

Abstract
The failure of normal hematopoiesis is observed in myeloid neoplasms. However, the precise mechanisms governing the replacement of normal hematopoietic stem cells in their niche by myeloid neoplasm stem cells have not yet been clarified. Primary acute myeloid leukemia and myelodysplastic syndrome cells induced aberrant expression of multiple hematopoietic factors including Jagged-1, stem cell factor and angiopoietin-1 in mesenchymal stem cells even in non-contact conditions, and this abnormality was reverted by extracellular vesicle inhibition. Importantly, the transfer of myeloid neoplasm-derived extracellular vesicles reduced the hematopoietic supportive capacity of mesenchymal stem cells. Analysis of extracellular vesicle microRNA indicated that several species, including miR-7977 from acute myeloid leukemia cells, were higher than those from normal CD34+ cells. Remarkably, the copy number of miR-7977 in bone marrow interstitial fluid was elevated not only in acute myeloid leukemia, but also in myelodysplastic syndrome, as compared with lymphoma without bone marrow localization. The transfection of the miR-7977 mimic reduced the expression of the posttranscriptional regulator, poly(rC) binding protein 1, in mesenchymal stem cells. Moreover, the miR-7977 mimic induced aberrant reduction of hematopoietic growth factors in mesenchymal stem cells, resulting in decreased hematopoietic-supporting capacity of bone marrow CD34+ cells. Furthermore, the reduction of hematopoietic growth factors including Jagged-1, stem cell factor and angiopoietin-1 were reverted by target protection of poly(rC) binding protein 1, suggesting that poly(rC) binding protein 1 could be involved in the stabilization of several growth factors. Thus, miR-7977 in extracellular vesicles may be a critical factor that induces failure of normal hematopoiesis via poly(rC) binding protein 1 suppression.
Introduction
In myeloid neoplasms (MNs) including acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS), a variety of mechanisms could be involved in the failure of normal hematopoiesis.1–3 In these disorders, neoplastic clones eventually take over the bone marrow (BM) niche even in lower-risk MDS and hypoplastic MDS.4 It has been suggested that normal hematopoiesis could be compromised in the development of AML/MDS as well as the growth advantage of AML/MDS cells.5–8 However, the precise molecular mechanisms governing the replacement of normal hematopoietic stem/progenitor cells by AML/MDS stem/progenitor cells have not yet been clarified.
Recently, it has been shown that BM stromal cells, including mesenchymal stem/stromal cells (MSCs), cooperate to maintain normal hematopoietic9–12 and leukemic stem cells via several molecules, including adhesion molecules, gap junction proteins, cytokines and morphogens.13 More recently, studies using mesenchymal progenitor-specific knockout mice demonstrated impaired microRNA (miRNA) biogenesis in BM MSCs and the development of MDS.14 In patients with AML/MDS, it has been shown by our group and others that abnormal protein expression, such as that of hedgehog-interacting protein15 or aurora kinase A/B,16 occurs in MSCs. These findings suggest that the dysfunction of MSCs could be associated with the development of AML/MDS.
Recently, extracellular vesicles (EVs) released from hematopoietic and BM stromal cells have been found and regarded as novel factors that modulate communication between stem cells and their niche.17 The EVs have been roughly classified into three types including apoptotic body, microvesicle and exosome, according to their size and production mechanism.18 EVs are extracellular nanoshuttles of RNA, protein and lipids that facilitate communication between cells and tissues. However, little is known about the precise molecular mechanisms and involvement of EVs that govern the induction of stromal abnormalities.19–21
In the present study, we first conducted comparative analyses between normal MSCs and those derived from AML/MDS patients to gain insight into the comprehensive changes in gene expression and cell function. We further attempted to identify effectors that were correlated with alterations in AML/MDS-derived MSCs. Consequently, we focused on EV miR-7977 released from AML/MDS cells. We found that the copy number of miR-7977 in the plasma of the BM cavity (BM fluid) was elevated not only in AML patients, but also in MDS patients. Moreover, transfection of a miR-7977 mimic induced the reduction of hematopoietic growth factors in BM MSCs, resulting in a decreased hematopoietic-supporting capacity of BM CD34+ cells.
Methods
Reagents and human BM MSCs
GW4869 (inhibitor of the neutral sphingomyelinase, SMPD2) was purchased from Cayman Chemical (Ann Arbor, MI, USA). Anti-Jagged 1 (JAG1) (ab109536) and anti- PCBP1 (poly(rC) binding protein 1) antibodies (ab168377) were purchased from Abcam® (Tokyo, Japan). StemPro®-34 (Life Technologies, Carlsbad, CA, USA) was used as a serum-free medium. This study was approved by the Institutional Review Board at our university and conducted according to the Declaration of Helsinki. The patients with lymphoma stage I/II and those with AML/MDS in this study were also fully informed of the experimental protocol. Human BM CD34+ cells and four different lot numbers of MSCs from healthy volunteers (HVs) were purchased from AllCells, LLC (Toronto, Canada) (HV-derived MSCs #1, #2, #3 and #4). Human primary MSCs and human BM CD34+ cells derived from lymphoma patients stage I/II without bone marrow localization (control MSCs) and patients with AML/MDS (Online Supplementary Table S1 and Table S2) were prepared as previously described.15, 22 The MSCs were cultured in MSCGM™ hMSC basal medium with the addition of supplements from the MSCGM hMSC SingleQuot Kit (Lonza Japan Ltd, Tokyo, Japan). The diagnostic criteria of AML/MDS were based according to the World Health Organization (WHO) 2008 Classification of MNs. MDS was further evaluated by the Revised International Prognostic Scoring System (IPSS-R).
Coculture of BM CD34+ cells with human MSCs
We modified our previous cord blood-based coculture system to a BM CD34+-based system.22,23 In this new coculture system, BM CD34+ cells were cocultured with human control- or AML/MDS-derived MSCs in serum-free StemPro®-34 medium in the presence of a cytokine cocktail consisting of 50 ng/mL human thrombopoietin (TPO), 10 ng/mL human stem cell factor (SCF), 50 ng/mL human Flk2/Flt3 ligand (FLT3LG) and 100 ng/mL human delta-like protein 4 (DLL4) (all from R&D Systems, Minneapolis, MN, USA). In this system, the primary MSC layer could be maintained for over 8 weeks even in a serum-free medium (Online Supplementary Methods).
Clonogenic analysis of cocultured hematopoietic cells
The clonogenic assay was performed using MethoCult GF H4434V (StemCell Technologies, Vancouver, Canada) as described previously.23
Contact and non-contact culture systems
Contact and non-contact culture systems were conducted using Polyester Membrane Transwell Clear Inserts and Companion Plates (BD Biosciences, San Jose, CA, USA; pore size: 0.4 μm, pore density: 1×108/cm2, 12 well) as reported previously (Online Supplementary Methods).24
EV transfer assay using cells labeled with GFP, PKH26 and SYTO RNAselect
Green fluorescent protein (GFP)-transduced leukemic cells were established using the retroviral vector, pRx-IRES-hrGFP, as described previously.23 The leukemic cells were stained with PKH26 (Sigma-Aldrich, St. Louis, MO, USA), a red fluorescent membrane cell linker, before coculture according to the manufacturer’s instructions as previously reported.25 Total RNA in leukemic cells was stained with SYTO RNAselect (Life Technologies). 2 × 104 to 2 × 105 labeled leukemic cells were added into the transwell insert and cocultured for 3 or 14 days with or without 10 μM GW4869 or nSMase2 siRNA (Stealth RNAi SMPD2 human (s13170), Life Technologies) to inhibit EV secretion. Target MSCs were transferred onto Lab-Tek II Chamber Slides (Thermo Scientific, Waltham, MA, USA), and visualized using ZEISS/ELYRAS 1LSM780 confocal microscope (ZEISS, Oberkochen, Germany).
EV preparation
EVs were isolated from the supernatant of hematopoietic cell lines or BM fluid by centrifugation, filtration and the Exosome Precipitation Solution (ExoQuick-TC; System Biosciences, Mountain View, CA, USA). Briefly, the supernatant of hematopoietic cells or the BM fluid was centrifuged at 3,000 g for 15 min to remove cells and apoptotic bodies.26 Subsequently, the sample was passed through a 0.45 μm pore size Millipore Hydrophilic Durapore filter (Merck Millipore, Tokyo, Japan).27,28 The larger-sized microvesicular particles were deposited onto the filter membrane. The resulting filtrate was transferred to a sterile vessel, and the appropriate volume of ExoQuick-TC was added. After incubation at 4°C for 2 hours, the mixed solution was centrifuged at 1,500 g for 30 minutes and the supernatant was removed. The pellet was kept on ice and used as the EV fraction after centrifugation at 1,500 g for 5 minutes to remove residual solution.
Microarray analysis for EV miRNA
To harvest EVs from 2 × 105 primary AML CD34+ cells, normal BM CD34+ cells and leukemic cell lines, including TF-1 and Kasumi-1, cells were cultured in serum-free StemPro®-34 medium with a cytokine cocktail on plates coated with FN fragments (Retronectin®: Takara Bio, Tokyo, Japan) instead of MSCs as reported earlier.29 EV miRNA from the supernatant of CD34+ hematopoietic and leukemic cells were prepared as reported previously.30 Microarray analysis of the miRNA profiles was done with the human miRNA Oligo chip (Human_miRNA_V20) and the 3D-Gene® miRNA labeling kit (TORAY, Kanagawa, Japan).
Statistical analysis
Each data set was first evaluated for normality of distribution by the Komolgorov-Smirnov test to decide whether a nonparametric rank-based analysis or a parametric analysis should be used. The significance of differences between groups was assessed by ANOVA, followed by Dunnett’s multiple comparison tests. Results are expressed as the mean ± standard deviation (SD). The significance of differences was assessed by either the Student’s t-test or the Mann-Whitney U-test, and a P-value < 0.05 was considered as statistically significant.
Results
Analysis of gene expression and hematopoietic-supporting capacity of MSCs derived from normal or AML/MDS BM
In the present study, we first screened for differences in gene expression between MSCs derived from control and AML/MDS BM by quantitative real-time PCR (qRT-PCR) array (Online Supplementary Methods). The expression of multiple hematopoietic factors was reduced in AML/MDS-derived MSCs as compared with control MSCs, although the expression of some genes such as toll-like receptor 4 (TLR4) and CD44 was elevated in AML/MDS-derived MSCs (Figure 1A). To assess the hematopoietic-supporting capacity of AML/MDS-derived MSCs, normal BM CD34+ cells were cocultured with human control- or AML/MDS-derived MSCs in serum-free StemPro®-34 medium in the presence of a cytokine cocktail consisting of human SCF, TPO, FLT3LG and DLL4.22 Using this system, we assessed the clonogenicity and in vivo repopulating activity of BM CD34+ cells 14 days after their coculture with primary MSCs derived from AML/MDS patients. The number of colony forming unit (CFU)-Mix was remarkably reduced as compared with control MSCs (Figure 1B). Moreover, BM CD34+ cells cocultured with AML/MDS-derived MSCs, but not control MSCs, lost in vivo repopulating activity in immunodeficient mice (Figure 1C). Furthermore, comparative analyses between control- (ID 11) and AML-derived MSCs (ID 2) using age-matched patients showed identical results (Online Supplementary Figure S1). These results indicated that AML/MDS-derived MSCs could not support normal hematopoietic progenitor/stem cells.
Figure 1.
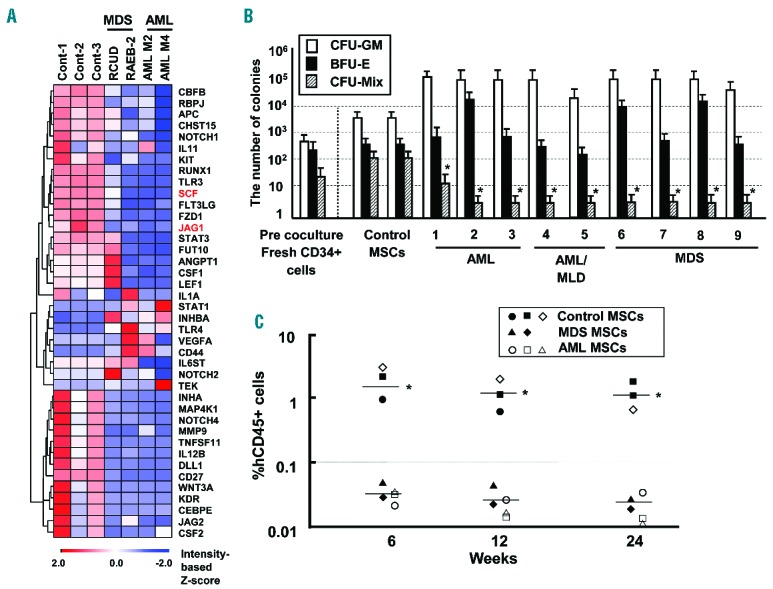
Comparative analysis of mRNA expression between HV- and AML/MDS-derived MSCs. (A) Analysis of hematopoietic factors in MSCs by qRT-PCR array. Results of cluster analysis in MSCs-derived from control (MSC ID 10, 11 and 12), RCUD (MSC ID 8), RAEB-2 (MSC ID 6), AML M2 (MSC ID 2) and AML M4 (MSC ID 1) patients (Online Supplementary Table S1). (B) Clonogenic assay after coculture with human stromal cells. Y-axis indicates the number of colonies after ex vivo coculture of 2×104 BM CD34+cells on MSC layer. X-axis indicates the individual MSCs derived from control and AML/MDS patients. Pre-coculture indicates the number of colonies derived from fresh BM CD34+ cells. MSCs derived from HV were used as normal control. *P<0.01, colony-forming units (CFU)-Mix: primary AML/MDS-derived MSCs vs. normal control-derived MSCs (Student’s t-test, two-tailed). BFU-E, burst forming units of erythroid; CFU-GM, CFU-granulocyte/monocyte; CFU-Mix, CFU-mixed. Results are expressed as means ± SD. Similar results were obtained in 3 independent experiments, performed in triplicate. AML/MLD: AML with multi-lineage dysplasia. (C) All hematopoietic cells that were cocultured with AML/MDS-derived MSCs were transplanted into immunodeficient mice. Representative results, each performed in triplicate of control MSCs (ID 10 (●), ID 11 (■), ID 12 (□)), MSCs derived from AML (ID 1 (○), ID 3 (□), ID 4 (△)) and MSCs derived from MDS (ID 9 (▲), ID 8 ()), are shown (Online Supplementary Table S1). Y-axis indicates the percentage of human specific CD45+ cells. X-axis indicates weeks after transplantation. *P<0.01 controls vs. AML/MDS. Similar results were obtained in two independent experiments.
Elucidation of the effectors that alter the hematopoietic factors derived from AML/MDS stromal cells
In an attempt to identify these effectors, we employed non-contact and contact coculture systems to determine whether the effectors are soluble or not (Figure 2A). In this analysis, we employed the change in stromal JAG1 expression as an indicator, and used AML cell lines such as TF-1 (AML M6) and Kasumi-1 (AML M2) in this screening. Unexpectedly, in both non-contact and contact coculture systems, decreased JAG1 and SCF expression in MSCs was observed with both leukemic cell lines (Figure 2B), indicating that the effectors were at least in part soluble or humoral.
Figure 2.
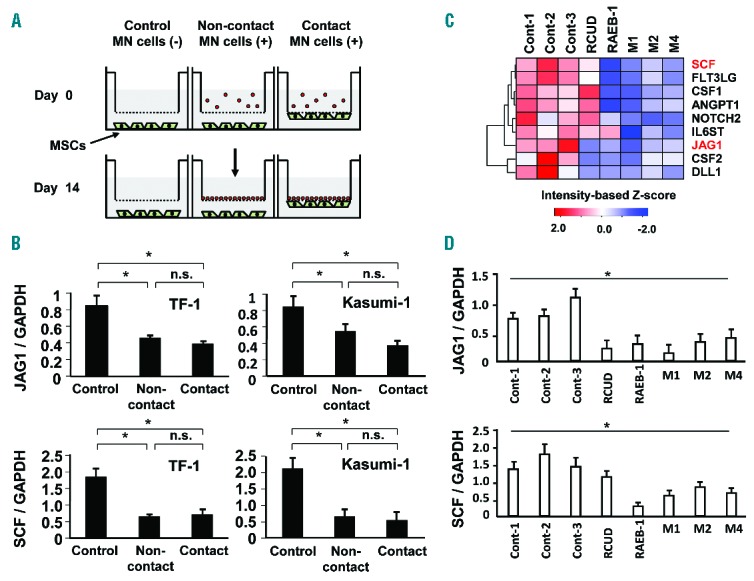
Analysis of the effect of MN cells on BM MSCs. (A) Non-contact and contact culture systems using transwell clear inserts with 0.4 μm pore membrane filter and companion plates in serum-free StemPro®-34 medium. (B) The effect of non-contact and contact coculture of TF-1 or Kasumi-1 leukemic cells with BM MSCs on JAG1 mRNA expression. Results are expressed as means ± SD. *P<0.05, normal MSCs cocultured with AML cells vs. normal MSCs without coculture (Control). (C) Effect of primary AML cells and MDS cells on the expression of hematopoietic factors in BM MSCs. CD34+ fraction of primary AML cells and MDS cells were cocultured with MSCs in the presence of cytokines including SCF, TPO and DLL4. The hematopoietic factors in MSCs were analyzed by qRT-PCR array. Sample ID: Cont-1, ID 1; Cont-2, ID 3; Cont-3, ID 5; RCUD, ID 11; RAEB-1, ID 23; AML M1, ID 35; AML M2, ID 37; AML M4, ID 40 (Online Supplementary Table S2). (D) The expression levels of JAG1 and SCF mRNAs were further confirmed by qRT-PCR. *P<0.01 controls vs. AML/MDS. Data represent three independent experiments, each done in triplicate.
We further investigated whether primary AML and MDS cells could induce a similar effect on primary BM MSCs cultured in a non-contact system using hematopoietic PCR array. Surprisingly, even primary MDS cells and AML cells induced altered mRNA expression of multiple hematopoietic growth factors in non-contact conditions (Figure 2C, Highlighted). Remarkably, key hematopoietic factors such as JAG1 and SCF in MSCs were significantly reduced (Figure 2D). Collectively, certain soluble/humoral factors may be involved in the reduction of JAG1 and SCF mRNA expression.
The release of EVs from leukemic cells and transfer to MSCs
It has been shown that hematopoietic cells, including those in MNs, release a variety of soluble/humoral factors such as cytokines, membrane-anchored mediators including Wnt/Hh,23,31 shed receptors and EVs.32 Among them, recent studies have focused on the involvement of EVs including exosomes in transcriptome alteration and cellular phenotype switching. Hence, we investigated whether leukemic cells secrete EVs in vitro into the supernatant. The fraction of EVs was examined by transmission electron microscopy, and 30–50 nm vesicles were mainly observed (Figure 3A). Subsequently, the tetraspanin, CD63, and endosomal markers, ALIX and TSG101, were examined by immunoblotting analysis. As a result, CD63, ALIX and TSG101 were detected in the fraction of EVs released from TF-1 and Kasumi-1 cells (Figure 3B), suggesting that EVs could contain exosomes as well as other microvesicles.
Figure 3.
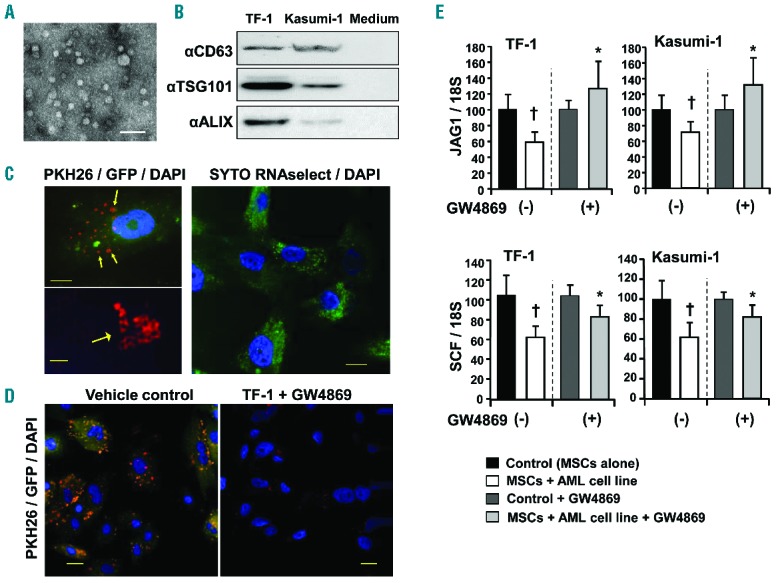
The effect of inhibition of EV biogenesis by the nSMase2 inhibitor, GW4869, on BM stromal cells cocultured with leukemic cells. (A) Transmission electron microscopy. The EV fraction was prepared from the supernatant of TF-1 cells. Scale bar: 100 nm. (B) The EV fraction was analyzed by immunoblotting using anti-CD63, anti-ALIX and anti-TSG101 antibodies. The EV fraction was prepared from 5 ml supernatant of TF-1 or Kasumi-1 cells, or serum-free medium. (C) EV transfer to MSCs was analyzed by super resolution confocal microscopy (ZEISS/ELYRAS 1LSM780). Left panels (upper and lower): Image of an MSC cocultured with Kasumi-1 leukemic cells labeled with GFP (green) and PKH26 (red) shows presence of microvesicles in the cell (yellow, arrows). Scale bar: 10 μm (left upper), 2.0 μm (left lower). Right panel: Image of an MSC cocultured with leukemic cells labeled with SYTO RNAselect™ (green) shows transfer of RNA from leukemic cells to the MSC. The signals were obtained by 5 rotations, and the pictures were reconstituted as a wide-field structured illumination microscopy (SIM) image. DAPI (blue) was used for nuclear staining. Scale bar: 10 μm. (D) EV transfer assay was conducted in the presence or absence of an EV inhibitor (GW4869) in MSCs cocultured with leukemic cells labeled with PKH26 (red) and GFP (green), and visualized by confocal microscopy. Merged images are shown, and DAPI (blue) was used for nuclear staining. Scale bar: 20 μm. (E) Changes in JAG1 and SCF mRNA levels in MSCs cocultured with TF-1 or Kasumi-1 cells in serum-free StemPro®-34 medium and treated with or without GW4869 were examined by qRT-PCR. Data shown are from one representative experiment of three showing similar results, each done in sextuplicate. Y-axis indicates fold change relative to the control or control + GW4869 (=1.0) after normalization to 18S expression level. Results are expressed as means ± SD. †P<0.05, HV-derived MSCs without coculture vs. HV-derived MSCs cocultured with AML cells. *P<0.05, HV-derived MSCs cocultured with AML cells in the absence of GW4869 vs. HV-derived MSCs cocultured with AML cells in the presence of GW4869 (Student’s t-test, two-tailed).
Subsequently, we determined whether EV transfer from leukemic cells to MSCs could be achieved. Leukemic cells labeled with GFP and PKH26 (cell membrane) or SYTO RNAselect (total RNA) were cocultured in a non-contact system with MSCs. Diffuse GFP (green) and multiple and sporadic PKH26 (red) signals were detected in MSCs by confocal microscopy (Figure 3C, upper left panel). Super-resolution analysis of these confocal images showed that PKH26 signals exhibited multiple aggregations of hollow shell pattern (Figure 3C, lower left panel). Further, sporadic signals for SYTO RNAselect (green) were observed in MSCs (Figure 3C, right panel). These findings clearly indicated that the EVs derived from leukemic cells were efficiently transferred into MSCs.
The effect of inhibition on microvesicular transfer and gene expression in MSCs
To confirm the transfer of EVs and determine its effect on MSCs, an EV inhibitor which functions through the suppression of neutral sphingomyelinases (GW4869), as well as siRNA and short hairpin (sh)RNA against the neutral sphingomyelinase, sphingomyelin phosphodiesterase 2 (SMPD2), were used to treat MSCs cultured in a non-contact system with leukemic cells. PKH26 (red) and GFP (green) signals were largely decreased in the presence of the inhibitor of EVs by confocal microscopic analysis, indicating a reduction in the transfer of EVs from leukemic cells to MSCs (Figure 3D and Online Supplementary Figure S2A,S2B). We subsequently examined whether the EV inhibitor protected against changes in mRNA expression by qRT-PCR array. Acute leukemic cell lines in non-contact coculture with MSCs reduced the gene expression of multiple hematopoietic factors including SCF and JAG1 in MSCs (Figure 3E). Importantly, the EV inhibitor reverted the reduction of a large number of genes including JAG1 and SCF (Figure 3E and Online Supplementary Figure S3A). We additionally confirmed the effect of EV inhibition on gene expression of SCF and JAG1 by qRT-PCR. The decrease in JAG1 expression was clearly inhibited by SMPD2 shRNA (Online Supplementary Figure S2B) and the EV inhibitor (Online Supplementary Figure S3A). SCF expression was partially restored by shRNA (Online Supplementary Figure S2C), suggesting that SCF expression was regulated by not only EVs, but other factors as well. We further examined whether these effects on MSCs could be induced by primary AML cells. Although normal CD34+ cells did not alter JAG1 expression in MSCs (Online Supplementary Figure S3B), myeloblastic leukemia (M2) and myelomonocytic leukemia (M4) cells induced the reduction in JAG1 expression, which was restored by EV inhibition. Collectively, the alterations in certain mRNA expressions such as JAG1 in MSCs were induced via EV transfer from leukemic cells.
Purification of EVs from primary AML and analysis of miRNA
It has been revealed that EVs contain multiple components including miRNA, mRNA, fragments of DNA, peptides and lipids. Moreover, our results demonstrated that detectable EV RNA from leukemic cells was transferred into MSCs (Figure 3C). Balakrishnan et al. have recently shown that miRNA interacts with mRNA and regulates gene expression in BM stromal cells.33 Therefore, we decided to analyze EV miRNA in our model. To obtain EV miRNA, we utilized a fibronectin (FN)-based, MSC-free culture system (Figure 4A). The CD34+ fraction of normal BM or AML cells was cultured on FN-coated dishes in 10 mL of serum-free medium. Subsequently, the EV fraction was enriched from the supernatant, and EV miRNA was extracted. We compared the contents of miRNA harvested from CD34+ cells derived from HV and AML patients using a human miRNA Oligo chip. Scatter plot and cluster analysis revealed that EVs derived from primary AML (M1 and M4) cells contained an elevated fraction of miRNAs as compared with normal BM CD34+ cells (Figure 4B,C). In particular, EV miR-4286, miR-7977 and miR-8073 from primary leukemic cells were more than 2-fold higher than those from normal CD34+ cells.
Figure 4.
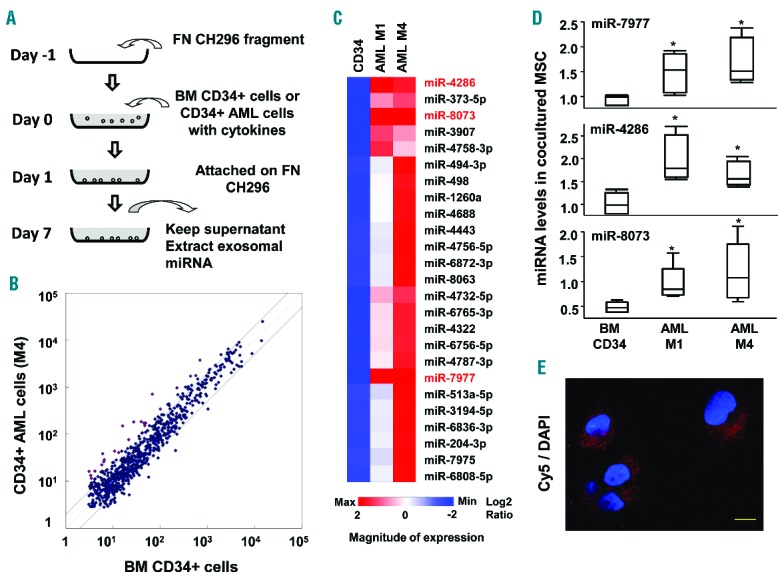
Comparison of miRNA between primary hematopoietic and leukemic CD34+ cells. (A) The culture system using normal BM or AML CD34+ cells plated on fibronectin substratum in the presence of cytokines including SCF, TPO, FLT3LG, IL-3 and DLL4. In this system, fibronectin fragment instead of MSCs was utilized to purify the EVs derived from hematopoietic cells. (B) miRNA was analyzed by miRNA chip and compared. Scattered plot demonstrates that EV miRNA is released from normal CD34+ and primary AML cells (M4, Sample ID 40). (C) Heatmap of elevated EV miRNAs derived from leukemic CD34+ cells as compared with those from normal CD34+ cells. Highlighted EV miRNAs show more than 2-fold elevation in both AML M1 (Sample ID 35) and AML M4 (Sample ID 40) as compared with normal CD34+ cells (Online Supplementary Table S2). CD34: commercially purchased normal CD34+ cells. (D) The levels of miR-7977, miR-4286 and miR-8073 were examined by qRT-PCR in MSCs cocultured with BM CD34+, primary AML M1 and AML M4 cells. SNORD61 was used as an internal standard. Results are expressed as means ± SD. *P<0.05, normal MSCs cocultured with AML cells vs. normal MSCs cocultured with CD34+ cells (Student’s t-test). (E) Cy5-labeled miR-7977 was transfected into AML M1 cells, which were cocultured with MSCs for 3 days. The labeled miR-7977 that was transferred into MSCs was analyzed by super resolution confocal microscopy (ZEISS/ELYRAS 1LSM780). Scale bar: 10 μm.
We examined whether these miRNAs could be transferred into MSCs after coculture with normal CD34+ cells and primary AML (M1 and M4) cells. Expectedly, the levels of miR-7977, miR-4286 and miR-8073 in MSCs after coculture with primary AML (M1 and M4) cells were significantly higher than in those cocultured with normal CD34+ cells (Figure 4D). Moreover, Cy5-labeled miR-7977 in AML M1 cells was transferred into MSCs 3 days after non-contact coculture (Figure 4E). These results indicated that miRNAs in AML cells could be transferred via EVs.
The level of miRNA in human BM cavity
We further investigated whether the level of EV miRNA was elevated in BM fluid in patients with AML/MDS obtained by BM aspiration. According to the Revised International Prognostic Scoring System (IPSS-R), MDS patients were classified into lower-risk MDS (very low and low) and higher-risk MDS (intermediate, high and very high). Electron microscopy revealed that multiple 30–50 nm vesicles could be observed in BM fluid (Figure 5A), and CD63, ALIX and TSG101 were detected in these vesicles (Figure 5B). Subsequently, we analyzed the copy number of miR-7977, miR-4286 and miR-8073 in EVs of BM from lymphoma stage I/II (as control), MDS and AML patients (Online Supplementary Table S2). We found that miR-7977 was significantly increased even in lower-risk MDS in addition to higher-risk MDS and AML patients (Figure 5C). Moreover, miR-7977 was significantly correlated with the percentage of blasts in BM (Figure 5D). However, miR-4286 and miR-8073 were not significantly increased in lower-risk MDS patients although miR-4286 was significantly increased in AML patients, and miR-8073 was significantly increased in higher-risk MDS and AML patients (Online Supplementary Figure S4A,S4B). Collectively, these results indicated that aberrant expression of miR-7977 in the BM cavity could be involved in the disturbance of normal hematopoiesis in patients with MDS and AML.
Figure 5.
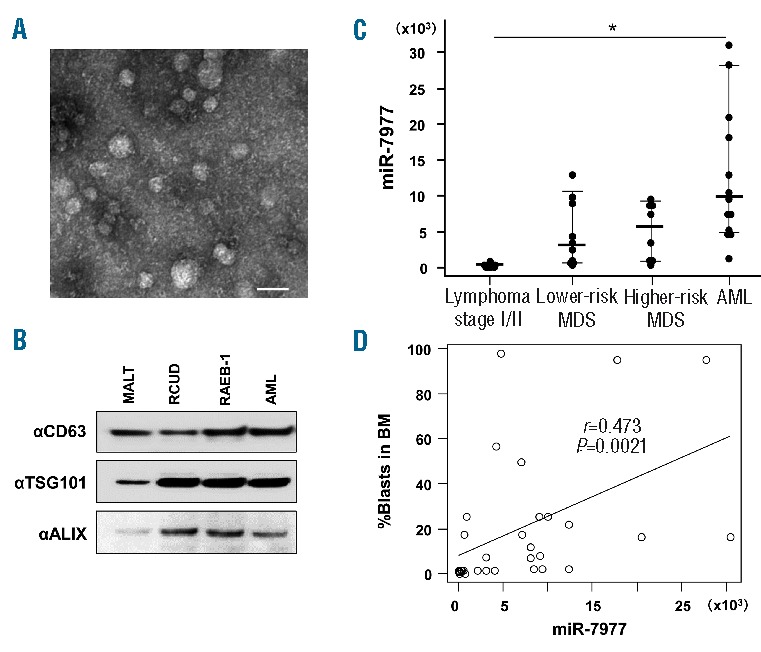
The copy number of EV miR-7977 in BM fluid. (A) The EVs in BM fluid after BM aspiration (Sample ID 3) were analyzed by transmission electron microscopy. Scale bar: 50 nm. (B) The EV fraction derived from patients with MALT lymphoma stage I (Sample ID 3), RCUD (ID 11), RAEB-1 (ID 23) and AML (ID 28) was analyzed by immunoblotting using anti-CD63, anti-ALIX and anti-TSG101 antibodies (Online Supplementary Table S2). (C) EV miR-7977 in BM fluid was analyzed by qRT-PCR. A miR-7977 mimic was used as internal positive control during miRNA quantification. Y-axis indicates the copy number of miR-7977 in 1 mL of BM fluid obtained by BM aspiration. Lymphoma stage I/II without BM localization as control (n=9), lower-risk MDS (n=10), higher-risk MDS (n=8) and AML (n=13) patients. Results are expressed as means ± SD. *P<0.05, the copy number of miR-7977 in lower-risk MDS, higher-risk MDS or AML vs. the copy number of miR-7977 in control (ANOVA, followed by Dunnett’s multiple comparison tests). (D) Correlation between the copy number of miR-7977 in BM fluid and percentage of blasts in BM among control, MDS and AML patients. Correlation coefficient: r=0.473. P=0.0021.
The effect of miR-7977 on MSCs
Using an online database for miRNA target prediction (miRDB), we found that miR-7977 can potentially interact with JAG1 mRNA and primarily interacts with PCBP1 mRNA, which is involved in posttranscriptional control.34 Hence, we employed a miR-7977 mimic to analyze the effect on BM MSCs. The levels of JAG1 and PCBP1 mRNA were decreased after transfection of the miR-7977 mimic (Online Supplementary Figure S5A), and target protection of JAG1 and PCBP1 reverted their reduction (Figure 6A,B). Moreover, luciferase assay with the 3′ untranslated region (3′UTR) of JAG1 and PCBP1 indicated that miR-7977 directly interacted with PCBP-1 and JAG1 mRNAs (Figure 6C,D). Unexpectedly, target protection of PCBP1 partially reverted the reduction in JAG1 mRNA after transfection with the miR-7977 mimic (Figure 6E). It has been revealed that the K-homologous (KH) domain of PCBP1 binds to the 3′UTR with a C-rich motif of mRNAs and enhances the efficiency of 3′ processing, thereby altering the levels of expression of subsets of mRNAs in the mammalian transcriptome.34 Thus, JAG1 mRNA may be a target of PCBP1. These findings indicated that miR-7977 regulated JAG1 expression at the translational and post-transcriptional levels via PCBP1. Importantly, the levels of mRNAs of multiple growth factors were reduced after transfection of the miR-7977 mimic into MSCs (Online Supplementary Figure S5B). Moreover, the reductions in SCF and ANGPT1 (Angiopoietin 1) proteins were reverted by target protection of PCBP1 (Figure 6F), suggesting that PCBP1 could be involved in the stabilization of multiple growth factors. Collectively, transfection of a miR-7977 mimic could induce disturbance of the expression of hematopoietic factors in BM MSCs.
Figure 6.
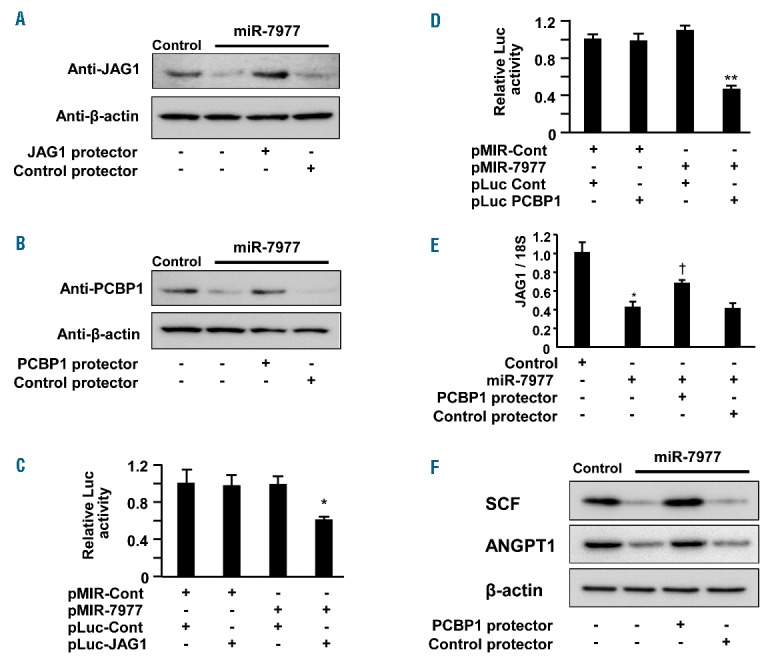
The effect of a miR-7977 mimic on MSCs. (A) The reduction in JAG1 was analyzed by western blotting analysis. Control, 5 nM negative control siRNA; miR-7977, 5 nM miR-7977 mimic; Control protector, 250 nM negative control miScript Target Protector; JAG1 protector, 250 nM Target Protector for JAG1. (B) The reduction in PCBP1 was analyzed by western blotting analysis. PCBP1 protector, Target Protector for PCBP1. Direct interaction between miR-7977 and target genes was evaluated by luciferase assay using pLuc-JAG1 (C) and pLuc-PCBP1 (D). Data are presented as the ratio of the normalized value to the light emission observed in the cells transduced with the control vector (pLuc-Cont). pMIR-Cont or pMIR-7977 was cotransfected with pLuc-JAG1 or pLuc-PCBP1 into MSCs 48 hrs before analysis (Online Supplementary Methods). Data shown are from one representative experiment of three showing similar results, each done in quadruplicate. Results are expressed as means ± SD. *P<0.05, pMIR-Cont with pLuc-Cont vs. pMIR-7977 with pLuc-JAG1. **P<0.01, pMIR-Cont with pLuc-Cont vs. miR-7977 with pLuc-PCBP1. (E) The expression of JAG1 mRNA in MSCs after transfer of 5 nM miR7977 mimic and 250 nM PCBP1 protector was examined by qRT-PCR. *P<0.05, HV-derived MSCs transduced with negative control vs. HV-derived MSCs transduced with miR-7977 mimic. †P<0.05 MSCs transfected with miR-7977 mimic vs. MSCs cotransfected with miR-7977 mimic and PCBP1 protector (Student’s t-test). Y-axis indicates fold change relative to the control or control + GW4869 (=1.0) after normalization to 18S expression level in MSCs. Data shown are from one representative experiment of three showing similar results, each done in quadruplicate. (F) The recovery of SCF and ANGPT1 expression after PCBP1 protector transfer into MSCs was analyzed by western blotting analysis.
Evaluation of hematopoietic-supporting capacity of MSCs after transfection of a miR-7977 mimic
In an attempt to assess the hematopoietic-supporting capacity of negative control or miR-7977 mimic-transfected MSCs, we analyzed the expression of surface markers on hematopoietic cells 7 days after coculture with BM CD34+ cells. The number of CD34+CD38− cells and CFU-Mix in coculture with miR-7977 mimic-transfected MSCs was significantly lower than that in coculture with negative control-transfected MSCs (Figure 7A,B and Online Supplementary Figure S6A). The percentage of CD34-CD38− and CD11b+ cells that were cocultured with miR-7977 mimic transfected MSCs was higher than that cocultured with negative control transfected MSCs (Figure 7A,B and Online Supplementary Figure S6B). These results suggested that the hematopoietic-supporting capacity of miR-7977 mimic transfected MSCs was reduced as compared with that of negative control transfected MSCs. Importantly, the reduction in the hematopoietic-supporting capacity of miR-7977 mimic transfected MSCs was reverted with the cotransfection of the PCBP1 protector, or ANGPT1 or SCF expression vector (Online Supplementary Figure S7A,S7B).35 Subsequently, we prepared EVs from 5 mL of BM fluid derived from HV or AML/MDS patients and labeled them with PKH26. AML/MDS-derived EVs contained abundant miR-7977 while control EVs had a drastically lower level of it (Figure 5C). The transfer efficiency of both control- and AML/MDS-derived EVs into MSCs was around 50% (Online Supplementary Figure S8). The percentage of CD34+ cells and the number of clonogenic cells in coculture with MSCs harboring AML/MDS-derived EVs were significantly lower than those in coculture with MSCs harboring control EVs (Figure 7C and Online Supplementary Figure S9). These results strongly suggest that miR-7977 modulates the hematopoietic-supporting capacity of BM MSCs via reduction of PCBP1.
Figure 7.
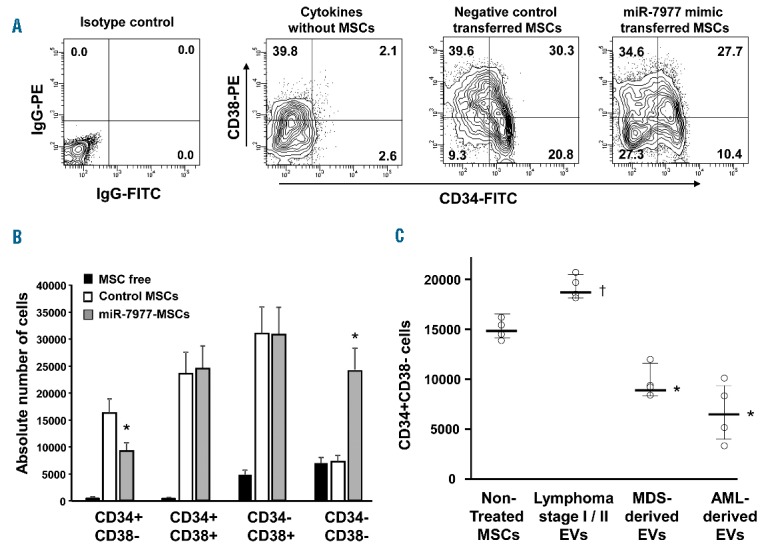
Hematopoietic-supporting capacity of miR-7977 mimic-transfected MSCs. (A) Expression of CD34 and CD38 in ex vivo cocultured hematopoietic cells. The X-axis indicates CD34 labeled with a FITC-conjugated monoclonal antibody (CD34-FITC). The Y-axis indicates CD38 labeled with a PE-conjugated monoclonal antibody (CD38-PE). Positivity for a surface antigen was defined using the isotype control monoclonal antibody. Left panel: Isotype control. Middle left panel: Expression of CD34/CD38 in ex vivo cultured hematopoietic cells without MSCs in the presence of SCF, TPO, FLT3LG and DLL4. Middle right panel: Expression of CD34/CD38 in cocultured hematopoietic cells with negative control-transfected MSCs. Right panel: Expression of CD34/CD38 in cocultured hematopoietic cells with miR-7977 mimic-transfected MSCs. Data shown are from one representative experiment of three showing similar results. (B) The summary of CD34/CD38 positive cells after culture with or without MSCs. *P<0.01 negative control vs. miR-7977 mimic. MSC free: CD34/CD38 positive cells among cultured hematopoietic cells without MSCs. (C) Expression of CD34 in ex vivo hematopoietic cells cocultured with HV-derived MSCs harboring EVs derived from lymphoma stage I/II (control), MDS or AML patients. Non-treated HV-derived MSCs (n=4, lot number #3), MSCs with control EVs (n=4, sample ID 1, 2, 3 and 4), MSCs with MDS-derived EVs (n=4, sample ID 10, 11, 22 and 23) and MSCs with AML-derived EVs (n=4, sample ID 28, 29, 31 and 33). MDS- and AML-derived EVs contained abundant miR-7977. The Y-axis indicates absolute number of CD34+CD38− cells. *P<0.01, Non-treated MSCs vs. MSCs with AML/MDS-derived EVs. †P<0.05, Non-treated MSCs vs. MSCs with HV-derived EVs (Student’s t-test, two-tailed). Data shown are from one representative experiment of three showing similar results.
Discussion
In the present study, we found that the expression of multiple growth factors was significantly reduced in AML/MDS-derived BM MSCs as compared with those from control BM MSCs. Functionally, AML/MDS-derived BM MSCs exhibited lower hematopoietic-supporting capacity of stem/progenitor cells. Moreover, EVs derived from the CD34+ fraction of AML/MDS cells could be transferred to MSCs, and EV inhibition partially restored aberrant expression of hematopoietic growth factors including JAG1, SCF and ANGPT1 in AML/MDS-derived MSCs. In addition, EV miR-7977 derived from AML/MDS cells was remarkably enriched in vitro and in BM cavity, and was found to be involved in aberrant expression of mRNAs and reduction in the hematopoietic-supporting capacity of BM MSCs.
Recently, mesenchymal progenitor-specific conditional Dicer1 knockout or osteoblast-specific activating β-catenin knock-in increased the frequency of genetic mutations in hematopoietic cells and eventually the development of AML/MDS.14,36 Further, activating β-catenin knock-in cells exhibited elevated JAG1 expression. Conversely, Dicer1 knockout cells exhibited a reduction in JAG1 expression (GDS3404 and GDS4504). These findings suggest that osteoblasts and MSCs play different roles in supporting normal hematopoiesis,37 and JAG1 expression could be regulated by β-catenin signaling and miRNA biogenesis. Consistent with these previous reports, we and others reported that mRNA expression of hematopoietic factors in MDS-derived MSCs was significantly disturbed.15,38 Moreover, we revealed in the present study that the mRNA expression of several hematopoietic factors in BM MSCs was decreased (Figure 1A), and these reductions correlated with the dysfunction of hematopoietic support in AML/MDS-derived MSCs (Figure 1B,C). Collectively, the disturbance of MSC function in AML/MDS could be involved in the failure of normal hematopoiesis.
From these results, an ensuing intriguing question is how the disturbance of stromal function is induced. One possible explanation is that stromal dysfunction occurs spontaneously with advancing age and/or genetic damage mediated by reactive oxygen species. Consistent with these notions, it was previously reported that chromosomal abnormalities such as loss of heterozygosity and uniparental disomy (UPD), which can result from double-stranded breaks, are sometimes observed in MSCs derived from AML/MDS patients.39–41 Another possible explanation is that certain factors can directly induce functional abnormality in MSCs. Recently, it was demonstrated that the transplantation of chronic myelogenous leukemia (CML) repopulating cells into immunodeficient mice altered the microenvironmental regulation of the stem cell niche.12,42,43 In accordance with these findings, we also found that primary CD34+ leukemic cells, but not normal CD34+ cells, induced a decrease in JAG1 and SCF expression. These findings indicated that leukemic cells could induce the dysfunction of BM stromal cells.
However, it is difficult to identify the effectors in leukemic cells which mediate these effects. To resolve this problem, we utilized the non-contact and contact culture systems using primary leukemic cells and MSCs. We found that even in a non-contact condition, primary leukemic cells induced the alteration of mRNA expression in MSCs, suggesting that the effectors are soluble or humoral factors (Figure 2). Recently, it was demonstrated that EVs derived from BM MSCs facilitated the progression of multiple myeloma,44 and those derived from CML and chronic lymphocytic leukemia cells facilitated the progression through an autocrine mechanism.19,21 Moreover, primary AML cells released EVs which were possibly enriched for both coding and non-coding RNAs.17,45 These findings led us to explore the possibility of EVs-mediated communication between leukemic cells and MSCs. In the present study, comparative analysis revealed that miRNA species, including miR-7977, were elevated in AML-derived EVs (Figure 4C), and miR-7977 was significantly elevated in MSCs cocultured with primary AML cells (Figure 4D) as well as BM fluid of AML, in lower-risk and higher-risk MDS patients (Figure 5C). These results suggested that BM EVs work as nanoshuttles to carry various biological elements including miRNA. Importantly, transfection of a miR-7977 mimic reduced the levels of JAG1 and PCBP1 (Figure 6A,B).
It has been revealed that the KH-domain of PCBP1 binds to a 3′UTR with C-rich motif of mRNAs and enhances the efficiency of 3′ processing, thereby altering the levels of expression of subsets of mRNAs in the mammalian transcriptome.34,46 In the present study, the mRNA levels of multiple growth factors were reduced after transfection of a miR-7977 mimic into MSCs (Figure 6C). Collectively, miR-7977 could alter the transcriptome in BM MSCs, suggesting that excess miR-7977 may have an impact on the hematopoietic function of MSCs. In fact, the hematopoietic-supporting capacity was significantly reduced in MSCs after transfection of a miR-7977 mimic and miR-7977-enriched EVs (Figure 7).
In the present study, although the regulation of EV miRNA levels, including miR-7977, miR-4286 and miR-8073, was not clarified, one plausible possibility could be that intracellular miRNAs were increased by differential regulation of the miRNA promoters via certain transcription factors.47,48 In order to determine this, biological databases were used, and several transcription factor binding sites, including those for Evi-1, GATA-2 and PAX-6, were detected in the miR-7977 promoter. Another possible explanation could be that the release of EV miRNAs was elevated.49 In fact, endosomal markers including TSG101 and ALIX were elevated in BM EVs derived from AML and MDS as compared with control EVs (Figure 5B). The third possibility is that certain unknown long non-coding RNAs such as HOTAIR or PCBP1-1:1 in hematopoietic cells may sponge several miRNAs.50 Further studies to investigate the miRNA promoter and level of non-coding RNAs in hematopoietic cells are required to understand the precise regulation of EV miRNAs in AML and MDS.
In conclusion, we found that EV miR-7977 derived from AML/MDS cells was transferred into BM MSCs and could reduce stem/progenitor cell-supporting capacity of MSCs via PCBP1 reduction. EV miR-7977 could be involved in the dysfunction of normal hematopoiesis in AML and MDS.
Acknowledgments
We would like to thank the staff at the division of electron microscopy in Sapporo Medical University for their technical support. We also thank Yumiko Kaneko for preparation of the BM MNSs from patient samples. The manuscript has been carefully reviewed by an experienced medical editor at NAI Inc. The extracellular vesicle miRNAs have been deposited into NCBI’s Gene Expression Omnibus under the accession code GSE64029.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/101/4/437
Funding
This work was supported in part by a grant from the Ministry of Health, Labour and Welfare of Japan to M.K. (ID: 15K09482).
References
- 1.Gupta P, Niehans GA, LeRoy SC, et al. Fas ligand expression in the bone marrow in myelodysplastic syndromes correlates with FAB subtype and anemia, and predicts survival. Leukemia. 1999;13(1):44–53. [DOI] [PubMed] [Google Scholar]
- 2.Contini P, Zocchi MR, Pierri I, Albarello A, Poggi A. In vivo apoptosis of CD8(+) lymphocytes in acute myeloid leukemia patients: involvement of soluble HLA-I and Fas ligand. Leukemia. 2007;21(2):253–260. [DOI] [PubMed] [Google Scholar]
- 3.Matsunaga T, Takemoto N, Sato T, et al. Interaction between leukemic-cell VLA-4 and stromal fibronectin is a decisive factor for minimal residual disease of acute myelogenous leukemia. Nat Med. 2003;9(9): 1158–1165. [DOI] [PubMed] [Google Scholar]
- 4.Tiu R, Gondek L, O’Keefe C, Maciejewski JP. Clonality of the stem cell compartment during evolution of myelodysplastic syndromes and other bone marrow failure syndromes. Leukemia. 2007;21(8):1648–1657. [DOI] [PubMed] [Google Scholar]
- 5.Guidetti F, Grazioli S, Capelli F, et al. Primitive hematopoietic stem cells shows a polyclonal pattern in myelodysplastic syndromes. Haematologica. 2004;89(1):21–28. [PubMed] [Google Scholar]
- 6.Karasawa M, Tsukamoto N, Sakai H, et al. Clinical outcome in three patients with myelodysplastic syndrome showing polyclonal hematopoiesis. Acta Haematol. 1999;101(1):46–49. [DOI] [PubMed] [Google Scholar]
- 7.Cancer Genome Atlas Research N. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Busque L, Patel JP, Figueroa ME, et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet. 2012;44(11): 1179–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Avecilla ST, Hattori K, Heissig B, et al. Chemokine-mediated interaction of hematopoietic progenitors with the bone marrow vascular niche is required for thrombopoiesis. Nat Med. 2004;10(1):64–71. [DOI] [PubMed] [Google Scholar]
- 10.Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature. 2012;481(7382):457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Asada N, Katayama Y. Regulation of hematopoiesis in endosteal microenvironments. Int J Hematol. 2014;99(6):679–684. [DOI] [PubMed] [Google Scholar]
- 12.Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. 2014;505(7483):327–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lanotte M, Allen TD, Dexter TM. Histochemical and ultrastructural characteristics of a cell line from human bone-marrow stroma. J Cell Sci. 1981;50:281–297. [DOI] [PubMed] [Google Scholar]
- 14.Raaijmakers MH, Mukherjee S, Guo S, et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature. 2010;464(7290):852–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kobune M, Iyama S, Kikuchi S, et al. Stromal cells expressing hedgehog-interacting protein regulate the proliferation of myeloid neoplasms. Blood Cancer J. 2012;2:e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oliveira FM, Lucena-Araujo AR, Favarin Mdo C, et al. Differential expression of AURKA and AURKB genes in bone marrow stromal mesenchymal cells of myelodysplastic syndrome: correlation with G-banding analysis and FISH. Exp Hematol. 2013;41(2):198–208. [DOI] [PubMed] [Google Scholar]
- 17.Huan J, Hornick NI, Shurtleff MJ, et al. RNA trafficking by acute myelogenous leukemia exosomes. Cancer Res. 2013;73(2):918–929. [DOI] [PubMed] [Google Scholar]
- 18.Gyorgy B, Szabo TG, Pasztoi M, et al. Membrane vesicles, current state-of-the-art: emerging role of extracellular vesicles. Cell Mol Life Sci. 2011;68(16):2667–2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raimondo S, Saieva L, Corrado C, et al. Chronic myeloid leukemia-derived exosomes promote tumor growth through an autocrine mechanism. Cell Commun Signal. 2015;13:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boyiadzis M, Whiteside TL. Information transfer by exosomes: A new frontier in hematologic malignancies. Blood Rev. 2015; 29(5):281–290. [DOI] [PubMed] [Google Scholar]
- 21.Yeh YY, Ozer HG, Lehman AM, et al. Characterization of CLL exosomes reveals a distinct microRNA signature and enhanced secretion by activation of BCR signaling. Blood. 2015;125(21):3297–3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kawano Y, Kobune M, Yamaguchi M, et al. Ex vivo expansion of human umbilical cord hematopoietic progenitor cells using a coculture system with human telomerase catalytic subunit (hTERT)-transfected human stromal cells. Blood. 2003;101(2):532–540. [DOI] [PubMed] [Google Scholar]
- 23.Kobune M, Ito Y, Kawano Y, et al. Indian hedgehog gene transfer augments hematopoietic support of human stromal cells including NOD/SCID-beta2m−/− repopulating cells. Blood. 2004;104(4): 1002–1009. [DOI] [PubMed] [Google Scholar]
- 24.Kobune M, Kawano Y, Takahashi S, et al. Interaction with human stromal cells enhances CXCR4 expression and engraft ment of cord blood Lin(−)CD34(−) cells. Exp Hematol. 2008;36(9):1121–1131. [DOI] [PubMed] [Google Scholar]
- 25.Kawano Y, Kobune M, Chiba H, et al. Ex vivo expansion of G-CSF-mobilized peripheral blood CD133+ progenitor cells on coculture with human stromal cells. Exp Hematol. 2006;34(2):150–158. [DOI] [PubMed] [Google Scholar]
- 26.Jeppesen DK, Hvam ML, Primdahl-Bengtson B, et al. Comparative analysis of discrete exosome fractions obtained by differential centrifugation. J Extracell Vesicles. 2014;3: 25011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grant R, Ansa-Addo E, Stratton D, et al. A filtration-based protocol to isolate human plasma membrane-derived vesicles and exosomes from blood plasma. J Immunol Methods. 2011;371(1–2):143–151. [DOI] [PubMed] [Google Scholar]
- 28.Channavajjhala SK, Rossato M, Morandini F, et al. Optimizing the purification and analysis of miRNAs from urinary exosomes. Clin Chem Lab Med. 2014;52(3):345–354. [DOI] [PubMed] [Google Scholar]
- 29.Kobune M, Xu Y, Baum C, Kelley MR, Williams DA. Retrovirus-mediated expression of the base excision repair proteins, formamidopyrimidine DNA glycosylase or human oxoguanine DNA glycosylase, protects hematopoietic cells from N,N′,N″-triethylenethiophosphoramide (thioTEPA)-induced toxicity in vitro and in vivo. Cancer Res. 2001;61(13):5116–5125. [PubMed] [Google Scholar]
- 30.Kosaka N, Iguchi H, Yoshioka Y, Takeshita F, Matsuki Y, Ochiya T. Secretory mechanisms and intercellular transfer of microRNAs in living cells. J Biol Chem. 2010;285(23): 17442–17452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chiba H, Kobune M, Kato J, et al. Wnt3 modulates the characteristics and cobblestone area-supporting activity of human stromal cells. Exp Hematol. 2004;32(12): 1194–1203. [DOI] [PubMed] [Google Scholar]
- 32.Yao Y, Wang C, Wei W, et al. Dendritic cells pulsed with leukemia cell-derived exosomes more efficiently induce antileukemic immunities. PLoS One. 2014;9(3):e91463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Balakrishnan I, Yang X, Brown J, et al. Genome-wide analysis of miRNA-mRNA interactions in marrow stromal cells. Stem Cells. 2014;32(3):662–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ji X, Wan J, Vishnu M, Xing Y, Liebhaber SA. alphaCP Poly(C) binding proteins act as global regulators of alternative polyadenylation. Mol Cell Biol. 2013;33(13):2560–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamauchi A, Ito Y, Morikawa M, et al. Pre-administration of angiopoietin-1 followed by VEGF induces functional and mature vascular formation in a rabbit ischemic model. J Gene Med. 2003;5(11):994–1004. [DOI] [PubMed] [Google Scholar]
- 36.Kode A, Manavalan JS, Mosialou I, et al. Leukaemogenesis induced by an activating beta-catenin mutation in osteoblasts. Nature. 2014;506(7487):240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boulais PE, Frenette PS. Making sense of hematopoietic stem cell niches. Blood. 2015;125(17):2621–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Medyouf H, Mossner M, Jann JC, et al. Myelodysplastic cells in patients reprogram mesenchymal stromal cells to establish a transplantable stem cell niche disease unit. Cell Stem Cell. 2014;14(6):824–837. [DOI] [PubMed] [Google Scholar]
- 39.Deeg HJ, Beckham C, Loken MR, et al. Negative regulators of hemopoiesis and stroma function in patients with myelodysplastic syndrome. Leuk Lymphoma. 2000;37(3–4):405–414. [DOI] [PubMed] [Google Scholar]
- 40.Blau O, Baldus CD, Hofmann WK, et al. Mesenchymal stromal cells of myelodysplastic syndrome and acute myeloid leukemia patients have distinct genetic abnormalities compared with leukemic blasts. Blood. 2011;118(20):5583–5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Santamaria C, Muntion S, Roson B, et al. Impaired expression of DICER, DROSHA, SBDS and some microRNAs in mesenchymal stromal cells from myelodysplastic syndrome patients. Haematologica. 2012;97(8): 1218–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schepers K, Pietras EM, Reynaud D, et al. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell. 2013;13(3):285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang B, Ho YW, Huang Q, et al. Altered microenvironmental regulation of leukemic and normal stem cells in chronic myelogenous leukemia. Cancer Cell. 2012;21(4):577–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roccaro AM, Sacco A, Maiso P, et al. BM mesenchymal stromal cell-derived exosomes facilitate multiple myeloma progression. J Clin Invest. 2013;123(4):1542–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guo WT, Wang XW, Wang Y. Micro-management of pluripotent stem cells. Protein Cell. 2014;5(1):36–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huo LR, Zhong N. Identification of transcripts and translatants targeted by overexpressed PCBP1. Biochim Biophys Acta. 2008;1784(11):1524–1533. [DOI] [PubMed] [Google Scholar]
- 47.Ruvolo PP. The Interplay between PP2A and microRNAs in Leukemia. Front Oncol. 2015;5:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang G, Wang Y, Shen C, et al. RNA polymerase II binding patterns reveal genomic regions involved in microRNA gene regulation. PLoS One. 2010;5(11):e13798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saito Y, Suzuki H, Taya T, et al. Development of a novel microRNA promoter microarray for ChIP-on-chip assay to identify epigenetically regulated microRNAs. Biochem Biophys Res Commun. 2012;426(1):33–37. [DOI] [PubMed] [Google Scholar]
- 50.Xing CY, Hu XQ, Xie FY, et al. Long non-coding RNA HOTAIR modulates c-KIT expression through sponging miR-193a in acute myeloid leukemia. FEBS Lett. 2015;589(15):1981–1987. [DOI] [PubMed] [Google Scholar]