

Abstract
MiR-98 expression was up-regulated in kidney in response to early diabetic nephropathy in mouse and down-regulated in muscle in type 2 diabetes in human. However, the expression prolife and functional role of miR-98 in human gestational diabetes mellitus (GDM) remained unclear. Here, we investigated its expression and function in placental tissues from GDM patients and the possible molecular mechanisms. The results showed that miR-98 was up-regulated in placentas from GDM patients compared with normal placentas. MiR-98 over-expression increased global DNA methylational level and miR-98 knockdown reduced global DNA methylational level. Further investigation revealed that miR-98 could inhibit Mecp2 expression by binding the 3′-untranslated region (UTR) of methyl CpG binding protein 2 (Mecp2), and then led to the expression dysregulation of canonical transient receptor potential 3 (Trpc3), a glucose uptake related gene. More importantly, in vivo analysis found that the expression level of Mecp2 and Trpc3 in placental tissues from GDM patients, relative to the increase of miR-98, was diminished, especially for GDM patients over the age of 35 years. Collectively, up-regulation of miR-98 in the placental tissues of human GDM is linked to the global DNA methylation via targeting Mecp2, which may imply a novel regulatory mechanism in GDM.
Gestational diabetes mellitus (GDM) is a condition in which women without previously diagnosed diabetes exhibit varying degrees of glucose intolerance during pregnancy1. In different studies, gestational diabetes mellitus affects approximately 1.1–14.3% of pregnant women2,3,4, and has 35.6% to 69% of recurrence risk5,6. GDM has adverse effects on the pregnant women and fetus including pre-eclampsia, caesarean section rates, perinatal mortality, birth defects, macrosomia, etc. In longitudinal studies with a duration of at least 5 years, 20% to 65% of women with GDM go on to develop type 2 diabetes (T2DM)7. The pathogenesis of GDM is not fully understood, the syndrome has many similarities to T2D that becomes manifest during the course of pregnancy. T2D, also called non-insulin-dependent diabetes (NIDDM), is characterized by hyperglycemia resulting from impairment of insulin secretion and/or defects in insulin action in peripheral tissues. GDM represents a combination of acquired and intrinsic abnormalities of insulin action. The precise mechanisms underlying gestational diabetes are still largely unknown.
MiRNAs are small 19–23 nucleotide RNA molecules that act as negative regulators of gene expression by mediating messenger RNAs degradation or translational arrest. They are potent drivers of differentiation and development in many biological processes. The evidence increasingly shows that miRNA dysregulation has been linked to diabetes in recent years. As is known, miRNAs play roles in type 1 and type 2 diabetes (T1D and T2D), focusing on β-cell biology, insulin resistance and diabetes complications8. MiR-98 expression is up-regulated in kidney in response to diabetes complications in mouse in and down-regulated in muscle in type 2 diabetes with insulin resistance in human9,10. Also, miR-98 is found to participate in embryo implantation during early pregnancy11. Although miR-98 is involved in T2DM and early pregnancy in the available literature9,10,11, the relationship between miR-98 and GDM remains unknown, and roles of miR-98 in GDM are still unclear.
In this study, we report the relationship between miR-98 and GDM and investigate the functional roles of miR-98 in GDM. Additionally, we also test the possible molecular mechanisms in which miR-98 is implicated.
Results
Up-regulation of miR-98 expression in placental tissues from patients with GDM
The distribution of miR-98 in GDM placental tissues and control tissues was determined by in situ hybridization (Fig. 1A). Old age is the risk factor for GDM. Therefore, the GDM tissues were grouped into four groups according to maternal age, including under 25 years old (Y < 25), 25~30 years old (Y25~30), 30~35 years old (Y30~35), over 35 years old (Y > 35). In different age groups, strong signals of miR-98 were found in GDM placental tissues. While in control group, there was only weak expression of miR-98 in placenta. The image analysis of optical densities showed that the mean optical densities (MODs) of positive signals were increased in GDM tissues, especially in the groups of Y25~30 (P < 0.01), Y30~35 (P < 0.01) and Y > 35 (P < 0.01), when compared with the corresponding controls.
Figure 1. Up-regulation of miR-98 in the placental tissues from patients with GDM.

The expression of miR-98 in the placental tissues from patients with GDM and normal pregnant women was detected by in situ hybridization using DIG-labeled LNA probes specific to miR-98 (A). The stain was developed with BCIP/NBT. Black arrows indicate hybridization signals and the positive signals of miR-98 are blue. The scale bar indicates a distance of 50 μm. The histogram represents the MODs of positive signals of miR-98 in placentas. The expression of miR-98 in the placental tissues was also detected by qRT-PCR (B). U6 serves as an internal reference to normalize the experimental error. *P < 0.05; **P < 0.01.
qRT-PCR was used to further confirm the results of in situ hybridization (Fig. 1B). The expression levels of miR-98 in GDM group were markedly higher than that in control group in Y30~35 (P < 0.05) and Y > 35 (P < 0.05). The overall expression trend of miR-98 in all placental tissues of GDM group was consistent with that in Y30~35 and Y > 35 age groups and significantly increased compared with control group (P < 0.05). These results imply that miR-98 is sensitive to occurrence of GDM.
Demethylation reduces the expression of miR-98
In order to analyze the effect of demethylation on the expression of miR-98, DNA methylation inhibitor 5-aza was used to treat JEG-3 cells, and then the expression level of miR-98 was detected by qRT-PCR (Fig. 2A). The results indicated that 0.1, 0.5, 1 μM 5-aza inhibited the expression of miR-98 in a dose-dependent manner. However, only 1 μM 5-aza significantly inhibited miR-98 expression (P < 0.01). These results suggest that miR-98 may be related with DNA methylation.
Figure 2. MiR-98 and DNA methylation level in the placental tissues from patients with GDM.

The miR-98 level was detected in JEG-3 cells treated with 5-aza by qRT-PCR (A). U6 serves as an internal reference to normalize the experimental error. Global DNA methylation level in miR-98 mimic or inhibitor-treated cells with or without 5-aza was detected by immunohistochemistry and estimated by the content of global 5-meC (B). The stain was developed with DAB and cell nuclei were stained with haematoxylin. Brown indicates the positive signals. Scale bar = 50 μm. The histogram represents the ratio of 5-meC positive cells (%). *P < 0.05; **P < 0.01.
MiR-98 enhances the DNA methylation level in vitro
To verify the relationship between miR-98 and DNA methylation, the global DNA methylation level in JEG-3 cells transfected by miR-98 mimic or inhibitor was estimated by the content of 5-methylcytosine (5-MeC) detected by cells immunohistochemistry (Fig. 2B). The ratio of 5-MeC positive cells was significantly increased in cells transfected by miR-98 mimic compared with mimic control (P < 0.05). MiR-98 inhibitor markedly reduced the ratio of 5-MeC positive cells (P < 0.05). DNA methylation inhibitor 5-aza can reduce the ratio of 5-MeC positive cells. However, transfection of miR-98 mimic into 5-aza-treated cells significantly enhanced the ratio of 5-MeC positive cells (P < 0.01). These results show that miR-98 can positively regulate the global DNA methylation level.
Mecp2 is a direct target of miR-98
To figure out the possible molecular mechanisms by which miR-98 may perform in DNA methylation, its target genes were researched. An online search of miR-98 targets by Targetscan, PicTar and miRanda provided a large number of putative miRNA targets. Among them, we focused on Mecp2 for the following reasons: (1) Targetscan, PicTar and miRanda prediction showed that there was a miR-98 responsive element in 3′-UTR of Mecp2, which is a highly conserved domain among different species (Fig. 3A). (2) It was reported that Mecp2 was associated with methylation12. (3) In this study, we found that the mRNA and protein level of Mecp2 was significantly up-regulated when miR-98 was down-regulated in 5-aza-treated cells (Fig. 3B,C). Thus, an evident inverse relationship is showed between miR-98 and Mecp2 expression levels.
Figure 3. The prediction of the miR-98 target gene.

MiR-98 binding site in the 3′-UTR region of Mecp2 was conserved in cross-species (A). 5-aza treatment enhanced Mecp2 mRNA level and protein level detected by qRT-PCR (B) and western blot (C). Gapdh and β-ACTIN serve as an internal reference for qRT-PCR and western blot, respectively. For western blot, the gels had been run under the same experimental conditions. The bands were analyzed using Quantity One analyzing system (Bio-Rad, Hercules, CA, USA). The histogram represents the optical densities of the signals quantified by densitometric analysis and expressed as MECP2 intensity/β-ACTIN intensity to normalize for gel loading and transfer. *P < 0.05; **P < 0.01.
To validate whether Mecp2 was the indeed target gene of miR-98 or not, a human Mecp2 3′-UTR fragment containing wild-type was cloned into the downstream of the firefly luciferase reporter gene in the pGL3 control vector (designated as Mecp2-pGL3) for the dual-luciferase assay (Fig. 4A). HEK-293T cells were co-transfected with Mecp2-pGL3 and miR-98 mimic or inhibitor. Compared with the mimic control, the luciferase activity was significantly suppressed by the miR-98 mimic, (P < 0.01). Furthermore, the luciferase activity was significantly enhanced by the miR-98 inhibitor compared with inhibitor control (P < 0.01; Fig. 5B). These results indicate that miR-98 affects the binding of miR-98 and 3′-UTR of Mecp2, leading to the change of Mecp2 translation.
Figure 4. The confirmation of the miR-98 target.

(A) The effects of miR-98 expression dysregulation on Mecp2 translation. HEK-293T cells were co-transfected with mimic control, miR-98 mimic, inhibitor control or miR-98 inhibitor and Mecp2-pGL3 for dual-luciferase assay. (B) Mutation analysis of the miR-98 binding site. Mutating the binding site of miR-98 in the 3′-UTR of Mecp2 was used as control (Mecp2-pGL3-Mut). PRL-TK containing Renilla luciferase was co-transfected for data normalization.*P < 0.05, **P < 0.01.
Figure 5. MiR-98 regulates Mecp2 expression in in vitro cell experiment.
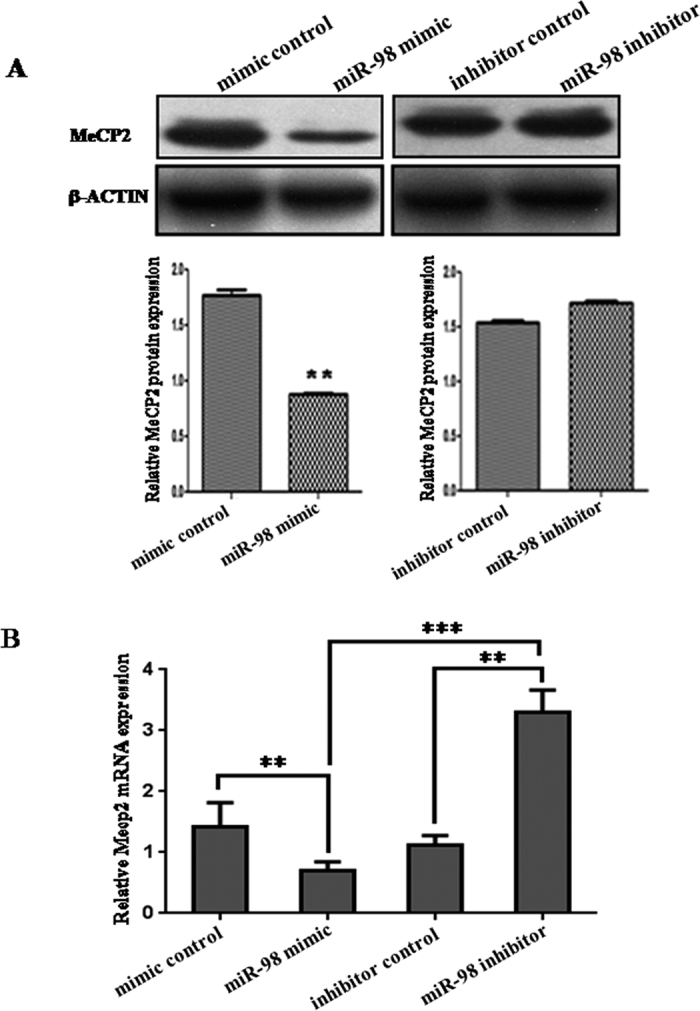
(A) MECP2 protein level in miR-98 mimic or inhibitor-treated JEG-3 cells was detected by western blot. The bands were analyzed using the Quantity One analyzing system (Bio-Rad Laboratory inc.). The gels had been run under the same experimental conditions. The expression of β-ACTIN serves as an internal control. (B) The mRNA level of Mecp2 in miR-98 mimic and inhibitor-treated JEG-3 cells was detected by qRT-PCR. Gapdh serves as an internal reference. **P < 0.01. ***P < 0.001.
To further confirm the binding site, base mutation of miR-98 targeting site in 3′-UTR of Mecp2 (designated as MeCP2-pGL3-Mut) was also conducted. The histogram in Fig. 4B showed that the enzyme activity was significantly reduced in cells co-transfected with miR-98 mimic and Mecp2-pGL3 compared with Mecp2-pGL3-Mut (P < 0.05). These data indicates that miR-98 may suppress Mecp2 expression through binding to miR-98 responsive element in the 3′-UTR of Mecp2, and Mecp2 may be a direct target of miR-98.
MiR-98 regulates endogenous Mecp2 expression in vitro
Although Mecp2 was identified as a target gene for miR-98, it was unknown whether miR-98 could regulate endogenous Mecp2 expression. To verify the endogenous effects of miR-98 expression dysregulation on Mecp2, JEG-3 cells were transfected with miR-98 mimic or inhibitor. Compared with corresponding control, the level of MECP2 protein was significantly down-regulated by miR-98 mimic and up-regulated by miR-98 inhibitor (Fig. 5A). Additionally, the mRNA level of Mecp2 detected by qRT-PCR was significantly decreased by miR-98 mimic (P < 0.01) and increased by miR-98 inhibitor (P < 0.01). Compared with miR-98 mimic, miR-98 inhibitor significantly enhanced Mecp2 mRNA level (P < 0.001; Fig. 5B). These results show that the mRNA and protein levels of endogenous Mecp2 are regulated by miR-98.
Mecp2 was down-regulated in GDM placental tissues
Then it was unclear whether miR-98 executed its effects by targeting Mecp2 in GDM in vivo? To verify the phenomenon, we tested the MECP2 expression in the placental tissues from patients with GDM by immunohistochemistry using rabbit anti-MECP2 antibody (Fig. 6A). The MODs of MECP2-positive signals were significantly decreased in the placental tissues from GDM patients in Y30–35 and Y > 35 age group compared with the corresponding controls (P < 0.05), which was further confirmed by qRT-PCR (Fig. 6B). All these facts show that Mecp2 expression level is down-regulated in GDM tissues, while miR-98 expression level is visibly increased in GDM placental tissues, suggesting that miR-98 may execute its effects by targeting Mecp2 in GDM in vivo.
Figure 6. The expression of Mecp2 in the placental tissues from patients with GDM.

(A) The protein level of MECP2 in the placental tissues from patients with GDM and normal pregnant women was detected by immunohistochemistry. The stain was developed with DAB and cell nuclei were stained with haematoxylin. Black arrow indicated positive signals and the positive signals of MECP2 are brown. The histogram represents the MODs of positive signals of MECP2 in placentas. Scale bar = 50 μm. (B) qRT-PCR was used to detected the mRNA level of Mecp2 in the placental tissues from patients with GDM and normal pregnant women. Gapdh serves as an internal reference. *P < 0.05.
MiR-98 and Mecp2 regulate the protein expression of DNA methyltransferase
Because miR-98 was involved in DNA methylation, we wondered whether miR-98 and its target gene would affect the protein expression of DNA methyltransferase or not (Fig. 7). MiR-98 mimic increased DNMT1 protein level (P < 0.05) and Mecp2 expression vector decreased DNMT1 protein level (P < 0.05) compared with corresponding control. DNMT1 protein level had a downward tendency in cells treated by miR-98 inhibitor and a obvious increase by Mecp2 siRNA (P < 0.05). However, miR-98 and Mecp2 had no significant effects on the protein levels of DNMT3a and DNMT3B.
Figure 7. The protein expression of DNA methyltransferase.

The effects of miR-98 (A) and Mecp2 (B) on the protein levels of DNA methyltransferase (DNMT1, DNMT3a and DNMT3b) were detected by western blot. The gels had been run under the same experimental conditions. The expression of β-ACTIN serves as an internal control to normalize for gel loading and transfer. The bands were analyzed using Quantity One analyzing system (Bio-Rad, Hercules, CA, USA). The protein level was represented as the relative ratio of the DNMT1, DNMT3a and DNMT3b signals vs β-ACTIN. *P < 0.05.
MiR-98 indirectly regulates the expression of Trpc3 by targeting Mecp2
Previous studies have identified that canonical transient receptor potential 3 (Trpc3) and secreted frizzled-related protein 4 (Sfrp4), which play roles in T2DM, are the target genes of Mecp213,14. Then it was unknown whether miR-98 affected the expression of Mecp2 target genes? Therefore, the expression of Trpc3 and Sfrp4 was detected by qRT-PCR (Fig. 8A,B). MiR-98 mimic significantly reduced the mRNA level of Trpc3 (P < 0.01). Mecp2 expression vector pMSCV-Mecp2 significantly increased Trpc3 expression (P < 0.01). When cells were co-transfected with miR-98 mimic and pMSCV-Mecp2, the mRNA level of Trpc3 were higher than transfection of miR-98 mimic (P < 0.01) and lower than transfection of pMSCV-Mecp2 (P < 0.01), implying that Trpc3 expression suppressed by miR-98 over-expression was partially rehabilitated by Mecp2 up-regulation. Additionally, miR-98 inhibitor significantly promoted the expression of Trpc3 (P < 0.01). Mecp2 siRNA significantly inhibited the expression of Trpc3 (P < 0.01). However, the mRNA level of Trpc3 in cells co-transfected with miR-98 inhibitor and Mecp2 siRNA was significantly weakened compared with miR-98 inhibitor alone (P < 0.01) and strengthened compared with Mecp2 siRNA alone (P < 0.01), displaying that miR-98 low expression-mediated the up-regulation of Trpc3 was partially attenuated by Mecp2 knockdown (Fig. 8A). MiR-98 mimic down-regulated Sfrp4 mRNA level, while miR-98 inhibitor, Mecp2 expression vector, Mecp2 siRNA also inhibited Sfrp4 expression. So, there was no distinct trend exhibiting the effects of miR-98 and Mecp2 on the expression of Sfrp4 (Fig. 8B).
Figure 8. The miR-98 indirectly regulates Trpc3 expression by Mecp2.

qRT-PCR analysis of Trpc3 (A) and Sfrp4 (B) mRNA levels and western blot analysis of TRPC3 protein level (C) in miR-98 mimic or miR-98 inhibitor-treated cells with Mecp2 expression vector or Mecp2 siRNA. Gapdh and β-ACTIN serve as an internal reference for qRT-PCR and western blot, respectively. *P < 0.05, **P < 0.01.
In order to further confirm that miR-98 affected the expression of Trpc3, the protein level of TRPC3 was detected by western blot (Fig. 8C). Comparable results were obtained by western blot and qRT-PCR. MiR-98 mimic significantly reduced the protein level of TRPC3 (P < 0.05), and pMSCV-Mecp2 significantly increased TRPC3 protein level (P < 0.05). When cells were co-transfected with miR-98 mimic and pMSCV-Mecp2, the protein level of TRPC3 was higher than transfection of miR-98 mimic lone (P < 0.05) and close to the control. MiR-98 inhibitor significantly enhanced TRPC3 protein level (P < 0.05). TRPC3 protein level had a downward trend in cells treated by Mecp2 siRNA. However, the protein level of TRPC3 in cells co-transfected with miR-98 inhibitor and Mecp2 siRNA was significantly weaker than transfected with miR-98 inhibitor alone (P < 0.05) and stronger than transfected with Mecp2 siRNA alone (P < 0.05). Taken together, these results indicate that miR-98 executes functions in GDM partially by targeting Mecp2-Trpc3 pathway.
Trpc3 expression in GDM placental tissues
In order to analyze Trpc3 expression in vivo, we tested the Trpc3 expression in the placental tissues from patients with GDM by qRT-PCR and western blot (Fig. 9). The Trpc3 mRNA level was significantly decreased in the placental tissues from GDM patients in Y > 35 age group compared with the normal control (P < 0.05; Fig. 9A). The TRPC3 protein level was significantly decreased in the placental tissues from GDM patients in Y30–35 and Y > 35 age group compared with the corresponding controls (P < 0.05; Fig. 9B).
Figure 9. Trpc3 expression in GDM placental tissues.

The Trpc3 expression in the placental tissues from patients with GDM was detected by qRT-PCR (A) and western blot (B). The gray histograms of B1–B4 represent the protein level of MECP2 in placental tissues in Y < 25, Y25~30, Y30~35 and Y > 35 age group, respectively. Gapdh and β-ACTIN serve as an internal reference for qRT-PCR and western blot, respectively. *P < 0.05.
Discussion
In this study, we found that miR-98 was significantly up-regulated in placental tissues from GDM patients compared with that in normal controls. The trend of miR-98 in four groups, including Y < 25, Y25~30, Y30~35 and Y > 35, is similar, suggesting that up-regulation of miR-98 may be associated with the occurrence of GDM.
Interestingly, DNA methylation inhibitor 5-azacytidine could decrease the miR-98 level in human choriocarcinoma cell line JEG-3. Emerging evidence indicates that GDM has epigenetic effects on genes through DNA methylation, with consequences on fetal growth and development15. So we speculate that miR-98 may be able to regulate DNA mathylation in GDM. In vitro cell experiment was used to confirm the relationship between miR-98 and DNA methylation, and found that over-expression of miR-98 increased global DNA methylational level, and partially recovered the inhibition of DNA methylation induced by 5-azacytidine. These facts show that enforced miR-98 expression promotes global DNA methylation level.
It is generally accepted viewpoint that miRNAs function via regulating the expression of their downstream target genes. An online search of miR-98 targets by Targetscan, PicTar and miRanda found that there was a miR-98 responsive element in 3′-UTR of Mecp2, which was a highly conserved domain among different species. When miR-98 mimic or inhibitor was co-transfected with the recombinant vector Mecp2-pGL3, luciferase activity was reduced by the miR-98 mimic and enhanced by the miR-98 inhibitor, suggesting that Mecp2 may be the target gene of miR-98. Mutation experiment further confirmed that the binding site in the 3′-UTR of Mecp2 was specific for miR-98. Additionally, over-expression of miR-98 reduced the protein and mRNA level of Mecp2 and knockdown of miR-98 enhanced the protein and mRNA level of Mecp2. These data further confirm that miR-98 not only directly targets Mecp2, but also regulates the endogenous MECP2 expression.
Previous studies have identified that Trpc3 and Sfrp4 are Mecp2 target gene respectively13,14. Trpc3 is involved in vasoconstriction and regulation of blood pressure in metabolic syndrome16. Sfrp4 reduces insulin secretion and may be a potential biomarker for islet dysfunction in T2D17. We found that miR-98 over-expression suppressed Trpc3 expression, which was partially rehabilitated by up-regulation of Mecp2. MiR-98 knockdown promoted Trpc3 expression, which was partially attenuated by depleting Mecp2 expression. There was no significant effect of miR-98 on Sfrp4 expression. A study analyzing insulin-mediated glucose uptake shows that Trpc3 interacts functionally and physically with GLUT4, and Ca(2+) influx and modulates insulin-mediated glucose uptake18. Therefore, miR-98 may indirectly regulate glucose up-take through targeting Mecp2-Trpc3 pathway.
To our knowledge, this is the first study to examine the relationship between the presence of maternal GDM and miR-98. Our findings demonstrate that miR-98 and its pathways in placental tissues are associated with GDM. MiR-98 not only directly targets Mecp2, but also indirectly regulates the target gene of Mecp2. These results imply that enhanced miR-98 expression may take part in the occurrence of GDM by Mecp2-Trpc3 pathway.
Materials and Methods
Patients samples and tissue preparation
Human placentas from 37 weeks~40 weeks (37w~40w) pregnancy were obtained from Maternal and Child Health Hospital, Haidian District, Beijing, from March 2012 to May 2013, including 193 GDM patients and 202 normal pregnant women as control. Basic characteristics of the population were displayed in Table 1. The investigation was conducted according to the principles expressed in the Declaration of Helsinki. The study was approved by Ethics Committee of Research Institute for Family Planning (2011-08) and informed consent was obtained from all participants. Placentas were fixed in 4% paraformaldehyde (PFA) diluted in 0.1 M phosphate-buffered saline (PBS) for immunohistochemical and in situ hybridization studies.
Table 1. Basic characteristics of study population according to GDM.
| Control, n = 202 | GDM, n = 193 | |||||
|---|---|---|---|---|---|---|
| Mean ± SD | 95% Confidence Interval for Mean | Mean ± SD | 95% Confidence Interval for Mean | |||
| Lower Bound | Upper Bound | Lower Bound | Upper Bound | |||
| Age (years) | 29.68 ± 3.66 | 29.17 | 30.19 | 30.93 ± 3.45 | 30.45 | 31.42 |
| Height (cm) | 161.69 ± 4.95 | 161.01 | 162.38 | 161.59 ± 4.81 | 160.91 | 162.27 |
| Body Weight (Kg) | 55.32 ± 8.87 | 54.09 | 56.55 | 59.82 ± 8.87** | 58.57 | 61.07 |
| BMI (Kg/m2) | 21.06 ± 2.90 | 20.66 | 21.46 | 22.97 ± 3.09** | 22.53 | 23.4 |
| Basic systolic (mmHg) | 114.99 ± 10.82 | 113.48 | 116.49 | 117.23 ± 11.34* | 115.63 | 118.83 |
| Foundation diastolic (mmHg) | 70.96 ± 9.04 | 69.71 | 72.21 | 72.90 ± 8.93 | 71.64 | 74.16 |
| Glu first detected | 4.52 ± 0.38 | 4.47 | 4.58 | 4.79 ± 0.58* | 4.7 | 4.87 |
| Fasting plasma glucose | 4.53 ± 0.37 | 4.48 | 4.58 | 5.40 ± 0.72** | 5.29 | 5.5 |
| OGTT-1 h | 7.54 ± 1.24 | 7.37 | 7.72 | 10.99 ± 1.44** | 10.78 | 11.2 |
| OGTT-2 h | 6.26 ± 0.97 | 6.12 | 6.39 | 9.07 ± 1.31** | 8.88 | 9.26 |
| Pregnancy Days | 274.41 ± 8.03 | 273.29 | 275.52 | 272.22 ± 6.39 | 271.32 | 273.12 |
| Newborn baby’s Weight | 3391.73 ± 433.08 | 3331.65 | 3451.82 | 3435.54 ± 475.04 | 3368.1 | 3502.99 |
P < 0.05; P < 0.01.
Plasmid construction and transfection
The Mecp2 3′-UTR and Mecp2 3′-UTR-mutant sequences were amplified by PCR from human genomic DNA using the primers in Table 2. After being double digested with SpeI and XbaI, the PCR products were cloned into pGL3 control vector (Invitrogen, Carlsbad, CA, USA). The coding region of Mecp2 sequence was amplified by RT-PCR from total mRNA of human JEG-3 cells using the primers in Table 2. After being double digested with BamHI and EcoRI, the PCR product was cloned into PMSCV-puro vector, designated as PMSCV-Mecp2. All the constructs were verified by DNA sequencing. Specific siRNAs for scramble and Mecp2 were synthesized as a duplex with the following sequence: scramble siRNA, 5′-CUUCUUAGGUGGUUUCUGC-dTdT-3′, Mecp2 siRNA, 5′-GCAGAAACCACCUAAGAAG- dTdT-3′.
Table 2. Primer sequences.
| Gene Name | Primer Sequence | Accession Number | Size and Location | Application |
|---|---|---|---|---|
| MeCP2 3′-UTR | Forward/SpeI:5′-GACTAGTCCCACCCTTGTTCCAGTTGT-3′ | NM_004992.3 | 365 bp (5105–5469) | PCR |
| Reverse/XbaI:5′-GCTCTAGATGCCACCCAACAGAAGATT-3′ | ||||
| MeCP2 3′-UTR (mutation) | Forward:5′-GTTCCAGTTGTTAGTTACTATATCCTCTCCTGACAATACT-3′ | NM_004992.3 | 365 bp (5105–5469) | PCR |
| Reverse:5′-TATAGTAACTAACAACTGGAACAAGGGTGGG-3′ | ||||
| MeCP2 (coding region) | Forward/kozak-BamHI: 5′-CGCGGATCGCCACCATGGTAGCTGGGATGTTAGGGCT-3′ | NM_004992.3 | 1461 bp (227–1687) | RT-PCR |
| Reverse/EcoRI: 5′-CCGGAATTCTCAGCTAACTCTCTCGGTCACGG-3′ | ||||
| MeCP2 | Forward: 5′-TGCAAAGAGGAGAAGATGCCCAGA-3′ | NM_004992.3 | 169 bp (1463–1631) | qRT-PCR |
| Reverse: 5′-GCCTTGGCATGGAGGATGAAACAA-3′ | ||||
| Trpc3 | Forward: 5′-CATTCCTGGCCATTGGCTACT-3′ | NM_001130698.1 | 117 bp (1375–1491 bp) | qRT-PCR |
| Reverse: 5′-GCAGACCCAGGAAGATGATGAA-3′ | ||||
| Gapdh | Forward: 5′-TGGTATCGTGGAAGGACTCA-3′ | NM_002046.4 | 129 bp (678–806) | qRT-PCR |
| Reverse: 5′-GTAGAGGCAGGGATGATGTTC-3′ |
The miR-98 mimic, mimic control, miR-98 inhibitor, inhibitor control, scramble siRNA control and Mecp2 siRNA were synthesized by GenePharma (GenePharma Co., Ltd, Shanghai, China), and transfected into cells by the lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacture’s instruction.
In situ hybridization
An in situ hybridization procedure was performed using the protocol developed in our laboratory. Placentas were arranged into tissue microarray and cut to a thickness of 6 μm. After deparaffinized in xylene, rehydrated in descending ethanol series, refixed in 4% PFA, deproteinized with 0.2 M HCl and digested with 20 μg/ml proteinase K (Tiangen, China), the sections were prehybridized with hybridization buffer (Roche, Mannheim, Germany) at 40 °C for 1 h and then hybridized with digoxigenin (DIG)-labeled LNA-MiR-98 probe (LNA-MiR-98 sequence: 5′–DIG-aAcaaTaCAaCttaCtAcCtCa-3′) overnight at 40 °C. After washed by descending saline-sodium citrate (SSC) and blocked by blocking buffer containing 5% bovine serum albumin (BSA), the sections were incubated with alkaline phosphatase (AP) labelled anti-DIG-antibody (Roche, Mannheim, Germany, 1:250) overnight at 4 °C, and developed with bromochloroindolyl phos-phate/nitro blue tetrazolium (BCIP/NBT; Promega, Madison, WI, USA). BCIP and NBT are the common substrate of alkaline phosphatase. Catalyzed by alkaline phosphatase, BCIP product will be hydrolyzed to produce a strong reactivity, and then the product will react with NBT and form an insoluble blue NBT-formazan. The probe was replaced by DIG-labeled LNA-scrambled probe (LNA-scrambled sequences: 5′-DIG-caTtaAtgTcGgaCaaCtcAat-3′) as negative control. Samples were viewed by Nikon TE 2000-U microscope (NIKON, Tokyo, Japan).
Immunohistochemistry
Sections of the tissues microarray were deparaffinized in xylene and rehydrated in descending ethanol series. Antigen retrieval was accomplished with 1 N HCl. Slides were incubated with rabbit anti-MECP2 polyclonal antibody (GeneTex, USA, 1:500) and mouse anti-5 methylcytosine monoclonal antibody (Santa Cruz, USA, 1:100), respectively, then incubated with HRP-conjugated goat anti-rabbit IgG and goat anti-mouse IgG (Jackson Immunoresearch Laboratories, West Grove, PA, USA). The antibody stains were developed by addition of diaminobenzidine (DAB; Sigma-Aldrich, St. Louis, MO, USA) and cell nuclei were stained with haematoxylin (Sigma-Aldrich, St. Louis, MO, USA). The sections were incubated with normal goat serum as negative control. Samples were viewed using under Nikon TE 2000-U microscope (NIKON, Tokyo, Japan). The MODs of positive signals were determined by NIS-Elements BR Image processing and analysis system (NIKON, Japan).
Quantitative reverse-transcriptase polymerase chain reaction (qRT-PCR)
Total RNAs from tissues and cells were extracted using Trizol (Invitrogen, Carlsbad, CA, USA) accordint to the manufacturer’s protocols. 1 μg of total RNA was subjected to reverse transcription of mRNAs using dT18 as primer and reverse transcription kit (TakaRa Biotechnology (Dalian) Co., Ltd. Dalian, Liaoning, China) to generate total cDNA. Then the mRNA quantitative PCR was carried using primers in Table 2 and FastStart Universal SYBR Green Master (Invitrogen, Carlsbad, CA, USA) using StepOneTM Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). Gapdh was used for normalization. The reverse transcription and quantitative PCR of miRNA were carried using special probes for miR-98 and U6 (Applied Biosystems, Foster City, CA, USA) with TaqMan MicroRNA Reverse Transcription Kit and Universal PCR Master Mix (Applied Biosystems, Foster City, CA, USA). The quantification was normalized to an endogenous control U6. Each sample in each group was detected in triplicate. The experiment was repeated at least three times.
Western blot analysis
Extracted protein was boiled in SDS/β-mercaptoethanol sample buffer, and 50 μg protein was run on 5–12% gradient polyacrylamide gel and transferred to PVDF membranes (Amersham, St Albans, Herts, UK). Membranes were incubated with rabbit anti-MECP2 polyclonal antibody (GeneTex, USA, 1:1000), mouse anti-β-ACTIN monoclonal antibody (Abcam, Cambridge, MA USA, 1:1000), rabbit anti-DNMT1, DNMT3a and DNMT3b polyclonal antibody (1:1000, SantCruz Biotechnology Inc., CA, USA) overnight at 4 °C. After that, the membranes were incubated with horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG or goat anti-mouse IgG (Jackson Immunoresearch Laboratories, Inc., West Grove, PA, USA, 1:10, 000) at room temperature for 1 h. ECL detection reagents (Millipore, Billerica, MA, USA) were added on the membranes for 1 min and were immediately exposed to X-ray film (Kodak, USA). The β-actin signal was used as a loading control. The experiment has been repeated at least three times. The bands were analyzed using Quantity One analyzing system (Bio-Rad, Hercules, CA, USA). The protein level was represented as the relative ratio of the MECP2, DNMT1, DNMT3a and DNMT3b signals vs the housekeeping gene (β-ACTIN).
Dual-luciferase activity assay
To generate 3′-UTR luciferase reporter, partial sequence of the 3′-UTR from Mecp2 were cloned into the downstream of the firefly luciferase gene in pGL3-Control Vector (Promega, Madison, WI, USA) using primers in Table 2. Mutating miR-98 target site in the 3′-UTR of Mecp2 was used as control. PRL-TK containing Renilla luciferase was co-transfected for data normalization. For luciferase reporter assay, HEK-293T cells were seeded in 48-well plates and allowed to attach overnight, then transfected using lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). Two days later, cells were harvested and assayed with the dual-luciferase assay (Promega, Madison, WI, USA). Each treatment was performed in triplicate in three independent experiments. The results were expressed as relative luciferase activity (Firefly LUC/Renilla LUC).
Statistical analyses
All statistical analysis were performed using the SPSS 13.0 statistical software package. Data are presented as mean ± SEM from at least three independent experiments. Multiple group comparisons were performed using one-way analysis of variance (ANOVA), and two group comparisons were performed using T-test. Differences were considered statistically significant at P < 0.05.
Additional Information
How to cite this article: Cao, J.-L. et al. Up-regulation of miR-98 and unravelling regulatory mechanisms in gestational diabetes mellitus. Sci. Rep. 6, 32268; doi: 10.1038/srep32268 (2016).
Acknowledgments
This work was funded by grants from National Natural Science Foundation of China (No. 81370720). With greatest thanks to my lab-mates Kun-Lun Yin, Xi-Meng Zhao and Wen-Hui Zhang for helping to embed the tissues with paraffin. Special thanks to Jing-Tao Mao in Longmaida company for helping to make tissues array and slides commercially.
Footnotes
Author Contributions J.-L.C. performed the experiments and drafted the manuscript. S.T. collected clinical tissues and patients information. L.Z. and X.-D.L. were responsible for qRT-PCR. X.-Q.W., Y.H. and J.L. were responsible for samples collecting and recording. X.S. and Y.L. were responsible for tissue sections. X.M. was responsible for institute management. H.-F.X. designed the research and revised the manuscript.
References
- Nielsen K. K., de Courten M. & Kapur A. Health system and societal barriers for gestational diabetes mellitus (GDM) services-lessons from World Diabetes Foundation supported GDM projects. BMC international health and human rights. 12, 33 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Society for Obstetricians and Gynecologists of Canada (SOGC) clinical practice guidelines. Screening for gestational diabetes mellitus. J Obstet Gynecol Can. 24, 894–903 (2002).12417905 [Google Scholar]
- Reece E. A., Leguizamón G. & Wiznitzer A. Gestational diabetes. the need for a common ground. Lancet. 373, 1789–1797 (2009). [DOI] [PubMed] [Google Scholar]
- Torloni M. R., Betrán A. P., Horta B. L., Nakamura M. U., Atallah A. N. et al. Prepregnancy BMI and the risk of gestational diabetes: a systematic review of the literature with meta-analysis. Obes Rev. 10, 194–203 (2009). [DOI] [PubMed] [Google Scholar]
- MacNeill S., Dodds L., Hamilton D. C., Armson B. A. & VandenHof M. Rates and risk factors for recurrence of gestational diabetes. Diabetes Care. 24, 659–662 (2001). [DOI] [PubMed] [Google Scholar]
- Major C. A., deVeciana M., Weeks J. & Morgan M. A. Recurrence of gestational diabetes: who is at risk? Am J Obstet Gynecol. 179, 1038–1042 (1998). [DOI] [PubMed] [Google Scholar]
- Chu C., Gui Y. H., Ren Y. Y. & Shi L. Y. The impacts of maternal gestational diabetes mellitus (GDM) on fetal hearts. Biomed Environ Sci. 25, 15–22 (2012). [DOI] [PubMed] [Google Scholar]
- Fernandez-Valverde S. L., Taft R. J. & Mattick J. S. MicroRNAs in beta-cell biology, insulin resistance, diabetes and its complications. Diabetes. 60, 1825–1831 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher I. J., Scheele C., Keller P., Nielsen A. R., Remenyi J. et al. Integration of microRNA changes in vivo identifies novel molecular features of muscle insulin resistance in type 2 diabetes. Genome medicine. 2, 9 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie S., Xie N., Li Y., Wang P., Zhang C. et al. Upregulation of TRB2 induced by miR-98 in the early lesions of large artery of type-2 diabetic rat. Molecular and cellular biochemistry 361, 305–314 (2012). [DOI] [PubMed] [Google Scholar]
- Xia H. F., Jin X. H., Cao Z. F., Shi T. & Ma X. MiR-98 is involved in rat embryo implantation by targeting Bcl-xl. FEBS Lett. 588, 574–583 (2014). [DOI] [PubMed] [Google Scholar]
- Fuks F., Hurd P. J., Wolf D., Nan X., Bird A. P. et al. The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J Biol Chem. 278, 4035–4040 (2003). [DOI] [PubMed] [Google Scholar]
- Li W., Calfa G., Larimore J. & Pozzo-Miller L. Activity-dependent BDNF release and TRPC signaling is impaired in hippocampal neurons of Mecp2 mutant mice. Proceedings of the National Academy of Sciences of the United States of America. 109, 17087–17092 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao C. G., Huang C., Huang Y., Yang Y. Y., He X. et al. MeCP2 modulates the canonical Wnt pathway activation by targeting SFRP4 in rheumatoid arthritis fibroblast-like synoviocytes in rats. Cellular signalling. 25, 598–608 (2013). [DOI] [PubMed] [Google Scholar]
- Ruchat S. M., Houde A. A., Voisin G., St-Pierre J., Perron P. et al. Gestational diabetes mellitus epigenetically affects genes predominantly involved in metabolic diseases. Epigenetics: official journal of the DNA Methylation Society. 8, 935–943 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D., Zhu Z. & Tepel M. The role of transient receptor potential channels in metabolic syndrome. Hypertens Res. 31, 1989–1995 (2008). [DOI] [PubMed] [Google Scholar]
- Mahdi T., Hänzelmann S., Salehi A., Muhammed S. J., Reinbothe T. M. et al. Secreted frizzled-related protein 4 reduces insulin secretion and is overexpressed in type 2diabetes. Cell Metab. 16, 625–633 (2012). [DOI] [PubMed] [Google Scholar]
- Lanner J. T., Bruton J. D., Assefaw-Redda Y., Andronache Z., Zhang S. J. et al. Knockdown of TRPC3 with siRNA coupled to carbon nanotubes results in decreased insulin-mediated glucose uptake in adult skeletal muscle cells. FASEB journal: official publication of the Federation of American Societies for Experimental Biology 23, 1728–1738 (2009). [DOI] [PubMed] [Google Scholar]