scholar]