

Abstract
Coated platelets, formed by collagen and thrombin activation, have been characterized in different ways: i) by the formation of a protein coat of α-granular proteins; ii) by exposure of procoagulant phosphatidylserine; or iii) by high fibrinogen binding. Yet, their functional role has remained unclear. Here we used a novel transglutaminase probe, Rhod-A14, to identify a subpopulation of platelets with a cross-linked protein coat, and compared this with other platelet subpopulations using a panel of functional assays. Platelet stimulation with convulxin/thrombin resulted in initial integrin αIIbβ3 activation, the appearance of a platelet population with high fibrinogen binding, (independently of active integrins, but dependent on the presence of thrombin) followed by phosphatidylserine exposure and binding of coagulation factors Va and Xa. A subpopulation of phosphatidylserine-exposing platelets bound Rhod-A14 both in suspension and in thrombi generated on a collagen surface. In suspension, high fibrinogen and Rhod-A14 binding were antagonized by combined inhibition of transglutaminase activity and integrin αIIbβ3. Markedly, in thrombi from mice deficient in transglutaminase factor XIII, platelet-driven fibrin formation and Rhod-A14 binding were abolished by blockage of integrin αIIbβ3. Vice versa, star-like fibrin formation from platelets of a patient with deficiency in αIIbβ3 (Glanzmann thrombasthenia) was abolished upon blockage of transglutaminase activity. We conclude that coated platelets, with initial αIIbβ3 activation and high fibrinogen binding, form a subpopulation of phosphatidylserine-exposing platelets, and function in platelet-dependent star-like fibrin fiber formation via transglutaminase factor XIII and integrin αIIbβ3.
Introduction
Platelet activation and blood coagulation are highly reciprocally interacting processes and both are essential for hemostasis and thrombosis. Activated platelets support and steer the coagulation process by at least four mechanisms: i) by releasing coagulation factors like factor V and XIII; ii) by exposing the procoagulant phospholipid phosphatidylserine (PS) at their outer surface to support thrombin generation; iii) by providing a scaffold for the formation of fibrin fibers; and iv) by causing retraction of the fibrin clot.1,2 In a growing thrombus, aggregated and procoagulant platelets form two distinct populations,3,4 which is at least partly explained by the high Ca2+ response required for PS exposure and coagulation factor binding, and by the calpain-dependent closure of active αIIbβ3 integrins after PS exposure, thus antagonizing inclusion of procoagulant platelets into a platelet aggregate.5,6
However, another platelet population has also been identified, usually referred to as coated platelets,7 which may partly overlap with the two other platelet populations described above.3 In the initial paper, Dale et al. describe COAT platelets (later renamed as coated platelets) as a population of platelets arising after combined stimulation with collagen and thrombin, which bind α-granule proteins, including factor V, fibrinogen, von Willebrand factor, thrombospondin, fibronectin and α2-antiplasmin, in a transglutaminase-dependent way via the formation of covalent serotonin conjugations.8
Since this first description, coated platelets have been invariably considered as platelets formed after combined stimulation of collagen receptors (e.g. with collagen, convulxin or collagen-related peptide) and thrombin receptors (e.g. with thrombin or thrombin receptor-activating peptides), but there is no uniform definition of this platelet population in the literature. The Dale group has been using the retention of secreted proteins, including platelet-derived serotonin-derivatized proteins, factor V and tissue factor pathway inhibitor on the platelet surface as a characteristic.9–11 Another definition has been used by the Jobe group, i.e. platelets containing high surface levels of fibrinogen, likely through cross-linking via the transglutaminase factor XIII.12 However, in recent years, it has become common practice to consider coated platelets more or less equivalent to fibrinogen binding platelets or PS-exposing platelets. For example, platelet subpopulations in patient studies have recently been characterized using biotin-fibrinogen.13–15 This ambiguity in definition and described properties raises questions as to whether coated platelets form a platelet subpopulation (after collagen/thrombin receptor stimulation) that is distinguishable from that of fibrinogen and/or PS-exposing platelets and whether they fulfill a specific function.
In the present paper, we used a specific transglutaminase substrate, i.e. the α2-antiplasmin-derived peptide Rhod-A14, as a tool to identify transglutaminase-active platelets. We compared the binding of Rhod-A14 to platelets, stimulated via the collagen and thrombin receptors, with other platelet activation markers. The results indicate that transglutaminase active platelets form as a subpopulation of PS exposing platelets. We also provide evidence that the transglutaminase activity along with integrin αIIbβ3 activation is required for fibrin anchoring at the platelet surface and star-like platelet-dependent fibrin formation.
Methods
Blood collection and platelet preparation
Experiments were approved by the local Medical Ethics Committees. Blood was taken from healthy volunteers and from a patient with Glanzmann thrombasthenia, with established deficiency in integrin αIIbβ3,16 after informed consent and in accordance with the Declaration of Helsinki. Platelet-rich plasma (PRP), defibrinated platelet-free plasma (PFP) and washed platelets were prepared from whole blood as described in the Online Supplementary Appendix.
Animal studies were approved by the local animal experimental committees. Mice deficient in factor XIII A1 subunit (F13a1tm1Gdi, abbreviated as F13a1−/−)17 were bred on a mixed 129Sv/CBA background and were compared to F13a1+/+ mice of the same background (Harlan Laboratories). Murine blood was taken on trisodium citrate for whole-blood flow experiments; other blood samples were taken on acid-citrate-dextrose anticoagulant to isolate washed platelets, as previously described.18
Flow cytometric platelet analyses
Washed human or mouse platelets (5×107/mL) were pre-incubated with indicated inhibitors or Me2SO vehicle for 10 min, and stimulated in the presence of 2 mM CaCl2. In the activations, 0.2 mM Gly-Pro-Arg-Pro (GPRP) was added to prevent formation of large fibrin fibers.19 Platelet sub-populations were distinguished by probing with Rhod-A14 (10 μg/mL), AF647-annexin A5 (1:200), AF488-factor V (20 nM), OG488-factor Xa (100 nM), AF647-fibrinogen (100 μg/mL) and FITC-PAC-1 (1.25 μg/mL). After staining for 5 min, samples were analyzed with a FACScan flow cytometer (BD Accuri Cytometer).6
In a separate set of experiments, reconstituted PRP was activated with tissue factor (2 pM) and CaCl2 (16.7 mM) in the presence of GPRP (2 mM) at 37°C, after which samples were taken for fluorescent labeling. Analysis was by flow cytometry as described above.
Thrombin generation
Thrombin generation was measured in citrate-anticoagulated human PRP as previously described.20 First-derivative curves were converted into curves of nanomolar thrombin concentrations using a calibrator for human α-thrombin.21 All analyses were in triplicate.
Thrombus formation on collagen under flow
Whole blood thrombus formation on collagen was assayed in the absence or presence of coagulation, as previously described.22 Thrombi formed on collagen were then stained with Rhod-A14 and AF647-annexin A5 in Hepes buffer, supplemented with CaCl2 (2 mM) and heparin (1 unit/mL). Phase-contrast and fluorescence images were captured and analyzed with Metamorph software v.7.5.0.0 (MDS Analytical Technologies), as detailed elsewhere.23 Statistical difference of quantitative colocalization of 2-color confocal images was determined using the ZEN software 2010 B SP1. Star-like fibrin formation was assessed as described in the Online Supplementary Appendix.
Statistical analysis
Significance of differences between control and experimental groups, as well as changes between groups over time, was determined by 1-way or 2-way analysis of variance followed by a Bonferroni post hoc test. Distribution of fibrin fibers was evaluated by χ2 analysis.24 P<0.05 was considered significant.
Further details on the materials and methods used are available in the Online Supplementary Appendix.
Results
Coated platelets as transglutaminase active platelets
First we assessed the appearance of a coated platelet population following the initial description of formation of a transglutaminase-dependent protein coat on the platelet surface.8 Therefore, we synthesized a fluorescent-labeled 14-amino acid transglutaminase peptide substrate, GNQEQVSPLTLLK(C-tetramethylrhodamine)W (Rhod-A14), derived from the N-terminal peptide sequence of α2-antiplasmin. This peptide contains a glutamine at Q3, which serves as a transglutamination site for cross-linking to available ε-amino lysine residues.25 The Rhod-A14 peptide was site-specifically labeled with a tetramethylrhodamine group, which did not interfere with its substrate properties. Control experiments indicated that the peptide could act as a substrate for both tissue-type transglutaminase and factor XIIIa (data not shown, but see below).
Flow cytometric analysis indicated that the Rhod-A14 label bound to a population of convulxin/thrombin-activated platelets, which was gradually formed after approximately 15 min of activation and increased until 60 min to approximately 50% Rhod-A14 positive platelets (Figure 1A and B). This differed markedly from the appearance of PS-exposing platelets analyzed by annexin A5 binding, which was already maximal after 15 min. Two-color flow cytometry indicated that the Rhod-A14 positive platelets represented a discrete subpopulation: the majority of these platelets were PS positive (binding of annexin A5), but had inactive αIIbβ3 integrins (no binding of PAC-1 mAb) (Online Supplementary Figure S1). When comparing various strong platelet agonists, we found that the Ca2+-ionophore ionomycin, while causing nearly complete PS exposure, resulted in Rhod-A14 labeling of a subpopulation of only approximately 25% platelets (Figure 1B). Dual platelet stimulation with convulxin and the thrombin receptor activating peptide SFLLRN resulted in a similar Rhod-A14 positive fraction, whereas stimulation with thrombin alone did not lead to either Rhod-A14 incorporation or to PS exposure.
Figure 1.
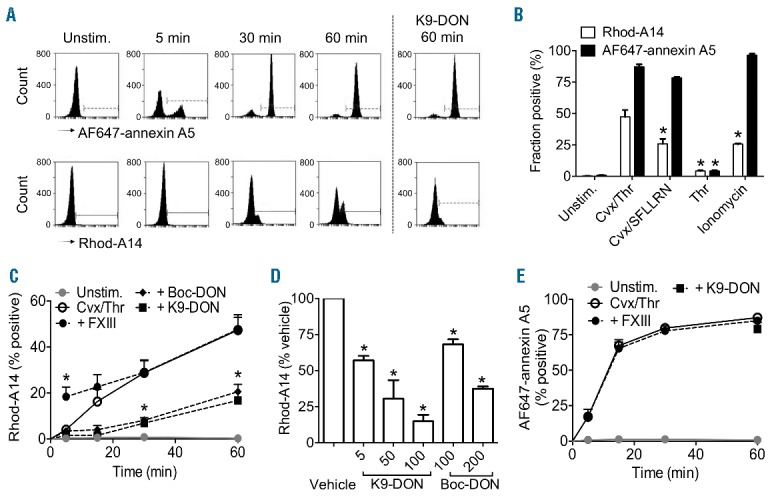
Transglutaminase activity confined to a subpopulation of phospholipid phosphatidylserine (PS)-exposing platelets. Washed human platelets (5×107/mL) were stimulated with convulxin (Cvx, 100 ng/mL) plus thrombin (Thr, 4 nM) in the presence of 0.2 mM GPRP and 2 mM CaCl2. Samples, taken at given time points, were labeled for 5 min with Rhod-A14 and AF647-annexin A5. Platelets were pre-treated with K9-DON (100 μM), where indicated. (A) Representative histograms obtained by flow cytometry. (B) Platelet subpopulations staining with Rhod-A14 (white bars) or AF647-annexin A5 (black bars) after 60-min stimulation with Cvx (100 ng/mL), Thr (4 nM), SFLLRN (15 μM) and/or ionomycin (10 μM), as indicated. (C–E) Effects of factor XIII and transglutaminase inhibitors on platelet subpopulations staining with Rhod-A14 (C) or AF647-annexin A5 (E) after stimulation with Cvx (100 ng/mL) plus Thr (4 nM). Platelets were pre-incubated with vehicle (○, solid line), 100 unit/mlLfactor XIII (●, dotted line), 100 μM K9-DON (■, dotted line), or 200 μM Boc-DON (♦, dotted line). Solid gray line, unstimulated platelets. (D) Dose-dependent inhibition of Rhod-A14 binding (60 min) by K9-DON or Boc-DON. Mean ± SEM (n=3–6); *P<0.05.
To specifically test the contribution of the transglutaminase factor XIII in Rhod-A14 binding to platelets, we analyzed platelets from F13a1−/− mice, which are deficient in the A-subunit of factor XIII. Washed platelets from F13a1−/− mice, when stimulated with convulxin/thrombin, showed greatly reduced Rhod-A14 binding in comparison to wild-type platelets (Online Supplementary Figure S2A). However, PS exposure, factor Xa, and fibrin(ogen) binding were unchanged in the F13a1−/− platelets (Online Supplementary Figure S2B-D). Vice versa, pre-treatment of human washed platelets with purified factor XIII prior to stimulation with convulxin/thrombin resulted in an enhanced appearance of the Rhod-A14 binding cells (Figure 1C). Additional control experiments again confirmed the involvement of transglutaminase activity, since Rhod-A14 binding to washed human platelets was antagonized in a dose-dependent way by the general transglutaminase inhibitor K9-DON and the tissue transglutaminase inhibitor Boc-DON (Figure 1D). In sharp contrast, neither the addition of factor XIII nor these inhibitors affected the PS exposure evoked by convulxin/thrombin (Figure 1E). Taken together, these results indicate that the transglutaminase-active Rhod-A14 positive platelets form as a subpopulation of PS-exposing platelets after collagen/thrombin receptor stimulation.
Coated platelets as high fibrinogen-binding platelets
Platelet activation with convulxin/thrombin has been demonstrated to result in a fraction of platelets displaying high fibrinogen binding, which has also been referred to as coated platelets.12 In the present study, after 5 min of stimulation with convulxin/thrombin, the binding of fluorescently labeled fibrinogen reached a maximal high level, and this occurred faster than the appearance of PS-exposing platelets (Online Supplementary Figure S3A–C). Other strong platelet agonists, ionomycin and convulxin/SFLLRN induced only low fibrinogen binding. However, thrombin alone induced similarly high fibrinogen binding as in combination with convulxin. This suggests that the high fibrinogen binding relies on the presence of thrombin, but not on PS exposure. Notably, the transglutaminase inhibitors K9-DON or Boc-DON did not affect either fibrinogen binding or PS exposure (Online Supplementary Figure S3A and D). Interestingly, the fibrinogen binding per platelet was slightly reduced upon prolonged incubation with convulxin/thrombin (Online Supplementary Figure S3B), while the fraction of fibrinogen-positive platelets remained stable (Figure 2A). This suggests that a portion of αIIbβ3 integrins on the surface Cvx/Thr activated platelets can close in time, which is in agreement with labeling studies using PAC-1 mAb (Online Supplementary Figure S1D). Dual-color flow cytometry indicated that Rhod-A14 binding platelet fraction was a subpopulation of the high fibrinogen-binding cells (Online Supplementary Figure S1C).
Figure 2.
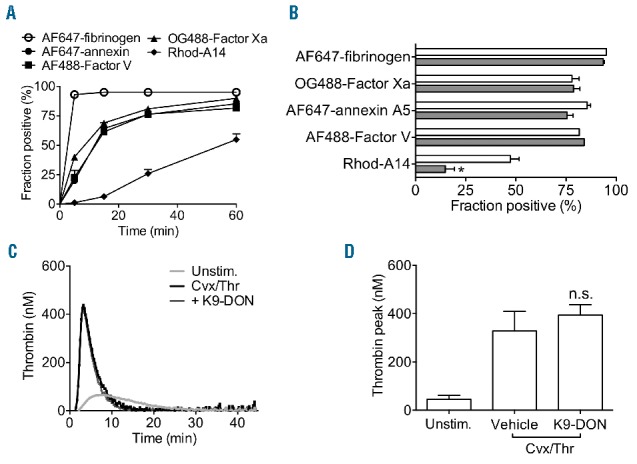
Transglutaminase-positive platelets are not identical to procoagulant platelets. (A and B) Washed platelets were stimulated with convulxin plus thrombin, as indicated for Figure 1. At given time points, samples were taken and labeled with AF647-fibrinogen (○), OG488-factor Xa (▲), AF647-annexin A5 (●), AF488-factor V(a) (■), or Rhod-A14 (♦). (A) Fractions of platelets binding the indicated probes. (B) Effect of platelet treatment with K9-DON (100 μM) (gray bars) on fractions of positive platelets. (C and D) Citrate-anticoagulated PRP (1×108 platelets/mL) was pre-incubated with vehicle or K9-DON (100 μM), and used for measurement of thrombin generation; coagulation was triggered with tissue factor/CaCl2. Representative thrombin generation curves (C) and quantification of peak height (D). Mean ± SEM (n=3–4), *P<0.05.
Coated platelets as procoagulant PS-exposing platelets
In recent papers, it has been proposed that coated platelets are equivalent to coagulation factor binding platelets or PS-exposing platelets.7,26 To investigate this in more detail, we compared the fractions of convulxin/thrombin-stimulated platelets that bind AF647-annexin A5, OG488-factor Xa, FITC-factor V or Rhod-A14. The appearance of a platelet population with binding sites for annexin A5, factor V and factor Xa occurred quickly with similar kinetics, whereas the formation of Rhod-A14-binding platelets was considerably slower (Figure 2A). Pre-incubation with transglutaminase inhibitor K9-DON did not influence the binding of annexin A5 nor the coagulation factors, but greatly impaired the binding of Rhod-A14 (Figure 2B). In agreement with this result, the presence of K9-DON did not influence the process of thrombin generation in PRP (Figure 2C and D). This indicated that the transglutaminase activity of platelets was independent of the interaction of factor Va and Xa, and of subsequent platelet-dependent thrombin generation, which relies on this interaction.20
Coated platelets as fibrin-forming platelets
Our earlier work had demonstrated that platelet stimulation with strong agonists leads to integrin αIIbβ3 activation, followed by PS exposure and calpain-dependent closure of the integrin.6 Furthermore, we found that PS exposure instigates the formation of a fibrin network linked to the platelet surface.27 In the present study, we investigated this again by assessing the determinants of high fibrin(ogen) binding and PS exposure of platelets in tissue factor-activated blood plasma. For this purpose, plasma samples were defibrinated to prevent massive clot formation,19 and then reconstituted with washed platelets. After triggering coagulation with tissue factor and calcium, the majority of platelets showed rapid and sustained high fibrinogen binding, followed by the appearance of a platelet subpopulation with PS exposure, as determined by dual-labeling with OG488-fibrinogen and AF647-annexin A5 (Figure 3A). Detailed analysis indicated that only a proportion of the high fibrinogen-binding platelets exposed PS, whereas the majority of PS-exposing platelets showed high fibrinogen binding (Figure 3B). Once again, the high fibrin(ogen) binding was dependent on the presence of thrombin, as these platelets stained with an anti-fibrin Ab and the thrombin inhibitor hirudin substantially suppressed fibrinogen binding (data not shown).
Figure 3.
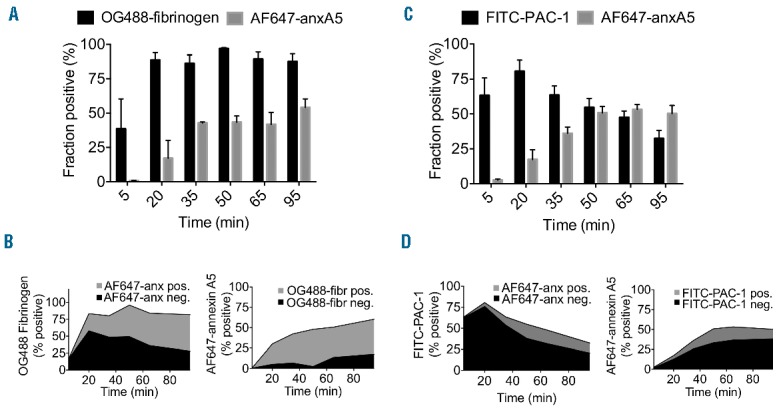
High fibrinogen binding of platelets is independent of αIIbβ3 activation and phospholipid phosphatidylserine (PS) exposure in tissue factor-activated plasma. Defibrinated PFP was reconstituted with platelets and triggered with tissue factor (2 pM) and CaCl2 (16.7 mM). Samples were taken for fluorescent labeling at indicated time points. (A and B) Populations of platelets with high binding of OG488-fibrinogen and binding AF647-annexin A5. Total fractions (A) and sub-fractions (B) of high-fibrinogen and annexin A5 binding platelets. (C and D) Populations of platelets binding FITC-PAC-1 mAb and AF647-annexin A5. Total fractions (C) and sub-fractions (D) of platelets positive for FITC-PAC-1 mAb and AF647-annexin A5. Mean ± SEM (n=3).
High fibrin(ogen) binding of a subpopulation of the PS-exposing cells was observed not only with labeled fibrinogen, but also with an antibody (FITC-WAK) recognizing platelet-bound fibrin(ogen) (Online Supplementary Figure S4A and B). This is in agreement with the results with washed platelets described above that showed that, in the presence of thrombin, the high fibrin(ogen) binding is regulated differently from PS exposure.
Staining of the tissue factor-triggered platelets in plasma with FITC-PAC-1 mAb, recognizing activated integrin αIIbβ3, pointed to rapid integrin activation that subsequently declined (Figure 3C). However, when these platelets were stained for the integrin β3-chain, fluorescence remained high (data not shown), indicating that the integrin was still present and accessible at the platelet surface. In comparison to integrin activation, exposure of PS rose more slowly over time (Figure 3C). Similar to our findings as reported before,6 the majority of PAC-1 mAb positive platelets were annexin A5 negative, while the annexin A5 positive platelets were mostly PAC-1 mAb negative (Figure 3D). Dot plots of platelets in plasma, double labeled with AF647-annexin A5 and FITC-PAC-1 mAb, confirmed that most PS-exposing cells were unable to bind PAC-1 mAb (Online Supplementary Figure S4C). In this plasmatic condition, a population of PS-positive platelets showed a mean 2.8-fold increase in Rhod-A14 fluorescence above background. Taken together, these results suggest that the high fibrin(ogen)-binding platelet fraction contains a fibrin coat, which precedes PS exposure and primes for binding or Rhod-A14.
Transglutaminase activity in platelet thrombus formation under flow conditions
To investigate where coated platelets localize in platelet thrombi, we performed whole blood perfusion experiments over a collagen surface under arterial shear conditions. Double staining with Rhod-A14 and AF657-annexin A5 indicated that, under non-coagulant conditions (with thrombin inhibitor present), Rhod-A14 binding was restricted to a subpopulation of PS-exposing platelets (Figure 4A). Co-localization analysis demonstrated that the majority of Rhod-A14 positive pixels also stained with AF647-annexin A5. Addition of K9-DON completely blocked the Rhod-A14 staining, but did not affect PS exposure. Zoomed images indicated that the Rhod-A14 labeling was confined to balloon-shaped, PS-exposing platelets, but often did not label the complete platelet (Figure 4A, overlay and bar graph). When thrombi were allowed to form on collagen under coagulant conditions (recalcification without thrombin inhibitor), this resulted in increased Rhod-A14 staining, which again was antagonized by K9-DON. Also a higher AF657-annexin A5 staining was found, which was unaffected by K9-DON (Figure 4B). Under these coagulant conditions, PS-exposing balloon-shaped platelets still bound Rhod-A14, while the majority of the Rhod-A14 label was located on fibrin fibers.
Figure 4.
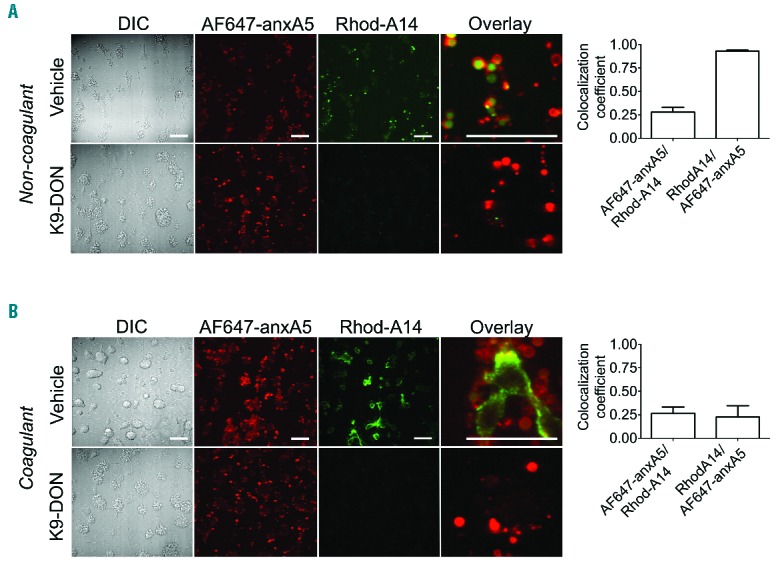
Subpopulation of phospholipid phosphatidylserine (PS)-exposing platelets in thrombi is transglutaminase positive. Human whole blood was perfused over collagen under anti-coagulated (A) or coagulated (B) conditions at shear rate of 1000 s−1. Blood samples were pre-incubated with vehicle (control) or K9-DON (100 μM). (A and B) Representative microscopic images of differential interference contrast (DIC), and confocal images of AF647-annexin A5 or Rhod-A14 fluorescence (bars, 20 μm). Right bar graphs indicate co-localization coefficient of AF647-annexin A5 with Rhod-A14 fluorescence and of Rhod-A14 with AF647-annexin A5 under vehicle-treated (non)coagulant conditions, as assessed with Zen software. Mean ± SEM (n=4).
Next, we performed flow experiments with F13a1+/+ or F13a1−/− mouse blood over collagen under both non-coagulant and coagulant conditions. In the non-coagulant condition, citrate anti-coagulated whole blood was recalcified in the presence of PPACK to block thrombin generation while physiological calcium and magnesium levels were restored. Under these conditions factor XIII activation is averted (no thrombin), whereas tissue type transglutaminase can be active (physiological calcium levels). The thrombi obtained with blood from F13a1+/+ or F13a1−/− mice were similar in size under these conditions with no more than low Rhod-A14 labeling (Figure 5A and B). These data suggest no major role for tissue type transglutaminase in thrombus formation and Rhod-A14 binding in the absence of coagulation, which is in line with the low expression of tissue-type transglutaminase in mouse platelets.28 Under coagulant conditions, large fibrin-containing thrombi were formed with both F13a1+/+ and F13a1−/− blood (Figure 5C and D). However, Rhod-A14 incorporated only into the fibrin fibers of wild-type thrombi and not of factor XIII-deficient thrombi. The Rhod-A14 staining again was antagonized by K9-DON, but not by tirofiban. These results hence indicate that the process of fibrin fiber formation can occur in the absence of transglutaminase activity.
Figure 5.
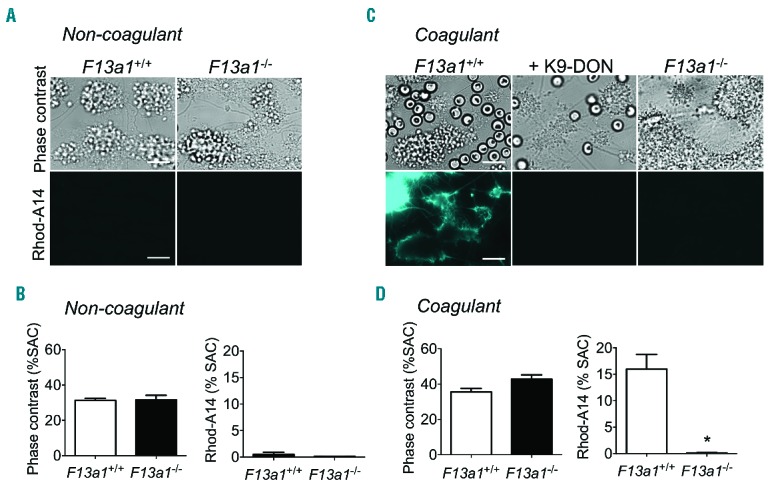
Absence of factor XIII results in fibrin formation without transglutaminase activity. Whole blood from F13a1+/+ or F13a1−/− mice was perfused over collagen under non-coagulant (A and B) or coagulant (C and D) conditions during 4 min at shear rate of 1000 s−1. Blood samples were pre-incubated with vehicle (control) or K9-DON (100 μM), and post-stained with Rhod-A14. Microscopic images of brightfield and Rhod-A14 fluorescence are shown (bars, 20 μm); also quantification of thrombus size [(% of surface area coverage, (SAC)] and Rhod-A14 fluorescence. Mean ± SEM (n=4); *P<0.05.
Synergistic roles of transglutaminase and integrin αIIbβ3 in platelet fibrin formation under flow conditions
We hypothesized that transglutaminase and integrin αIIbβ3 could be involved in the high fibrin(ogen) binding to coated platelets. Given this, we investigated the contribution of both of these in human and mouse systems. We used inhibitors against transglutaminases (K9-DON) and αIIbβ3 integrin (tirofiban) in concert with blood from F13a1−/− mice and a Glanzmann thrombasthenia patient with established deficiency in αIIbβ3. In washed platelets from control subjects stimulated with convulxin/thrombin, the combined presence of the K9-DON and tirofiban inhibited the binding of fibrin(ogen) and Rhod-A14 (P<0.05) (Figure 6A and B). A similar reduction of Rhod-A14 binding was observed when pre-treating Glanzmann platelets with K9-DON (P<0.05) (data not shown).
Figure 6.
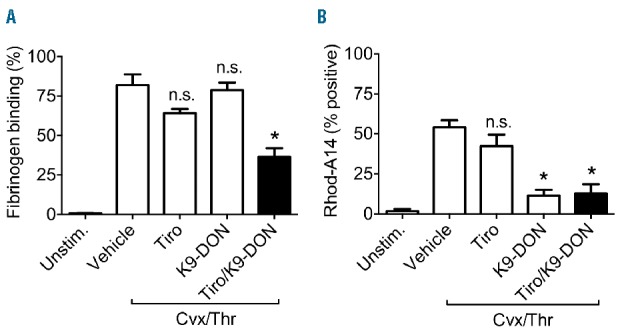
High fibrinogen binding of platelets requires both transglutaminase and αIIbβ3 activities. Washed platelets were stimulated with convulxin (100 ng/mL) and thrombin (4 nM) (see Figure 1). Pre-incubation was with vehicle, K9-DON (100 μM) and/or tirofiban (5 μg/mL). Flow cytometric data were assessed in the presence of Rhod-A14 and AF647-fibrinogen. Percentages of mean fluorescence intensity of fibrinogen binding (A) and platelets positive for Rhod-A14 are shown (B). Mean ± SEM (n=3); *P<0.05 versus vehicle.
To investigate the formation of fibrin fibers, blood from F13a1+/+ and F13a1−/− mice was pre-labeled with AF647-fibrinogen and perfused over a collagen surface under coagulating conditions. Detailed examination by confocal fluorescence microscopy indicated that, for wild-type thrombi, fibrin fibers were connected to the platelet surface and were oriented in all directions (Figure 7A and B). This staining pattern resembled the star-like formation of fibrin at the platelet surface that had been previously observed.27 In wild-type thrombi treated with the integrin blocker tirofiban, and also in factor XIII-deficient thrombi, the orientation of fibrin fibers from platelets was typically more frequent in the direction of the blood flow (Figure 7A and B). A similar shift in orientation of fibrin fibers was observed on human thrombi formed with whole blood from a Glanzmann thrombasthenia patient (Online Supplementary Figure S5). The addition of tirofiban to factor XIII-deficient blood markedly and nearly completely blocked the star-like orientation of fibrin fibers, but still allowed the plasmatic formation of fibrin fibers rectilinear with the blood flow.
Figure 7.
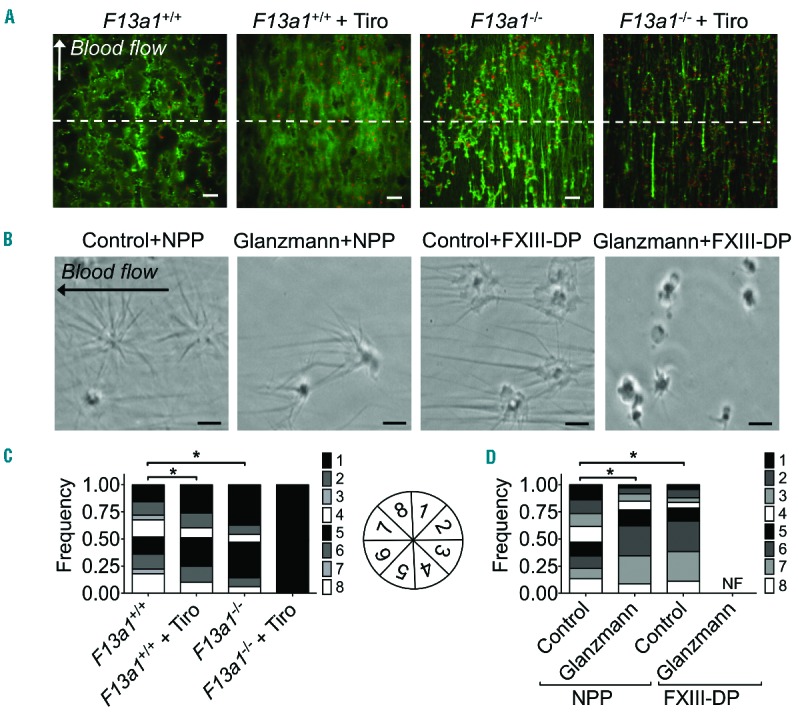
Absence of factor XIII and blockage/absence of integrin αIIbβ3 impairs platelet-derived fibrin formation. (A and C) Blood from F13a1+/+ or F13a1−/− mice was perfused over collagen under coagulant conditions (4 min, shear rate 1000 s−1). Tirofiban (5 μg/mL) was added where indicated. (A) Confocal images of AF647-fibrinogen fluorescence (bars, 20 μm). (C) Bar graph with distribution of number of fibrin fibers per region (region 1=0–45°, 2=45–90°, 3=90–135°, 4=135–180°, 5=180–225°, 6=225–270°, 7=270–315°, 8=315–360°), indicating the orientation of the formed fibrin fibers relative to the flow direction. The orientation of fibrin fibers was determined from thrombi traversing the arbitrarily drawn dotted white line (n=4); *P<0.001. (B and D) Washed platelets from a control subject or a patient with Glanzmann thrombasthenia were allowed to adhere to a glass coverslip, blocked and then post-perfused under coagulating conditions with normal pool plasma (NPP) or FXIII-deficient plasma (FXIII-DP). (B) Representative phase contrast images taken during the first minute of fibrin formation; bar is 5 μm. Note that no fibrin formation was observed with Glanzmann platelets during 20 min of post-perfusion with FXIII-DP. (D) Bar graph with distribution of number of fibrin fibers per region relative to the flow direction (region 1=0–45°, 2=45–90°, 3=90–135°, 4=135–180°, 5=180–225°, 6=225–270°, 7=270–315°, 8=315–360°), indicating the orientation of the formed fibrin fibers on the level of individual platelets. Independent experiments for control (n=4) and Glanzmann (n=1, triplicate); *P<0.001.
In subsequent experiments, platelet-dependent star-like fibrin formation was examined on the level of single platelets by using a previously established method.27 Whereas on the surface of control platelets a star-like fibrin network was formed upon post-perfusion with normal pool plasma, the orientation of the fibrin fibers was significantly altered upon post-perfusion with factor XIII-deficient plasma (Figure 7C and D). A similar shift in orientation of fibrin fibers was observed on the surface of Glanzmann thrombasthenia platelets. Post-perfusion of Glanzmann thrombasthenia platelets with factor XIII-deficient plasma completely blocked fibrin formation during more than 20 min. However, rectilinear plasmatic fibrin formation did occur after approximately 30 min of plasma perfusion, but no platelet-dependent fibrin formation could be observed. A similar absence of platelet-dependent fibrin formation was seen upon addition of K9-DON to Glanzmann thrombasthenia platelets (P<0.001). Taken together, these data indicate a non-redundant and synergistic role of the transglutaminase factor XIII and integrin αIIbβ3 in platelet-dependent fibrin formation.
Discussion
The original paper by Dale et al. describes COAT (later renamed coated) platelets as a population of platelets that result from combined stimulation with collagen and thrombin, and that can bind secreted α-granule proteins in a transglutaminase-dependent way.8 In the present paper, we used a novel fluorescent-labeled transglutaminase peptide substrate, Rhod-A14, to identify transglutaminase-active platelets and characterize the formation and functional responses of these platelets.
In washed human platelets, stimulated by convulxin/thrombin, we identified a slowly formed (1-h) subpopulation of PS-exposing platelets capable of binding Rhod-A14 by flow cytometry. The general transglutaminase inhibitor K9-DON and the tissue-type transglutaminase inhibitor Boc-DON prevented Rhod-A14 incorporation, whereas the transglutaminase factor XIII enhanced incorporation. Rhod-A14-positive platelets appeared as a discrete population of platelets with high fibrin(ogen) binding and PS exposure, while only a subpopulation of PS-positive and high fibrin(ogen)-binding platelets bound Rhod-A14. We further found that PS exposure, but not Rhod-A14 binding, was accompanied by the binding of coagulation factors Va and Xa to platelets. This is in agreement with results from other groups, also reporting high similarity of the annexin A5- and the coagulation factor-binding platelet populations.29,30 Furthermore, both in time of appearance and in subpopulation, the high fibrin(ogen)-binding platelets differed from the Rhod-A14-binding platelets. Hence, in contrast to others,13,14 we consider (transglutaminase-active) coated platelets as non-identical to fibrinogen-binding platelets.
Whereas convulxin/thrombin stimulated washed platelets showed persistently high fibrin(ogen) binding and slowly increasing RhodA14 binding, PAC-1 mAb binding gradually decreased after an initial peak. This is indicative of secondary inactivation of integrin αIIbβ3, which is in agreement with earlier results.6,31 One explanation for the persistent fibrin(ogen) binding to platelets with apparently inactive integrins, is that the activated αIIbβ3 displays distinct binding sites for fibrinogen and fibrin, allowing interaction with fibrin, independently of the classical integrin activation domains.32 However, in the present study, we observed that the high fibrin(ogen) binding of washed platelets was antagonized by combined inhibition of αIIbβ3 and transglutaminases. This suggests initial binding of fibrinogen to the activated integrin, followed by secondary binding of a growing fibrin fiber to platelet surface proteins via transglutaminase activity.
In contrast to washed platelets, Rhod-A14 binding platelets were formed much faster (minutes) during human whole-blood thrombus formation on collagen. This can be explained by a rapid availability of transglutaminases like human factor XIII in the blood plasma, but it may also point to a relatively slow externalization and/or activation of platelet-derived transglutaminases in the absence of plasma. Given that transglutaminases are broad-spectrum enzymes, cross-linking ε-(γ-glutamyl)lysine isopeptide bonds of multiple proteins,33,34 it can safely be assumed that Rhod-A14-binding platelets are covered (coated) with a large set of cross-linked proteins at their surface.
In our previous work, we had shown that platelets act as a scaffold for star-like fibrin formation.27 Here we provide evidence of a combined role of integrin αIIbβ3 and factor XIII in this process. In agreement with the results from others,12 we find that murine factor XIII is not essential for high fibrinogen binding and PS exposure. Although platelet-dependent fibrin fiber formation could still occur in blood from F13a1−/− mice (which points to the availability of factor XIII for the formation of fibrin fibers as such), we did observe a significant change in the attachment of fibrin fibers formed on the thrombus surface. Additional blockage of integrin αIIbβ3 led to a further shift from platelet-dependent star-like fibrin formation to platelet-independent fiber orientation rectilinear to the blood flow. In the human system, the star-like fibrin fiber formation on the platelet surface was also synergistically mediated by integrin αIIbβ3 activation (Glanzmann thrombasthenia patient) and factor XIII activity. This functional redundancy of integrin αIIbβ3 and factor XIII might explain the poor correlation of the defect in αIIbβ3 with the bleeding tendency observed in Glanzmann thrombasthenia patients.35
Although earlier studies found tissue type transglutaminase to be present in human platelets,8,36 more recent studies showed it could not be detected on the platelet protein level.37,38 Interestingly, in mouse platelets, tissue type transglutaminase (Tgm2) could be detected, albeit with a 25-fold lower expression in comparison to factor XIII (F13a).28 Our present findings point to a predominant role of factor XIII, and not of tissue type transglutaminase, in both the human and murine systems, which is in line with the recent studies. A role for platelet-derived FXIII was recently proposed in regulating anti-fibrinolysis by cross-linking α2-antiplasmin to fibrin.38 Our present findings significantly extend this study by demonstrating a role for factor XIII earlier in the process of hemostasis and thrombosis, i.e. by mediating the formation of a star-like coat of fibrin fibers on the platelet surface.
In summary, our study shows that the mechanism for formation of coated Rhod-A14-binding platelets is that initial collagen/thrombin receptor-induced αIIbβ3 activation leads to fibrin(ogen) binding, which on PS-exposing platelets can be consolidated by factor XIII transglutaminase activity, and act as a scaffold for star-like platelet-driven fibrin fiber formation. Since many of these coated platelets still bind coagulation factors, they could also have a role in stimulating prothrombinase activity and thrombin generation, leading to stabilization of the platelet-fibrin clot.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/101/4/427
Funding
This work was supported by research funding from ZonMW MKMD (114021004) to JWMH and JMEMC; the Dutch Heart Foundation (2011T6) to JMEMC; the Dutch Landsteiner Foundation for Blood Transfusion Research (1006) to FS, PEJvdM and JWMH; the Cardiovascular Center Maastricht to PEJvdM; the National Institutes of Health (R01HL101972) to OJTM; the American Heart Association (13EIA12630000) to OJTM; the Whitaker International Fellows Program (MAB-L); and Cyttron II (LSH framework: FES0908) to TMH.
References
- 1.Cosemans JM, Angelillo-Scherrer A, Mattheij NJ, Heemskerk JW. The effects of arterial flow on platelet activation, thrombus growth, and stabilization. Cardiovasc Res. 2013;99(2):342–352. [DOI] [PubMed] [Google Scholar]
- 2.de Witt SM, Swieringa F, Cavill R, et al. Identification of platelet function defects by multi-parameter assessment of thrombus formation. Nat Commun. 2014;5:4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Munnix IC, Cosemans JM, Auger JM, Heemskerk JW. Platelet response heterogeneity in thrombus formation. Thromb Haemost. 2009;102(6):1149–1156. [DOI] [PubMed] [Google Scholar]
- 4.Versteeg HH, Heemskerk JW, Levi M, Reitsma PH. New fundamentals in hemostasis. Physiol Rev. 2013;93(1):327–358. [DOI] [PubMed] [Google Scholar]
- 5.Heemskerk JW, Mattheij NJ, Cosemans JM. Platelet-based coagulation: different populations, different functions. J Thromb Haemost. 2013;11(1):2–16. [DOI] [PubMed] [Google Scholar]
- 6.Mattheij NJ, Gilio K, van Kruchten R, et al. Dual mechanism of integrin αIIbβ3 closure in procoagulant platelets. J Biol Chem. 2013;288(19):13325–13336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dale GL. Coated-platelets: an emerging component of the procoagulant response. J Thromb Haemost. 2005;3(10):2185–2192. [DOI] [PubMed] [Google Scholar]
- 8.Dale GL, Friese P, Batar P, et al. Stimulated platelets use serotonin to enhance their retention of procoagulant proteins on the cell surface. Nature. 2002;415(6868):175–179. [DOI] [PubMed] [Google Scholar]
- 9.Alberio LJ, Clemetson KJ. All platelets are not equal: COAT platelets. Curr Hematol Rep. 2004;3(5):338–343. [PubMed] [Google Scholar]
- 10.Szasz R, Dale GL. Thrombospondin and fibrinogen bind serotonin-derivatized proteins on COAT-platelets. Blood. 2002;100(8):2827–2831. [DOI] [PubMed] [Google Scholar]
- 11.Maroney SA, Haberichter SL, Friese P, et al. Active tissue factor pathway inhibitor is expressed on the surface of coated platelets. Blood. 2007;109(5):1931–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jobe SM, Leo L, Eastvold JS, et al. Role of FcRγ and factor XIIIA in coated platelet formation. Blood. 2005;106(13):4146–4151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kirkpatrick AC, Tafur AJ, Vincent AS, Dale GL, Prodan CI. Coated-platelets improve prediction of stroke and transient ischemic attack in asymptomatic internal carotid artery stenosis. Stroke. 2014;45(10):2995–3001. [DOI] [PubMed] [Google Scholar]
- 14.Prodan CI, Stoner JA, Cowan LD, Dale GL. Higher coated-platelet levels are associated with stroke recurrence following nonlacunar brain infarction. J Cereb Blood Flow Metab. 2013;33(2):287–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prodan CI, Vincent AS, Dale GL. Coated-platelet levels are persistently elevated in patients with mild traumatic brain injury. J Head Trauma Rehabil. 2014;29(6):522–526. [DOI] [PubMed] [Google Scholar]
- 16.Rosado JA, Meijer EM, Hamulyak K, Novakova I, Heemskerk JW, Sage SO. Fibrinogen binding to the integrin αIIbβ3 modulates store-mediated calcium entry in human platelets. Blood. 2001;97(9):2648–2656. [DOI] [PubMed] [Google Scholar]
- 17.Lauer P, Metzner HJ, Zettlmeissl G, et al. Targeted inactivation of the mouse locus encoding coagulation factor XIII-A: hemostatic abnormalities in mutant mice and characterization of the coagulation deficit. Thromb Haemost. 2002;88(6):967–974. [PubMed] [Google Scholar]
- 18.Gilio K, Harper MT, Cosemans JM, et al. Functional divergence of platelet protein kinase C (PKC) isoforms in thrombus formation on collagen. J Biol Chem. 2010;285(30):23410–23419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van der Meijden PE, Feijge MA, Swieringa F, et al. Key role of integrin αIIbβ3 signaling to Syk kinase in tissue factor-induced thrombin generation. Cell Mol Life Sci. 2012;69(20):3481–3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vanschoonbeek K, Wouters K, van der Meijden PE, et al. Anticoagulant effect of dietary fish oil in hyperlipidemia: a study of hepatic gene expression in APOE2 knock-in mice. Arterioscler Thromb Vasc Biol. 2008;28(11):2023–2029. [DOI] [PubMed] [Google Scholar]
- 21.Hemker HC, Giesen P, Al Dieri R, et al. Calibrated automated thrombin generation measurement in clotting plasma. Pathophysiol Haemost Thromb. 2003;33(1):4–15. [DOI] [PubMed] [Google Scholar]
- 22.Gilio K, van Kruchten R, Braun A, et al. Roles of STIM1 and Orai1 in glycoprotein VI- and thrombin-dependent procoagulant activity and thrombus formation. J Biol Chem. 2010;285:23629–23638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van Kruchten R, Cosemans JMEM, Heemskerk JWM. Measurement of whole blood thrombus formation using parallel-plate flow chambers: a practical guide. Platelets. 2012;23(31):229–242. [DOI] [PubMed] [Google Scholar]
- 24.Cosemans JM, Munnix IC, Wetzker R, Heller R, Jackson SP, Heemskerk JW. Continuous signaling via PI3K isoforms β and γ is required for platelet ADP receptor function in dynamic thrombus stabilization. Blood. 2006;108(9):3045–3052. [DOI] [PubMed] [Google Scholar]
- 25.Miserus RJ, Herías MV, Prinzen L, et al. Molecular MRI of early thrombus formation using a bimodal alpha2-antiplasmin-based contrast agent. JACC Cardiovasc Imaging. 2009;2(8):987–996. [DOI] [PubMed] [Google Scholar]
- 26.Abaeva AA, Canault M, Kotova YN, et al. Procoagulant platelets form an α-granule protein-covered “cap” on their surface that promotes their attachment to aggregates. J Biol Chem. 2013;288(41):29621–29632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cosemans JM, Schols SE, Stefanini L, et al. Key role of glycoprotein Ib/V/IX and von Willebrand factor in platelet activation-dependent fibrin formation at low shear flow. Blood. 2011;117(2):651–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zeiler M, Moser M, Mann M. Copy number analysis of the murine platelet proteome spanning the complete abundance range. Mol Cell Proteomics. 2014;13(12):3435–3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.London FS, Marcinkiewicz M, Walsh PN. A subpopulation of platelets responds to thrombin- or SFLLRN-stimulation with binding sites for factor IXa. J Biol Chem. 2004;279(19):19854–19859. [DOI] [PubMed] [Google Scholar]
- 30.Panteleev MA, Ananyeva NM, Greco NJ, Ataullakhanov FI, Saenko EL. Two sub-populations of thrombin-activated platelets differ in their binding of the components of the intrinsic factor X-activating complex. J Thromb Haemost. 2005;3(11): 2545–2553. [DOI] [PubMed] [Google Scholar]
- 31.Kulkarni S, Jackson SP. Platelet factor XIII and calpain negatively regulate integrin αIIbβ3 adhesive function and thrombus growth. J Biol Chem. 2004;279(29):30697–30706. [DOI] [PubMed] [Google Scholar]
- 32.Podolnikova NP, Yakovlev S, Yakubenko VP, Wang X, Gorkun OV, Ugarova TP. The interaction of integrin αIIbβ3 with fibrin occurs through multiple binding sites in the αIIbβ-propeller domain. J Biol Chem. 2014;289(4):2371–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nikolajsen CL, Dyrlund TF, Poulsen ET, Enghild JJ, Scavenius C. Coagulation factor XIIIa substrates in human plasma: identification and incorporation into the clot. J Biol Chem. 2014;289(10):6526–6534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Siebenlist KR, Meh DA, Mosesson MW. Protransglutaminase (factor XIII) mediated crosslinking of fibrinogen and fibrin. Thromb Haemost. 2001;86(5):1221–1228. [PubMed] [Google Scholar]
- 35.Nurden AT, Nurden P. Inherited disorders of platelet function: selected updates. J Thromb Haemost. 2015;13 Suppl 1:S2–9. [DOI] [PubMed] [Google Scholar]
- 36.Puszkin EG, Raghuraman V. Catalytic properties of a calmodulin-regulated transglutaminase from human platelet and chicken gizzard. J Biol Chem. 1985;260(29):16012–16020. [PubMed] [Google Scholar]
- 37.Burkhart JM, Vaudel M, Gambaryan S, et al. The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood. 2012;120(15):e73–82. [DOI] [PubMed] [Google Scholar]
- 38.Mitchell JL, Lionikiene AS, Fraser SR, Whyte CS, Booth NA, Mutch NJ. Functional factor XIII-A is exposed on the stimulated platelet surface. Blood. 2014;124(26):3983–3990. [DOI] [PMC free article] [PubMed] [Google Scholar]