

Over the last decades, allogeneic hematopoietic stem cell transplantation (allo-HSCT) has considerably improved the outcome of acute myeloid leukemia (AML). Unfortunately, disease relapse remains a frequent occurrence, and a major cause of post-transplant mortality.1 Most salvage treatments do not provide encouraging results when given in morphological relapse. This means that efforts are aimed at anticipating relapse detection and treatment to the minimal residual disease (MRD) stage, and, as a consequence, to identify the optimal markers and techniques for post-transplantation followup disease monitoring.2,3
In the present study, we considered three genes that have recently been identified as recurrently mutated in AML, sharing a common involvement in the regulation of DNA methylation: DNMT3A, IDH1, and IDH2.4,5 Mutations in these genes, and in DNMT3A in particular, have been reported to occur very early during the stepwise process of leukemogenesis. These possibly represent disease founder mutations,6 shared by all disease subclones and stably maintained throughout the patient longitudinal history until the time of relapse,7,8 features which would make them interesting candidate molecular markers. However, it should also be considered that these mutations are not specific hallmarks of transformed cells, and have frequently been detected in the peripheral blood (PB) of healthy individuals where they are considered to represent pre-leukemic alterations, acting as a fertile soil for subsequent development of hematologic malignancies.9,10 In line with this model, patients achieving hematologic complete remission (CR) after sole chemotherapy have been reported to frequently recover a hematopoiesis verging on DNMT3A-mutated, but otherwise healthy, progenitors.11,12 This suggests that a cautionary approach should be adopted in the use of these mutations as MRD markers in the post-chemotherapy setting.
Based on these considerations, we focused our attention on myeloablative allo-HSCT, a clinical context in which persistence of a host-derived hematopoiesis, either normal or pre-leukemic, is not warranted.
An innovative technology, droplet digital polymerase chain reaction (ddPCR), was used to track mutations of interest over time. Features of this approach are ultrahigh sensitivity (up to the 0.001% mutated allele frequency) and higher precision than conventional quantitative PCR (qPCR) assays,13 which can achieve similar results only through the use of multiple replicates.14
To select our study population, conventional sequencing techniques were used to characterize the exonic regions of interest of DNMT3A, IDH1 and IDH2 in bone marrow (BM) samples harvested at diagnosis or pre-transplant active disease from 89 patients who had received myeloablative allo-HSCT for high-risk myeloid malignancies at the San Raffaele Scientific Institute, Milan, between January 2009 and November 2014. Details of patients’ and transplant characteristics are summarized in Table 1 (see also Online Supplementary Appendix). Primer sequences and sequencing conditions are provided in the Online Supplementary Appendix.
Table 1.
Patients’ and transplant characteristics.
Of the 89 patients screened, 30 (33.7%) resulted positive for at least one mutation in the three genes of interest, 16 (18%) carrying DNMT3A mutations, 5 (5.6%) IDH1 mutations, and 13 (14.6%) IDH2 mutations (Table 1 and Figure 1A). The lower frequency of DNMT3A and IDH mutations in our cohort compared to other studies4 might be explained, at least in part, by the fact that our cohort included not only cases of de novo AML, but also secondary and complex cytogenetics AMLs, in which these mutations are less frequent.15
Figure 1.

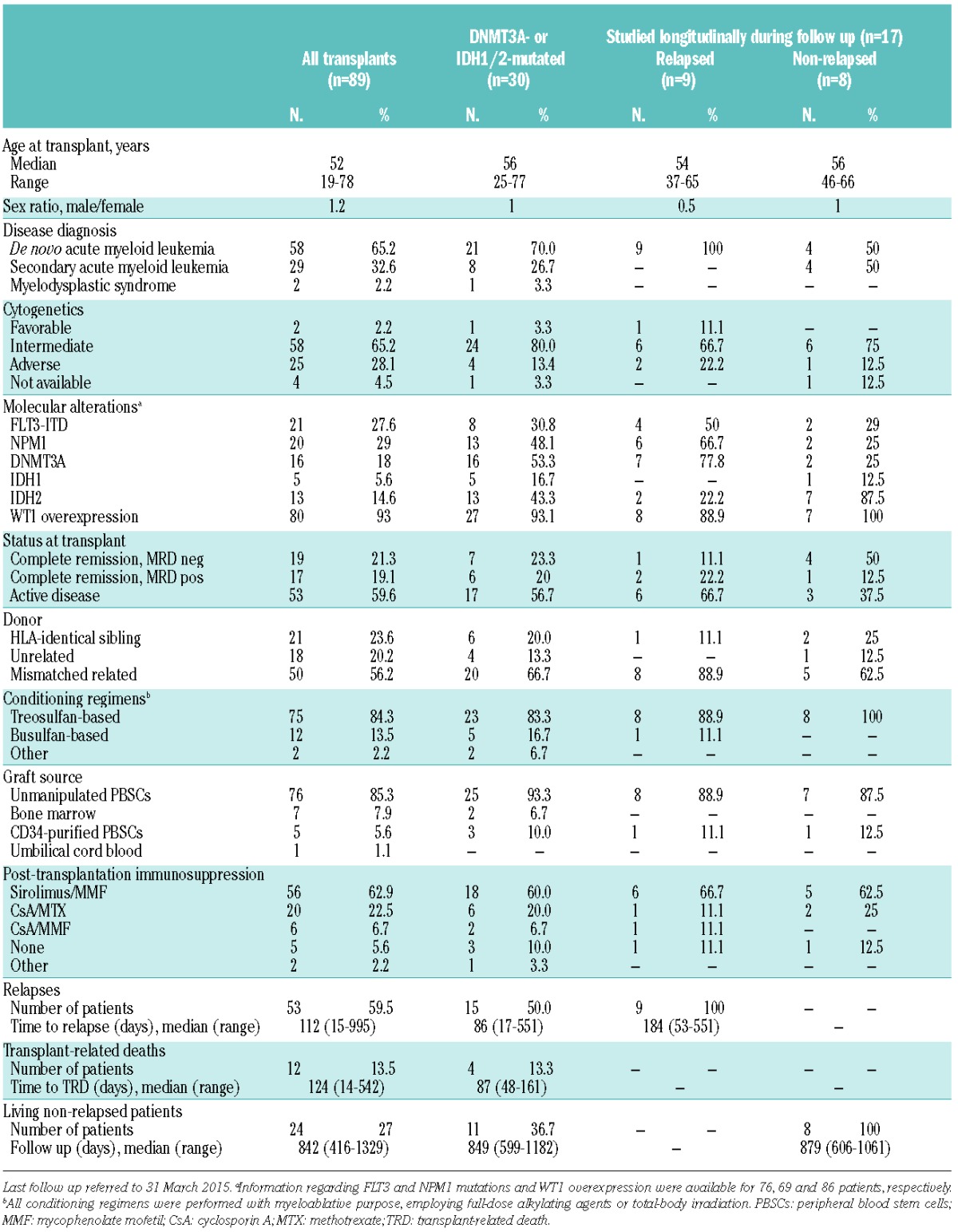
Longitudinal ddPCR evaluation of DNMT3A and IDH1/2 after allogeneic hematopoietic stem cell transplantation (HSCT). (A) The dendrogram summarizes presence and association of mutations in DNMT3A, IDH1/2, FLT3 and NPM1 in the study cohort (n=89), clustered based on disease diagnosis and cytogenetics. Red boxes indicate presence of the mutations, dashed boxes indicate data not available. AML: acute myeloid leukemia; MDS: myelodysplastic syndrome. (B) Correlation between the leukemic blast percentage assessed by morphological BM evaluation (on the X axis) and the percentage of mutated cells in the same sample inferred from ddPCR results (on the Y axis) in samples harvested at disease diagnosis or pre-transplant disease persistence (n=26). The red dashed line indicates the expected 1:1 linear correlation, dots above the line indicate samples for which the mutated frequency of mutated cells exceeds the percentage of leukemic blasts. (C) Correlation between the leukemic blast percentage assessed by morphological BM evaluation (on the X axis) and the percentage of mutated cells in the same sample inferred from ddPCR results (on the Y axis) in samples harvested at disease relapse (n=14). The red dashed line indicates the expected 1:1 linear correlation, dots above the line indicate samples for which the mutated frequency of mutated cells exceeds the percentage of leukemic blasts. (D) ddPCR monitoring of DNMT3A and IDH1/2 mutations in patients who experienced post-transplantation relapse (n=9). Red diamonds indicate time points in which BM morphological evaluation evidenced disease remission, but mutation-specific ddPCR resulted positive. The dashed red line indicates the 0.1% mutant allele frequency positivity threshold. (E) ddPCR monitoring of DNMT3A and IDH1/2 mutations in patients who did not experience post-transplantation relapse (n=8). Green diamonds indicate time points in which mutation-specific ddPCR resulted positive. The dashed red line indicates the 0.1% mutant allele frequency positivity threshold.
In particular, 27 patients carried at least one mutation for which commercial or custom-designed ddPCR assays were available (DNMT3A R882H and R882C, IDH1 R132H and R132C, IDH2 R140Q and R172K); of these, 5 patients were studied only before transplant (either at time of diagnosis or at pre-transplant relapse), one only at post-transplant relapse, 4 both before transplant and at post-transplantation relapse, and 17 at serial time points during follow up, including at relapse in the 9 cases in which it occurred.
ddPCR assays were performed using as template genomic DNA extracted from patient BM and employing the Bio-Rad QX100 system with 6 different assays, each designed to simultaneously quantify the mutation of interest and its wild-type counterpart in the same sample. The mutant allele frequency was then calculated using a Poisson distribution model as the fraction of positive droplets divided by total droplets containing a target. Technical validation of these assays set the sensitivity threshold at 0.1% mutated allele frequency, which also corresponded to the maximal background positivity detected in mutation-negative controls (details on ddPCR assays and validation experiments are provided in the Online Supplementary Appendix).
All the 26 pre-transplant leukemia samples that typed positive for the mutations of interest by sequencing also resulted positive for the corresponding ddPCR assays (median mutated allele frequency 43.2%, range 0.124%–48.9%). In the 4 cases with mutations in more than one gene, the allelic frequencies of the two alterations showed impressive similarity (r2=0.9964; P=0.0018), suggesting that both were present in the same leukemic clones.
Interestingly, when we employed the mutated allele frequency to estimate the percentage of BM cells carrying the corresponding genomic alteration, in 23 of 26 cases (88.5%) the population carrying the mutant allele, estimated by ddPCR, consistently exceeded the morphological count of leukemic blasts (Figure 1B), implying presence of the mutations also in morphologically normal BM cells. This observation suggests either the presence at disease diagnosis of apparently healthy progeny of mutated pre-leukemic cells, or a conserved, although limited, ability of leukemic blasts to differentiate into mature offspring. However, since at post-transplantation relapse the percentage of leukemic blasts and the estimated percentage of mutated cells were highly correlated (r2=0.4251; P=0.0115) (Figure 1C), the first explanation appears more likely.
Notably, all 14 post-transplantation relapses tested still carried the DNMT3A and IDH1/2 mutations present at diagnosis, except for one case originally carrying both DNMT3A and IDH2 mutations and typing negative for the latter at relapse. This observation further suggests that DNMT3A mutations represent an earlier event in leukemogenesis than IDH mutations, and thus, when present, could represent more stable markers. Of interest, in the same patient series, stability of NPM1 and FLT3 mutations were 86% and 75%, respectively. Next, we analyzed ddPCR samples collected longitudinally over time during the clinical follow up of 17 transplanted patients who carried the mutations of interest and for whom samples harvested at post-transplant hematologic remission were available. We evaluated the potential usefulness of monitoring this eventual persistence over time. All the patients had achieved hematologic complete remission at the BM evaluation performed one month after the transplant, and underwent morphological and molecular BM evaluation monthly in the first three months, then once every three months for the first year, and yearly after that.
Nine of the 17 patients (53%) eventually experienced post-transplantation relapse (median time to relapse 184 days, range 53–551 days). Seven of these 9 patients were positive by ddPCR at at least one pre-relapse time point (78% sensitivity for these assays in relapse prediction) (Figure 1D). The BM evaluation immediately preceding relapse resulted positive by ddPCR in 6 of these cases, and median time from first positivity to relapse was 60 days (range 20–491 days); this could represent a feasible time frame in which to attempt a pre-emptive treatment.
When we analyzed BM samples harvested from the 8 patients who remained disease-free over the entire course of their follow up (median follow up 29 months, range 20–35 months), 6 resulted negative for the mutations of interest at all time points tested (an overall 75% specificity for ddPCR assays in relapse prediction) (Figure 1E). A total of 4 false positive determinations were seen; 3 of these were accounted for by the early post-transplant time points of a single patient (UPN#68). Interestingly, when the clinical history of that patient was retrospectively reviewed, the time point in which mutations became negative coincided with immunosuppressive therapy tapering (Figure 2).
Figure 2.
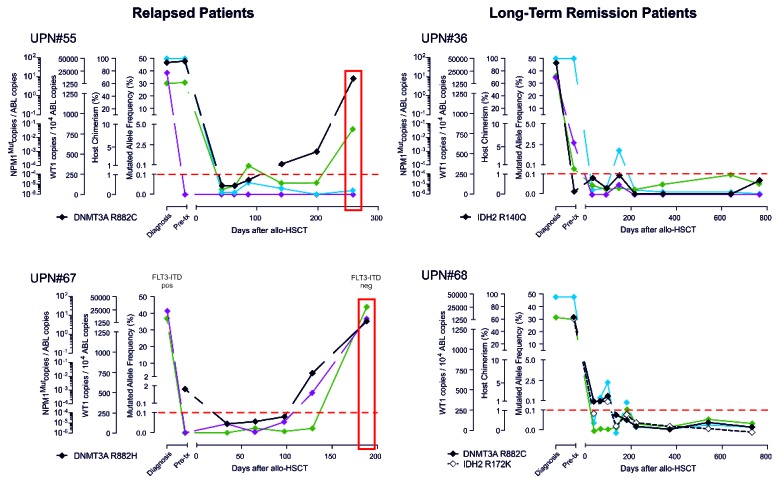
Post-transplantation minimal residual disease (MRD) monitoring using different molecular assays. Shown are results obtained during the longitudinal follow up of 2 representative relapsed patients (left panels) and of 2 representative non-relapsed patients (right panels) employing ddPCR assays specific for mutations in DNMT3A and IDH1/2 (in black) or qPCR assays specific for the WT1 gene transcript (in green), for host-specific chimerism markers (in cyan), or for NPM1 mutation A (in purple). The dashed red line indicates the positivity threshold for all four assays. Time of relapse is boxed in red. Results from longitudinal monitoring of an additional 7 relapsed patients and 6 non-relapsed patients are provided in the Online Supplementary Appendix.
Results from DNMT3A and IDH1/2 ddPCR monitoring were compared with those obtained from the same BM samples employing quantitative PCR for NPM1 mutations, for Wilms Tumor gene 1 (WT1) transcript, and for host-specific hematopoietic chimerism (see Online Supplementary Appendix for details). Longitudinal kinetics of the different markers appeared to be largely concordant, with mutation-specific assays (qPCR for NPM1 mutations and ddPCR for DNMT3A and IDH1/2) providing the most reliable results in detecting disease persistence and predicting relapse (Figure 2 and Online Supplementary Figures S3 and S4). On the other hand, it should be remembered that mutation-specific assays are informative in only a fraction of patients, and their targets might be lost upon disease clonal evolution. In this context, a multi-gene panel based on each specific mutation of each patient, including leukemia founder events, might represent the optimal tool to monitor MRD and provide a clinically meaningful estimate of the risk of relapse. Moreover, with the highly sensitive techniques now available, MRD monitoring in PB rather than BM samples appears worthy of future investigation.
Taken together, our results show that ddPCR monitoring of DNMT3A and IDH1/2 mutations during clinical follow up was feasible and enhanced routine qPCR-based MRD techniques in tracing leukemia dynamics after allo-HSCT. In conclusion, our findings suggest that longitudinal monitoring of these mutations can be extremely useful in the allotransplantation setting, a context in which these alterations can be considered markers of undesired residual pre-leukemic host hematopoiesis. They further support the pivotal role of DNMT3A and IDH1/2 mutations in AML biology.
Footnotes
Funding: this work was supported by the Italian Ministry of Health (RF-2011-02351998), by the Associazione Italiana per la Ricerca sul Cancro (Start-Up Grant #14162), and by the Conquer Cancer Foundation (2014 Young Investigator Award).
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
The online version of this letter has a SupplementaryAppendix.
References
- 1.Pavletic SZ, Kumar S, Mohty M, et al. NCI First International Workshop on the Biology, Prevention, and Treatment of Relapse after Allogeneic Hematopoietic Stem Cell Transplantation: report from the Committee on the Epidemiology and Natural History of Relapse following Allogeneic Cell Transplantation. Biol Blood Marrow Transplant. 2010;16(7):871–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Christopeit M, Kroger N, Haferlach T, Bacher U. Relapse assessment following allogeneic SCT in patients with MDS and AML. Ann Hematol. 2014;93(7):1097–1110. [DOI] [PubMed] [Google Scholar]
- 3.Grimwade D, Freeman SD. Defining minimal residual disease in acute myeloid leukemia: which platforms are ready for “prime time”¿ Blood. 2014;124(23):3345–3355. [DOI] [PubMed] [Google Scholar]
- 4.Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Im AP, Sehgal AR, Carroll MP, et al. DNMT3A and IDH mutations in acute myeloid leukemia and other myeloid malignancies: associations with prognosis and potential treatment strategies. Leukemia. 2014;28(9):1774–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Welch JS, Ley TJ, Link DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150(2):264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Welch JS. Mutation position within evolutionary subclonal architecture in AML. Semin Hematol. 2014;51(4):273–281. [DOI] [PubMed] [Google Scholar]
- 8.Kronke J, Bullinger L, Teleanu V, et al. Clonal evolution in relapsed NPM1-mutated acute myeloid leukemia. Blood. 2013;122(1):100–108. [DOI] [PubMed] [Google Scholar]
- 9.Genovese G, Kahler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371(26):2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shlush LI, Zandi S, Mitchell A, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014; 506(7488):328–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Corces-Zimmerman MR, Hong WJ, Weissman IL, Medeiros BC, Majeti R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc Natl Acad Sci USA. 2014;111(7):2548–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gaidzik VI, Weber D, Paschka P, et al. DNMT3A mutations in acute myeloid leukemia (AML): monitoring of minimal residual disease (MRD). A study of the AML study group (AMLSG). Meeting Abstract; 20th Congress European Hematology Association. Vienna, 2015. [Google Scholar]
- 13.Hindson CM, Chevillet JR, Briggs HA, et al. Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat Methods. 2013;10(10):1003–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koren-Michowitz M, Shimoni A, Daraio F, et al. Sensitive Replicate Real-Time Quantitative PCR of BCR-ABL Shows Deep Molecular Responses in Long-Term Post-Allogeneic Stem Cell Transplantation Chronic Myeloid Leukemia Patients. Biol Blood Marrow Transplant. 2015;21(10):1852–1855. [DOI] [PubMed] [Google Scholar]
- 15.Lindsley RC, Mar BG, Mazzola E, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood. 2015; 125(9):1367–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]