

A short history of precision medicine
Precision medicine (PM) is certainly not a novel concept, but recent technological improvements have given it a huge boost. Although immunochemotherapy has advanced results of lymphoma treatment enormously, it remains a toxic therapy that does not benefit every patient and may result in serious lasting side-effects. Current clinical research tries to address this conundrum by striving to develop targeted therapies with significant efficacy and reduced side-effects, and to identify biomarkers which can accurately predict therapeutic responsiveness. Since the approval of the molecularly targeted agent all-trans retinoic acid for acute promyelocytic leukemia and the tyrosine kinase inhibitor imatinib for chronic myeloid leukemia, the therapeutic arsenal for hematologic malignancies has expanded considerably.
Here, we intend to briefly discuss available targeted therapies for diffuse large B-cell lymphoma (DLBCL), current issues in clinical research, and challenges in the implementation of novel techniques and targeted agents in routine patient management.
Improvement of therapy is warranted
The World Health Organization (WHO) lymphoma classification recognizes more than 50 different subtypes, with DLBCL being the most common one.1 DLBCL is a biologically and clinically heterogeneous entity.1–3 Since the 1970s, the front-line chemotherapy regimen for DLBCL has consisted of CHOP (cyclophosphamide, doxorubicin, vincristine and prednisone). A significant improvement in overall survival (OS) was achieved with the incorporation of the CD20 monoclonal antibody rituximab in front-line chemotherapy regimens.4,5 Today, modern rituximab-containing chemotherapy regimens cure approximately 60% of patients with DLBCL. Therefore still a considerable percentage of patients will have refractory disease or will relapse.6 Standard treatment for refractory or relapsed DLBCL (RR-DLBCL) is salvage with platinum-based immunochemotherapy followed by high-dose chemotherapy and autologous stem cell transplantation.4 However, the results in R-CHOP pre-treated patients, especially in those relapsing early (<1 year) after first-line treatment are disappointing. In addition, this intensive regimen is only feasible in younger, fit patients. There are no curative options for elderly RR-DLBCL patients.7 Improving current treatment strategies and the development of novel therapeutic approaches are imperative to improve the outcome of these patients.8
Lymphomagenesis: is it all about genetics?
Lymphoma, like any other type of cancer, is characterized by genetic alterations leading to a disturbed balance between proliferation and apoptosis. B lymphocytes are particularly prone to DNA damage during the physiological germinal center reaction, as the immunoglobulin genes undergo somatic hypermutation and class-switch recombination.7 The biological heterogeneity of DLBCL has been comprehensively investigated, resulting in further sub-categorization. Based on gene expression profiling (GEP), DLBCLs are often classified according to the cell-of-origin principle into the germinal center B-cell-like (GCB, 50%), activated B-cell like (ABC, 30%), primary mediastinal B-cell lymphoma and unclassifiable subtypes, with ABC-type DLBCL clearly carrying an inferior prognosis.2,5,9 Cytogenetic studies have furthermore identified MYC translocation in a subset of (mostly GCB-type) DLBCL patients, which is associated with a poor prognosis, especially in the presence of a concurrent BCL2 translocation (“double-hit” lymphoma).6 Overexpression of MYC and BCL2 by other mechanisms is also seen in ABC-type DLBCL (“double-expressors”).
Recent advances in molecular high-throughput methodologies have elucidated multiple genetic aberrations in lymphoma such as single gene mutations, translocations, copy number alterations, amplifications, rearrangements and loss of functions.10 These genetic changes affect cell surface receptors, intracellular oncogenic and non-oncogenic signaling pathways, transcription factors, and epigenetic proteins, thereby influencing a whole range of cellular mechanisms (Figure 1): cell growth, metabolism, proliferation, differentiation, apoptosis, survival, immune regulation, DNA damage response and angiogenesis.2,5,8–10 In DLBCL, pathways involving the B-cell receptor (BCR), Toll-like receptor (TLR), growth factor receptor (GFR) and cytokine receptor are most commonly affected.11,12 The complex interactive network of intracellular cascades, including crosstalk between parallel pathways and negative or positive feedback loops, has been extensively reviewed elsewhere.2,6,8,9
Figure 1.
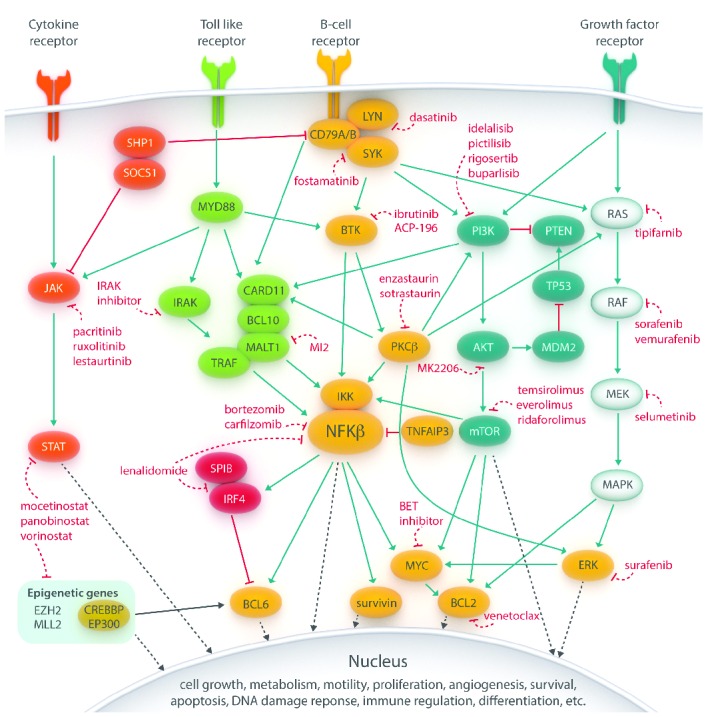
Main pathways involved in lymphomagenesis in B-cell non-Hodgkin lymphoma. These signaling cascades can be inhibited by novel drugs at several levels, from cell surface receptors, intracellular proteins to transcription factors. Importantly, crosstalk between the different pathways is present precluding black-and-white conclusions from pathway-driven analyses of targeted therapies and offering an explanation for some of the drug resistance which can be encountered when using targeted agents. (Blue lines implicate activation, red (dashed) lines indicate inhibition and black (dashed) lines denote direct effects in the nucleus).
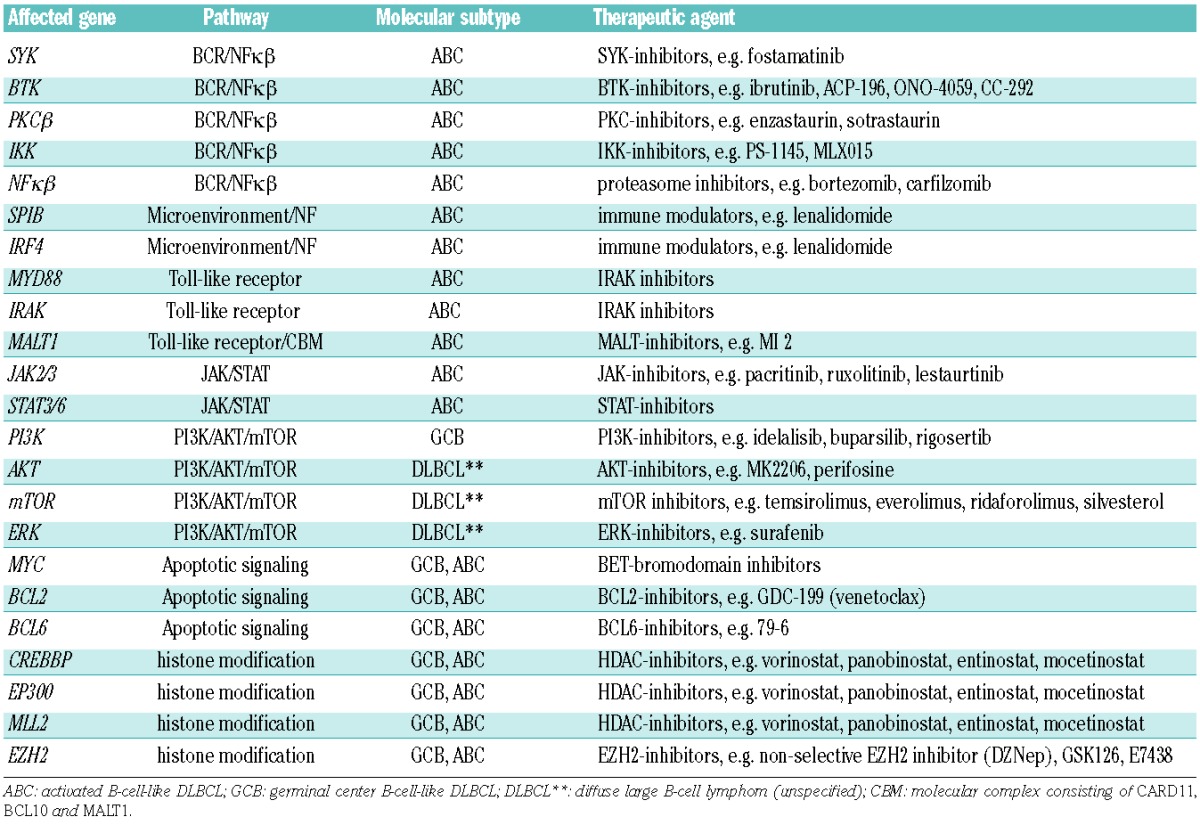
Importantly, DLBCL demonstrates high genetic complexity and heterogeneity with on average 90 alterations per patient.11,13 Consequently, several signaling pathways are altered, making the development of customized targeted therapy in DLBCL challenging.1,11,14–16 Identifying which mutations are driver mutations is of the greatest importance. The landscape of mutations found in the GCB and ABC/non-GCB subtypes has recently been reported by several independent groups. ABC-type lymphomas are enriched for mutations in TNFAIP3, CD79A/B, CARD11, MYD88, BCL10, MALT1 and BCL6, associated with active BCR and/or TLR signaling pathways and resulting in activation of the transcription factor NFκβ, a pivotal player in lymphomagenesis (Table 1). GCB subtypes have been shown to predominantly contain genetic alterations implicated in histone modification such as MLL2, EZH2, CREBBP, EP300 together with PTEN, BCL2 and BCL6.2,6 Notably, the majority of these genetic alterations are not exclusive to either of the subtypes. Furthermore, several oncogenic signaling pathways, such as the JAK/STAT, RAS/MAPK and the PI3K/AKT/mTOR, can be activated in both subtypes by inflammatory cytokines or growth factors produced by cells present in the tumor microenvironment in the absence of genetic alterations in the tumor cells.8 A brief summary of possible affected genes, involved pathways and potential drugs in DLBCL is presented in Table 1.
Table 1.
Overview of affected genes and associated pathways and their corresponding putative therapeutic agents in diffuse large B-cell lymphoma (DLBCL).
Development of novel targeted therapies
The availability of a large number of signal transduction inhibitors that can selectively target unique cellular proteins and kinases has provided an unprecedented opportunity to improve the precision of cancer therapy. Based on the above mentioned dissimilarities between GCB- and non-GCB type DLBCL, the outcome of targeted strategies are also expected to be different.2 Both the BCR- and the TLR pathway, important in non-GCB DLBCL and leading to NFκβ activation, are amenable to therapeutic interventions at almost every level in the intracellular signaling cascade.7,11 The BCR pathway can be blocked upstream by dasatinib (LYN-inhibitor), ibrutinib (BTK-inhibitor), fostamatinib (SYK-inhibitor) or enzastaurin (PKCβ-inhibitor). So far, BTK seems to be the most promising target; in a phase I/II study ibrutinib showed an ORR of 41% in ABC-type, whereas it was only 5% in GCB-DLBLC.1,2,17 Currently, ibrutinib is being investigated as addition to first-line R-CHOP treatment in a randomized phase III trial for which only patients with non-GCB DLBCL are eligible. Inhibitors targeting the IRAK kinases, downstream of MYD88 are being developed. Downstream of these pathways, indirect NFκβ-inhibition with the proteasome inhibitor bortezomib is also expected to be more effective in ABC-subtype DLBCL. The same holds true for the immune modulator lenalidomide, which affects IRF4 and has demonstrated more efficacy in non-GCB RR-DLBCL compared to GCB-subtype [overall response rate (ORR) 53% and 9%, respectively].17 Promising targets in GCB-DLBCL include the PI3k/AKT/mTOR pathway (activated through PTEN inactivation), BCL6 and the methyltransferase EZH2. Overexpression of MYC (through translocation in GCB-type DLBCL or by other mechanisms in ABC-type DLBCL) can be indirectly targeted by epigenetic manipulation with a BET bromodomain inhibitor. Histone modifying agents (e.g. vorinostat) hold promise for lymphomas with CREBBP/EP300 mutations, with an ORR in unselected RR-DLBCL of 20%–30%.7,8 Finally, overexpression of the anti-apoptotic protein BCL2 can be targeted with BH3 mimetics, such as GDC-199. Overall, these examples justify the importance of developing prospective clinical trials based on DLBCL subtype and/or mutational status.18
Because lymphoma cells frequently utilize several oncogenic signaling pathways to promote their growth and survival, rational mechanism-based combinations of targeted agents will be needed to improve treatment efficacy; however, this will probably also lead to more toxicity.
Challenges and pitfalls of precision medicine
Progress in targeted therapies in DLBCL will shift treatment paradigms from broad-spectrum poly-chemotherapy towards more mechanism-based therapeutic combinations. The highest unmet need in treatment now lies in high-risk DLBCL patients, especially RR-DLBCL. To make the next step possible, several hurdles need to be overcome.
Translational research is needed to further elucidate lymphomagenesis and to identify driver mutations in lymphoma subtypes.
The problem of clonal diversity and clonal evolution needs to be addressed. The selective evolutionary processes can lead to different (sub)clones each with their own characteristic profile of mutated genes.16 This complex heterogeneity is reflected by differences amongst DLBCL patients, between the primary disease and the relapse, and by differences in mutational profiles within patients when synchronously sampled at different lymphoma localizations. The enormous capacity of NGS technology blueprinting of all lymphoma-associated genes, thereby disentangling the entire ‘DLBCL mini-genome’ has become feasible.1 Moreover, the genetic defects in the microenvironment contributing to lymphomagenesis and therapeutic resistance can likewise be evaluated.10
As NGS is increasingly being used in clinical practice, there is an urgent need to standardize the sequencing methods used in clinical assays so results can be compared across different clinical trials and centers. Recommendations and the advantages and disadvantages of different NGS platforms for whole-exome sequencing or targeted sequencing strategies have recently been discussed elsewhere.2,14,15,19–22
As presented in Figure 2, we anticipate the possibility of a continuous loop of adaptive PM in which all DLBCL patients both at diagnosis and at relapse can enter the cycle of advanced tumor profiling (with NGS, immunohistochemistry, GEP and proteomics), risk stratification and subsequent therapy adjustment based on druggable genes and pathways. Hundreds of compounds have so far been identified that have the potential to inhibit cancer cells at virtually every level of entire signaling pathways.4,5 The majority of tested compounds have demonstrated low response rates (<30%) and little durability of responses (<6 months) in phase I/II studies in unselected patients. In addition, the majority of lymphomas will not harbor only one genetic defect, which partly explains the modest efficacy of monotherapy with targeted drugs. New rationally designed biomarker-driven clinical trials with pre-selected patient populations are warranted to investigate the true value of novel targeted drugs.2,4,5,8
Pharmacogenomics, -kinetics, -dynamics, and therapeutic drug monitoring will be necessary to optimize the dose and dosing schedule of these novel drugs. Studies are ongoing to determine whether it is possible to combine targeted therapies in order to impede signaling cascades at several levels, for example also blocking negative/positive feedback loops.2
Predictive biomarkers are warranted to pre-select patients to improve ORR.1 Prior to incorporation of biomarkers in clinical settings it is necessary to adequately define sensitivity and specificity. With the substantial reduction of costs of NGS over the past years, this technique can be applied in the search for predictive biomarkers.19,20,22 Predicting response or resistance to a specific therapy will not only expedite the introduction of the most effective therapy to the patients, but will also likely reduce the overall treatment costs.1
How to manage the sheer quantity of data yielded by high-throughput technological studies is in itself an emerging field (Figure 3). Interpretation and integration of the data, followed by translation into potential druggable targets applicable for clinical use, are crucial steps in PM.23 If not adequately dealt with, this increasing complexity of information could result in an escalating divergence of science, drifting away from the patient. Pathway-driven analysis by computational systems biology methods might be the logical step to do this.24 However, crosstalk between pathways might complicate black-and-white conclusions also from pathway-driven analyses and to overcome drug resistance mechanisms it might be important to parallel ‘co-target’ accessory pathways.25
Finally, lymphomagenesis is not only explained by genetics. Therefore, to obtain a global overview and comprehensive understanding of tumor evolution, thereby optimizing treatment strategies, it will be necessary to integrate genomic data together with proteomic, metabolomics knowledge and functional studies using an integrative systems biology approach.23,25
Figure 2.
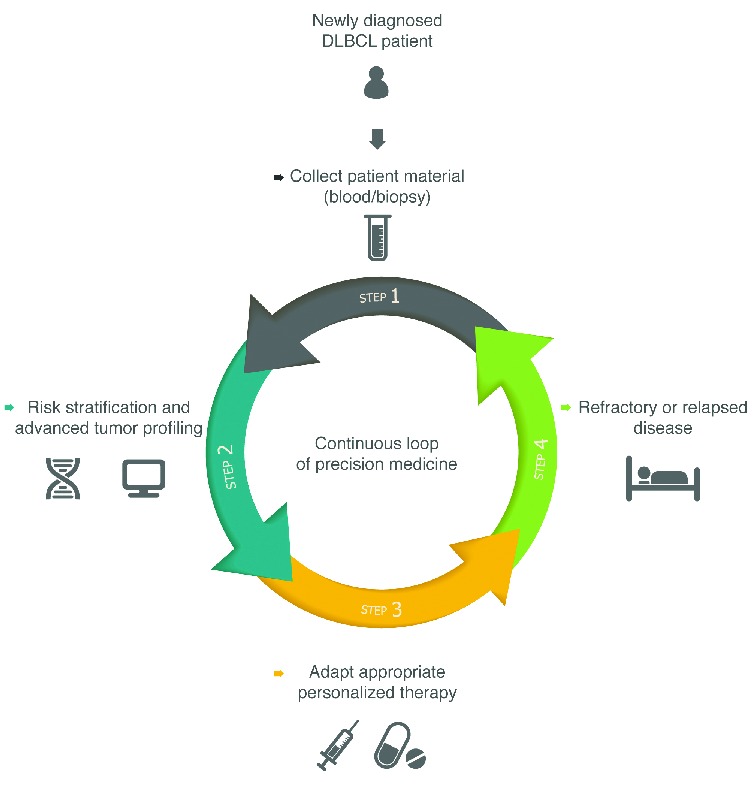
Scheme representing the circle (continuous loop) of adaptive precision cancer medicine. For newly diagnosed diffuse large B-cell lymphoma (DLBCL) patients, after collection of patient material specific tumor profiles acquired by several high-throughput technologies together with risk stratification will guide precision therapeutic strategies. When patients are confronted with refractory or relapsed DLBCL disease, tumor profiling and risk stratification will be redone, resulting in redirection of the original therapeutic approaches. As DLBCL is an evolutionary genetic disease, this procedure can be repeatedly used to serially repress the lymphoma in view of specific evolved subclones, thereby optimizing PM at every disease stage of the patient.
Figure 3.
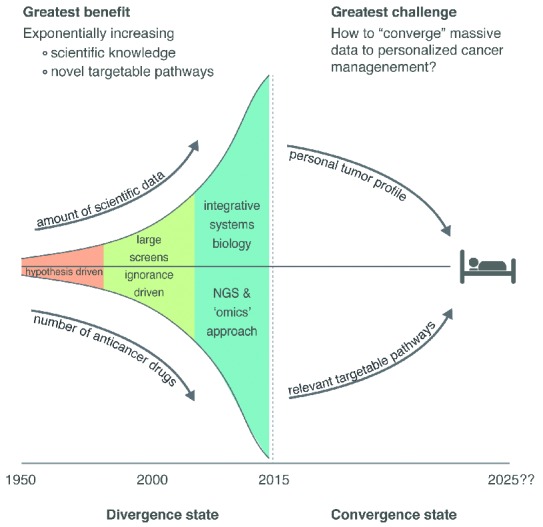
Schematic view of the development of cancer research, high-throughput technologies and precision cancer medicine. The amount of data these studies yield is exponentially increasing and the major challenge in the near future is to converge these data to precision cancer management. As mentioned, improvement of precision medicine is a multifactorial process, which includes: 1) Increasing knowledge of lymphomagenesis and 2) clonal evolution, 3) incorporating high-throughput technologies into clinical standard care, 4) adapting the circle of precision medicine by advanced tumor profiling for every patient (Figure 2), 5) optimizing therapy by pharmacological competence, 6) propelling investigation of predictive biomarkes, 7) using pathway-driven analysis and system biology methods to process the quantity of data, and 8) integrating data produced by diverse research platforms (genomics, protemics, metabolomics, immunohistochemistry, etc.).
Conclusions
With high-throughput technological improvements, genetic cancer research has moved beyond single gene analysis to the investigation of hundreds of genes and entire signaling pathways simultaneously. We anticipate that this will lead to identification of druggable targets and to predictive biomarkers in DLBCL. PM will hopefully improve the outcome of lymphoma patients. The main challenges, however, will be to design and execute innovative biomarker-driven clinical trials demonstrating efficacy of PM, to implement these high-throughput techniques in clinical practice, and to keep this approach financially viable.
Acknowledgments
The authors wish to acknowledge Sanne Tonino and Arnon Kater for critically reviewing the manuscript.
Footnotes
Financial and other disclosures provided by the author using the ICMJE (www.icmje.org) Uniform Format for Disclosure of Competing Interests are available with the full text of this paper at www.haematologica.org.
References
- 1.Intlekofer AM, Younes A. Precision therapy for lymphoma–current state and future directions. Nat Rev Clin Oncol. 2014;11(10):585–596. [DOI] [PubMed] [Google Scholar]
- 2.Roschewski M, Staudt LM, Wilson WH. Diffuse large B-cell lymphoma-treatment approaches in the molecular era. Nat Rev Clin Oncol. 2014;11(1):12–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martelli M, Ferreri AJ, Agostinelli C, Di Rocco A, et al. Diffuse large B-cell lymphoma. Crit Rev Oncol Hematol. 2013;87(2):146–71. [DOI] [PubMed] [Google Scholar]
- 4.Younes A. XIV. The rationale for combining targeted and biological anti-lymphoma drugs. Hematol Oncol. 2013;31(Suppl 1):81–83. [DOI] [PubMed] [Google Scholar]
- 5.Younes A, Berry DA. From drug discovery to biomarker-driven clinical trials in lymphoma. Nat Rev Clin Oncol. 2012;9(11):643–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.King RL, Bagg A. Genetics of diffuse large B-cell lymphoma: paving a path to personalized medicine. Cancer J. 2014;20(1):43–47. [DOI] [PubMed] [Google Scholar]
- 7.Young RM, Staudt LM. Targeting pathological B cell receptor signalling in lymphoid malignancies. Nat Rev Drug Discov. 2013;12(3):229–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leslie LA, Younes A. Targeting oncogenic and epigenetic survival pathways in lymphoma. Leuk Lymphoma. 2013;54(11):2365–2376. [DOI] [PubMed] [Google Scholar]
- 9.Sehn LH, Gascoyne RD. Diffuse large B-cell lymphoma: optimizing outcome in the context of clinical and biologic heterogeneity. Blood. 2015;125(1):22–32. [DOI] [PubMed] [Google Scholar]
- 10.Puvvada S, Kendrick S, Rimsza L. Molecular classification, pathway addiction, and therapeutic targeting in diffuse large B cell lymphoma. Cancer Genet. 2013;206(7–8):257–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blombery PA, Dickinson M, Westerman DA. Molecular lesions in B-cell lymphoproliferative disorders: recent contributions from studies utilizing high-throughput sequencing techniques. Leuk Lymphoma. 2014;55(1):19–30. [DOI] [PubMed] [Google Scholar]
- 12.Rossi D, Ciardullo C, Gaidano G. Genetic aberrations of signaling pathways in lymphomagenesis: revelations from next generation sequencing studies. Semin Cancer Biol. 2013;23(6):422–30. [DOI] [PubMed] [Google Scholar]
- 13.Zhang J, Grubor V, Love CL, et al. Genetic heterogeneity of diffuse large B-cell lymphoma. Proc Natl Acad Sci USA. 2013;110(4):1398–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnsen JM, Nickerson DA, Reiner AP. Massively parallel sequencing: the new frontier of hematologic genomics. Blood. 2013;122(19):3268–3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mullighan CG. Genome sequencing of lymphoid malignancies. Blood. 2013;122(24):3899–3907. [DOI] [PubMed] [Google Scholar]
- 16.Landau DA, Carter SL, Getz G, Wu CJ. Clonal evolution in hematological malignancies and therapeutic implications. Leukemia. 2014;28(1):34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cai Q, Westin J, Fu K, et al. Accelerated therapeutic progress in diffuse large B cell lymphoma. Ann Hematol. 2014;93(4):541–556. [DOI] [PubMed] [Google Scholar]
- 18.Barton S, Hawkes EA, Wotherspoon A, Cunningham D. Are we ready to stratify treatment for diffuse large B-cell lymphoma using molecular hallmarks¿ Oncologist. 2012;17(12):1562–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Macconaill LE, Garraway LA. Clinical implications of the cancer genome. J Clin Oncol. 2010;28(35):5219–5228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Badalian-Very G. Personalized medicine in hematology - A landmark from bench to bed. Comput Struct Biotechnol J. 2014;10(17):70–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Simon R, Roychowdhury S. Implementing personalized cancer genomics in clinical trials. Nat Rev Drug Discov. 2013;12(5):358–369. [DOI] [PubMed] [Google Scholar]
- 22.Garraway LA. Genomics-driven oncology: framework for an emerging paradigm. J Clin Oncol. 2013;31(15):1806–1814. [DOI] [PubMed] [Google Scholar]
- 23.Hawkins RD, Hon GC, Ren B. Next-generation genomics: an integrative approach. Nat Rev Genet. 2010;11(7):476–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Medvedev P, Stanciu M, Brudno M. Computational methods for discovering structural variation with next-generation sequencing. Nat Methods. 2009;6(11 Suppl):S13–20. [DOI] [PubMed] [Google Scholar]
- 25.Ocana A, Pandiella A. Personalized therapies in the cancer “omics” era. Mol Cancer. 2010;9:202. [DOI] [PMC free article] [PubMed] [Google Scholar]