

Abstract
Microglia are the resident immune cells in the spinal cord and brain. Mounting evidence suggests that activation of microglia plays an important role in the pathogenesis of chronic pain, including chronic orofacial pain. In particular, microglia contribute to the transition from acute pain to chronic pain, as inhibition of microglial signaling reduces pathologic pain after inflammation, nerve injury, and cancer but not baseline pain. As compared with inflammation, nerve injury induces much more robust morphologic activation of microglia, termed microgliosis, as shown by increased expression of microglial markers, such as CD11b and IBA1. However, microglial signaling inhibitors effectively reduce inflammatory pain and neuropathic pain, arguing against the importance of morphologic activation of microglia in chronic pain sensitization. Importantly, microglia enhance pain states via secretion of proinflammatory and pronociceptive mediators, such as tumor necrosis factor α, interleukins 1β and 18, and brain-derived growth factor. Mechanistically, these mediators have been shown to enhance excitatory synaptic transmission and suppress inhibitory synaptic transmission in the pain circuits. While early studies suggested a predominant role of microglia in the induction of chronic pain, further studies have supported a role of microglia in the maintenance of chronic pain. Intriguingly, recent studies show male-dominant microglial signaling in some neuropathic pain and inflammatory pain states, although both sexes show identical morphologic activation of microglia after nerve injury. In this critical review, we provide evidence to show that caspase 6—a secreted protease that is expressed in primary afferent axonal terminals surrounding microglia—is a robust activator of microglia and induces profound release of tumor necrosis factor α from microglia via activation of p38 MAP kinase. The authors also show that microglial caspase 6/p38 signaling is male dominant in some inflammatory and neuropathic pain conditions. Finally, the authors discuss the relevance of microglial signaling in chronic trigeminal and orofacial pain.
Keywords: orofacial pain, sex-related, spinal cord, trigeminal pain, tumor necrosis factor, microglia
Introduction
The mechanisms underlying chronic pain states, such as inflammatory pain, neuropathic pain, and cancer pain, have been extensively studied in last several decades. Chronic pain is characterized by peripheral sensitization (sensitization of primary sensory neurons) and central sensitization (sensitization of spinal cord and brain neurons; Dubner and Ruda 1992; Ji et al. 2003; Hucho and Levine 2007). Loss of inhibitory synaptic transmission in the pain pathway has been implicated in the transition from acute pain to chronic pain (Moore et al. 2002; Coull et al. 2003). A major advance in the previous decade in pain research is the demonstration of a critical role of glial cells, such as microglial cells and astrocytes, in the pathogenesis of chronic pain (Ren and Dubner 2010, 2016; Ji et al. 2013; Grace et al. 2014). A growing body of evidence indicates that microglial cells play an important role in the development of central sensitization and neuropathic pain via neuron-glia interactions (Tsuda et al. 2003; Tsuda et al. 2005; Ji et al. 2013). Activated microglia can secrete proinflammatory and pronociceptive mediators, such as tumor necrosis factor (TNF), interleukins 1β and 18 (IL-1β and IL-18), and brain-derived growth factor (BDNF), to interact with neurons, thereby modulating synaptic transmission and pain states (Coull et al. 2005; Miyoshi et al. 2008; Berta et al. 2014). Peripheral or dental nerve injury induces marked proliferation and morphologic activation of microglia associated with upregulations of microglial markers (e.g., IBA1, CD11b) in the central nervous system (CNS). However, these morphologic changes are not essential for pain sensitization (Colburn et al. 1997; Sorge et al. 2015; Taves et al. 2015). After nerve injury, the injured sensory neurons produce colony-stimulating factor 1 (CSF1) to induce microglial activation and neuropathic pain (Guan et al. 2016). CSF1 upregulates microglial membrane adaptor protein DAP12, which is required for nerve injury and CSF1-induced upregulation of pain-related microglial genes, and it contributes to mechanical allodynia but not microglial proliferation (Guan et al. 2016). In some inflammatory pain conditions without obvious morphologic changes, spinal microglia can still drive inflammatory pain by secreting the proinflammatory cytokine TNF-α. Microglia have also been shown to be involved with orofacial pain (Fan et al. 2010; Ma et al. 2015); activation of the P2X7 receptor in microglia of trigeminal nucleus facilitates extraterritorial facial pain via TNF-α secretion following transection of the mental nerve, the third branch of the trigeminal nerve (Murasaki et al. 2013). Recent studies have found that the role of microglia in pain also depends on sex (Sorge et al. 2015; Taves et al. 2015).
Clinically, it is well established that chronic pain, such as chronic orofacial pain and temporomandibular disorders, occurs more frequently in women (Slade et al. 2013; Horst et al. 2015), although acute dental pain is not significantly worse in women (Law et al. 2015). However, despite the fact that 70% of pain clinic patients are women and clinical trials must incorporate women into their trial designs, the majority of animal pain studies, including those related to microglia, were exclusively done in males (Mogil 2012). Surprisingly, basal mechanical and thermal pain sensitivity is very similar in animals as function of sex (Callahan et al. 2008). As an early effort to characterize pain processing in both sexes, Mogil’s group found that hyperalgesia resulting from inflammation or nerve injury depends on male but not female Toll-like receptor 4 (TLR4) in the spinal cord (Sorge et al. 2011). Since TLR4 is known to regulate spinal microglial activation and neuropathic pain (Tanga et al. 2005), this study indicated possible sex difference in spinal microglia.
In a follow-up study, Sorge et al. (2015) found equivalent morphologic activation of microglia in the spinal cords of male and female mice in response to peripheral nerve injury, accompanied by similar levels of allodynia. However, spinal inhibition of microglial function with minocycline, P2X4 blocker, or microglial BDNF signaling or by elimination of spinal microglia with a toxin reduced mechanical allodynia in male but not female mice. Interestingly, the researchers also observed male-specific upregulation of P2X4 receptors in the spinal cord after nerve injury. This male-specific response requires testosterone, as minocycline fails to inhibit allodynia in castrated males but reduces allodynia in testosterone-treated females. In contrast, female mice have higher basal numbers of T cells in the blood and increased T-cell marker expression in the spinal cord after injury. Apparently, female rodents switch from microglial cells to T cells for neuropathic pain sensitization.
p38 MAPK Is Required for Pain Sensitization in Males via Microglial Signaling
Another line of evidence to support sex-dependent microglial regulation of pain states came from a study on p38 mitogen-activating kinase MAPK (Taves et al. 2015). Previous studies have shown that activation of p38 MAPK in spinal microglia participates in the generation of inflammatory and neuropathic pain in various rodent models (Jin et al. 2003; Tsuda et al. 2004; Ji and Suter 2007). However, these studies focused on male mice to avoid confounding effects of the estrous cycle of females. We investigated the sex-dependent role of spinal p38 in inflammatory and neuropathic pain using a highly selective p38 inhibitor: skepinone. We found that intrathecal injection of skepinone prevented formalin-induced inflammatory pain in male but not female mice (Taves et al. 2015). Notably, application of skepinone outside the CNS via intraperitoneal or local perineural administration inhibited mechanical allodynia in both sexes of mice in the chronic constriction injury (CCI) model of neuropathic pain. Therefore, it is suggested that this sex dependence is restricted to the spinal cord (Taves et al. 2015).
We also compared nerve injury–induced microglial proliferation and microglial activation markers in both sexes. CCI induced comparable microglial proliferation, microgliosis, and expression of the microglial markers CX3CR1 and IBA-1 in both sexes. However, nerve injury induced spinal p38 phosphorylation (p-p38), a marker of microglia activation in the spinal cord (Ji and Suter 2007), predominantly in male mice. As expected, p-p38 is coexpressed with the microglial marker CX3CR1 in the spinal cord dorsal horn of male transgenic mice expressing CX3CR1-GFP fluoresce protein (Taves et al. 2015). Together, these data suggest that only some changes in microglia (p-p38 but not IBA-1) are sex dependent. Upon activation, microglia can induce spinal cord synaptic plasticity, a driving force for central sensitization and chronic pain (Ji et al. 2003). As expected, CCI induced comparable increases in the frequency of spontaneous excitatory postsynaptic currents (sEPSCs) in lamina IIo interneurons of the spinal cord pain circuit in both sexes. Strikingly, skepinone suppressed CCI-induced sEPSC increases in the nociceptive neurons of spinal cord slices from only males (Taves et al. 2015). Therefore, the sex-specific p38 activation and signaling appear to be confined to the spinal cord in inflammatory and neuropathic pain conditions.
Caspase 6 Contributes to Pain Sensitization in Males via Microglial Signaling
Caspases are a family of intracellular cysteine proteases and are well known for their roles in regulating apoptosis and neurodegeneration. Among different caspases, caspase 6 (CASP6) is of particular interest because it is localized in axons and involved in axonal degeneration (Graham et al. 2011). CASP6 is highly expressed in the neuronal axons of primary sensory neurons (Nikolaev et al. 2009), suggesting a potential role of this caspase in pain control. We recently showed that CASP6 can modulate microglial activation and inflammatory pain hypersensitivity in male mice (Berta et al. 2014).
Immunohistochemistry shows intense CASP6 immunoreactivity in nerve fibers but not neuronal cell bodies and glial cells in the superficial dorsal horn (laminae I-II) of the spinal cord (Berta et al. 2014). Since CASP6 is also expressed in primary sensory neurons in dorsal root ganglia (DRG), it can be transported to the spinal cord central terminal of primary afferents. This notion was supported by an ablation experiment: ablation of the TRPV1 (transient receptor potential subtype V1 ion channel) expressing primary afferents with resiniferatoxin very effectively abolished CASP6 immunostaining in the spinal cord. The result strongly suggests that CASP6 in the spinal cord is derived from C-fiber nociceptive neurons in DRG. As expected, the spinal cord primary afferent terminals coexpress CASP6 and CGRP (calcitonin gene–related peptide), a neuropeptide that is derived only from primary afferents in the spinal cord. Notably, CASP6/CGRP-coexpressing primary afferents exhibit close contacts with microglial cell bodies and processes in the dorsal horn of CX3CR1-GFP mice (Fig. 1a). Furthermore, we have shown the CASP6 levels in cerebrospinal fluid significantly increase after inflammation, suggestive of inflammation-evoked CASP6 secretion (Berta et al. 2014).
Figure 1.

Caspase 6 (CASP6) induces mechanical allodynia via spinal microglial signaling in male mice. (a) Triple staining of CASP6, CGRP, and CX3CR1 (GFP) in the ipsilateral dorsal horn. Note close contacts between CASP6/CGRP-expressing axonal terminals and microglial cell body and processes. Scale bar, 10 µm. (b–d) Recombinant CASP6 (rCASP6) induces tumor necrosis factor α (TNF-α) release in primary microglial cultures via p38 activation. (b) Release of TNF-α and interleukins 1β and 6 (IL-1β and IL-6; ELISA analysis) in microglial culture medium after stimulation of rCASP6 (5 U/mL, 3 h). *P < 0.05, compared with control, n = 4 cultures. (c) Expression of p38 phosphorylation (p-p38) and TNF-α, revealed by Western blot analysis, in microglial cultures after rCASP6 treatment (5 U/mL, 3 h). Note a robust increase in the secreted form of TNF-α (17 kDa). (d) Effects of p38 inhibitor SB203580 (50 µM) on rCASP6-induced TNF-α release in microglial cultures. *P < 0.05, compared with vehicle (1% DMSO), n = 4 cultures. (e–g) Intrathecal injection of rCASP6- or CASP6-activated microglia induces mechanical allodynia via TNF-α secretion. BL, baseline. (e) rCASP6-induced mechanical allodynia (intrathecal [i.t.], 5 U) is reduced by minocycline pretreatment (i.t., 50 µg). *P < 0.05, n = 5 mice. (f) rCASP6-induced mechanical allodynia (i.t., 5 U) is abrogated in Tnfr double-knockout (Tnfr1/2 DKO) mice. *P < 0.05, n = 7 mice. WT, wild type. (g) Spinal (i.t.) injection of rCASP6-stimulated microglia, but not control microglia, induces mechanical allodynia. *P < 0.05, n = 5 to 7 mice. Modified from Berta et al. (2014) with permission from the journal. GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Stimulation of primary cultures of microglia with recombinant CASP6 (rCASP6) elicited a profound and dose-dependent TNF-α release in microglial culture medium. In contrast, rCASP6 had mild effects on IL-6 release and no effects on IL-1β release (Fig. 1b). Western blot analysis revealed that rCASP6 increased the expression of both the membrane-bound TNF-α (precursor, 26 kDa) and the secreted TNF-α (17 kDa), with a much greater increase in secreted TNF-α (Fig. 1c), suggesting an important role of rCASP6 in eliciting TNF-α secretion. Treatment of microglia with inhibitor of p38 (SB203580) suppressed the rCASP6-induced TNF-α release (Fig. 1d). This result confirms an important role of p38 in CASP6-triggered TNF-α release.
Single-cell polymerase chain reaction analysis demonstrated predominant TNF-α expression in spinal cord microglia with limited expression in astrocytes and no expression in neurons in lamina II of spinal cord slices (Berta et al. 2014). Intrathecal injection of rCASP6 elicited rapid and persistent mechanical allodynia in wild-type mice, which was suppressed by intrathecal injection of the microglial inhibitor minocycline and abrogated in Tnfr1/2 double-knockout mice (Fig. 1e, f). Furthermore, spinal injection of rCASP6-activated microglia is sufficient to induce mechanical allodynia (Fig. 1g), which can be transiently reduced by intrathecal injection of the TNF-α neutralizing antibody (Berta et al. 2014). Therefore, rCASP6-activated microglia can cause pain hypersensitivity via TNF-α release. Mechanistically, rCASP6 is sufficient to enhance spinal cord synaptic transmission via TNF-α signaling, and inflammation-induced spinal cord synaptic plasticity can be reduced by CASP6 inhibitor. Minocycline blocked only the rCASP6-induced synaptic plasticity (i.e., increase in sEPSC frequency) but had no effect on basal synaptic transmission (Berta et al. 2014).
The results mentioned above were obtained entirely from male mice. To determine a sex-dependent role of microglia and CASP6, we further compared different involvement of CASP6 in neuropathic pain in male and female mice using both a genetic approach (CASP6 knockout [KO] mice) and a pharmacologic approach (CASP6 inhibitor). Notably, CCI induced marked mechanical allodynia in male and female mice (Fig. 2a, b). However, CCI-induced mechanical allodynia was reduced in only male but not female CASP6-KO mice (Fig. 2a, b). Similarly, intrathecal injections of the CASP6 inhibitor ZVEID, 1 wk after nerve injury, inhibited CCI-induced allodynia in only male but not female mice (Fig. 2c, d). Together, these genetic and pharmacologic approaches suggest that spinal CASP6 predominantly regulates neuropathic pain in male mice.
Figure 2.

Spinal caspase 6 (CASP6) contributes to mechanical allodynia in male mice after nerve injury. (a, b) Chronic constriction injury (CCI)–induced mechanical allodynia in wild-type and CASP6-knockout (CASP6-KO) mice of males (a) and females (b). *P < 0.05, 2-way analysis of variance, followed by post hoc Bonferroni test, n = 5 mice/sex/group. BL, baseline. (c, d) CCI-induced mechanical allodynia in wild-type mice of males (c) and females (b) before and after intrathecal injections (arrows) of CASP6 inhibitor ZVEID (10 or 30 µg). *P < 0.05, 2-way analysis of variance, followed by post hoc Bonferroni test, n = 6 mice/sex/group. ZVEID was given 7 and 8 d after CCI. All the animal procedures were approved by the Institutional Animal Care and Use Committee of Duke University. CCI was conducted as we previously demonstrated, and mechanical allodynia was tested blindly with von Frey hairs analyzed with the up-down method (Chen et al. 2015). Mechanical allodynia was also tested by 10 stimuli with a low-threshold force (0.4 g) of von Frey hair (a, b). The doses of ZVEID were based on our previous study (Berta et al. 2014).
To further explore how CASP6 controls microglial signaling and neuropathic pain in both sexes, we compared CCI-induced gene expression in DRG and spinal cord tissues of male and female mice using quantitative real-time polymerase chain reaction analysis. Nerve injury increased CASP6 expression in DRG, but this increase is identical between males and females (Fig. 3a). CCI also induced comparable increases in the DRG expression of ATF3 (a marker of axonal injury) and the spinal cord dorsal horn expression of IBA1 (a microglial marker) in both sexes (Fig. 3a). Since neuropathic pain is reduced in only male mice, we further examined the expression of several genes in DRG and spinal cord dorsal horn tissues of wild-type and CASP6-KO male mice. We found that CCI-induced ATF3 expression in DRG is partially reduced in KO mice. Of great interest, CCI-induced spinal cord expression of BDNF and TNF-α was also substantially reduced in KO mice (Fig. 3b). These results suggest that, in addition to TNF-α release, CASP6 may regulate the expression of BDNF and TNF-α in the spinal microglia of male mice.
Figure 3.
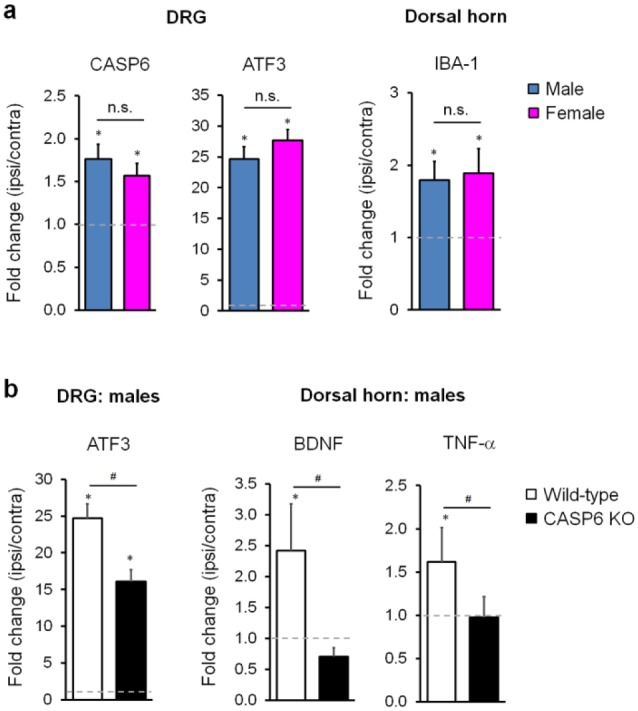
Caspase 6 (CASP6) is required for nerve injury–induced tumor necrosis factor (TNF) and brain-derived growth factor (BDNF) expression in spinal cords of male mice. (a) Chronic constriction injury (CCI) induces comparable expression of CASP6 and ATF3 in dorsal root ganglia (DRG) and IBA1 expression in the spinal cord dorsal horns of male and female mice. Gene expression was revealed by real-time quantitative polymerase chain reaction as fold change (ipsilateral side [ipsi] vs. contralateral side [contra] of the same animals). *P < 0.05, ipsi vs. contra, Student’s t test, n = 4 mice/group/sex. n.s., no significance. (b) CCI-induced expression of ATF3 in DRG and BDNF and TNF-α in dorsal horns is reduced in male mice with CASP6 deficiency. *P < 0.05, ipsi vs. contra; #P < 0.05, Student’s t test, n = 4 mice/group. DRG and spinal cord dorsal horn tissues were collected 1 week after CCI, and polymerase chain reaction analysis was conducted as previously demonstrated (Chen et al. 2015; Sorge et al. 2015).
In summary, we have revealed here a previously unrecognized extracellular role of CASP6 in modulating spinal cord synaptic transmission and persistent pain, in sharp contrast to the traditional roles of intracellular CASP6 in inducing axonal degeneration and neuronal apoptosis (Nikolaev et al. 2009; Graham et al. 2011). Given the unique distribution of CASP6 in spinal cord axonal terminals of C-fibers, we now propose a novel form of axon-microglia interaction in the spinal cord for the induction of inflammatory and neuropathic pain (Fig. 4). Tissue and nerve injury causes axonal release of CASP6, which acts on microglia to trigger TNF-α release, leading to enhanced synaptic transmission and pain states. Our data suggest that targeting the CASP6/p38 MAPK/TNF-α pathway may offer a new approach for the management of chronic pain by modulating microglial signaling. Given the well-known role of CASP6 in axonal degeneration (LeBlanc 2013), targeting CASP6 may also alleviate neuropathic pain by protecting against neuropathy after nerve trauma, diabetes, and chemotherapy. Additionally, p38 is implicated in the synthesis and secretion of BDNF in microglia, and BDNF plays a critical role in central sensitization and disinhibition in chronic pain (Coull et al. 2005; Trang et al. 2009). Finally, given the function of sex that we observed in male mice (Taves et al. 2015), we also postulate that in some persistent pain conditions, the CASP6/p38/TNF-α pathway or the CASP6/p38/BDNF pathway can promote pain in males (Fig. 4).
Figure 4.

Schematic illustration of axon-microglia interactions and the CASP6/p38/TNF-α signaling pathway in the spinal cord dorsal horn of male rodents. CASP6 is synthesized by C-fiber neurons of DRG and uniquely localized in axonal terminals in the superficial dorsal horn. CASP6-expressing axonal terminals form synapses with lamina IIo excitatory neurons, which in turn synapse with lamina I projection neurons to form a pain circuit (Todd 2010). These CASP6-expressing axonal terminals also have close contacts with microglial cell bodies and processes. Peripheral tissue and nerve injury results in CASP6 release from axonal terminals, and the secreted CASP6 then acts on microglial cells to trigger p38 activation. Upon activation, p38 not only induces synthesis of TNF-α and BDNF in microglia but also causes release of TNF-α and BDNF, which then act on nociceptive neurons to induce central sensitization and transition from acute pain to chronic pain. TNF-α and BDNF induce pain hypersensitivity (e.g., mechanical allodynia) via regulating both excitatory and inhibitory synaptic transmission in spinal cord pain circuits (Coull et al. 2005; Kawasaki et al. 2008; Zhang et al. 2010). It remained to be tested whether this mechanism also applies to the trigeminal system and orofacial pain. Modified from Berta et al. (2014) with permission from the journal. BDNF, brain-derived growth factor; CASP6, caspase 6; DRG, dorsal root ganglia; TNF-α, tumor necrosis factor α.
Limitations and Future Directions
While a PubMed search for the keywords “microglia and pain” shows 1,185 publications, a search for “microglia in trigeminal pain” shows only 41 publications. The limited data are likely due to the difficulties inherent in studying orofacial pain, as behavioral phenotyping in rodents is far simpler in the hind limb than in the face. However, the data do suggest that microglia are likely as important to chronic pain states in the trigeminal nucleus as in the spinal cord (Chiang et al. 2011). Although rodent hind paws are easier to study than teeth, in humans the opposite holds true, as dental nerves are readily accessible to the application of microglial inhibitors after dental procedures.
Although we have not examined the role of CASP6 in microglia activation in the trigeminal system, somatic pain and trigeminal pain may share similar mechanisms of microglial regulation. For instance, injury or compression of particular trigeminal nerve roots in an animal model of trigeminal neuralgia also induces p38 activation in microglia of the medullary dorsal horn, which is essential for the pathogenesis of trigeminal neuropathic pain via regulation of TNF-α (Piao et al. 2006; Ma et al. 2015). It was also shown that the chemokine fractalkine (CX3CL1) signaling in microglia contributes to ectopic orofacial pain following muscle inflammation via IL-1b released from activated microglia (Kiyomoto et al. 2013). Whether a sex difference based on these mechanisms exists in orofacial pain is still an open question.
It is critical to appreciate that although there are differences between human and rodent microglia (Smith and Dragunow 2014), human studies have found glial activation in chronic pain states using functional magnetic resonance imaging (Loggia et al. 2015) and in postmortem spinal cords (Shi et al. 2012). As a microglial inhibitor, minocycline (a tetracycline antibiotic) is approved for use in humans; some clinical trials have been performed showing limited promise for targeting the microglial pathway in the management of orofacial pain (Stavropoulos et al. 2006; Gelesko et al. 2011). However, propentofylline (a CNS glial modulator) does not decrease pain in postherpetic neuralgia patients (Landry et al. 2012). This failure argues that it is critical to target specific proinflammatory signaling pathways within microglia rather than block the overall function of these cells. Notably, microglia have different phenotypes, and M2-like microglia (anti-inflammatory) can also be protective and play a role in the resolution of inflammation and chronic pain (Milligan and Watkins 2009).
The sex difference in microglial effect also gives us reason to pause to consider the ramifications for human trials and clinical practice. As recently demonstrated (Sorge et al. 2015; Taves et al. 2015), microglial proliferation occurs similarly in male and female rodents, despite the differences in their phenotypic response to inhibition of microglial signaling or ablation of microglia. These results definitively highlight the importance of using both sexes in basic science studies, as mandated by the Food and Drug Administration for human studies. To some, the microglia’s lack of a phenotypic role in pain in female rodents may suggest that they are a poor target for further study, as females are more likely to suffer from chronic pain states. This would be shortsighted because insights into this mechanism may help us understand why females are more likely to be afflicted by chronic pain states. It is also possible that microglia may play different roles in different chronic pain conditions as well as in different phases of chronic pain development. For example, in a breast cancer pain condition, spinal microglia were shown to play an important role in the maintenance of bone cancer pain in female rats (Yang et al. 2015). Of interest, naltrexone, an opioid receptor antagonist, may inhibit the activity of microglia at low doses. A pilot study showed that fibromyalgia symptoms were reduced by low-dose naltrexone in female patients (Younger and Mackey 2009). Future study is needed to monitor microglial activity following the treatment.
One of the interesting predictors of orofacial pain is the role of intrauterine hormonal exposure as measured by digit ratio. Individuals exposed to more estrogen in utero were more likely to develop temporomandibular disorders (Sanders et al. 2013). This is critical to note, as gonadal hormones appear to modulate the effect of microglia on the developing brain, with a critical period where the effect of testosterone on microglia can permanently alter adult behavior (Lenz et al. 2013). Exposure of adult female mice to testosterone allows their microglia to show a male-like sensitivity to microglial inhibitors (Sorge et al. 2015). This effect of hormones on altering the responsiveness of microglia may be clinically useful and also highly relevant in the setting of aging and chronic opioid use, which are both known to cause changes in the hypothalamic-pituitary-gonadal axis (Bale and Epperson 2015; Gudin et al. 2015). It should be appreciated that difference in sex on microglia is just a part of the broader differences found in the immune system as a function of sex (Giefing-Kroll et al. 2015). Better understanding and harnessing these differences in the pain phenotype may help in the development of novel treatments for preventing and resolving chronic pain states.
Finally, it is critical to appreciate that, in addition to microglia, astrocytes—the most abundant type of glial cells in the CNS—play an important role in inflammatory and neuropathic pain in somatic and trigeminal areas (Gao and Ji 2010; Chiang et al. 2012). In particular, activation of astrocytic hemichannel connexin 43 (Cx43), also known as a gap junction protein, in persistent pain conditions results in increased secretion of chemokines and cytokines (e.g., CXCL1 and IL-1β) from astrocytes. CXCL1 and IL-1β can interact with nociceptive neurons in the spinal cord and trigeminal nucleus to induce central sensitization and enhance pain states (Guo et al. 2007; Chen et al. 2014). It is of great interest to study whether there is sex-dependent astrocytic signaling in chronic pain and, furthermore, how microglia, astrocytes, and neurons interact in chronic pain states in males and females. We should point out that these interactions are much more complicated than we thought, but mechanistic insights into these interactions will shed light on future therapeutics for preventing the transition from acute pain to chronic pain and for treating existing chronic pain.
Author Contributions
T. Berta, G. Chen, contributed to design, data acquisition, and analysis, drafted and critically revised the manuscript; Y.J. Qadri, contributed to design and data analysis, drafted and critically revised the manuscript; R.R. Ji, contributed to conception, design, and data interpretation, drafted and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Footnotes
This study was supported by grants from the National Institutes of Health (DE17794, DE22743, NS67686) to R.R.J. Y.J.Q. was supported by the Foundation for Anesthesia Education and Research Fellowship Grant.
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
- Bale TL, Epperson CN. 2015. Sex differences and stress across the lifespan. Nat Neurosci. 18(10):1413–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berta T, Park CK, Xu ZZ, Xie RG, Liu T, Lü N, Liu YC, Ji RR. 2014. Extracellular caspase-6 drives murine inflammatory pain via microglial TNF-alpha secretion. J Clin Invest. 124(3):1173–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan BL, Gil AS, Levesque A, Mogil JS. 2008. Modulation of mechanical and thermal nociceptive sensitivity in the laboratory mouse by behavioral state. J Pain. 9(2):174–184. [DOI] [PubMed] [Google Scholar]
- Chen G, Park CK, Xie RG, Berta T, Nedergaard M, Ji RR. 2014. Connexin-43 induces chemokine release from spinal cord astrocytes to maintain late-phase neuropathic pain in mice. Brain. 137(Pt 8):2193–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Park CK, Xie RG, Ji RR. 2015. Intrathecal bone marrow stromal cells inhibit neuropathic pain via TGF-beta secretion. J Clin Invest. 125(8):3226–3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang CY, Dostrovsky JO, Iwata K, Sessle BJ. 2011. Role of glia in orofacial pain. Neuroscientist. 17(3):303–320. [DOI] [PubMed] [Google Scholar]
- Chiang CY, Sessle BJ, Dostrovsky JO. 2012. Role of astrocytes in pain. Neurochem Res. 37(11):2419–2431. [DOI] [PubMed] [Google Scholar]
- Colburn RW, DeLeo JA, Rickman AJ, Yeager MP, Kwon P, Hickey WF. 1997. Dissociation of microglial activation and neuropathic pain behaviors following peripheral nerve injury in the rat. J Neuroimmunol. 79(2):163–175. [DOI] [PubMed] [Google Scholar]
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. 2005. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 438(7070):1017–1021. [DOI] [PubMed] [Google Scholar]
- Coull JA, Boudreau D, Bachand K, Prescott SA, Nault F, Sik A, De Koninck P, De Koninck Y. 2003. Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature. 424(6951):938–942. [DOI] [PubMed] [Google Scholar]
- Dubner R, Ruda MA. 1992. Activity-dependent neuronal plasticity following tissue injury and inflammation. Trends Neurosci. 15(3):96–103. [DOI] [PubMed] [Google Scholar]
- Fan W, Huang F, Zhu X, Dong W, Gao Z, Li D, He H. 2010. Involvement of microglial activation in the brainstem in experimental dental injury and inflammation. Arch Oral Biol. 55(9):706–711. [DOI] [PubMed] [Google Scholar]
- Gao YJ, Ji RR. 2010. Targeting astrocyte signaling for chronic pain. Neurotherapeutics. 7(4):482–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelesko S, Long L, Faulk J, Phillips C, Dicus C, White RP., Jr. 2011. Cryotherapy and topical minocycline as adjunctive measures to control pain after third molar surgery: an exploratory study. J Oral Maxillofac Surg. 69(11):e324–e332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giefing-Kroll C, Berger P, Lepperdinger G, Grubeck-Loebenstein B. 2015. How sex and age affect immune responses, susceptibility to infections, and response to vaccination. Aging Cell. 14(3):309–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace PM, Hutchinson MR, Maier SF, Watkins LR. 2014. Pathological pain and the neuroimmune interface. Nat Rev Immunol. 14(4):217–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham RK, Ehrnhoefer DE, Hayden MR. 2011. Caspase-6 and neurodegeneration. Trends Neurosci. 34(12):646–656. [DOI] [PubMed] [Google Scholar]
- Guan Z, Kuhn JA, Wang X, Colquitt B, Solorzano C, Vaman S, Guan AK, Evans-Reinsch Z, Braz J, Devor M, et al. 2016. Injured sensory neuron-derived CSF1 induces microglial proliferation and DAP12-dependent pain. Nat Neurosci. 19(1):94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudin JA, Laitman A, Nalamachu S. 2015. Opioid related endocrinopathy. Pain Med. 16 Suppl 1:S9–S15. [DOI] [PubMed] [Google Scholar]
- Guo W, Wang H, Watanabe M, Shimizu K, Zou S, LaGraize SC, Wei F, Dubner R, Ren K. 2007. Glial-cytokine-neuronal interactions underlying the mechanisms of persistent pain. J Neurosci. 27(22):6006–6018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horst OV, Cunha-Cruz J, Zhou L, Manning W, Mancl L, DeRouen TA. 2015. Prevalence of pain in the orofacial regions in patients visiting general dentists in the northwest practice-based research collaborative in evidence-based dentistry research network. J Am Dent Assoc. 146(10):721–728.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hucho T, Levine JD. 2007. Signaling pathways in sensitization: toward a nociceptor cell biology. Neuron. 55(3):365–376. [DOI] [PubMed] [Google Scholar]
- Ji RR, Berta T, Nedergaard M. 2013. Glia and pain: is chronic pain a gliopathy? Pain. 154 Suppl 1:S10–S28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Kohno T, Moore KA, Woolf CJ. 2003. Central sensitization and LTP: do pain and memory share similar mechanisms? Trends Neurosci. 26(12):696–705. [DOI] [PubMed] [Google Scholar]
- Ji RR, Suter MR. 2007. p38 MAPK, microglial signaling, and neuropathic pain. Mol Pain. 3:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SX, Zhuang ZY, Woolf CJ, Ji RR. 2003. p38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. J Neurosci. 23(10):4017–4022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki Y, Zhang L, Cheng JK, Ji RR. 2008. Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in regulating synaptic and neuronal activity in the superficial spinal cord. J Neurosci. 28(20):5189–5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyomoto M1, Shinoda M, Okada-Ogawa A, Noma N, Shibuta K, Tsuboi Y, Sessle BJ, Imamura Y, Iwata K. 2013. Fractalkine signaling in microglia contributes to ectopic orofacial pain following trapezius muscle inflammation. J Neurosci. 33(18):7667–7680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry RP, Jacobs VL, Romero-Sandoval EA, DeLeo JA. 2012. Propentofylline, a CNS glial modulator does not decrease pain in post-herpetic neuralgia patients: in vitro evidence for differential responses in human and rodent microglia and macrophages. Exp Neurol. 234(2):340–350. [DOI] [PubMed] [Google Scholar]
- Law AS, Nixdorf DR, Aguirre AM, Reams GJ, Tortomasi AJ, Manne BD, Harris DR; National Dental PBRN Collaborative Group. 2015. Predicting severe pain after root canal therapy in the National Dental PBRN. J Dent Res. 94(3 Suppl):37S–43S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBlanc AC. 2013. Caspase-6 as a novel early target in the treatment of Alzheimer’s disease. Eur J Neurosci. 37(12):2005–2018. [DOI] [PubMed] [Google Scholar]
- Lenz KM, Nugent BM, Haliyur R, McCarthy MM. 2013. Microglia are essential to masculinization of brain and behavior. J Neurosci. 33(7):2761–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loggia ML, Chonde DB, Akeju O, Arabasz G, Catana C, Edwards RR, Hill E, Hsu S, Izquierdo-Garcia D, Ji RR, et al. 2015. Evidence for brain glial activation in chronic pain patients. Brain. 138 Pt 3:604–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma F, Zhang L, Oz HS, Mashni M, Westlund KN. 2015. Dysregulated TNFalpha promotes cytokine proteome profile increases and bilateral orofacial hypersensitivity. Neuroscience. 300:493–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan ED, Watkins LR. 2009. Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci. 10(1):23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi K, Obata K, Kondo T, Okamura H, Noguchi K. 2008. Interleukin-18-mediated microglia/astrocyte interaction in the spinal cord enhances neuropathic pain processing after nerve injury. J Neurosci. 28(48):12775–12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogil JS. 2012. Sex differences in pain and pain inhibition: multiple explanations of a controversial phenomenon. Nat Rev Neurosci. 13(12):859–866. [DOI] [PubMed] [Google Scholar]
- Moore KA, Kohno T, Karchewski LA, Scholz J, Baba H, Woolf CJ. 2002. Partial peripheral nerve injury promotes a selective loss of GABAergic inhibition in the superficial dorsal horn of the spinal cord. J Neurosci. 22(15):6724–6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murasaki K, Watanabe M, Takahashi K, Ito G, Suekawa Y, Inubushi T, Hirose N, Uchida T, Tanne K. 2013. P2X7 receptor and cytokines contribute to extra-territorial facial pain. J Dent Res. 92(3):260–265. [DOI] [PubMed] [Google Scholar]
- Nikolaev A, McLaughlin T, O’Leary DD, Tessier-Lavigne M. 2009. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 457(7232):981–989. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Piao ZG, Cho IH, Park CK, Hong JP, Choi SY, Lee SJ, Lee S, Park K, Kim JS, Oh SB. 2006. Activation of glia and microglial p38 MAPK in medullary dorsal horn contributes to tactile hypersensitivity following trigeminal sensory nerve injury. Pain. 121(3):219–231. [DOI] [PubMed] [Google Scholar]
- Ren K, Dubner R. 2010. Interactions between the immune and nervous systems in pain. Nat Med. 16(11):1267–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren K, Dubner R. 2016. Activity-triggered tetrapartite neuron-glial interactions following peripheral injury. Curr Opin Pharmacol. 26:16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders AE, Slade GD, Bair E, Fillingim RB, Knott C, Dubner R, Greenspan JD, Maixner W, Ohrbach R. 2013. General health status and incidence of first-onset temporomandibular disorder: the OPPERA prospective cohort study. J Pain. 14(12 Suppl):T51–T62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Gelman BB, Lisinicchia JG, Tang SJ. 2012. Chronic-pain-associated astrocytic reaction in the spinal cord dorsal horn of human immunodeficiency virus-infected patients. J Neurosci. 32(32):10833–10840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slade GD, Fillingim RB, Sanders AE, Bair E, Greenspan JD, Ohrbach R, Dubner R, Diatchenko L, Smith SB, Knott C, et al. 2013. Summary of findings from the OPPERA prospective cohort study of incidence of first-onset temporomandibular disorder: implications and future directions. J Pain. 14(12 Suppl):T116–T124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AM, Dragunow M. 2014. The human side of microglia. Trends Neurosci. 37(3):125–135. [DOI] [PubMed] [Google Scholar]
- Sorge RE, Lacroix-Fralish ML, Tuttle AH, Sotocinal SG, Austin JS, Ritchie J, Chanda ML, Graham AC, Topham L, Beggs S, et al. 2011. Spinal cord Toll-like receptor 4 mediates inflammatory and neuropathic hypersensitivity in male but not female mice. J Neurosci. 31(43):15450–15454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorge RE, Mapplebeck JC, Rosen S, Beggs S, Taves S, Alexander JK, Martin LJ, Austin JS, Sotocinal SG, Chen D, et al. 2015. Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat Neurosci. 18(8):1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavropoulos MF, Shugars DA, Phillips C, Conrad SM, Fleuchaus PT, White RP., Jr. 2006. Impact of topical minocycline with third molar surgery on clinical recovery and health-related quality of life outcomes. J Oral Maxillofac Surg. 64(7):1059–1065. [DOI] [PubMed] [Google Scholar]
- Tanga FY, Nutile-McMenemy N, DeLeo JA. 2005. The CNS role of Toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proc Natl Acad Sci U S A. 102(16):5856–5861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taves S, Berta T, Liu DL, Gan S, Chen G, Kim YH, Van de, Ven T, Laufer S, Ji RR. 2015. Spinal inhibition of p38 MAP kinase reduces inflammatory and neuropathic pain in male but not female mice: sex-dependent microglial signaling in the spinal cord. Brain Behav Immun. 55:70–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd AJ. 2010. Neuronal circuitry for pain processing in the dorsal horn. Nat Rev Neurosci. 11(12):823–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trang T, Beggs S, Wan X, Salter MW. 2009. P2X4-receptor-mediated synthesis and release of brain-derived neurotrophic factor in microglia is dependent on calcium and p38-mitogen-activated protein kinase activation. J Neurosci. 29(11):3518–3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda M, Inoue K, Salter MW. 2005. Neuropathic pain and spinal microglia: a big problem from molecules in “small” glia. Trends Neurosci. 28(2):101–107. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Mizokoshi A, Shigemoto-Mogami Y, Koizumi S, Inoue K. 2004. Activation of p38 mitogen-activated protein kinase in spinal hyperactive microglia contributes to pain hypersensitivity following peripheral nerve injury. Glia. 45(1):89–95. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, Inoue K. 2003. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. 424(6950):778–783. [DOI] [PubMed] [Google Scholar]
- Yang Y, Li H, Li TT, Luo H, Gu XY, Lü N, Ji RR, Zhang YQ. 2015. Delayed activation of spinal microglia contributes to the maintenance of bone cancer pain in female Wistar rats via P2X7 receptor and IL-18. J Neurosci. 35(20):7950–7963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younger J, Mackey S. 2009. Fibromyalgia symptoms are reduced by low-dose naltrexone: a pilot study. Pain Med. 10(4):663–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Nei H, Dougherty PM. 2010. A p38 mitogen-activated protein kinase-dependent mechanism of disinhibition in spinal synaptic transmission induced by tumor necrosis factor-alpha. J Neurosci. 30(38):12844–12855. [DOI] [PMC free article] [PubMed] [Google Scholar]