

Abstract
With the deaths of Janet Rowley and John Goldman in December 2013, the world lost two pioneers in the field of chronic myeloid leukemia. In 1973, Janet Rowley, unraveled the cytogenetic anatomy of the Philadelphia chromosome, which subsequently led to the identification of the BCR-ABL1 fusion gene and its principal pathogenetic role in the development of chronic myeloid leukemia. This work was also of major importance to support the idea that cytogenetic changes were drivers of leukemogenesis. John Goldman originally made seminal contributions to the use of autologous and allogeneic stem cell transplantation from the late 1970s onwards. Then, in collaboration with Brian Druker, he led efforts to develop ABL1 tyrosine kinase inhibitors for the treatment of patients with chronic myeloid leukemia in the late 1990s. He also led the global efforts to develop and harmonize methodology for molecular monitoring, and was an indefatigable organizer of international conferences. These conferences brought together clinicians and scientists, and accelerated the adoption of new therapies. The abundance of praise, tributes and testimonies expressed by many serve to illustrate the indelible impressions these two passionate and affable scholars made on so many people’s lives. This tribute provides an outline of the remarkable story of chronic myeloid leukemia, and in writing it, it is clear that the historical triumph of biomedical science over this leukemia cannot be considered without appreciating the work of both Janet Rowley and John Goldman.
Introduction: the power of targeted therapy
The biology and treatment of patients with chronic myeloid leukemia (CML), a rare heterogeneous clonal hematopoietic stem cell disorder characterized by a consistent cytogenetic abnormality (the Philadelphia chromosome) and the presence of the BCR-ABL1 fusion gene, must surely be ranked as one of the most successful cancer medicine stories of the past century. The BCR-ABL1 fusion gene encodes the oncoprotein BCR-ABL1 (also referred to as p210 or BCR-ABL) with a constitutive active tyrosine kinase activity that is the primary cause of the chronic phase of CML.1,2 The discovery in 1996 that this kinase activity could be pharmacologically inactivated by a modified 2-phenylaminopyrimidine paved the way for the successful introduction of imatinib (also known as STI571, glivec, or gleevec) as an initial oral treatment for newly diagnosed CML patients.3 Imatinib, now termed a 1st-generation tyrosine kinase inhibitor (TKI), substantially and durably reduces the number of CML cells in the chronic phase at a daily oral dose of 400 mg, and has improved the 10-year survival rates from less than 20% to around 83% (Figure 1).4 The greatest advance is in those patients who achieve a complete cytogenetic response (CCyR) within two years of starting imatinib leading to life spans indistinguishable from the general population.5 These impressive results with imatinib therapy have had profound effects on the natural history of CML and its prevalence. Current estimates suggest that in the USA, where about 5500 new cases are diagnosed annually, the prevalence will increase to about 120,000 by 2020 and to about 200,000 by 2050.6
Figure 1.
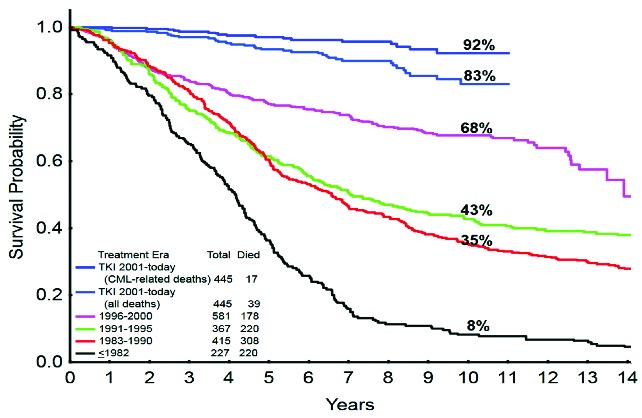
Survival with chronic myeloid leukemia over time (1993–2013): the German CML-Study Group experience. Courtesy of Prof H Kantarjian; adapted, with permission, from Harrison’s Principles of Internal Medicine, 2014.
However, imatinib is far from perfect, with only approximately 60% of patients remaining on the standard daily dose of 400 mg after six years due to either lack of drug tolerance or drug resistance.7 Imatinib is inducing responses also in the more advanced phases of CML, but these responses are not durable. There are now four newer TKIs, three so-called 2nd-generation inhibitors and one 3rd-generation inhibitor, all of which are more potent than imatinib in in vitro assays. Of the 2nd-generation drugs, nilotinib (also known as AMN107) and dasatinib (also known as BMS-354825) are licensed in the US and many other parts of the world for patients with CML in the chronic phase as first-line and subsequent therapies, while bosutinib (also known as SKI-606), is currently licensed for CML patients resistant or refractory to first-line drugs and is anticipated to be approved for first-line use in the near future. The 3rd-generation inhibitor ponatinib (also known as AP24534), is the newest and is licensed for CML patients who either have a T315I mutation or who fail to respond to any of the other currently approved TKIs. Current experience suggests both nilotinib and dasatinib achieve deeper and faster molecular responses than imatinib, but the precise benefits of such responses remain an enigma. Thus far, there is little evidence of a statistically significant improvement in overall survival (OS), though long-term follow up confirmed a superior rate of freedom from progression compared with patients with less deep molecular responses at the same time points.8
The advent of TKIs in the treatment of CML has opened a new era of precision medicine for diverse malignancies in which relatively non-specific and often toxic drugs are gradually being replaced by safer and better tolerated agents whose mechanism of action is precisely defined, and for which the treatment algorithm is guided by individual patient genomic information.9 Indeed, many TKIs have activity against other tyrosine kinases and could, therefore, be useful in treating patients whose malignancies harbor these gene mutations. In this review, we discuss the various milestones in the study, diagnosis, monitoring and treatment of CML, and speculate on the notion of cure and candidates for future therapy.10
Cytogenetics and molecular biology
Claims of priority can almost always be challenged but it is generally agreed that Alfred Velpeau in France be credited with the first detailed description of what must have been leukemia in 1827.11 As a result of astute clinical observations, he described a 63-year old florist and lemonade salesman who presented with gross hepatosplenomegaly and was noted to have “globules of pus” in his blood. The precise diagnosis, however, remained elusive. The first plausible story of what we now know as CML probably began in 1845 almost simultaneously by John Bennett in Edinburgh and Rudolph Virchow in Berlin.12,13 They both published accurate case reports and probably neither were aware of the other’s publication until later. Major progress in both the therapy and, indeed, the understanding of the disease did not occur until 1960. Figure 2 depicts the principal milestones in the study and treatment of CML.
Figure 2.

Milestones in the study and treatment of chronic myeloid leukemia.

Janet Rowley and John Goldman.
Janet Rowley defines the cytogenetics of the Philadelphia chromosome
Following the discovery by Joe Tjo and Albert Levan in 1956 that humans have 46 chromosomes, many efforts were directed to the study of chromosomal abnormalities in human cancers.14 By 1959, reports pertaining to the presence of constitutional abnormalities related to particular phenotypes began to appear, the most well known being the association of the gain of chromosome 21 in patients with Down syndrome.15 The work of Peter Nowell and David Hungerford led in 1960 to the discovery of the Philadelphia (Ph) chromosome.16 These investigators were tinkering with cytological techniques, which revealed metaphase spreads in bone marrow by accidentally rinsing slides with water. Among a series of bone marrow samples from patients with leukemia were 2 males with CML, in which they observed a “minute” chromosome. From cutting out the chromosomes from photographs of metaphases and laying them in rows according to centromere position and size, they deduced that this abnormal chromosome was a deletion of the Y chromosome. As these 2 patients had received therapy, there was some debate that the chromosomal abnormality had resulted from chromosomal damage induced by the treatment. Following additional work, they speculated that the chromosomal abnormality was probably not constitutive and may well be causally associated to CML.
At around the same time, Balkie and colleagues made the same discovery in Edinburgh, Scotland.17 They showed the presence of the same “small” chromosome in bone marrow and blood samples, but not in skin cells. With this observation, they were able to conclude that the abnormal chromosome was an acquired abnormality associated with the leukemia, particularly as the bone marrow and blood samples contained a high level of myeloblasts. In addition, a number of their patients were untreated, thus refuting the claim that the abnormality was therapy induced. They concluded that this small chromosome was derived from the group of small acrocentric chromosomes, known as chromosomes 21 and 22. As Down syndrome is associated with an increased risk of developing leukemia, although not CML, they made the assumption that the small chromosome must have arisen from a chromosome 21 and that the most likely explanation was a deletion. The Ph chromosomal abnormality was heralded as the first consistent cytogenetic abnormality in a human malignancy and the superscript ‘1’ was added, Ph1, on the premise that additional abnormalities would be discovered. This did not occur and the superscript had been dropped by 1990. The formal recognition that a human cancer might be caused by an acquired chromosomal aberration, vindicated the hypothesis postulated by Theodore Boveri in Germany in 1914 that cancer may be caused by acquired chromosomal abnormalities.18 The next important observations which established that CML was a stem cell-derived clonal disease came from Phillip Fialkow and colleagues in 1967.19 They applied a genetic technique developed by Ohno et al. based on X chromosome mosaicism in females, and by demonstrating polymorphism in the X-linked glucose-6-phosphatase dehydrogenase locus, established the clonal nature of CML.20
With the advent of the new chromosomal banding techniques in the early 1970s, it became possible to accurately identify the individual chromosome for the first time. Janet Rowley from Chicago used these techniques, which she had learnt whilst on a visit to Oxford, UK. Among samples from patients with hematologic malignancies collected over previous years were those from patients with CML in whom the Ph chromosome was present. Whilst laboriously comparing the chromosomal preparations made in the conventional manner with those prepared using these novel approaches, she noted on chromosome 9 “a duly fluorescein segment resembling the end of chromosome 22, but equally other chromosomes”. This remarkable observation of the balanced reciprocal translocation of genetic material between the long arms of chromosomes 9 and 22, t(9;22)(q34;q11) was published with difficulty in Nature in June 1973 (Figure 3).21 There were some valuable quotations in that paper which remain unchanged to this day: “the mechanism for the production of such a specific chromosomal translocation (if this is the correct explanation of these findings) is not clear”; “this would constitute the only specific translocation in humans that has been identified”; “this abnormality is involved in initiation rather than a consequence”.
Figure 3.
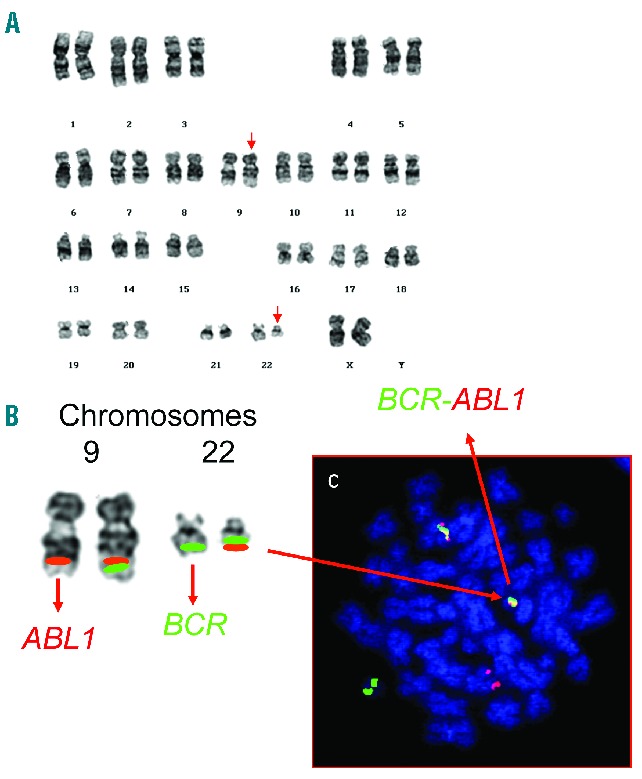
Detection of the t(9;22)(q34;q11) chromosomal translocation. (A) Karyotype from a patient with chronic myeloid leukemia depicting the translocation, t(9;22)(q34;q11) (abnormal chromosomes arrowed). (B) A partial karyotype of the same chromosomes 9 and 22 with the relevant FISH probes for BCR and ABL1 is shown. The red green fusion signals of the BCR-ABL1 and ABL1-BCR on chromosomes 22 and 9, respectively, are also shown. A metaphase counterstained with DAPI (blue) indicates their appearance under the fluorescent microscope (C).
The Molecular Biology Story
Janet Rowley’s seminal work in deciphering the Ph chromosome provided the framework for the unraveling of the genomic architecture, structure and function of the oncogene driving CML, which would become known as BCR-ABL1. These molecular events began in 1982, when Nora Heisterkamp et al. in Rotterdam, the Netherlands, observed that c-Abl, the human homolog of v-Abl, the oncogene of a murine leukemia virus first described by Abelson in 1970, localized to human chromosome 9.21–24 This discovery rekindled interest in a possible role of c-Abl in Ph-positive leukemia, even after attempts to demonstrate transforming capacity for c-Abl had proven unsuccessful.
The proof that c-abl was implicated in the Ph translocation was achieved on the basis of somatic cell hybrids generated by fusions of murine or hamster cell lines with cells from CML patients and healthy controls. These lines contained the rearranged chromosomes from the Ph translocation or their normal counterparts. Southern blot analysis was performed on DNA from the various hybridoma lines using human c-Abl probes and unequivocally demonstrated the translocation of c-Abl sequences to the Ph chromosome,25 and was confirmed at the cytogenetic level.26 While the breakpoints on chromosome 9 spanned a large genomic region, breakpoints on chromosome 22 localized to a relatively small genomic region that was hence called “breakpoint cluster region” or BCR.23 This name was later used to designate the previously unknown gene on chromosome 22 that serves as the 5’ fusion partner for ABL1. Thereafter, the BCR-ABL fusion mRNA was demonstrated and the proof that it gave rise to the p210 BCR-ABL1 protein followed. By the mid-1980s the molecular anatomy of the BCR-ABL1 oncogene had been unraveled (Figure 4).1,27,28
Figure 4.

The structure of the normal BCR and ABL1 genes and the fusion transcripts found in Ph-positive leukemias. The ABL1 gene contains two alternative 5′ exons (named 1b and 1a) followed by 10 ‘common’ exons numbered a2–a11 (green boxes). Breakpoints in CML and Ph-positive ALL usually occur in the introns between exons 1b and 1a or between exons 1a and a2 (as shown by vertical arrows). The BCR gene comprises a total of 23 exons, 11 exons upstream of the M-BCR region, five exons in the M-BCR that were originally termed b1–b5 and now renamed e12–e16, and seven exons downstream of M-BCR (orange boxes). For convenience, only exons e1, e12–e16 and e23 are shown. Breakpoints in CML usually occur between exons e13 (b2) and e14 (b3) or between exons e14 (b3) and e15 (b4) of the M-BCR (as shown by two vertical arrows placed centrally). The majority of patients with Ph-positive ALL have breakpoints in the first intron of the gene, between e1 and e2 (arrow at left). Three possible BCR–ABL1 mRNA transcripts are shown below. The first two (e13a2 and e14a2, respectively) are characteristic of CML. The bottom mRNA (e1a2) is found in the majority of patients with Ph-positive ALL.
The next major step forward in our understanding of CML was the demonstration that BCR-ABL1 was a tyrosine kinase and that tyrosine kinase activity was critical to its ability to transform cells. v-Abl had been recognized as a tyrosine kinase in 1980 and subsequent deletion mutagenesis revealed that the sequences containing the tyrosine kinase were critical to cellular transformation.29,30 As early as 1984, the Witte lab had identified an altered c-Abl protein in K562 cells and suspected that a structural alteration present in the 210 kD protein had unmasked c-Abl’s tyrosine kinase activity, leading to cellular transformation.31 This was subsequently substantiated by experiments that convincingly showed a correlation between the tyrosine kinase activity of BCR-ABL1 proteins and their transforming capacity.32,33
In 1990, George Daley and Rick van Etten, working with the Nobel laureate David Baltimore, showed that transplantation of bone marrow infected with a BCR-ABL1 retrovirus into lethally irradiated syngeneic recipient mice induced a disease that resembled human CML, providing a causal connection between the BCR-ABL1 cDNA and the clinical disease phenotype of CML.34 This was confirmed by work by Elephanty et al. in Australia and Kelliher et al. in Los Angeles.35,36 The notion that the BCR-ABL1 fusion gene could have a central role in CML was thereafter generally accepted and established a scientific rationale to target BCR-ABL1 kinase activity for the treatment of ABL-related leukemias.37 The 1990s saw the elucidation of the complex signaling network operated by the BCR-ABL1 kinase, with contributions from many laboratories. Myc, Ras, phosphatidyl inotisol 3′ kinase (PI3K), JAK/STAT and cytoskeletal proteins were identified as pathways activated by BCR-ABL1 or as important downstream mediators (Figure 5).38–43 However, what proved to be very difficult was identifying transformation critical molecules downstream of BCR-ABL1, testimony to a high level of redundancy in the signaling network. Moreover, the more became known about signal transduction in BCR-ABL1 transformed cells, the more it became evident that fundamental differences exist between leukemia cell lines and primary leukemia cells, limiting the applicability of conclusions derived from in vitro studies. Experiments on primary cells and murine models identified additional molecules important for BCR-ABL1 transformation, including β-catenin, Hedgehog, PP2A, BCL-6 and Alox5, amongst others.44–48 Involvement of these pathways in CML stem cell survival suggests they may be excellent therapeutic targets, but their role in the sustenance of normal hematopoiesis and/or normal development could also limit their utility.49 Since CML stem cells are not addicted to BCR-ABL1, unlike progenitor cells, the search for specific molecular vulnerabilities in leukemic founder cells continues, as does the molecular story of CML.50
Figure 5.

Cytoplasmic BCR-ABL1 activates a myriad of signal pathways. BCR-ABL1 domain structure and simplified representation of molecular signaling pathways activated in chronic myeloid leukemia (CML) cells. Following dimerization of BCR-ABL1, autophosphorylation generates docking sites on BCR-ABL1 that facilitate interaction with intermediary adapter proteins (brown) such as GRB2. CRKL and CBL are also direct substrates of BCR-ABL1 that are part of a multimeric complex. These BCR-ABL1-dependent signaling complexes in turn lead to activation of multiple pathways whose net result is enhanced survival, inhibition of apoptosis, and perturbation of cell adhesion and migration. A subset of these pathways and their constituent transcription factors (blue), serine/threonine-specific kinases (purple), cell cycle regulatory protein (yellow) and apoptosis-related proteins (red) are shown. Also included are a few pathways that have been more recently implicated in CML stem cell maintenance and BCR-ABL1-mediated disease transformation (orange). However, it is important to note that this is a simplified diagram and that many more associations between BCR-ABL and signaling proteins have been reported.
Treatment options
Historical perspectives
Efforts to improve the quality of life by controlling the symptoms attributed to CML probably began with the use of arsenicals by Thomas Fowler in 1865, and Arthur Doyle in 1882, and continued in the first half of the 20th century with radiation therapy to the spleen in 1902, antileukocyte sera in 1932, benzene in 1935, urethane in 1950 and leukapheresis in the 1960s.51 There were a number of other notable treatment attempts, but most, if not all, were unsuccessful. Busulfan, an alkylating agent, was introduced by David Galton in London in 1953.52 Galton then carried out the first prospective randomized study in CML, comparing busulfan and splenic radiation, and showed improved survival in the busulfan cohort. In the mid-1960s, busulfan was replaced by hydroxycarbamide (previously hydroxyurea), a ribonucleotide reductase inhibitor, following recognition that busulfan is mutagenic, and a randomized study confirming the superiority of hydroxycarbamide, though neither drug was able to reduce the proportion of Ph positive cells or prolong overall survival.53,54 Interferon alpha (IFN-α) was introduced into the clinics in the mid-1980s and proved popular, despite frequent side-effects such as flu-like symptoms and fatigue.55 In the early 1990s, several randomized studies comparing IFN-α or interferon-α n1 (wellferon) with hydroxycarbamide or busulfan were undertaken and demonstrated an improvement in overall survival by about 2–3 years with IFN-α.56–58 In addition, a French study testing the addition of cytarabine to IFN-2b found this to result in an increased proportion of patients achieving a cytogenetic response.59 Thereafter, interferon, either alone or in combination with cytarabine, replaced hydroxycarbamide as the preferred treatment for CML in the chronic phase.60 The precise mode of action of IFN-α remains unclear, but is probably related to its immunomodulatory properties. IFN-α was replaced by imatinib as the preferred treatment for patients with CML in the chronic phase in the summer of 2001 following a randomized study comparing imatinib with IFN-α plus cytarabine.61–63 The results were very impressive and established the firm position of imatinib, and also constituted the final proof of the importance of the BCR-ABL1 oncoprotein to CML. The introduction of imatinib was rapidly followed by the development of the next generation tyrosine kinase inhibitors (TKIs).
Prognostic and predictive factors
Various efforts have been made to establish criteria definable at diagnosis that may help to predict response to therapy and survival for individual patients. Historically, the Sokal score was developed in 1984 for patients treated with busulfan, and the Hasford (also known as the Euro) score in 1998 for patients treated with IFN-α.64,65 Both scoring systems have since been confirmed to be useful in the TKI era. Stratifying patients into good-, intermediate-, and poor-risk categories may assist in the decision-making process regarding appropriate treatment options. In 2011, a simpler and TKI-specific score, the European Treatment and Outcome Study (EUTOS), was proposed but requires further confirmation before it can be widely used.66 More recently, the response to TKIs at a given time point, disease risk and stage, BCR-ABL1 genotype, the presence of comorbidities, financial aspects and local monitoring capabilities are all being increasingly used to personalize treatment.67
The Imatinib Story
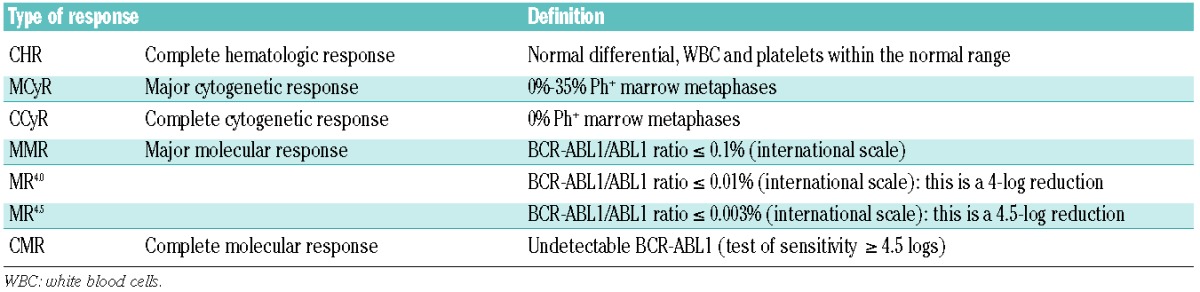
It is remarkable how, in 1994, against a background of considerable skepticism about any possible clinical value of TKIs, Brian Druker in Portland, Oregan, and collaborating scientists at Ciba-Geigy (now Novartis) in Basel, Switzerland, developed a compound, imatinib, that could reverse the clinical and hematologic features of CML.3,68 Imatinib, a 2-phenylaminopyrimidine, inhibits the enzymatic action of the activated BCR-ABL1 tyrosine kinase by occupying the ATP-binding pocket of the tyrosine kinase component of the BCR-ABL1 oncoprotein, thereby blocking the capacity of the enzyme to phosphorylate and activate downstream effector molecules that cause the leukemic phenotype. It also binds to an adjacent part of the kinase domain in a manner that holds the ABL-activation loop of the oncoprotein in an inactive configuration (Figure 6).69 The International Randomized Study of Interferon and STI571 (IRIS) demonstrated that imatinib induced ‘cumulative best’ CCyR, equivalent to a 2-log reduction in BCR-ABL1 transcripts level, in 82% of all previously untreated patients with CML in the chronic phase.60,70 About 2% of all patients in the chronic phase progress to advanced-phase disease each year, which contrasts with estimated annual progression rates of more than 15% for patients treated with hydroxycarbamide and about 10% for patients receiving IFN-α, either with or without cytarabine.4,71 The 8-year event-free survival was 83% and the estimated overall survival was 93% (corrected for CML-related deaths only), confirming the notion that imatinib substantially prolongs overall survival compared with historical patients who received IFN-α or hydroxycarbamide.72 A substantial proportion of the patients in CCyR also achieve a 3-log reduction or more in BCR-ABL1 transcripts (referred to as MMR, or MR3), and this proportion seems to have continued to increase steadily with time on imatinib. A minority of patients achieve a deeper molecular response with more than 4-log or 4.5-log reduction in BCR-ABL1 transcripts [referred to as MR4.0 and MR,4.5 respectively; MR4.5 was previously referred to as a complete molecular response (CMR)] (Table 1).73,74 These results were confirmed by independent single centers as well as company-led registration studies.75 It should also be said that the success of these and other CML treatment studies epitomize the critical importance of an optimal molecular monitoring methodology (see below).
Figure 6.
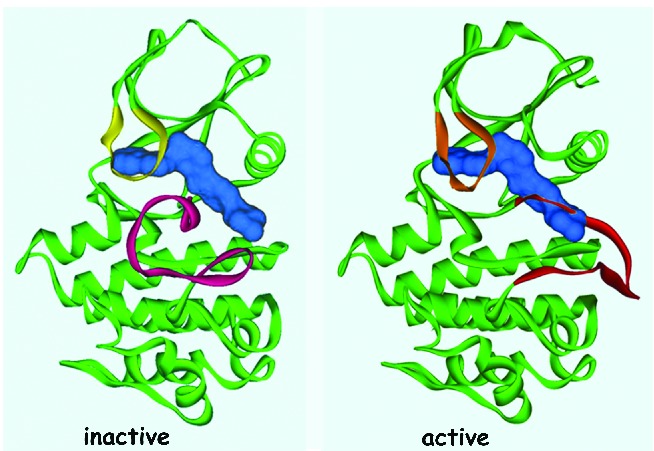
Imatinib binds an Inactive ABL1 conformation. Adapted, with permission, from Schindler et al. Science 2000.
Table 1.
Definitions of response.
The standard starting daily dose of imatinib is 400 mg for newly diagnosed patients in the chronic phase, but the optimal dose is not known and no maximum tolerated dose was established in the initial phase I study.60 Several single-arm studies suggest that higher doses, up to 800 mg daily, might give better results with a greater proportion of patients achieving CCyR and MMR.76,77 Such studies also suggest better PFS and transformation-free survival, but with potentially more side-effects, particularly myelosuppression. Amongst randomized studies, the TOPS (Tyrosine Kinase Inhibitor Optimization and Selectivity) study showed imatinib 800 mg to induce MR3 more rapidly than imatinib 400 mg, but at one year there was no statistically significant difference.78 In contrast, there is persuasive evidence from the recent randomized German (CML IV) study that optimized high-dose imatinib allows most patients to achieve MR4.5, and this may provide an improved therapeutic basis for treatment discontinuations.79 A subset analysis from this randomized study also showed a greater benefit for patients over 65 years of age.80 Another recent randomized intergroup phase II study also demonstrated deeper molecular responses in the 800 mg daily arm compared with 400 mg daily, with MR4 of 25% and 10%, respectively, with a trend for improved progression-free and overall survival, but with substantially more grade 3 and 4 side-effects.81 There is also some evidence that imatinib 600 mg daily is tolerated in more than 80% of CML patients and results in superior cytogenetic and molecular responses at 12 and 24 months compared to the conventional 400 mg daily dose.82 It is also of interest to note that in the German CML IV study, the median daily dose of imatinib was actually 628 mg, lending additional support to the 600 mg dose strategy.
Regardless of the dose of imatinib, the current safety analysis of imatinib is quite impressive, with very few potentially serious long-term side-effects noted after ten years or more continuous use.83 Table 1 depicts the relative toxicities of all currently available TKIs for CML. When the drug is used at the standard starting daily dose of 400 mg, most adverse effects occur within the first two years of starting therapy, and are generally mild to moderate (grades 1 and 2). Most of these include lethargy, nausea, headache, various skin reactions (including Steven-Johnson syndrome), infraorbital edema, bone pains, and sometimes, generalized fluid retention. In general these effects are easily manageable and potentially reversible. Significant cytopenias, in particular neutropenia and/or thrombocytopenia and sometimes anemia occur less commonly and usually in the first 6–12 months of therapy. Liver chemistry can also be abnormal, and this may, on rare occasions, progress to liver failure. Rare incidences of prolongation of the QTc interval on the electrocardiograph have also been reported. It is possible that some patients, such as older patients and those with other co-morbidities such as impaired cardiac function, might be more susceptible to toxicity. The longer-term follow-up studies do, however, indicate an adverse effect on the quality of life, particularly in younger female patients, and other unique effects, such as effects on bone growth and mineralization and gynecomastia.84–86 Finally, although there appears to be no definitive evidence to suggest exposure to imatinib increases the risk of developing a second malignancy, it is reasonable for specialists to remain cautious and follow patients on long-term treatment carefully.87
The next generation-TKI story
Further research into the imatinib story has shown that only about 60% of CML patients remain on the standard dosages of imatinib after six years, implying that about 40% have required an alternative treatment or higher doses of imatinib.88 In addition, a population-based report found that only half of newly diagnosed patients with CML in chronic phase were in CCyR and receiving imatinib at two years after starting treatment.89 The main reasons for this are secondary (acquired) resistance which in most cases results from the expansion of subclones with point mutations in the BCR-ABL1 kinase domain (KD).90–92 A variety of other resistance mechanisms have also been described, including poor adherence, amplification of the BCR-ABL1 fusion gene, relative overexpression of BCR-ABL1 protein, and overexpression of the multidrug resistant P-glycoprotein (MDR1).
Point mutations in the kinase domain of BCR-ABL1 that confer resistance to imatinib code for amino acid substitutions that may preclude entry of imatinib into the ATP-binding pocket or, in general, the inhibitory action of imatinib. The precise position of the mutation appears to dictate the degree of resistance to the drug. Some mutations are associated with minor degrees of drug resistance, while the T315I (also referred to as the gatekeeper) mutation confers a very high level of resistance.93 The precise significance, and indeed the kinetics, of the over 100 currently well-characterized mutations have only partially been characterized (Figure 7).94 It is also possible, though not confirmed in vivo, that resistant mutant clones could enhance the fitness of sensitive clones by altering their microenvironment by generating paracrine factors, such as IL-3.95
Figure 7.

Mutations in the kinase domain of ABL1 identified in tyrosine kinase inhibitors (TKI) resistant chronic myeloid leukemia cells. The 10 most frequent mutations, accounting for approximately 70% of TKI-resistant CML patients are highlighted in red.
Primary resistance to imatinib appears to be very rare, and when observed may be related to poor drug compliance, poor gastrointestinal absorption, p450 cytochrome polymorphisms, interactions with other medications, or abnormal drug efflux and influx at the cellular level due to low drug influx transporter (OCT1).96 The recognition of imatinib’s qualified success led efforts to develop the next-generation TKIs and other alternative treatments. Initial efforts focused on two 2nd-generation TKIs: nilotinib and dasatinib (Figure 8).97
Figure 8.
Chemical structures of imatinib, nilotinib, dasatinib, bosutinib and ponatinib.
Nilotinib was designed as a chemical modification of imatinib in an effort to increase its selectivity and activity. Indeed, nilotinib has little activity against other kinases inhibited by imatinib, such as KIT and PDGFRA/B.98 Nilotinib is taken orally twice daily with food restrictions due to its bioavailibity being affected adversely by high fat intake. Like imatinib, it acts as an ATP-competitive inhibitor by binding to the closed (inactive) conformation of the ABL1 kinase domain, but with a much higher affinity. In vitro studies suggest that nilotinib is approximately 30- to 50-fold more potent than imatinib. Nilotinib is also active in 32 of the currently 33 imatinib-resistant cell lines with mutant BCR-ABL1, but like imatinib has no activity against the T315I mutation.99
Dasatinib is a thiazole-carboxamide structurally unrelated to imatinib which binds to the ABL1 kinase domain regardless of the conformation of the activation loop (i.e. whether open or closed).100,101 It also inhibits some of the Src family kinases that are involved in signal transduction in lymphoid cells and results in NK-cell expansion. Preclinical studies showed that dasatinib is 300-fold more potent than imatinib and is active against 18 of 19 tested imatinib-resistant BCR-ABL1 mutants, with the notable exception of the T315I mutant.99
In 2004, both drugs entered studies of patients who were resistant or intolerant to imatinib at standard dosages. The efficacy, but not the toxicity, of both drugs was fairly similar, with about 45% of the imatinib-resistant patients achieving CCyR and a 4-year overall survival of 78%. The results for the imatinib-intolerant group were slightly better for both drugs.99–109 All responses were similar in patients with or without mutations, except for the cohort with T315I mutation, where no responses were noted with either drug. Though these results are impressive, it is interesting that only one-third of the responding patients remained on nilotinib or dasatinib at five years, which means that two-thirds of patients required a further change of therapy. In an analysis of a sub-set receiving dasatinib for imatinib-resistant/intolerant disease, it was noted that dasatinib maintained durable efficacy irrespective of the presence or absence of pre-existing KD mutations.110
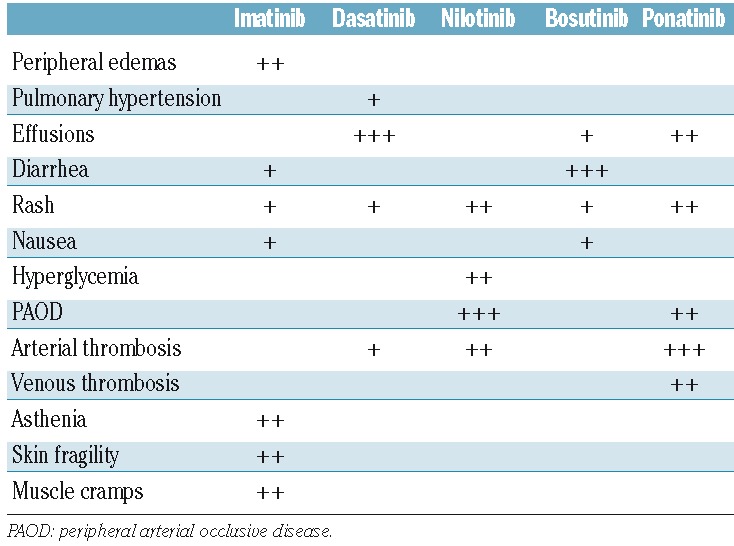
The most common nilotinib treatment-related toxicity was myelosuppression (although this was less pronounced than that observed with most other TKIs) followed by headaches, pruritus, and rashes (Table 2). Overall, 22% of the patients in the chronic phase experienced thrombocytopenia, with 19% having either grade 3 or 4 severity; 16% had neutropenia and a further 16% had anemia; metabolic effects included hyperglycemia. Following longer follow up, an increased incidence of cardiovascular events, in particular peripheral arterial disease, was noted, although many affected patients had predisposing risk factors.111 About 19% of all patients experience arthralgia, and about 14% experience fluid retention, particularly pleural effusions, and rarely pericardial effusions and other unique effects, such as panniculitis.104,112 In the dasatinib-treated cohort, hematologic toxicity was more common, with neutropenia and/or thrombocytopenia occurring in one-half of all patients and anemia in 20%.108 Non-hematologic toxicity includes diarrhea, headaches, superficial edema, pleural effusions, and occasional pericardial effusions. Grades 3/4 side effects were rare; grades 3/4 pleural effusions occurred in 6% of patients.
Table 2.
Adverse events related to tyrosine kinase inhibitors in patients with chronic myeloid leukemia.
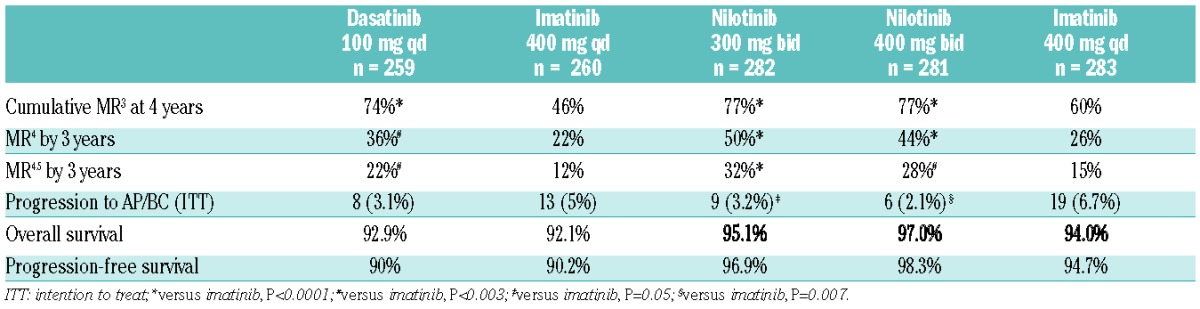
Following these encouraging results, both nilotinib and dasatinib entered clinical trials for first-line therapy of newly diagnosed patients in 2006. Nilotinib at two dosages, either 300 mg/day or 400 mg/day, was tested against imatinib 400 mg/day in the Evaluating Nilotinib Efficacy and Safety in Newly Diagnosed Patients (ENESTnd) randomized study, with the rate of MR3 at 12 months as the primary end point.113 Dasatinib was tested at a dose of 100 mg/day in a trial known as Dasatinib versus Imatinib Study in Treatment-Naïve CML Patients (DASISION), with confirmed CCyR at 12 months as the primary end point.114 Both of these primary end points were met: ENESTnd accorded higher rates of MR3 at 12 months for both doses of nilotinib compared with imatinib (44% and 43% vs. 22%; P<0.001) and DASISION showed that dasatinib resulted in more frequent confirmed CCyR at 12 months compared with imatinib (77 vs. 66%; P=0.007). Both drugs were licensed for first-line use in patients with CML in the chronic phase in 2010. Table 3 summarizes the latest updates from both trials.
Table 3.
DASISION and ENESTnd: summary of data from different studies.
Many of the secondary end points were also met in both trials, and the overall results suggested that front-line therapy with dasatinib or nilotinib (at either dose) achieves earlier and higher molecular response rates, in particular faster and deeper molecular responses (MR3, MR4 and beyond), that in turn appear to decrease the rates of progression to the advanced phases of CML.115–117 Nilotinib was associated with hyperglycemia, hypercholesterolemia, increased triglycerides, and an increased incidence of cardiovascular, cerebrovascular and peripheral arterial occlusive disease.118,119 Dasatinib was associated with substantial hematologic toxicity, pleural effusions and, infrequently, pericardial effusions and pulmonary hypertension (Table 2).120 Discontinuation rates for disease progression or treatment failure for any cause appears to be similar at around 33%–38% at three years for both drugs, with the caveat that the definitions of progression and the duration of follow up prior to censoring in these two large studies were not uniform. A recent independent North American consortia trial comparing daily dasatinib 100 mg to daily imatinib 400 mg produced very similar results to DASISION in terms of efficacy and safety.121 Collectively, neither of these two studies, nor the ENESTnd or the companion ENESTcmr studies, demonstrated a survival advantage for a 2nd-generation TKI being used for first-line therapy of a newly diagnosed patient with CML in chronic phase, despite the superior early molecular responses and the subsequent MR4.5 responses.122 In addition, the associated cardiovascular toxicity in all three trials has been higher than that seen with imatinib.
The third and newest of the 2nd-generation TKIs, bosutinib, an oral dual ABL1 and SRC inhibitor, is chemically different from both dasatinib and nilotinib, and appears to be able to overcome binding impediments conferred by several kinase domain mutations to imatinib, nilotinib, or dasatinib (Figure 8).123 Phase II studies of once daily bosutinib 500 mg/day in CML patients who were either resistant or intolerant to imatinib demonstrated a CCyR of 47%, an overall survival at two years of 88% in the imatinib-resistant cohort, and a remarkable 98% in the imatinib intolerant cohort; three years later, 40% of patients remained on bosutinib.124–127 The principal side-effects included diarrhea, abnormal liver chemistry and various skin rashes, all of which were easily manageable with dose reduction and/or concomitant medications (Table 2). Based on these results, the drug was approved in 2012 for the treatment of adult CML patients with chronic phase or advanced phase disease who were resistant to prior TKI therapy. The drug was then tested in the phase III Bosutinib Efficacy and safety in CML (BELA) study, in newly diagnosed patients with CML in the chronic phase.128 Since the primary end point was not met, and with the CCyR results being similar in both arms of the study (70% for bosutinib and 68% for imatinib) the drug was not approved for first-line use. It is, however, of interest that the MR3 rates at 12 months were significantly improved at 41% with bosutinib compared to 27% with imatinib, and the drug discontinuation rate was 37% at two years for bosutinib and 29% for imatinib. Furthermore, the risk of transformation to the advanced phases was significantly lower for bosutnib. These latest molecular results lend some support for the drug’s future candidacy as first-line therapy.
Ponatinib is a 3rd-generation TKI which has an interesting chemical structure based on a modification of a purine scaffold and a central triple carbon-carbon bond with a substructure beyond the bond that is similar to imatinib (Figure 8).129 The drug inhibits ABL1, SRC and a variety of other kinases, including KIT, PDFGRA, FGFR1 and FLT3.130,131 It was developed initially for patients who were considered to have become resistant to TKIs as a result of a T315I subclone. This feature is attributed to the compound’s unique structure which allows it to bind and inhibit ABL1 with no steric hindrance due to the T315I mutation.132,133 The drug was tested in the Ponatinib Phpositive acute lymphoblastic leukemia (ALL) and CML Evaluation (PACE) phase II study in which 449 patients with CML in the chronic and advanced phases and Phpositive ALL with resistant to or intolerance from dasatinib or nilotinib or with the T315I mutation were enrolled. The patients received once daily 45 mg ponatinib, and the results indicated that the drug had considerable activity in all patients, including those in advanced phase disease, regardless of baseline kinase domain mutations and the responses seemed to be durable.134,135 The study results showed that there were 46% CCyR (40% without T315I; 66% with T315I) and 34% MR3 (27% without T315I; 56% with T315I). The most common side-effect was thrombocytopenia, rash, dry skin and abdominal pain, and platelet dysfunction was also noted (Table 2).136 Serious thrombotic events were observed in 9%, but considered to be treatment-related in 3%. The study drug-discontinuation rate due to toxicity was 12%.
The Evaluation of Ponatinib versus Imatinib in CML (EPIC) phase III randomized study of ponatinib and imatinib in newly diagnosed patients began in 2012 and preliminary results suggest that the drug accords high rates of early molecular response and MR3 compared with imatinib.137 The drug was licensed in December 2012 for patients with CML in the chronic or advanced phases resistant or intolerant to prior TKI therapy and Ph-chromosome positive ALL resistant or intolerant to prior TKI therapy. This approval constituted ponatinib to be the only licensed TKI with activity against the T315I subclone. Unfortunately, in October 2013, concerns about excessive arterial vascular events led to the suspension of the drug and the manufacturer elected to discontinue the EPIC study.118,119 In early 2014, despite these serious risks, ponatinib was re-licensed exclusively for the treatment of adult patients with T315I-positive CML in all phases or T315I-positive Ph-chromosome positive ALL and adult patients with CML in all phases or Ph-chromosome positive ALL for whom no other TKI therapy was indicated. The precise mechanisms of ponatinib-related arterial vascular events, and indeed those associated with nilotinib, which seem to occur at a considerably lower frequency, still remain elusive.
There is continuing interest in developing effective treatments for T315I-positive CML, which are of additional interest given the challenges with ponatinib. Omecetaxine (formerly called homoharringtonine) is a semi-synthetic plant alkaloid that enhances apoptosis of CML cells. It has actually been under investigation in CML and other myeloid malignancies since the 1970s.138 The results of recent studies were encouraging, with modest activity noted in patients with CML in the chronic and advanced phases, including some with the T315I subclone. The drug was licensed in 2014 for use in patients with CML (all phases) who were resistant or intolerant to two or more TKIs. Another candidate drug that has shown some activity in T315I subclone is HS-438, which has been tested in clinical trials.139
The allogeneic stem cell transplantation story
“Thy bones are marrowless, thy blood is cold” (The Tragedy of Macbeth: William Shakespeare, 1606).
Though the original concept of bone marrow transplantation was probably first advocated by Thomas Fraser in 1894, when he famously recommended that patients eat bone marrow “sandwiches; flavored by port-wine” (to improve taste), sporadic attempts at marrow transplantation were taken much earlier.140 The modern era of bone marrow transplantation [now stem cell transplantation (SCT)] did not begin until a basic understanding of the histocompatibility system was gained in 1958. Much of the pioneering work thereafter was carried out by the Nobel laureate E. Donnall Thomas in Seattle, resulting in the first successful allo-SCT using syngeneic donors in 1979.141,142 In 1978, John Goldman in London showed that marrow-populating stem cells were present in the peripheral blood of untreated CML patients.143 This led to the use of an autograft for patients ineligible for an allo-SCT, and though in some patients Ph-negative hematopoiesis was restored, very few patients remained Ph-negative for extended periods.
Subsequent efforts in allo-SCT using sibling and volunteer unrelated donors were increasingly successful, as a result of the recognition that the graft-versus-leukemia (GvL) effect plays a major role in eradicating CML after allo-SCT, and improvements in the conditioning regimens.144,145 This coupled with the availability of hematopoietic stem cells from a variety of sources, improvements in SCT technology, and a better understanding and treatment of the alloimmmune-mediated graft-versus-host disease (GvHD) led to significantly improved results for the majority of patients transplanted in the chronic phase, and indeed made SCT more widely available to higher risk and also older patients.146–148 The potential to accord long-term survival and probable cure for patients with CML in the chronic phase was firmly established by the early 1990s, and an allograft was then considered the first-line treatment for all eligible patients in the chronic phase.149 The use of donor lymphocyte infusions to treat early relapse after allograft by exploiting the GvL effect became popular in the mid-1990s, and confirmed the importance of the donor derived immune system to overcome residual leukemic cells.150,151
The major factors influencing survival are patient age, disease phase at time of SCT, disease duration, degree of histocompatibility between donor and recipient, and gender of donor.152 The best results are achieved following a full intensity (conventional) allograft, using an HLA-identical sibling donor or a suitable matched unrelated donor; the 5-year leukemia-free survival is 80% and 60%, respectively (Figure 9).149 The results following a reduced intensity regiment are generally inferior.153 There is a roughly 20% chance of transplant-related mortality and a 15% chance of relapse. The possible major complications include GvHD, reactivation of infection with cytomegalovirus or other viruses, idiopathic pneumonitis and sinusoidal obstruction syndrome (previously known as veno-occlusive disease of the liver). Post-transplant molecular monitoring studies suggest that most, but not all, patients who are negative for BCR-ABL1 transcripts at five years following the allograft, remain negative for long periods, and it is likely that, in the majority of these patients, the CML may truly have been eradicated.154,155
Figure 9.
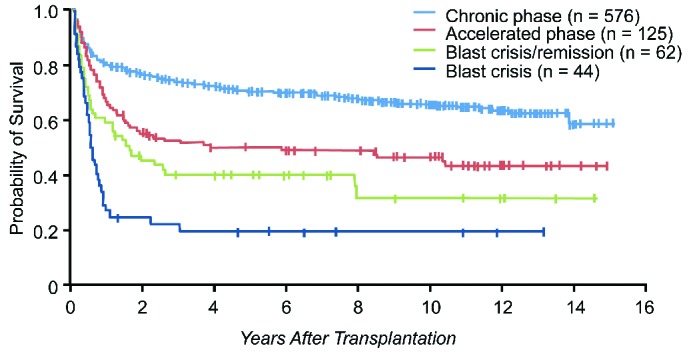
Chronic myeloid leukemia survival after allo-stem cell transplantation. Data from the Fred Hutchinson Cancer Research Center, Seattle. *Includes both matched related and unrelated donors. Patients receiving allografts at the Fred Hutchinson Cancer Research Center from 1995 to the present. Figure is courtesy of Dr Ted Gooley. SCT: stem cell treatment.
Since 1999, the numbers of allografts performed for CML have dramatically decreased, interestingly, some three years prior to the licensing of imatinib for CML. This trend has continued, and the use of allo-SCT is now increasingly being restricted to patients who have failed multiple lines of TKIs, or harbor a T315I mutation and are either ineligible for or have failed ponatinib. Earlier experience in patients who proceed to a transplant after treatment with imatinib did suggest a higher relapse incidence compared with historical patients, but more recent experience did not confirm this.156,157 Current data also suggest that prior treatment with any TKI does not increase the probability of transplant-related mortality. Moreover, patients with kinase domain mutations appear to fare as well post transplant as those lacking such mutations.158 Current challenges include the development of clinical and biological predictors of outcome following relapse post allo-SCT as well as earlier recognition of TKI failures.159 The value of using a TKI after a successful allograft is unknown, particularly as most patients come to transplant having failed 3 or more TKIs. In this regard, it is of interest that the National Comprehensive Cancer Network (NCCN) recommends considering 12 months of standard dose imatinib following allo-SCT.160,161 Finally, it is also reasonable to consider an allo-SCT for patients in the advanced phases of CML, in particular for those who show an initial response to TKI with or without conventional chemotherapy.162,163 In general, responses to TKIs for such patients tend to be short term and the probability of relapse to blast crisis high.
How to stop TKI treatment: the problem of the leukemia stem cells
The great success story of the treatment of CML patients has also brought several related translational and clinical research issues sharply into focus.164 The notion of CML stem cells, while not perfect, has become fairly convincing, and the 15 years of TKI use has confirmed our inability to eliminate them, even with the most potent TKIs.165 Seminal studies demonstrate how these stem cells survive despite virtually complete inhibition of the BCR-ABL1 kinase activity, indicating that they are probably not dependent on BCR-ABL1 for survival.166 It, therefore, begs the question as to whether it is necessary to eliminate CML stem cells for a conventional cure, or whether we should simply accept the ‘operational cure’ offered at present. Clearly a principal goal in cancer medicine is to provide cure and discontinue medication safely and effectively. An operational cure in CML can be defined by sustained molecular remission upon stopping medication. This would be of additional interest due to the impact of TKIs on quality of life, the high cost of these drugs, and of course, many other issues, such as pregnancy and nursing.167 Our best insights are probably provided by the preliminary results from clinical studies of stopping TKIs in patients with CML who were in MR4 or MR4.5 for at least two years. The French Stop Imatinib (STIM), the European Stop Kinase Inhibitor (EURO-SKI) and the Australian CML8 trials probably represent the best efforts so far and have yielded similar results with molecular relapse rates of about 60% within the first six months of stopping TKIs. Results of a smaller study of stopping first-line nilotinib or dasatinib indicate similar findings.168,169 The efforts so far have identified patients with a low Sokal risk score, male sex, and longer duration of imatinib treatment as potential prognostic factors for the maintenance of MR4 or MR4.5 after stopping medication.170
It is, therefore, reasonable to speculate that, for patients with CML, irrespective of achieving a deeper molecular response, additional treatment approaches targeting pathways that regulate the survival and maintenance of CML stem cells might be required to eliminate residual CML stem cells that might contribute to relapse.171 Candidate pathways that appear to be activated by BCR-ABL1 include the JAK-STAT, mTOR, PI3K/AKT and autophagy signaling pathways, and the mechanisms by which CML stem cells interact with their microenvironment (Figure 10).172,173 Studies combining JAK 1/2 inhibitors with TKIs are ongoing, specifically in patients with CML in chronic phase who appear not to have achieved an optimal response to TKIs alone.174 When considering bone marrow microenvironment, it is particularly important to consider the marrow niche, a physico-chemical space that not only protects the stem cells, but also appears to play a major role in the trafficking and retention of these cells via the chemokine receptor CXCR4 and its ligand CXCL12.175–177
Figure 10.

Targeting chronic myeloid leukemia cells at different levels.
The monitoring story
Today, we are able to monitor the quantity of leukemia in an individual patient, following starting treatment with TKI, with considerable precision. First by an examination of the peripheral blood we confirm normalization of the blood count, second by bone marrow metaphase cytogenetic we confirm CCyR and finally by measuring numbers of BCR-ABL1 transcripts in the blood or marrow by quantitative reverse transcriptase PCR (RQ-PCR) we confirm a molecular response (MR). The use of fluorescence in situ hybridization (FISH) to detect a BCR-ABL1 fusion gene in interphase cells is more sensitive than metaphase cytogenetics but much less sensitive than RQ-PCR.
Molecular monitoring was initially developed in 1988 as a qualitative assay to detect early relapse following an allograft and was thereafter replaced by quantitative PCR, which is now generally considered the optimal method for monitoring patients with CML during treatment.178–181 Unfortunately, there remains widespread inconsistency in RQ-PCR results. This is mainly due to interlaboratory differences in technology and methodology employed since molecular monitoring was popularized in the early imatinib era. The RQ-PCR standardization project was started by John Goldman in 2006 in Bethesda, Maryland, to address some of these challenges.182 The results are expressed as a ratio of BCR-ABL1 copy numbers to copy numbers of a control gene (× 100% on a log scale) or as a log10 reduction from standardized value of 0 for untreated patients. In practice, the recommended way of expression is to use a laboratory-specific conversion factor to convert the value obtained in a given laboratory to a value of the International Scale (IS), where 100% is the value for a specific cohort of untreated patients studied in 2002, based on 30 newly diagnosed patients with CML in the chronic phase, tested in three laboratories.183 Patients who achieve a transcript number of 0.1% on the IS, which is equivalent to a 3-log reduction from the baseline for untreated patients, are said to have achieved a MR3, and those without detectable transcripts have achieved a MR4.5, as discussed in the section on treatment (Table 3).184
Despite the many efforts towards harmonization of molecular methods, widespread inconsistency remains.185 It is likely that some of the intrinsic difficulties related to the complex time consuming methodology which requires specialized skills and knowledge may be overcome by the new automated BCR-ABL1 assay that is contained within a single-use microfluidic cartridge, using a specialized instrument, such as the Cepheid GeneXpert.186 It is of interest that this specialized equipment was initially developed for bioterrorism assays following the anthrax attacks in the USA soon after the September 11 attacks in 2001. In this system, RNA extraction and real-time PCR is prepared. This system incorporates conversion to the BCR-ABL1 international reporting scale and has the same sensitivity as usual quantitative methods. This system is especially attractive for hospitals where only sporadic CML cases are treated.
Further improvements include the introduction of digital PCR; in particular with regards to the assessment of the impact of deep molecular response, which is increasingly recognized as an effective clinical strategy to allow for discontinuing TKI therapy safely in some patients, even in the presence of minimal residual disease.187 This is a conceptual approach where a sample is partitioned into thousands of separate reactions. This partitioning can be performed either by sorting into different reaction wells by pumps and valves (Fluidigm), or by diluting the sample into separate micelles (BioRad). Either method seems to increase sensitivity by over a log compared to conventional RQ-PCR. This powerful digital tool appears to be particularly attractive to help improve efforts to discontinue TKI therapy safely in candidate patients who have been in MR4, MR4.5 or MR5. It is likely that further improvements will be made by the application of the next generation DNA sequencing approaches.188 Conversely, many efforts are addressing suitable methodology and technology for wider and, importantly, more affordable use of RQ-PCR.189
Another important test in molecular monitoring of CML patients is BCR-ABL1 kinase domain mutation analysis in those who have acquired TKI resistance and who might require an alternative treatment, or those who progress to advanced phase disease. This test also helps to determine the clinical consequences of clonal diversity of BCR-ABL1 and the co-existence of subclones. Next generation sequencing (NGS) techniques appear to be superior to the current Sanger sequencing, in particular for the identification of compound mutations, which might be more frequent in acquired resistance to 2nd- and 3rd-generation TKIs.190 Compound mutations are two or more mutations in the same BCR-ABL1 allele, and not multiple clones with different mutations. It is of interest that while over 100 different point mutations have now been described, only 12 positions appear to be involved in compound mutations, many of which include the T315I mutation. New technologies incorporating computer modeling help us understand how the leukemic cells develop clever tactics to evade selective pressures of the more potent TKIs, in particular ponatinib, and result in structural changes which limit or exclude TKI binding.132,133
Expert panel definition of response and failure to respond to TKI treatment
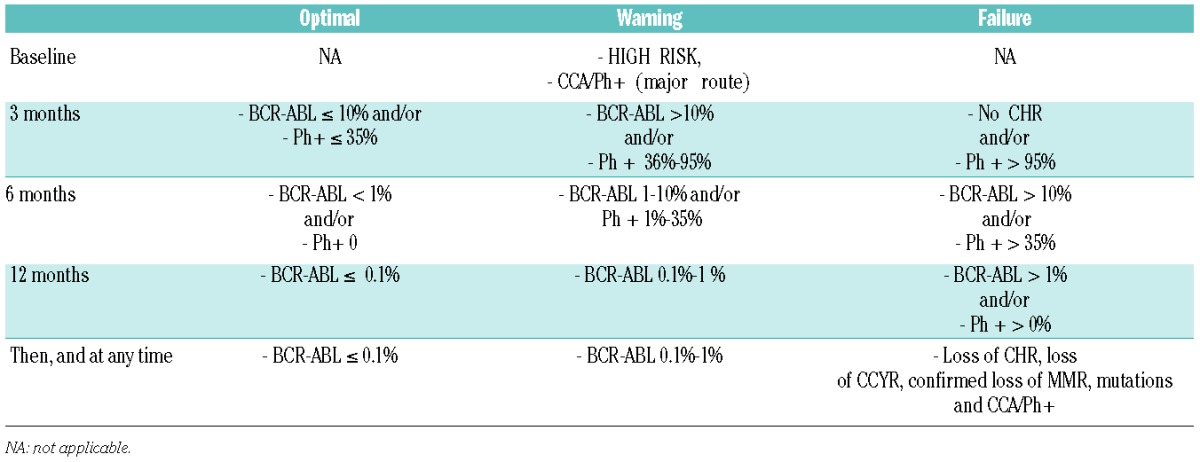
The recommendations of an expert panel of hematologists convened under the aegis of the European LeukemiaNet (ELN) up-dated a series of recommendations in 2013, designed to help the clinician in optimal management of CML and to benefit from the 15 years of experience with TKI treatment in patients with CML (Table 4).191 The panel had been providing such recommendations since 2005 when the original consensus report was collated. They recommended as initial treatment with imatinib, nilotinib or dasatinib, with response being assessed by RQ-PCR and/or conventional cytogenetics at three, six and 12 months. Optimal response was defined as BCR-ABL1 transcript levels of less than 10% at three months, less than 1% at six months, and less than 0.1% from 12 months onwards, whereas more than 10% at six months and more than 1% from 12 months onward define failure and required alternative treatment. Interestingly, the panel also considered a partial cytogenetic response (PCyR) at three months and a CCyR from six months onwards as comprising optimal response, whereas no cytogenetic response at three months, less than PCyR at six months, and less than CCyR from 12 months onward define failure. This reflects the opinions of several other CML experts, highlighting the notion of a CCyR, rather than a deep molecular response, being associated with survival.5,192 The panel felt that despite the notion of an early molecular response being a clear predictor of outcome and impending risk, a change of therapy was not mandatory since current studies do not suggest that a change of therapy at three months changes the outcome. In tandem, some experts consider that halving BCR-ABL1 transcripts within 76 days together with the achievement of a BCR-ABL1 less than 10% by six months, in addition to a CCyR at one year and beyond, may be reasonable milestones for change of therapy.193–196
Table 4.
European LeukemiaNet 2013 Guidelines: response to first-line treatment with imatinib, nilotinib or dasatinib.
Future prospects and conclusions
The CML success story has unfolded over a relatively short period of time and the efforts of Janet Rowley and John Goldman have been crucial to our understanding of the biology of what is now considered a genetically simple cancer. Their work has provided vital insights that have resulted in the success of molecularly targeted therapy, not only for CML patients, but also for other malignancies.197 Two decades since Brian Druker’s initial studies with imatinib, a personalized treatment algorithm is available for the newly diagnosed patient with CML. Treatment involves a choice of three first-line orally administered drugs and two effective next-line therapies that should be used based on risk stratification, co-morbidities, the side-effects profile and the BCR-ABL1 genotype. Furthermore, drug access and the cost of TKI therapy are significant issues on the agenda of world-wide healthcare, given the increased prevalence of CML across the globe.165,198 Currently, there is little difference in the pricing structure of the licensed first-line drugs, but this should change dramatically now that generic imatinib becomes available as the patent for Gleevec (imatinib mesylate) expired in the US in 2015 and in Europe expires in 2016. Regardless of the initial choice of TKI, the vast majority of patients achieve a durable CCyR, with a lifespan approaching that of the general population. In most instances the medication must be continued indefinitely, and a principal challenge now is to develop strategies to stop TKIs safely and effectively. For the moment, it is probably best to discontinue the TKI therapy only within the framework of a clinical trial.
It seems crucial to improve our understanding of the various resistance mechanisms, in particular the emerging role of the bone marrow microenvironment and stem cell niche, and to assess the importance of the persistence of BCR-ABL1 by PCR, even in patients who have confirmed MR4.5 and beyond.199 Challenges also remain in the optimal monitoring of patients with CML on treatment, in particular with regard to the interlaboratory discrepancy in results, and indeed, harmonizing results to the international standard. The monitoring technology would also benefit from being further simplified, and importantly, by being more affordable. Last, but not least, the ELN 2013 recommendations should be up-dated to harmonize expert opinions. A recent French report expressed some concern in not being able to validate the current recommendations with regards to identifying optimal response, though treatment failure was confirmed for a cohort of 180 patients being treated with imatinib.200 Our understanding of the mechanisms and treatment of patients with advanced phase disease remains limited. In addition, questions remain with regard to the initiating biological event, at least in some patients with CML in chronic phase.201 Clearly, the CML story is richly studded with insight, innovation and scientific breakthroughs. Arguably, however, there is much work to be done in order to pay tribute to, and to continue the story that was initiated by Janet Rowley and John Goldman.
Acknowledgments
The authors wish to thank Alpine Oncology Foundation, in particular Dr. Alpa Parmar, for organizing the Janet Rowley and John Goldman Special Colloquium during the 19th EHA Meeting and Novartis Oncology Global for their support. TIM wishes to thank Professor Bob Lowenberg for his mentorship and help in arranging this event, Professor Christine Chomienne (President of EHA-2014) for her opening address and the International CML Foundation. JFA acknowledges the support of NIHR Biomedical Research Centre funding
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/101/5/541
References
- 1.Ben-Neriah Y, Daley GQ, Mes-Masson AM, et al. The chronic myelogenous leukemia-specific P210 protein is the product of the bcr/abl hybrid gene. Science. 1986;233(4760):212–214. [DOI] [PubMed] [Google Scholar]
- 2.Goldman JM, Melo JV. Targeting the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344(14):1084–1086. [DOI] [PubMed] [Google Scholar]
- 3.Druker BJ, Tamura S, Buchdunger E, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2(5):561–566. [DOI] [PubMed] [Google Scholar]
- 4.Druker BJ, Guilhot F, O’Brien SG, et al. Five-year Follow-up of Patients Receiving Imatinib for Chronic Myeloid Leukemia. N Engl J Med. 2006;355(23):2408–2417. [DOI] [PubMed] [Google Scholar]
- 5.Kantarjian H, Cortes JE. Complete cytogenetic response, not deep molecular response, is associated with survival in chronic myeloid leukemia. J Clin Oncol. 2014;32(27):3077. [DOI] [PubMed] [Google Scholar]
- 6.Huang XL, Cortes J, Kantarjian H. Estimations of the increasing prevalence and plateau prevalence of chronic myeloid leukemia in the era of tyrosine kinase inhibitor therapy. Cancer. 2012;118(12): 3123–3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mughal TI, Cortes J, Cross NCP, et al. Chronic myeloid leukemia – some topical issues. Leukemia. 2007;21(7):1347–1352. [DOI] [PubMed] [Google Scholar]
- 8.Falchi L, Kantarjian HM, Wang X, et al. Significance of deeper molecular responses in patients with chronic myeloid leukemia in early chronic phase treated with tyrosine kinase inhibitors. Am J Hematol. 2013;88(12):1024–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mughal TI, Goldman JM. An essay in cancer medicine: Lessons learned from patients with chronic myeloid leukaemia. Clin Med. 2005;1:2–7. [Google Scholar]
- 10.Cheah CY, Burbury K, Apperley JF, et al. Patients with myeloid malignancies bearing PDGFRB fusion genes achieve durable long-term remissions with imatinib. Blood. 2014;123(23):3574–3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Velpeau A. [Sur la resorption du puseat sur l’alteration du sang dans les maladies Clinique de persection nenemant.] Premier observation. Rev Med. 1827;2:216. [Google Scholar]
- 12.Bennett JH. Case of hypertrophy of the spleen and liver in which death took place from suppuration of the blood. Edinb Med Surg J. 1845;64:413–423. [Google Scholar]
- 13.Virchow R. [Weisses Blut]. Froriep’s Notzien 1845;36:151–156. [Google Scholar]
- 14.Tjio JH, Levan A. The chromosome number of man. Hereditas. 1956;42:1–6. [Google Scholar]
- 15.Lejeune J, Turpin R, Gautier M. [Chromosomic diagnosis of mongolism]. Archives Francaises de Pediatrie. 1959;16: 962–963. [PubMed] [Google Scholar]
- 16.Nowell PC, Hungerford DA. A minute chromosome in human granulocytic leukemia. Science. 1960;132:1497. [Google Scholar]
- 17.Baikie AG, Court-Brown WM, Buckton KE, Harnden DG, Jacobs PA, Tough IM. A possible specific chromosome abnormality in human chronic myeloid leukaemia. Nature. 1960;188:1165–1166. [DOI] [PubMed] [Google Scholar]
- 18.Boveri T. [Frage der Entstehung maligner Tumoren.] Jena: Gustav Fischer;1914. [Google Scholar]
- 19.Fialkow PJ, Garler SM, Yoshida A. Clonal origin of chronic myelocytic leukemia in man. Proc Natl Acad Sci USA. 1967;58(4): 1468–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ohno S, Makino S. The single X-nature of sex chromatin in man. Lancet. 1961;1(7168):78–79. [DOI] [PubMed] [Google Scholar]
- 21.Rowley JD. Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973;243(5405):290–293. [DOI] [PubMed] [Google Scholar]
- 22.Abelson HT, Rabstein LS. Lymphosarcoma: virus-induced thymic-independent disease in mice. Cancer Res. 1970;30(8):2213–2222. [PubMed] [Google Scholar]
- 23.Groffen J, Stephenson JR, Heisterkamp N, de Klein A, Bartram CR, Grosveld G. Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome 22. Cell. 1984;36(1):93–99. [DOI] [PubMed] [Google Scholar]
- 24.Heisterkamp N, Groffen J, Stephenson JR, et al. Chromosomal localization of human cellular homologues of two viral oncogenes. Nature. 1982;299(5885):747–749. [DOI] [PubMed] [Google Scholar]
- 25.de Klein A, van Kessel AG, Grosveld G, et al. A cellular oncogene is translocated to the Philadelphia chromosome in chronic myelocytic leukaemia. Nature. 1982;300(5894): 765–767. [DOI] [PubMed] [Google Scholar]
- 26.Bartram CR, de Klein A, Hagemeijer A, et al. Translocation of c-abl oncogene correlates with the presence of a Philadelphia chromosome in chronic myelocytic leukaemia. Nature. 1983;306(5940):277–280. [DOI] [PubMed] [Google Scholar]
- 27.Stam K, Heisterkamp N, Grosveld G, et al. Evidence of a new chimeric bcr/c-abl mRNA in patients with chronic myelocytic leukemia and the Philadelphia chromosome. N Engl J Med. 1985;313(23):1429–1433. [DOI] [PubMed] [Google Scholar]
- 28.Shtivelman E, Lifshitz B, Gale RP, et al. Alternative splicing of RNAs transcribed from the human abl gene and from the bcr-abl fused gene. Cell. 1986;47(2):277–284. [DOI] [PubMed] [Google Scholar]
- 29.Witte ON, Dasgupta A, Baltimore D. Abelson murine leukaemia virus rotein is phosphorylated in vitro to form phosphotyrosine. Nature. 1980;283(5750):826–831. [DOI] [PubMed] [Google Scholar]
- 30.Prywes R, Foulkes JG, Baltimore D. The minimum transforming region of v-abl is the segment encoding protein-tyrosine kinase. J Virol. 1985;54(1):114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Konopka JB, Watanabe SM, Witte ON. An alteration of the human c-abl protein in K562 leukemia cells unmasks associated tyrosine kinase activity. Cell. 1984;37(3): 1035–1042. [DOI] [PubMed] [Google Scholar]
- 32.Daley GQ, Baltimore D. Transformation of an interleukin 3-dependent hematopoetic cell line by chronic myelogenous leukemia-specific P210 bcr/abl protein. Proc Natl Acad Science. 1988;85(23):9312–9316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lugo TG, Pendergast AM, Muller AJ, et al. Tyrosine kinase activity and transformation potency of bcr-abl oncogene products. Science. 1990;247(4946):1079–1082. [DOI] [PubMed] [Google Scholar]
- 34.Daley GQ, van Etten, Baltimore D. Induction of chronic myelogenous leukemia in mice by P210 BCR/ABL gene of the Philadelphia chromosome. Science. 1990; 247(4944):824–830. [DOI] [PubMed] [Google Scholar]
- 35.Elephanty AG, Hariharan IK, Cory S. Bcr-abl, the hallmark of chronic myeloid leukeima in man, induces multiple hemopoietic neoplasms in mice. EMBO J. 1990;9(4):1069–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kelliher MA, McLaughlin J, Witte ON, Rosenberg N. Induction of a Chrnic myeloid leukemia-like Syndrome in Mice with v-abl and BCR/ABL. Proc Natl Acad Sci USA. 1990;87(17):6649–6653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anafi M, Gazit A, Gilon C, et al. Selective interactions of transforming and normal abl proteins with ATP, tyrosine-copolymer substrates, and tyrphostins. J Biol Chem. 1992;267(7):4518–4523. [PubMed] [Google Scholar]
- 38.Sawyers CL, Callahan W, Witte ON. Dominant negative MYC blocks transformation by ABL oncogenes. Cell. 1992; 70(6):901–910. [DOI] [PubMed] [Google Scholar]
- 39.Pendergast AM, Quilliam LA, Cripe LD, et al. BCR-ABL-induced oncogenesis is mediated by direct interaction with the SH2 domain of the GRB-2 adaptor protein. Cell. 1993;75(1):175–185. [PubMed] [Google Scholar]
- 40.Neshat MS, Raitano AB, Wang HG, et al. The survival function of the Bcr-Abl oncogene is mediated by Bad-dependent and -independent pathways: roles for phosphatidylinositol 3-kinase and Raf. Mol Cell Biol. 2000;20(4):1179–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sillaber C, Gesbert F, Frank DA, et al. STAT5 activation contributes to growth and viability in Bcr/Abl- transformed cells. Blood. 2000;95(6):2118–2125. [PubMed] [Google Scholar]
- 42.Salgia R, Sattler M, Pisick E, et al. p210BCR/ABL induces formation of complexes containing focal adhesion proteins and the protooncogene product p120c-Cbl. Exp Hematol. 1996;24(2):310–313. [PubMed] [Google Scholar]
- 43.Sattler M, Mohi MG, Pride YB, et al. Critical role for Gab2 in transformation by BCR/ABL. Cancer Cell. 2002;1(5):479–492. [DOI] [PubMed] [Google Scholar]
- 44.Zhao C, Blum J, Chen A, et al. Loss of beta-catenin impairs the renewal of normal and CML stem cells in vivo. Cancer Cell. 2007;12(6):528–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dierks C, Beigi R, Guo GR, et al. Expansion of Bcr-Abl-positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer Cell. 2008;14(3):238–249. [DOI] [PubMed] [Google Scholar]
- 46.Neviani P, Harb JG, Oaks JJ, et al. PP2A-activating drugs selectively eradicate TKI-resistant chronic myeloid leukemic stem cells. J Clin Invest. 2013;123(10):4144–4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hurtz C, Hatzi K, Cerchietti L, et al. BCL6-mediated repression of p53 is critical for leukemia stem cell survival in chronic myeloid leukemia. J Exp Med. 2011;208(11):2163–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen Y, Hu Y, Zhang H, et al. Loss of the Alox5 gene impairs leukemia stem cells and prevents chronic myeloid leukemia. Nat Genet. 2009;41(7):783–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.O’Hare T, Zabriskie MS, Eiring AM, et al. Pushing the limits of targeted therapy in chronic myeloid leukaemia. Nat Rev Cancer. 2012;12(8):513–526. [DOI] [PubMed] [Google Scholar]
- 50.Corbin AS, Agarwal A, Loriaux M, et al. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J Clin Invest. 2011;121(1):396–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mughal T, Goldman J. Chronic myeloid leukemia: a historical perspective. In: Mughal T, Goldman J, (eds.) Chronic Myeloproliferative Disorders. Paul Street, London: Informa Healthcare; 2008: pp 1–16. [Google Scholar]
- 52.Dalton DAG. Myelran in chronic myeloid leukaemia. Lancet. 1953;1:208. [DOI] [PubMed] [Google Scholar]
- 53.Kennedy BJ, Yarbo JW. Metabolic and therapeutic effects of hydroxyurea in chronic myelogenous leukemia. Trans Assoc Am Physicians. 1965;78:391–399. [PubMed] [Google Scholar]
- 54.Hehlmann R, Heimpel H, Hasford J, et al. Randomized comparison of busulfan and hydroxyurea in chronic myelogenous leukemia: prolongation of survival by hydroxyurea. The German CML Study Group. Blood. 1993;82(2):398–407. [PubMed] [Google Scholar]
- 55.Talpaz M, Kantarjian H, McCredie K, et al. Hematologic remission and cytogenetic improvement induced by recombinant human interferon alpha A in chronic myelogenous leukemia. N Engl J Med. 1986;314(17):1065–1069. [DOI] [PubMed] [Google Scholar]
- 56.Hehlmann R, Heimpel H, Hasford J, et al. ; on behalf of the German CML Study Group. Randomized comparison of interferon-alpha with busulphan and hydroxyurea in chronic myelogenous leukemia. Blood. 1994;84(12):4064–4077. [PubMed] [Google Scholar]
- 57.The Italian Cooperative Study Group on Chronic Myeloid Leukemia. Interferon alfa-2a as compared with conventional chemotherapy for the treatment of chronic myeloid leukemia. N Engl J Med. 1994; 330(12):820–825. [DOI] [PubMed] [Google Scholar]
- 58.Allan NC, Richards SM, Shepherd PC. UK Medical Research Council randimised, multicentre trial of interferon-alpha n1 for chronic myeloid leukaemia: improved survival irrespective of cytogenetic response. The UK Medical Research Council’s Working Parties for Therapeutic Trials in Adult Leukaemia. Lancet. 1995;345(8962): 1392–1397. [DOI] [PubMed] [Google Scholar]
- 59.Guilhot F, Chastang C, Michallet M, et al. ; on behalf of the French Chronic Myeloid Leukemia Study Group. Interferon alfa-2b combined with cytarabine versus interferon alone in chronic myelogenous leukemia. N Engl J Med. 1997;337(4):223–329. [DOI] [PubMed] [Google Scholar]
- 60.Silver RT, Woolf SH, Hehlmann R, et al. An evidence-based analysis of the effect of busulfan, hydoxyurea, interferon, and allogeneic bone marrow transplantation in treating the chronic phase of chronic myeloid leukemia: developed for the American Society of Hematology. Blood. 1999;94(5):1517–1536. [PubMed] [Google Scholar]
- 61.Druker BJ, Talpaz M, Resta DJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344(14):1031–1037. [DOI] [PubMed] [Google Scholar]
- 62.Mughal TI, Goldman JM. Chronic myeloid leukaemia: A therapeutic challenge. Ann Oncol. 1995;6(7):637–644. [DOI] [PubMed] [Google Scholar]
- 63.Mughal TI, Goldman JM. Chronic Myeloid leukemia: Current Status and Controversies. Oncology (Williston Park). 2004;18(7):837–847. [PubMed] [Google Scholar]
- 64.Sokal JE, Cox EB, Baccarani M, et al. Prognostic discrimination in “good-risk” chronic granulocytic leukemia. Blood. 1984;63(4):789–799. [PubMed] [Google Scholar]
- 65.Hasford J, Pfirrmann M, Hehlmann R, et al. A new prognostic score for survival of patients with chronic myeloid leukaemia treated with interferon alpha. J Natl Cancer Inst. 1998;90(11):850–858. [DOI] [PubMed] [Google Scholar]
- 66.Hoffman VS, Baccarani M, Lindoerfer D, et al. The EUTOS prognostic score: review and validation in 1288 patients with CML treated frontline with imatinib. Leukemia. 2013;27(10):2016–2022. [DOI] [PubMed] [Google Scholar]
- 67.Mughal TI, Barbui T, Abdel-Wahab O, et al. Novel insights into the biology and treatment of chronic myeloproliferative neoplasms. Leuk Lymphoma. 2015;56(7):1938–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Druker BJ, Lydon NB. Lessons learned from the development of an abl tyrosine kinase inhibitor for chronic myelogenous leukemia. J Clin Invest. 2000;105(1):3–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Deininger MW, Buchdunger E, Druker BJ. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood. 2005;105(7):2640–2653. [DOI] [PubMed] [Google Scholar]
- 70.Kantarjian H, Sawyers C, Hochhaus A, et al. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N Engl J Med. 2002;346(9):645–652. [DOI] [PubMed] [Google Scholar]
- 71.Hochhaus A, Druker B, Sawyers C, et al. Favorable long-term follow-up results over 6 years for response, survival, and safety with imatinib mesylate therapy in chronic-phase chronic myeloid leukemia after failure of interferon-alpha treatment. Blood. 2008;111(3):1039–1043. [DOI] [PubMed] [Google Scholar]
- 72.Deininger MW, O’Brien SG, Guilhot F, et al. International Randomized Study of Interferon vs STI571 (IRIS) 8 years Followup: Sustained Survival and Low Risk for Progression or Events in Patients with Newly Diagnosed Chronic Myeloid Leukemia in Chronic Phase Treated with Imatinib. ASH Annual Meeting Abstracts 2009;114:1126–. [Google Scholar]
- 73.Baccarani M, Soverini S. Molecular Response in CML: Where is the bar? Blood. 2014;124(4):469–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mughal TI, Vannucchi AM, Soverini S, et al. Preclinical and clinical issues in chronic myeloproliferative neoplasms. Haematologica. 2014;99(5):797–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kantarjian H, O’Brien S, Jabbour E, et al. Improved survival in chronic myeloid leukemia since the introduction of imatinib: a single-institution historical experience. Blood. 2012;119(9):1981–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kantarjian H, Talpaz M, O’Brien S, et al. High-dose imatinib mesylate therapy in newly diagnosed Philadelphia chromosome-positive chronic phase chronic myeloid leukemia. Blood. 2004;103(8):2873–2878. [DOI] [PubMed] [Google Scholar]
- 77.Cortes JE, Kantarjian HM, Goldberg SL, et al. High-dose imatinib in newly diagnosed chronic-phase chronic myeloid leukemia: high rates of rapid cytogenetic and molecular responses. J Clin Oncol. 2009;27(28): 4754–4759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cortes JE, Baccarani M, Guilhot F, et al. Phase III, randomized. Open-label study of daily imatinib mesylate 400mg versus 800mg in patients with newly diagnosed, previously untreated chronic myeloid leukemia in chronic phase using molecular end points: tyrosine kinase inhibitor optimization and selectivity study. J Clin Oncol. 2010;28(3):424–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hehlmann R, Muller MC, Lauseker M, et al. Deep Molecular Response Is Reached by the Majority of Patients Treated With Imatinib, Predicts Survival, and Is Achieved More Quickly by Optimized High-Dose Imatinib: Results From the Randomized CML-Study IV. J Clin Oncol. 2014;32(5):415–423. [DOI] [PubMed] [Google Scholar]
- 80.Proetel U, Pletsch N, Lauseker M, et al. Older patients with chronic myeloid leukemia (>65 years) profit more from higher imatinib doses than younger patients: a subanalysis of the randomized CML-Study IV. Ann Hematol. 2014;93(7):1167–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Deininger MW, Kopecky KJ, Radich JP, et al. Imatinib 800mg daily induces deeper molecular responses than imatinib 400mg daily: results of SWOG S0325, an intergroup randomized PHASE II trial in newly diagnosed chronic phase chronic myeloid leukaemia. Br J Haematol. 2014;164(2):223–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Preudhomme C, Guilhot J, Nicolini FE, et al. Imatinib plus peginterferon alfa-2a in chronic myeloid leukemia. N Engl J Med. 2010;363(26):2511–2521. [DOI] [PubMed] [Google Scholar]
- 83.Gambacorti-Passerini C, Antolini L, Mahon FX, et al. Multicenter independent assessment of outcomes in chronic myeloid leukemia patients treated with imatinib. J Nat Cancer Inst. 2011;103(7):553–561. [DOI] [PubMed] [Google Scholar]
- 84.Mughal TI, Schreiber A. Long term toxicity of imatinib when used as a first-line therapy in chronic myeloid leukemia. Biologics. 2010;4:315–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Efficace F, Baccarani M, Breccia M, et al. Chronic fatigue is the most important factor limiting health-related quality of life of chronic myeloid leukemia patients treated with imatinib. Leukemia. 2013;27(7):1511–1519. [DOI] [PubMed] [Google Scholar]
- 86.Gambacorti-Passerini C, Tornaghi L, Cavagnini F, et al. Gynaecomastia in men with chronic myeloid leukaemia after imatinib. Lancet. 2003;361(9373):1954–1956. [DOI] [PubMed] [Google Scholar]
- 87.Verma D, Kantarjian H, Strom SS, et al. Malignancies occurring during therapy with tyrosine kinase inhibitors (TKIs) for chronic myeloid leukemia (CML) and other hematologic malignancies. Blood. 2011;118(16): 4353–4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.de Lavallade H, Apperley JF, Khorashad JS, et al. Imatinib for newly diagnosed patients with chronic myeloid leukemia: incidence of sustained responses in an intention-to-treat analysis. J Clin Oncol. 2008;26(20):3358–3363. [DOI] [PubMed] [Google Scholar]
- 89.Lucas CM, Wang L, Austin GM, et al. A population study of imatinib in chronic myeloid leukemia demonstrates lower efficacy than in clinical trials. Leukemia. 2008;22(10): 1963–1966. [DOI] [PubMed] [Google Scholar]
- 90.Apperley JF. Chronic myeloid leukaemia. Lancet. 2015;385(9976):1447–1459. [DOI] [PubMed] [Google Scholar]
- 91.Gorre ME, Mohammed M, Ellwood K, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293(5531):876–680. [DOI] [PubMed] [Google Scholar]
- 92.Apperley JF. Part I: mechanisms of resistance to imatinib in chronic myeloid leukemia. Lancet Oncol. 2007;8(11):1018–1029. [DOI] [PubMed] [Google Scholar]
- 93.Khorashad JS, de Lavallade H, Apperley JF, et al. Finding of kinase domain mutations in patients with chronic phase chronic myeloid leukemia responding to imatinib may identify those at risk of disease progression. J Clin Oncol. 2008;26(29):4806–4813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Soverini S, Branford S, Nicolini FE, et al. Implications of BCR-ABL1 kinase domain-mediated resistance in chronic myeloid leukemia. Leuk Res. 2014;38(1):10–20. [DOI] [PubMed] [Google Scholar]
- 95.Eiring AM, Deininger MW. Individualizing kinase-targeted cancer therapy: the paradigm of chronic myeloid leukemia. Genome Biol. 2014;15(9):461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Watkins DB, Hughes TP, White DL. OCT1 and imatinib transport in CML: is it clinically relevant? Leukemia. 2015;29(10):1960–1969. [DOI] [PubMed] [Google Scholar]
- 97.Goldman JM. How I treat chronic myeloid leukemia in the imatinib era. Blood. 2007;110(8):2828–2837. [DOI] [PubMed] [Google Scholar]
- 98.Weisberg E, Manley PW, Breitenstein W, et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell. 2005;7(2):129–141. [DOI] [PubMed] [Google Scholar]
- 99.Bradeen HA, Eide CA, O’Hare T, et al. Comparison of imatinib mesylate, dasatinib (BM-354825) and nilotinib (AMN107) in an N-ethyl-N-nitrosourea (ENU)-based mutagenesis screen: high efficacy of drug combinations. Blood. 2006;108(7):2332–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tokarski JS, Newitt JA, Chang CYJ, et al. The structure of dasatinib (BMS-354825) bound to activated ABL kinase domain elucidates its inhibitory activity against imatinib-resitant ABL mutants. Cancer Res. 2006;66(11):5790–5797. [DOI] [PubMed] [Google Scholar]
- 101.Schittenhelm MM, Shiraga S, Schroeder A, et al. Dasatinib (BMS-354825), a dual SRC/ABL kinase inhibitor inhibits the kinase activity of wild-type, juxtamembrane, and activation loop mutant KIT isoforms associated with human malignancies. Cancer Res. 2006;66(1):473–481. [DOI] [PubMed] [Google Scholar]
- 102.Milojkovic D, Apperley JF, Gerrad G, et al. Responses to second-line tyrosine kinase inhibitors are durable: an intention-to-treat analysis in chronic myeloid leukemia patients. Blood. 2012;119(8):1838–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kantarjian H, Giles F, Wunderle L, et al. Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N Engl J Med. 2006;354(24):2542–2551. [DOI] [PubMed] [Google Scholar]
- 104.Giles FJ, le Coutre PD, Pinilla-Ibarz J, et al. Nilotinib in imatinib-resistant or imatinib-intolerant patients with chronic myeloid leukemia in chronic phase: 48-month follow-up results of a phase II study. Leukemia. 2013;27(1):107–112. [DOI] [PubMed] [Google Scholar]
- 105.Talpaz M, Shah NP, Kantarjian H, et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med. 2006;354(24):2531–2541. [DOI] [PubMed] [Google Scholar]
- 106.Hochhaus A, Baccarani M, Deininger M, et al. Dasatinib induces durable cytogenetic responses in patients with chronic myelogenous leukemia in chronic phase with resistance or intolerance to imatinib. Leukemia. 2008;22(6):1200–1206. [DOI] [PubMed] [Google Scholar]
- 107.Shah NP, Kantarjian HM, Kim DW, et al. Intermittent target inhibition with dasatinib with dasatinib 100mg once daily preserves efficacy and improves toerability in imatinib-resistant and – intolerant chronic-phase chronic myeloid leukemia. J Clin Oncol. 2008;26(19):3204–3212. [DOI] [PubMed] [Google Scholar]
- 108.Shah NP, Guilhot F, Cortres JE, et al. Long-term outcome with dasatinib after imatinib failure in chronic-phase chronic myeloid leukemia: follow-up of a phase 3 study. Blood. 2014;123(15):2317–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Brümmendorf TH, Shah NP, Cortes JE, et al. Long-term efficacy and safety of dasatinib 100mg once daily (QD) in patients with imatinib resistant/intolerant chronic-phase chronic myeloid leukemia (CML-CP): 5-year folow-up from CA180-034. Onkologie. 2011;34:263. [Google Scholar]
- 110.Müller MC, Cortes JE, Kim DW, et al. Dasatinib treatment of chronic-phase chronic myeloid leukemia: analysis of responses according to preexisting BCR-ABL mutations. Blood. 2009;114(24):4944–4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Le Coutre P, Rea D, Abruzzese E, et al. Severe peripheral arterial disease during nilotinib therapy. J Natl Cancer Inst. 2011;103(17):1347–1348. [DOI] [PubMed] [Google Scholar]
- 112.Assouline S, Laneuville P, Gambacorti-Passerini C. Panniculitis during dasatinib therapy for imatinib-resistant chronic myelogenous leukemia. N Engl J Med. 2006;354(24):2623–2624. [DOI] [PubMed] [Google Scholar]
- 113.Saglio G, Kim DW, Issaragrisil S, et al. Nilotinib versus imatinib for newly diagnosed chronic mye;loid leukemia. N Engl J Med. 2010;362(24):2251–2259. [DOI] [PubMed] [Google Scholar]
- 114.Kantarjian H, Shah NP, Hochhaus A, et al. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2010;362(24):2260–2270. [DOI] [PubMed] [Google Scholar]
- 115.Larson RA, Hochhaus A, Clark RE, et al. Nilotinib vs imatinib in patients with newly diagnosed Philadelphia chromosome-positive chronic myeloid leukemia in chronic phase: ENESTnd 3-year follow-up. Leukemia. 2012;26(10):2197–2203. [DOI] [PubMed] [Google Scholar]
- 116.Jabbour E, Kantarjian HM, Saglio G, et al. Early response with dasatinib or imatinib in chronic myeloid leukemia: 3-year follow-up from a randomized phase 3 trial (DASISION). Blood. 2014;123(4):494–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tefferi A. Nilotinib treatment-associated accelerated atherosclerosis: when is the risk justified? Leukemia. 2013;27(9):1939–1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Giles FJ, Mauro MJ, Hong F, et al. Rates of peripheral arterial occlusive disease in patients with chronic myeloid leukemia in the chronic phase treated with imatinib, nilotinib, or non-tyrosine kinase therapy: a retrospective cohort analysis. Leukemia. 2013;27(6):1310–1315. [DOI] [PubMed] [Google Scholar]
- 119.Montani D, Bergot E, Gunther S, et al. Pulmonary arterial hypertension in patients treated with dasatinib. Circulation. 2012;125(17):2128–2137. [DOI] [PubMed] [Google Scholar]
- 120.Radich JP, Kopecky KJ, Appelbaum FR, et al. A randomized trial of dasatinib 100mg versus imatinib 400mg in newly diagnosed chronic-phase chronic myeloid leukemia. Blood. 2012;120(19):3898–3905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hughes TP, Saglio G, Kantarjian HM, et al. Early molecular response predicts outcomes in patients with chronic myeloid leukemia in chronic phase treated with frontline nilotinib or imatinib. Blood. 2014;123(9):494–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hughes TP, Lipton JH, Spector N, et al. Deep molecular responses achieved in patients with CML-CP who are switched to nilotinib after long-term imatinib. Blood. 2014;124(5):729–736. [DOI] [PubMed] [Google Scholar]
- 123.Remsing LL, Rix U, Colinge J, et al. Global target profile of the kinase inhibitor bosutinib in primary chronic myeloid cells. Leukemia. 2009;23(3):477–480. [DOI] [PubMed] [Google Scholar]
- 124.Cortes JE, Kantarjian HM, Brummendorf TH, et al. Safety and efficacy of bosutinib (SKI-606) in chronic phase Philadelphia chromosome-positive chronic myeloid leukemia patients with resistance or intolerance to imatinib. Blood. 2011;118(17):4567–4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kantarjian HM, Cortes JE, Kim DW, et al. Bosutinib safety and management of toxicity in leukemia patients with resistance or intolerance to imatinib and other tyrosine kinase inhibitors. Blood. 2014;123(9):1309–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gambacorti-Passerini C, Brummendorf TH, Kim DW, et al. Bosutinib efficacy and safety in chronic phase chronic myeloid leukemia after imatinib resistance or intolerance: minimum 24-month follow-up. Am J Hematol. 2014;89(7):732–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lipton JH, Cortes JE, Khoury HJ, et al. Long-term bosutinib in patients with chronic phase chronic myeloid leukemia after prior imatiniib failure. J Clin Oncol. 2015;33(15): 7076. [Google Scholar]
- 128.Brümmendorf TH, Cortes JE, de Souza CA, et al. Bosutinib versus imatinib in newly diagnsoed chronic-phase chronic myeloid leukaemia: results from the 24-month follow-up of the BELA trial. Br J Haemtol. 2015;168(1):69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.O’Hare T, Shakespeare WC, Zhu X, et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potentially inhibits the T315I mutant and overcomemutation-based resistance. Cancer Cell. 2009; 16(5):401–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Goldman JM. Ponatinib for Chronic Myeloid Leukemia. N Engl J Med 2012; 367(22):2148–2149. [DOI] [PubMed] [Google Scholar]
- 131.Lierman E, Smits S, Cools J, et al. Ponatinib is active against imatinib-resistant mutants of FIP1L1-PDGFRA and KIT, and against FGFR1-derived fusion kinases. Leukemia. 2012;26(7):1693–1695. [DOI] [PubMed] [Google Scholar]
- 132.Zabriskie M, Eide CA, Tantravahisk, et al. BCR-ABL1 compound mutations combining key kinase domain positions confer clinical resistance to ponatinib in Ph chromosome-positive leukemia. Cancer Cell. 2014;26(3): 428–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Deininger MW, Hodgson JG, Shah NP, et al. Compound mutations in BCR-ABL1 are not major drivers of primary or secondary resistance to ponatinib in CP-CML patients. Blood. 2015. November 24 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cortes JE, Kantarjian HM, Shah NP, et al. Ponatinib in refractory Philadelphia chromosome-positive leukemias. N Engl J Med. 2012;367(22):2075–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Cortes JE, Kim D-W, Pinella-Ibarz J, et al. A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N Engl J Med. 2013;369(19):1783–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Neelkantan P, Marin D, Laffan M, et al. Platelet dysfunction associated with ponatinib, a new pan BCR-ABL inhibitor with efficacy for CML resistant to multiple tyrosine kinase inhibitor therapy. Haematologica. 2012;97(9):1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lipton JH, Chuah C, Guerci-Bresler A, et al. EPIC: A phase III trial of ponatinib (PON) versus imatinib (IM) in patients with newly diagnosed CR-CML. Blood. 2014;124(21): 519.24740813 [Google Scholar]
- 138.Gandhi V, Plunkett W, Cortes JE, et al. Omacetaxine: A protein translation inhibitor for treatment of chronic myelogenous leukemia. Clin Cancer Res. 2014;20(7):1735–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Yun SM, Jung KH, Kim SJ, et al. HS-438, a new inhibitor of imatinib-resistant BCR-ABL T315I mutation in chronic myeloid leukemia. Cancer Lett. 2014;348(1–2):50–60. [DOI] [PubMed] [Google Scholar]
- 140.Mughal TI, Kavita R. Port-wine flavoured sandwiches and haematopoetic stem cell transplantation. Lancet Oncology. 2009;10(9):926. [DOI] [PubMed] [Google Scholar]
- 141.Appelbaum FR. Hematopoietic-cell transplantation at 50. N Engl J Med. 2007; 357(15):1472–1475. [DOI] [PubMed] [Google Scholar]
- 142.Mughal T. Chronic leukemias. Classic papers in Hematologic Malignancies. Ed Thomas, Nathan, Goldman Pub: Taylor Francis, London and New York: 2001;139–44. [Google Scholar]
- 143.Goldman JM, Catovsky D, Galton DA. Reversal of blast-cell crisis in C.G.L. by transfusion of stored autologous buffy-coat cells. Lancet. 1978;1(8061):437–438. [DOI] [PubMed] [Google Scholar]
- 144.Goldman JM, Baughan AS, McCarthy DM, et al. Marrow transplantation for patients in the chronic phase of chronic granulocytic leukaemia. Lancet. 1982;2(8299):623–625. [DOI] [PubMed] [Google Scholar]
- 145.Hansen JA, Gooley TA, Martin PJ, et al. Bone marrow transplants from unrelated donors for patients with chronic myeloid leukemia. N Engl J Med. 1998;338(14):962–968. [DOI] [PubMed] [Google Scholar]
- 146.Gluckman E, Broxmeyer HA, Auerbach AD, et al. Hematopoietic reconstitution in a patient with Fanconi’s anemia by means of umbilical-cord blood from an HLA-identical sibling. N Engl J Med. 1989;321(17):1174–1178. [DOI] [PubMed] [Google Scholar]
- 147.Goldman JM, Apperley JF, Jones L, et al. Bone Marrow Transplantation for patients with Chronic Myeloid Leukemia. N Engl J Med. 1986;314(4):202–207. [DOI] [PubMed] [Google Scholar]
- 148.Apperley JF, Jones J, Hale G, et al. Bone marrow transplantation for patients with chronic myeloid leukaemia: T-cell depletion with Campath-1 reduces the incidence of graft-versus-host disease but may increase the risk of leukaemic relapse. Bone Marrow Transplant. 1986;1(1):53–66. [PubMed] [Google Scholar]
- 149.Innes AJ, Milojkovic D, Apperley JF. Allogeneic transplantation for CML in the TKI era: striking the right balance. Nat Rev Clin Oncol. 2015. November 17 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 150.Kolb HJ, Mittermüller J, Clemm C, et al. Donor leukocyte transfusions for treatment of recurrent chronic myelogenous leukemia in marrow transplant patients. Blood. 1990;76(12):2462–2465. [PubMed] [Google Scholar]
- 151.McKinnon S, Papadopoulos EB, Carabasi MH, et al. Adoptive immunotherapy evalutaing escalating doses of donor leukocytes for relapse of chronic myeloid leukemia after bone marrow transplantation: separation of graft-versus-leukemia responses from graft-versus-host disease. Blood. 1995; 86(4):1261–1268. [PubMed] [Google Scholar]
- 152.Gratwohl A, Hermans J, Goldman JM, et al. Risk assessment for patients with chronic myeloid leukemia before allogeneic blood or marrow transplantation. Lancet. 1998;352(9134):1087–1082. [DOI] [PubMed] [Google Scholar]
- 153.Crawley C, Szydlo R, Lalancette M, et al. Outcomes of reduced-intensity transplantation for chronic myeloid leukemia: an analysis of prognostic factors from the Chronic Leukemia Working Party of the EBMT. Blood. 2005;106(9):2969–2976. [DOI] [PubMed] [Google Scholar]
- 154.Mughal TI, Yong A, Szydlo R, et al. The probability of long-term leukemia free survival for patients in molecular remission 5 years after allogeneic stem cell transplantation for chronic myeloid leukemia in chronic phase. Br J Haematol. 2001;115(3):569–574. [DOI] [PubMed] [Google Scholar]
- 155.Radich JP, Gooley T, Bryant E, et al. The significance of bcr-abl molecular detection in chronic myeloid leukemia patients “late,” 18 months or more after transplantation. Blood. 2001;98(6):1701–1707. [DOI] [PubMed] [Google Scholar]
- 156.Sauselle S, Lauseker M, Gratwohl A, et al. Allogeneic hematopoietic stem cell transplantation (allo SCT) for chronic myeloid leukemia in the imatinib era: evaluation of its impact within a subgroup of the randomized German CML Study IV. Blood. 2010;115(10):1880–1885. [DOI] [PubMed] [Google Scholar]
- 157.Oyekunle A, Zander A, Binder M, et al. Outcome of allogeneic SCT in patients with chronic myeloid leukemia in the era of tyrosine kinase inhibitor therapy. Ann Hematol. 2013;92(4):487–496. [DOI] [PubMed] [Google Scholar]
- 158.Jabbour E, Cortes JE, Santos FP, et al. Results of allogeneic hematopoietic stem cell transplantation for chronic myelogenous leukemia patients who failed tyrosine kinase inhibitors after developing BCR-ABL1 kinase domain mutations. Blood. 2011;117(13):3641–3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Jain NA, Ito S, Tian X, et al. Clinical and biological predictors of outcome following relapse of CML post-allo-SCT. Bone Marrow Transplant. 2015;50(8):1138–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Shimoni A, Volchek Y, Koren-Michowitz M, et al. Phase 1/2 study of nilotinib prophylaxis after allogeneic stem cell transplantation in patients with advanced chronic myeloid leukemia or Philadelphia chromosome-positive acute lymphoblastic leukemia. Cancer. 2015;121(6):863–871. [DOI] [PubMed] [Google Scholar]
- 161.O’Brien S, Abboud CN, Akhtari M, et al. Clinical Practice Guidelines in Oncology. Chronic Myelogenous Leukemia, Version 3. National Comprehensive Cancer Network; [Last accessed June 2 2014]. [Google Scholar]
- 162.Sawyers CL, Hochhaus A, Feldman E, et al. Imatinib induces hematologic and cytogenetic responses in patients with chronic myelogenous leukemia in myeloid blast crisis: results of a phase II study. Blood. 2002;99(10):3530–3539. [DOI] [PubMed] [Google Scholar]
- 163.Apperley JF, Cortes JE, Kim DW, et al. Dasatinib in the treatment of chronic myeloid leukemia in accelerated phase after imatinib failure: the START-A trial. J Clin Oncol. 2009;27(21):3472–3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Bjorkholm M, Ohm L, Eloranta S, et al. Success story of targeted therapy in chronic myeloid leukemia: a population-based study of patients diagnosed in Sweden from 1973 to 2008. J Clin Oncol. 2011;29(18):2514–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Gallipoli P, Abraham SA, Holyoake TL. Hurdles Toward a Cure for CML: The CML Stem Cell. Hematol Oncol Clin North Am. 2011;25(5):951–966. [DOI] [PubMed] [Google Scholar]
- 166.Copland M, Hamilton A, Elrick LJ, et al. Dasatinib (BMS-354825) targets an earlier progenitor population than imatinib in primary CML but does not eliminate the quiescent fraction. Blood. 2006;107(11):4532–4539. [DOI] [PubMed] [Google Scholar]
- 167.CML Experts. The price of drugs for chronic myeloid leukemia (CML) is a reflection of the unsustainable prices of cancer drugs: from the perspective of a large group of CML experts. Blood. 2013;121(22):4439–4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Mahon F, Rea D, Guilhot J, et al. Discontinuation of imatinib in patients with chronic myeloid leukemia who have maintained complete molecular remission for at least 2 years: the prospective multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010;11(11):1029–1035. [DOI] [PubMed] [Google Scholar]
- 169.Ross DM, Branford S, Seymour JF, et al. Safety and efficacy of imatinib cessation for CML patients with stable undetectable minimal residual disease: results from the TWISTER study. Blood. 2013;122(4):515–522. [DOI] [PubMed] [Google Scholar]
- 170.Branford S, Yeung DT, Ross DM, et al. Early molecular response and female sex strongly predict stable undetectable BCR-ABL1, the criteria for imatinib discontinuation in patients with CML. Blood. 2013;121(19): 3818–3824. [DOI] [PubMed] [Google Scholar]
- 171.Ahmed W, van Etten RA. Alternative approaches to eradicating the malignant clone in chronic myeloid leukemia: tyrosine-kinase inhibitor combinations and beyond. Hematology Am Soc Hematol Educ Program. 2013;2013:189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.O’Hare T, Zabriske MS, Eiring AM, Deininger MW. Pushing the limits of targeted therapy in chronic myeloid leukemia. Nat Rev Cancer. 2012;12(8):513–526. [DOI] [PubMed] [Google Scholar]
- 173.Bhatia R. Altered microenvironment regulation of CML stem cells. Leuk Supplements. 2014;S1–S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Gallipoli P, Cook A, Rhodes S, et al. JAK2/STAT5 inhibition by nilotinib with ruxolitinib contributes to the elimination of chronic phase CML CD34+ cells in vitro and in vivo. Blood 2014;124(9):1492–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zhang B, Ho YW, Maeda T, et al. Altered microenvironmental regulation of leukemic and normal stem cells in chronic myelogenous leukemia. Cancer Cell. 2012;21(4):577–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Weisberg E, Azab AK, Manley PW, et al. Inhibition of CXCR4 in CML cells disrupts their interaction with the bone marrow microenvironment and sensitizes them to nilotinib. Leukemia. 2012;26(5):985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Krause DS, Scadden DT. A hostel for the hostile: the bone marrow niche in hematologic neoplasms. Haematologica. 2015;100(11):1376–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Kawasaki ES, Clark SS, Coyne MY, et al. Diagnosis of chronic myeloid and acute lymphocytic leukemias by detection of leukemia-specific mRNA sequences amplified in vitro. Proc Natl Acad Sci USA. 1988;85(15):5698–5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Sawyers CL, Timpson L, Kawasaki ES, Clark SS, Witte ON, Champlin R. Molecular relapse in chronic myelogenous leukemia patients after bone marrow transplantation detected by polymerase chain reaction. Proc Natl Acad Sci. 1990;87(2):563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Hughes TP, Morgan GJ, Martiat P, Goldman JM. Detection of residual leukemia after bone marrow transplant for chronic myeloid leukemia: role of polymerase chain reaction in predicting relapse. Blood. 1991;77(4):874–878. [PubMed] [Google Scholar]
- 181.Cross NC, Feng L, Chase A, Bungey J, Hughes TP, Goldman JM. Competitive polymerase chain reaction to estimate the number of BCR-ABL transcripts in chronic myeloid leukemia patients after bone marrow transplantation. Blood. 1993;82(6): 1929–1936. [PubMed] [Google Scholar]
- 182.Hughes T, Deininger M, Hochhaus A, et al. Monitoring CML patients responding to treatment with tyrosine kinase inhibitors: review and recommendations for harmonizing current methodology for detecting BCR-ABL transcripts and kinase domain mutations and for expressing results. Blood. 2006;108(1):28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Hughes TP, Kaeda J, Branford S, et al. Frequency of major molecular responses to imatinib or interferon alfa plus cytarabine in newly diagnosed chronic myeloid leukemia. N Engl J Med. 2003;349(15):1423–1432. [DOI] [PubMed] [Google Scholar]
- 184.Cross NCP, White HE, Müller MC, Saglio G, Hochhaus A. Standardized definitions of molecular response in chronic myeloid leukemia. Leukemia. 2012;26(10):2172–2175. [DOI] [PubMed] [Google Scholar]
- 185.Cross NCP, White HE, Colomer D, et al. Laboratory recommendations for scoring deep molecular responses following treatment for chronic myeloid leukemia. Leukemia 2015;29(5):999–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Winn-Deen ES, Helton B, Van Atta R, et al. Development of an integrated assay for detection of Bcr-Abl RNA. Clin Chem. 2007;53(9):1593–1600. [DOI] [PubMed] [Google Scholar]
- 187.Oehler VG, Wuin J, Ramakrishnan R, et al. Absolute quantitative detection of ABL tyrosine kinase domain point mutations in chronic myeloid leukemia using a novel nanofluidic platform and mutation-specific PCR. Leukemia. 2009;23(2):396–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Ross D, Branford S, Seymour JF, et al. Patients with chronic myeloid leukemia who maintain a complete molecular remission after stopping imatinib treatment have evidence of persistent leukemia by DNA PCR Leukemia. 2010;24(10):1719–1724. [DOI] [PubMed] [Google Scholar]
- 189.LaBarre P, Hawkins KR, Gerlach J, et al. A simple, inexpensive device for nucleic acid amplification without electricity-toward instrument-free molecular diagnostics in low-resource settings. PLoS One. 2011;6(5): e19738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Soverini S, Hochhaus A, Nicolini FE, et al. BCR-ABL kinase domain mutation analysis in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors: recommendations from an expert panel on behalf of European LeukemiaNet. Blood. 2011;118(5): 1208–1215. [DOI] [PubMed] [Google Scholar]
- 191.Baccarani M, Deininger MW, Rosti G, et al. European LeukemiaNet Recommendations for the management of Chronic Myeloid Leukemia. Blood. 2013;122(6):872–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.San-Miguel JF, Kantarjian HM. Improved understanding of disease biology and treatment: Multiple Myeloma and Chronic Myeloid Leukaemias in 2014. Nat Rev Clin Oncol. 2015;12(2):71–72. [DOI] [PubMed] [Google Scholar]
- 193.Branford S, Yeung DT, Parker WT, et al. Prognosis for patients with CML and >10%BCR-ABL1 after 3 months of imatinib depends on the rate of BCR-ABL1 decline. Blood. 2014;124(4):511–518. [DOI] [PubMed] [Google Scholar]
- 194.Branford S, Roberts N, Yeung DT, et al. Any BCR-ABL reduction below 10% at 6 months of therapy significantly improves outcome for CML patients with a poor response at 3 months. Blood. 2013;122(21): (Abstract 254). [Google Scholar]
- 195.Kim DD, Hamad N, Lee HG, et al. BCR/ABL level at 6 months identifies good risk CML subgroup after failing early molecular response at 3 months following imatinib therapy for CML in chronic phase. Am J Hematol. 2014;89(6):626–632. [DOI] [PubMed] [Google Scholar]
- 196.Neelakantan P, Gerrad C, Lucas C, et al. Combining BCR-ABL1 transcript levels at 3 and 6 months in chronic myeloid leukemia: implications for early intervention strategies. Blood. 2013;121(14):2739–2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.The Lancet Haematology. Chronic myeloid Leukaemia: time to push for a cure? Lancet. 2015;2(5):e175. [DOI] [PubMed] [Google Scholar]
- 198.Mathisen MS, Kantarjian HM, Cortes JE, Jabbour E. Practical issues surrounding the explosion of tyrosine kinase inhibitors for the management of chronic myeloid leukemia. Blood Rev. 2014;28(5):179–187. [DOI] [PubMed] [Google Scholar]
- 199.Holyoake Tl, Helgason GV. Do we need more drugs for CML? Immunol Rev. 2015;263(1):106–123. [DOI] [PubMed] [Google Scholar]
- 200.Etienne G, Dulucq S, Lascaux A, et al. ELN 2013 response status criteria: Relevance for de novo imatinib chronic phase chronic myelpoid leukemia patients? Am J Hematol. 2015;90(1):37–41. [DOI] [PubMed] [Google Scholar]
- 201.Ross TS, Mgbemena VE. Re-evaluating the role of BCR/ABL in chronic myelogenous leukemia. Mol Cell Oncol. 2014;1(3): e963450. [DOI] [PMC free article] [PubMed] [Google Scholar]